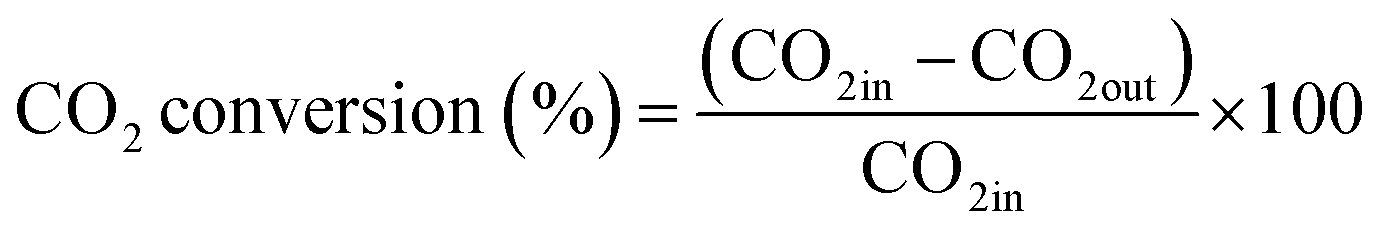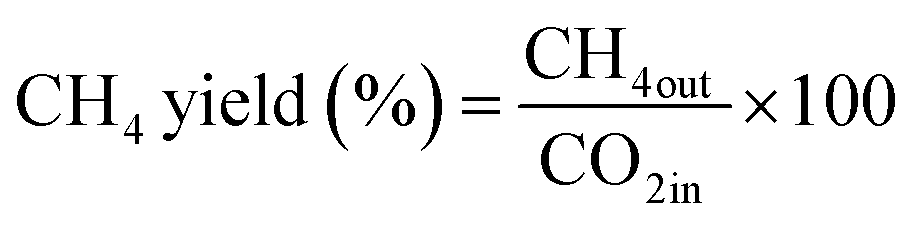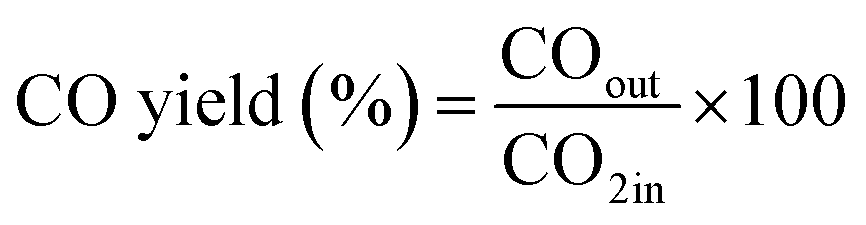DOI:
10.1039/D1RA02327F
(Paper)
RSC Adv., 2021,
11, 17648-17657
Role of oxide support in Ni based catalysts for CO2 methanation
Received
24th March 2021
, Accepted 7th May 2021
First published on 14th May 2021
Abstract
The CO2 methanation reaction of reduced and unreduced Ni based CeO2, Al2O3, TiO2 and Y2O3 supported catalysts was investigated. The Ni/CeO2 and Ni/Y2O3 catalysts exhibited similar CO2 conversions at all reaction temperatures. The catalysts were studied by X-ray diffraction (XRD), H2 chemisorption, H2 temperature-programmed reduction (TPR), and in situ diffuse reflection infrared Fourier transform spectroscopy (DRIFTS); the results suggested that the reducibility of both metal and support at low temperature, strong metal support interaction and small Ni particle size are important factors for low-temperature CO2 methanation. Based on the DRIFT studies, the difference in the CO2 adsorption properties and reaction pathway depending on the reduced and unreduced Ni based supported catalysts was discussed.
1 Introduction
The CO2 methanation reaction is important in industry because it produces methane and removes CO2. Among the CO2 conversion reactions using catalysts, the CO2 methanation reaction or so-called Sabatier reaction, is the most advantageous reaction.1 CO2 methanation is a simple reaction which makes it possible to integrate the transformation of methane into biogas through the process of anaerobic digestion to generate power, or into other industrial plants with CO2-rich exhaust gases.2 CO2 methanation is a highly exothermic reaction, and it can produce the heat. CO2 methanation also has a large equilibrium constant at a lower temperature, so that methane can be generated with a high conversion at lower temperatures; however, the conversion was still low due to kinetic limitation and catalyst performance. Catalytic performance is dependent on various parameters, such as the kinds of support, preparation methods, and the addition of promoters.3 Especially, many researchers have studied the effect of support to enhance the performance of the CO2 methanation reaction. The support has a significant effect on the redox properties, metal–support interaction, metal dispersion and adsorption properties.4–6 Many efforts have been made to develop Ni-based supported catalysts for low-temperature CO2 methanation.7–11 The CO2 methanation activity has been investigated using Ni-based catalysts deposited on various supports such as SiO2,12 α-Al2O3,13 MgO,14 ZrO2,15 Y2O3,16 CeO2,17 TiO2,18 β-zeolite,19 and their composite supports such as Ni/CeO2–ZrO2,20 Ni/γ-Al2O3–ZrO2–TiO2–CeO2,21 Ni/CeO2–Al2O3.22 Frontera et al.23 reported that the catalytic activity strongly effects on the characteristics of the support for Ni based catalysts. Le et al.24 founded that the high Ni dispersion and strong CO2 adsorption plays an important role in the high CO and CO2 methanation activities for Ni/CeO2 catalyst. Tada et al.13 reported that the Ni/CeO2 catalyst showed a high conversion compare to Ni/α-Al2O3. A large CO2 adsorption amount and high CO2 reduction ability could result in high CO2 conversion at a low temperature using a Ni/CeO2 catalyst. Muroyama et al.16 found that the Ni/Y2O3 catalyst exhibited a high CO2 conversion and CH4 yield compare to Ni/CeO2, Ni/Al2O3, Ni/ZrO2, Ni/La2O3 and Ni/Sm2O3 catalysts. They expected that the promotion of the decomposition of formate species would lead to high catalytic activity. Abello et al.25 have reported that the Ni–Al-activated catalyst prepared by a co-precipitation method exhibited a high CO2 conversion and stability at a high space velocity and highly loaded and dispersed small Ni nanoparticles (ca. 6 nm) dispersed over NiO–alumina by partial reduction of the mixed oxide. Vogt et al.26 reported that CO2 hydrogenation over Ni is considered to follow two steps; direct dissociation and H-mediated and they proved how structure sensitivity effects the mechanism of CO2 hydrogenation over Ni/SiO2. Zeolite and TiO2 supports are also used as Ni catalysts during CO2 methanation. Liu et al.18 reported that the 15 wt% Ni/TiO2 catalyst prepared by deposition–precipitation method showed excellent CO2 methanation activity (conversion: 96%; selectivity: 99%) at 260 °C. The good dispersion of Ni particles with high exposure of active sites, which may lead to enhanced exposure of active sites that facilitate the generation of surface-dissociated hydrogens. Recent reports have shown substantial improvement in the CO2 methanation rate by changing the support's properties and/or by the addition of promoters such as Ni–W–Mg,27 Ni–La,28 Ni–Cu or Fe/Al2O3 (ref. 29) catalysts. Many studies have examined the performance of highly active Ni-based catalysts supported on various metal oxides for CO2 methanation at low temperature. However, the effect of metal–support interaction and role of support on catalytic performances during Ni based CO2 methanation reaction is yet to be unraveled. In this work, CeO2, Y2O3, TiO2 and Al2O3 supports were selected as representative Ni catalysts and prepared by wet impregnation method. We investigated the catalytic activities of all catalysts for CO2 methanation and examined their physicochemical properties. Moreover, the adsorbed species on the reduced and unreduced catalyst surface were compared.
2 Experimental
2.1 Preparation of catalysts
Ni-Based catalysts supported on CeO2 (Sigma Aldrich Co.), α-Al2O3 (Alfa Aesar Co.), Y2O3 (Sigma Aldrich Co., St. Louis, MO, USA) and TiO2 (G-5, Cristal Global Co.) were prepared by a wet impregnation method. Nickel nitrate hexahydrate (Ni(NO3)2·6H2O; Sigma Aldrich Co.) was dissolved in distilled water at 80 °C. After impregnation, the moisture was removed at 80 °C using a rotary vacuum evaporator and then dried at 103 °C oven for 12 h. The samples were calcined at 400 °C for 2 h and then samples were reduced at 420 °C for 2 h with a 30% H2/N2. The obtained samples were grounded and sieved using a 40–50 mesh. Ni loading was fixed at 10 wt%.
2.2 Catalytic activity
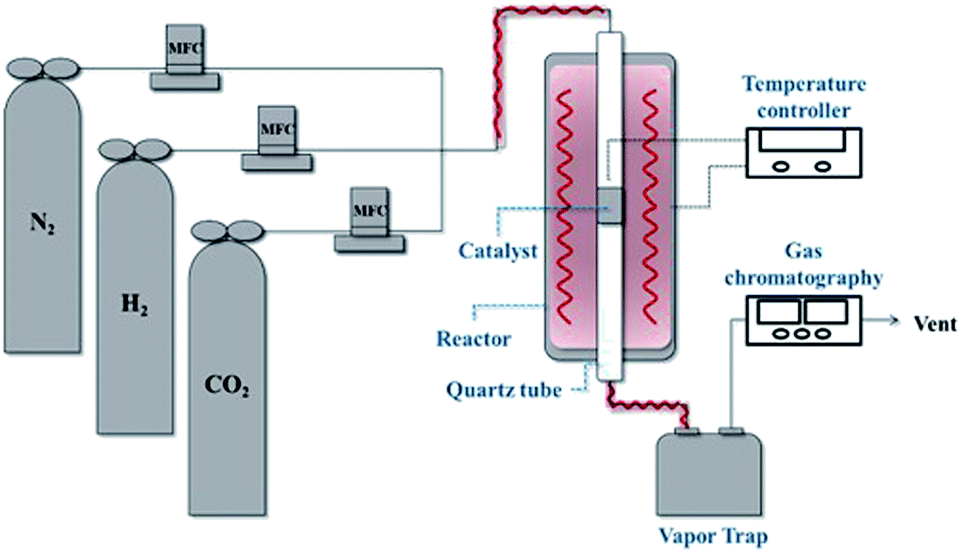
The CO2 methanation experimental apparatus consisted of a continuous flow-type fixed-bed reactor comprising a quartz tube (inner diameter: 8 mm; height: 600 mm) and a catalytic bed (Fig. 1). To measure the gas temperature, another K-type thermocouple was installed at the top of the catalytic bed. Prior to the experiment, the catalysts were pretreated at 420 °C for 1 h with a 30% H2/N2 at a flow rate of 100 cm3 min−1. The feed gases comprised 16.67% CO2, 66.66% H2 and 16.67% N2. The total flow through the reactor was 120 cm3 min−1 and a space velocity of 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 l kg−1 h−1 was obtained. The outlet gas-supply pipe was made of stainless steel and wrapped with a heating band set at 180 °C to prevent water condensation. The concentrations of the reactants and products were measured as follows: the inlet and outlet gas concentration were analyzed using a gas chromatograph with a thermal conductivity detector (GOW-MAC, series 580). The CO2 conversion and yield can be calculated as the following:
400 l kg−1 h−1 was obtained. The outlet gas-supply pipe was made of stainless steel and wrapped with a heating band set at 180 °C to prevent water condensation. The concentrations of the reactants and products were measured as follows: the inlet and outlet gas concentration were analyzed using a gas chromatograph with a thermal conductivity detector (GOW-MAC, series 580). The CO2 conversion and yield can be calculated as the following:
 |
| | Fig. 1 Schematic of the experimental fixed bed catalytic reactor for CO2 methanation. | |
2.3 Characterization
The surface areas of Ni based catalysts were obtained by the Brunauer–Emmett–Teller (BET) equation using an ASAP 2010 instrument (Micrometrics). The Ni dispersion was evaluated by H2 chemisorption at 35 °C using a Micrometrics ASAP 2020 instrument (Micrometrics). All catalysts were reduced under a H2 airflow at 300 °C for 1 h and then cooled into 35 °C. X-Ray diffraction (XRD) analysis was measured on an X'Pert PRO MRD instrument (PANalytical) with a Cu Kα (λ = 1.5056 Å) radiation. Field emission-transmission electron microscope (TEM) analysis was carried out on a JEM-2100 F (JEOL) microscope (200 keV voltage). All samples were prepared by suspending an ultrasonicated catalyst powder in ethanol and placing the suspension on a Cu grid. For H2 chemisorption analysis, the catalysts were activated with 10% H2 at 300 °C for 0.5 h and then cooled to 50 °C and saturated with H2 pulses. The temperature-programmed reduction (TPR) of H2 was measured by 10% H2/Ar using 0.3 g of the catalyst at a total flow rate of 50 cm3 min−1. Before the H2 TPR measurement, the catalyst was pretreated in a flow of air at 400 °C for 0.5 h, followed by cooling to 50 °C. The catalyst was placed in dilute hydrogen, and the consumption of hydrogen was monitored using Autochem 2920 (Micrometrics) by increasing the temperature to 900 °C at a rate of 10 °C min−1. X-ray photoelectron spectroscopy (XPS) analysis was performed using an ESCALAB 210 (VG Scientific), and Al Kα monochromate (1486.6 eV) was used as an excitation source. Fourier-transform infrared (FT-IR) spectroscopy experiments were conducted in a diffuse reflection cell equipped with a CaF2 window using an FT-IR spectrometer (Nicolet IS 10, ThermoFisher), and diffuse reflectance (DR) 400 accessory was used. The spectra included 30 accumulated scans at resolutions of 4 cm−1, which were obtained using a mercury–cadmium–telluride (MCT) detector. To investigate the characteristics of CO2 adsorption and CO2 methanation reaction, the gas flowing over the samples pretreated by H2 and air were switched to CO2 or CO2 + H2 for 20 min at 200 and 300 °C.
3 Results and discussion
3.1 Catalytic activities
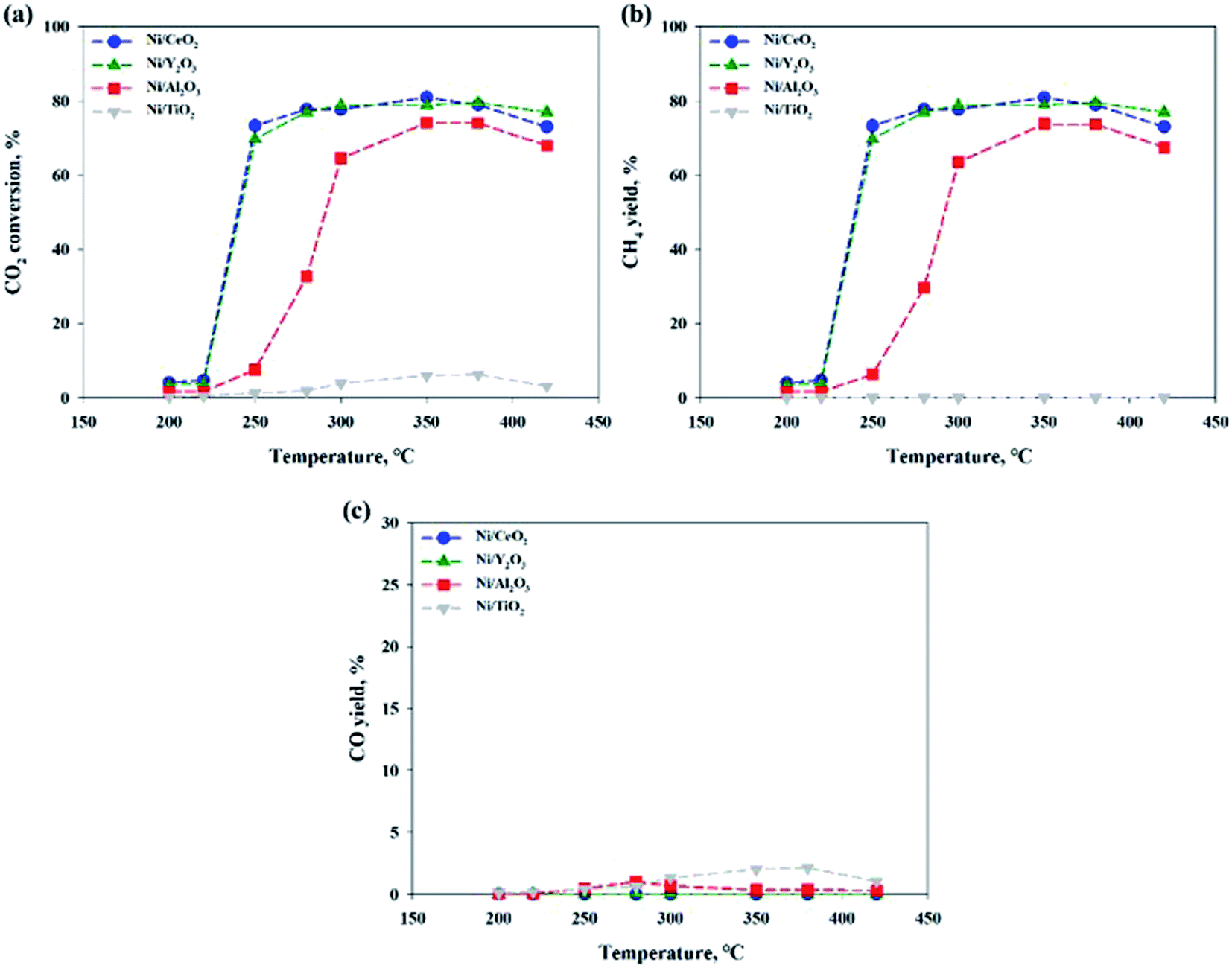
Fig. 2 shows the CO2 conversions of Ni-based catalysts supported on CeO2, TiO2, Y2O3 and Al2O3 at different reaction temperatures for the CO2 methanation reaction. Among the catalysts, the reduced Ni/CeO2 and Ni/Y2O3 catalysts exhibited the similar CO2 conversion over a range from 250 to 400 °C. For the reduced Ni/Al2O3 catalyst, the CO2 conversion decreased at 250–280 °C and then reached the maximal value at 350–380 °C. Especially, the Ni/TiO2 catalyst exhibited a very poor CO2 conversion at all temperature range. The CO2 to CH4 and CO conversions are also shown in Fig. 2(b) and (c). CO2 was fully converted to the CO (0.1–2.1%) in all temperature range for Ni/TiO2 catalyst. From the properties of the equilibrium conversion, the reverse water gas shift reaction is not favored at a low temperature. Thus, it is indicated that the CO2 should be able to convert CO on the reduction sites on a TiO2 support. Except for the Ni/TiO2 catalyst, CO yields were nearly zero at a reaction temperature under 300 °C, indicating that all CO2 conversion is mostly concordant with CO2 conversion to CH4. The order of catalytic activity regardless of the reaction temperature was as follows: Ni/CeO2 ≈ Ni/Y2O3 > Ni/Al2O3 > Ni/TiO2. It could be concluded that the reduced Ni/CeO2 and Ni/Y2O3 catalysts showed superior activity at a low temperature. Although catalytic activity of the Ni-based catalyst differs with operation conditions such as temperature, pressure, catalyst loading, and gas component, the general results showed similarities to previously reported studies by other researchers.11,30,31 Cai et al.30 reported that CO2 conversion of Ni/CexZr1−xO2 catalyst showed the 72.21% at 390 °C. The higher reducibility of the Ce-rich supported highly-dispersed Ni catalyst was considered to be an important factor for long-term stability.30,32,33 However, the role of Ni dispersion in the stability of the Ni/CeO2 catalyst is difficult to elucidate in this work, and further studies will be necessary to fully address this point.
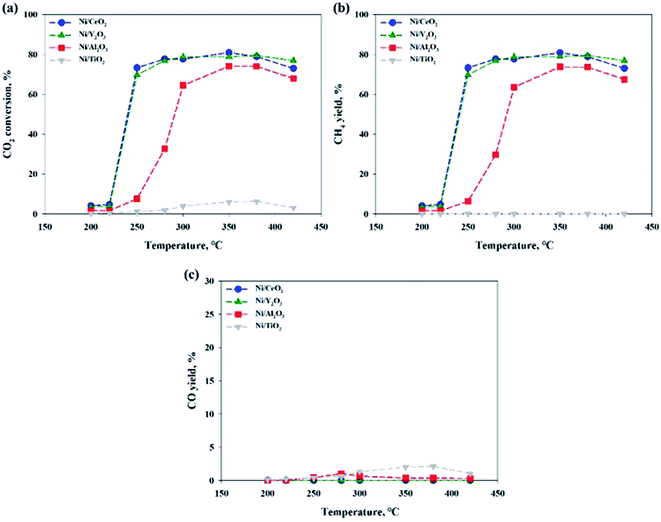 |
| | Fig. 2 (a) CO2 conversions, (b) CH4 and (c) CO yield in CO2 methanation over 10 wt% Ni/support catalysts. CO2:H2:N2 = 1:4:1, GHSV: 14400 l kg−1 h−1. | |
3.2 Physicochemical properties
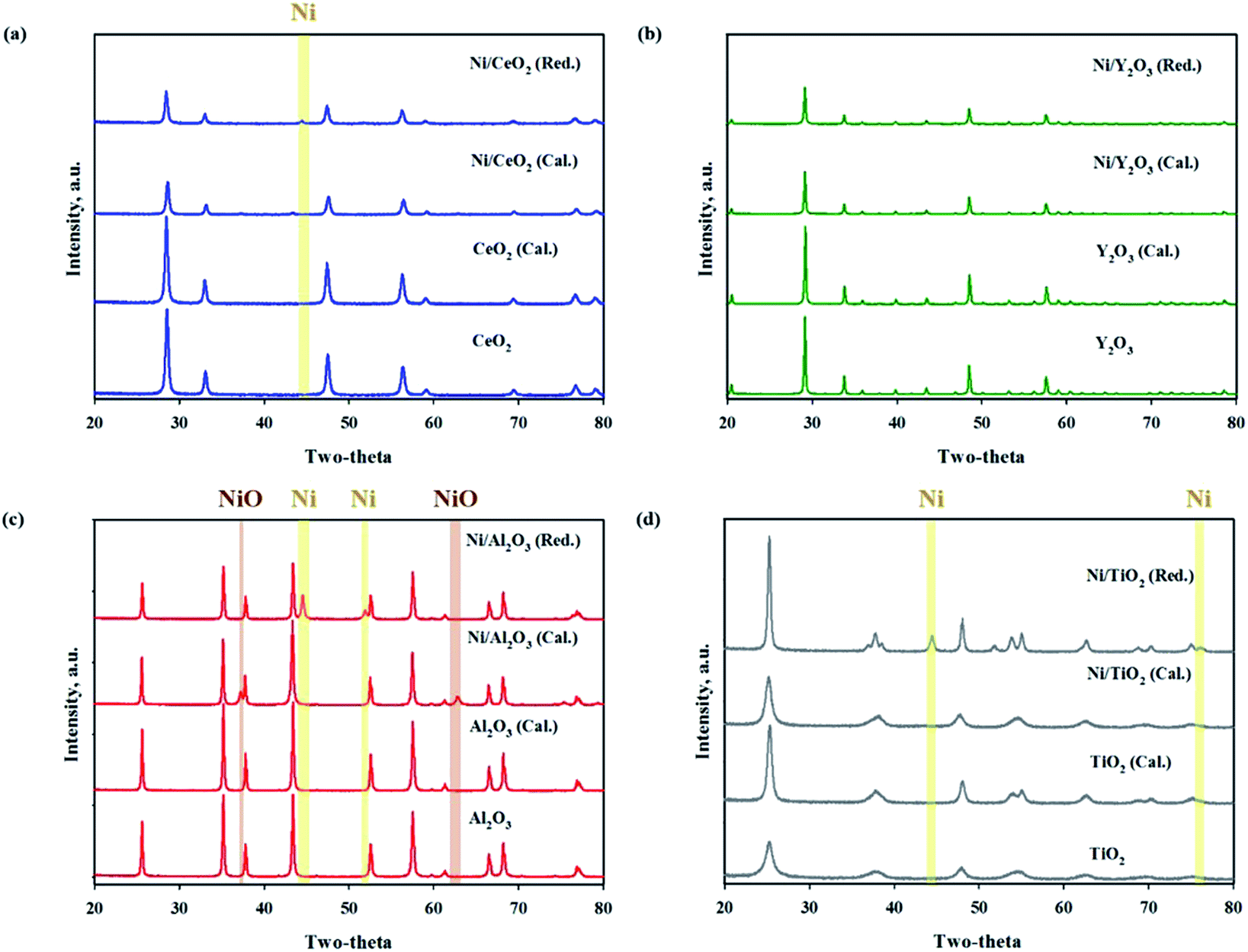
The BET surface areas and metal dispersions of the reduced and unreduced samples are summarized in Table 1. The order of support's surface area was as follows: TiO2 > CeO2 > Al2O3 > Y2O3 (TiO2: 316.226, CeO2: 29.557, Al2O3: 7.375, Y2O3: 2.615 m2 g−1; not shown in the figure). It can be observed that the Ni/TiO2 catalyst had a much larger specific surface area than other catalysts. Zhang et al.34 showed that the catalyst's specific surface area is not directly related to the catalytic activity in CO2 methanation for Ni-based catalysts. Our results were in good agreement with previously mentioned evidence from the literature. The reduced Ni/TiO2 catalyst had the lowest value of Ni dispersion (0.0116%). The reduced Ni/Y2O3 catalyst showed the highest metal dispersion (2.7883%) of all the unreduced Ni-based catalysts. Reduction degree of surface area and metal dispersion for Ni based catalysts with heat treatment by air or H2 can be attributed to complex interdependency on metal–support interactions. This would suggest that a decrease of metal dispersion by reduction treatment could be explained by a strong interaction between Ni and oxide support. The order of strength of the metal and support interaction was as follows: Ni/Al2O3 < Ni/TiO2 < Ni/CeO2 < Ni/Y2O3. Fig. 3(a)–(d) shows the XRD patterns of raw supports, calcined supports, calcined Ni based catalysts and calcined Ni based catalysts. The XRD pattern of Ni consists of three main peaks at 45°, 53°, and 76° corresponding to (111), (200), and (220) planes, respectively. The XRD pattern of NiO consists of two main peaks at 37° and 62° corresponding to (111) and (220) planes, respectively.35 For the TiO2 powder, the main peaks were observed to be at 2θ = 25.3, 37.1 and 47.5°, corresponding to typical anatase TiO2 peaks. The sharp peaks were observed for the crystallite TiO2 structure when the amorphous TiO2 powder was calcined at a temperature of 400 °C. NiO peaks were not observed in calcined Ni/TiO2 catalyst. It is interesting to note that Ni crystallite main peaks were detected at 2θ = at 44.5, 51.8 when using the reduced Ni/TiO2 catalyst. The release of bonding oxygen atoms within Ni–O–Ti by H2 reduction will help to move the Ni particles, then the Ni particles are easily agglomerated. Ni and NiO peaks were observed in both reduced and unreduced Ni/Al2O3 catalysts. Unlike the Ni/TiO2 catalyst, it confirmed that Ni and NiO particles were agglomorated by heat treatment regardless of the migration of oxygen atom for Ni supported on irreducible Al2O3. CeO2 and Y2O3 support had quite low surface area. Nevertheless, the Ni and NiO peaks were not observed in reduced and unreduced Ni/Y2O3 catalyst and very low Ni and NiO peaks were observed in reduced and unreduced Ni/CeO2 catalyst, which is related to strong metal–support interactions (SMSI effect). This phenomenon is in agreement with results from previous metal dispersion. TEM analysis was conducted to estimate the Ni particle sizes of Ni based catalysts. The TEM and mapping images of the reduced Ni catalysts are presented in Fig. 4(a)–(l). It could be seen that the Ni/Y2O3 mean particle size of Ni was 12.8 nm, while that of Ni/CeO2, Ni/Al2O3 and Ni/TiO2 were 16.5 nm, 19.9 nm and 21 nm, respectively. Vogt et al.25 investigated the particle size effect of Ni/SiO2 catalysts prepared by homogeneous deposition precipitation and co-precipitation with different Ni loadings (1–60 wt%). It was concluded that the Ni based CO2 methanation is structure sensitive from 1–7 nm for Ni/SiO2 catalyst. Many studies have attempted to enhance the Ni dispersion by increasing H2 adsorption as active sites.11,36–39 In this study, the 10 wt% Ni-based catalysts prepared by the impregnation method have relatively large Ni particle size of 12.8–21 nm, but it has low temperature CO2 methanation activity. In the case of Ni/Al2O3 and Ni/TiO2 catalyst, it was confirmed that the difference in activity at high temperature (280–420 °C) was clearly displayed despite the similar particle size and metal dispersion. The fact that the Ni particles play an important role in the adsorption of hydrogen as active sites, but other factors such as CO2 adsorption characteristics and oxygen transfer by hydrogen can influence the catalytic performance.
Table 1 BET surface area and metal dispersion of reduced and unreduced Ni/metal oxide catalysts
| |
Surface area (m2 g−1) |
Metal dispersiona (%) |
| Unreduced |
Reduced |
Unreduced |
Reduced |
| Calculated by H2 pulse chemisorption. |
| Ni/CeO2 |
29.907 |
22.845 |
4.932 |
1.823 |
| Ni/Y2O3 |
16.794 |
21.233 |
3.438 |
2.788 |
| Ni/Al2O3 |
9.867 |
8.698 |
2.323 |
0.027 |
| Ni/TiO2 |
121.74 |
43.606 |
0.067 |
0.012 |
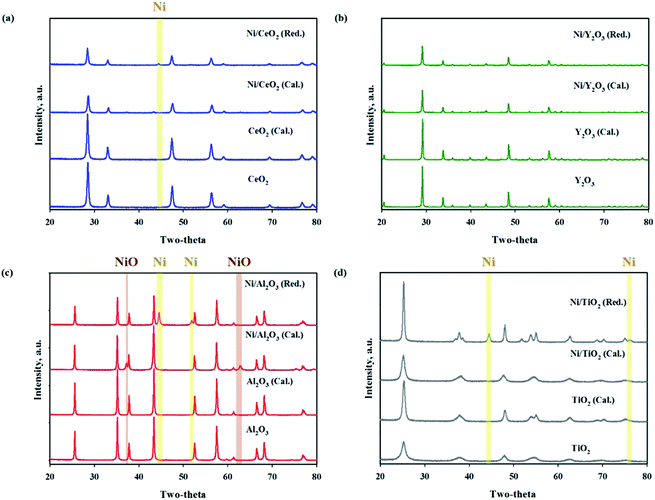 |
| | Fig. 3 XRD patterns of metal oxide supports and reduced (red.) and unreduced (cal.) Ni supported catalysts ((a) Ni/CeO2, (b) Ni/Y2O3, (c) Ni/Al2O3, (d) Ni/TiO2). | |
 |
| | Fig. 4 TEM and mapping images of Ni/support catalysts ((a) Ni/CeO2, (b) Ce, (c) Ni, (d) Ni/Y2O3, (e) Y2O3, (f) Ni, (g) Ni/Al2O3, (h) Al2O3, (i) Ni, (j) Ni/TiO2, (k) TiO2, (l) Ni). | |
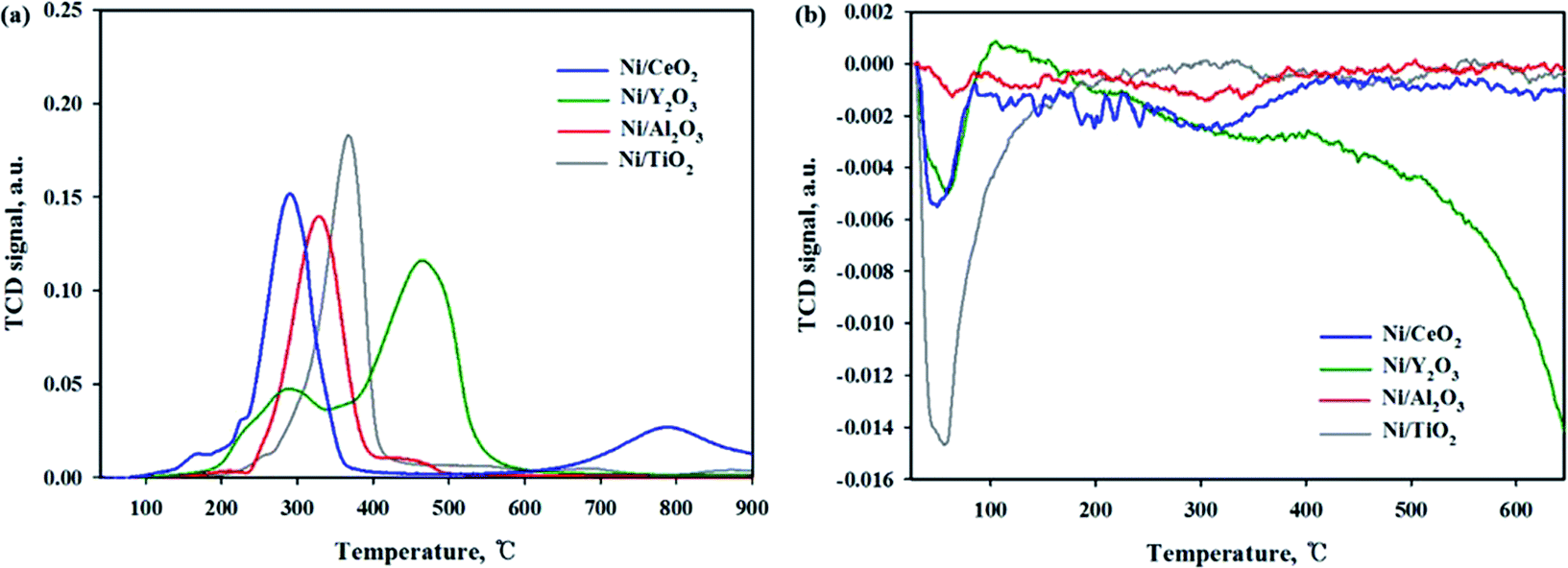
The reducibility of Ni-based catalysts was investigated by a H2-temperature programmed reduction, and the profiles are shown in Fig. 5(a). A similar H2-TPR result for the Ni/CeO2 catalyst was previously reported.13,17,40 Zhou et al.17 reported that three reduction peaks can be seen at around 220 °C, 280–330 °C and 380 °C. The first low temperature peak attributed to the reduction of highly dispersed NiO species. The second peak attributed to the reduction of NiO species on the subsurface of the Ni/CeO2 catalyst and highly dispersive NiO species. The last peak can be assigned to the reduction of bulk NiO species. Tada et al.13 reported that the two reduction peaks can be observed at 340 and 420 °C, which were attributed to the reduction of NiO on CeO2 at about 400 °C. Ding et al.40 also reported that the main reduction peaks appeared at 350–450 °C; these peaks were attributed to the reduction of NiO. In this study, the Ni/CeO2 catalyst exhibited four-hydrogen-consumption maximum peaks at 165 °C, 225 °C, 290 °C and 785 °C, which were attributed to highly dispersive NiO species or Ni(OH)2, NiO species on the subsurfaces, Ni–O–Ce species, and bulk CeO2 reduction peaks, respectively. It should be noted that all the reduction peaks are shifted to a lower temperature compared to previously reported studies. It can be suggested that, although the catalytic composition was the same, the reduction trend can differ, depending on the preparation conditions e.g., the kinds of precursor, calcination temperature/time, and metal loadings. For the Ni/Al2O3 catalyst, two reduction peaks were observed at 250–400 °C and 400–500 °C. The first peak is assigned to the free Ni species and second peak is attributed to the Ni species combined with the Al2O3 support (Ni–O–Al).40 Evidence from the literature showed that the last reduction peak can be observed for the Ni/Al2O3 catalyst in the high-temperature region (750–850 °C), suggesting a stronger interaction between NiO and the Al2O3 support.21 But this peak was not observed in this sample; this might be due to a low calcination temperature. For the Ni/Y2O3 catalyst, the two broad reduction peaks were observed in the 200–350 °C and 370–600 °C ranges, respectively. The first peak indicate the existence of a interaction between NiO and the Y2O3 support and the second reduction peak is attributed to the bulk Y2O3 support. The Ni/TiO2 catalyst exhibited only one broad hydrogen consumption peak at 250–420 °C, which was attributed to the Ni species combined with the TiO2 support (Ni–O–Ti). It is expected that the 10 wt% Ni based catalysts may have a metallic Ni form at 420 °C of reduction temperature. According to the H2 TPR and activity test results of all catalysts, enhancement of the CO2 conversion could be mainly due to the higher amount of NiO species able to be reduced at low temperature. CO2 temperature-programmed oxidation (TPO) was performed to observe the CO2 conversion for reduced catalysts at 420 °C as shown in Fig. 5(b). For the reduced Ni/TiO2 catalyst, the one huge CO2 consumption peak was observed in the 50–150 °C. This result indicated that the reduced Ni/TiO2 catalyst was able to accept oxygen by introduction of CO2 on reducible sites. However, on the Ni surface H, adsorption is difficult due to low Ni dispersion, which makes it difficult to react the CO2 methanation. The reduced Ni/CeO2 and Ni/Y2O3 catalysts exhibited the similar CO2 consumption peak at 50–100 °C. For the reduced Ni/Al2O3 catalyst, the CO2 consumption peak was not observed at all temperature range, it may be concluded that the CO2 molecules cannot be dissociated on reduced Ni/Al2O3 catalyst. Among the catalysts used in this study, the Ni/CeO2 and Ni/Y2O3 catalyst had the higher dispersion and enhanced reducibility of Ni particles, as well as strong metal and support interactions, which could become an important factor for the catalytic activity during low-temperature CO2 methanation.
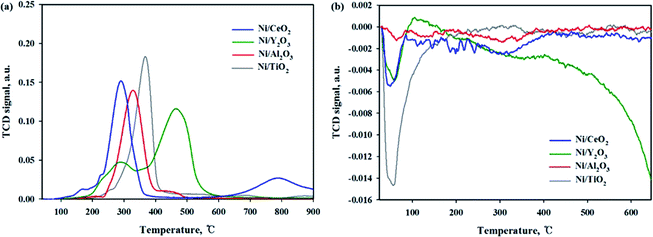 |
| | Fig. 5 (a) H2-TPR and (b) CO2 TPO profiles of Ni/metal oxide catalysts. | |
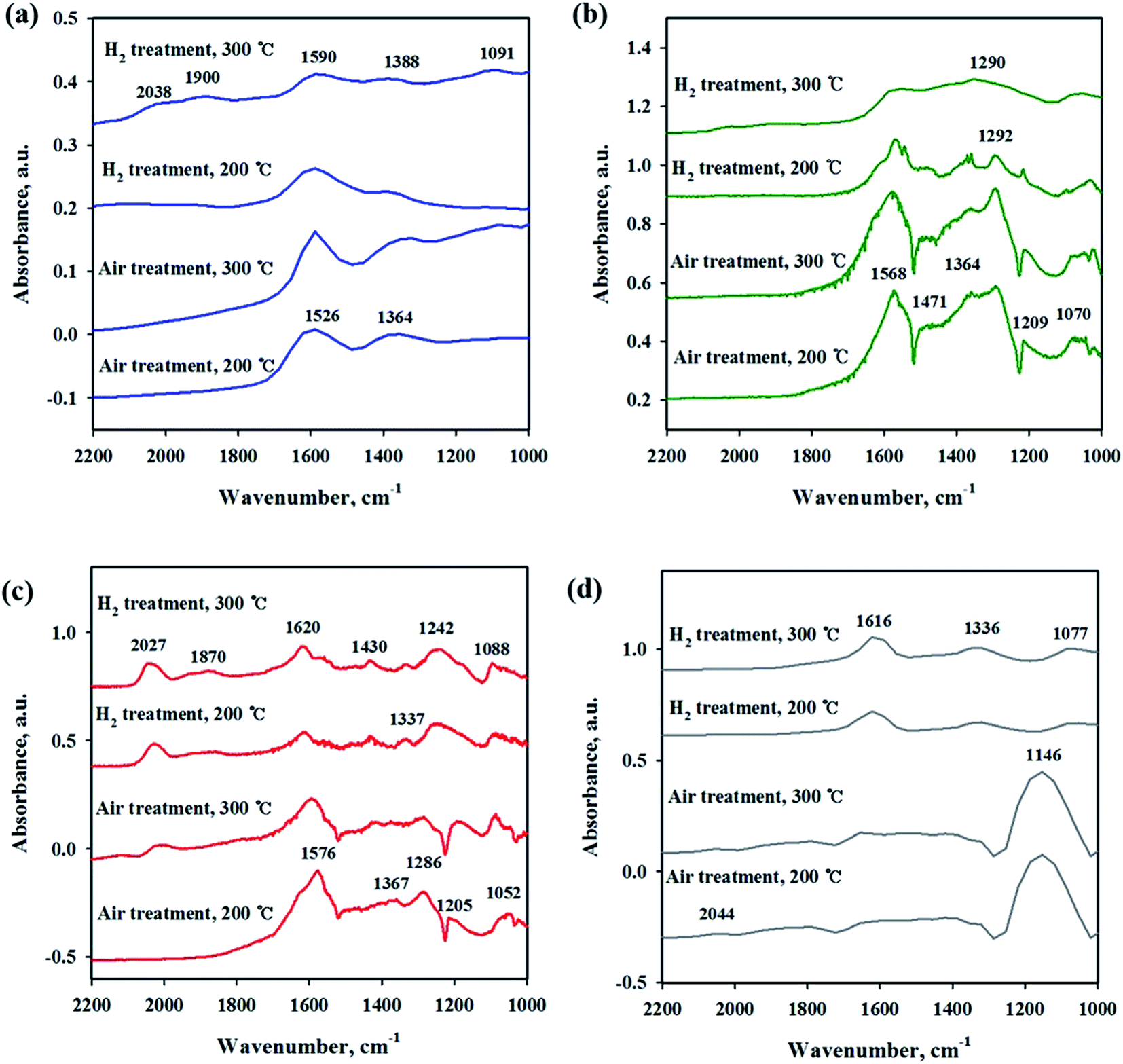
3.3 In situ DRIFTS
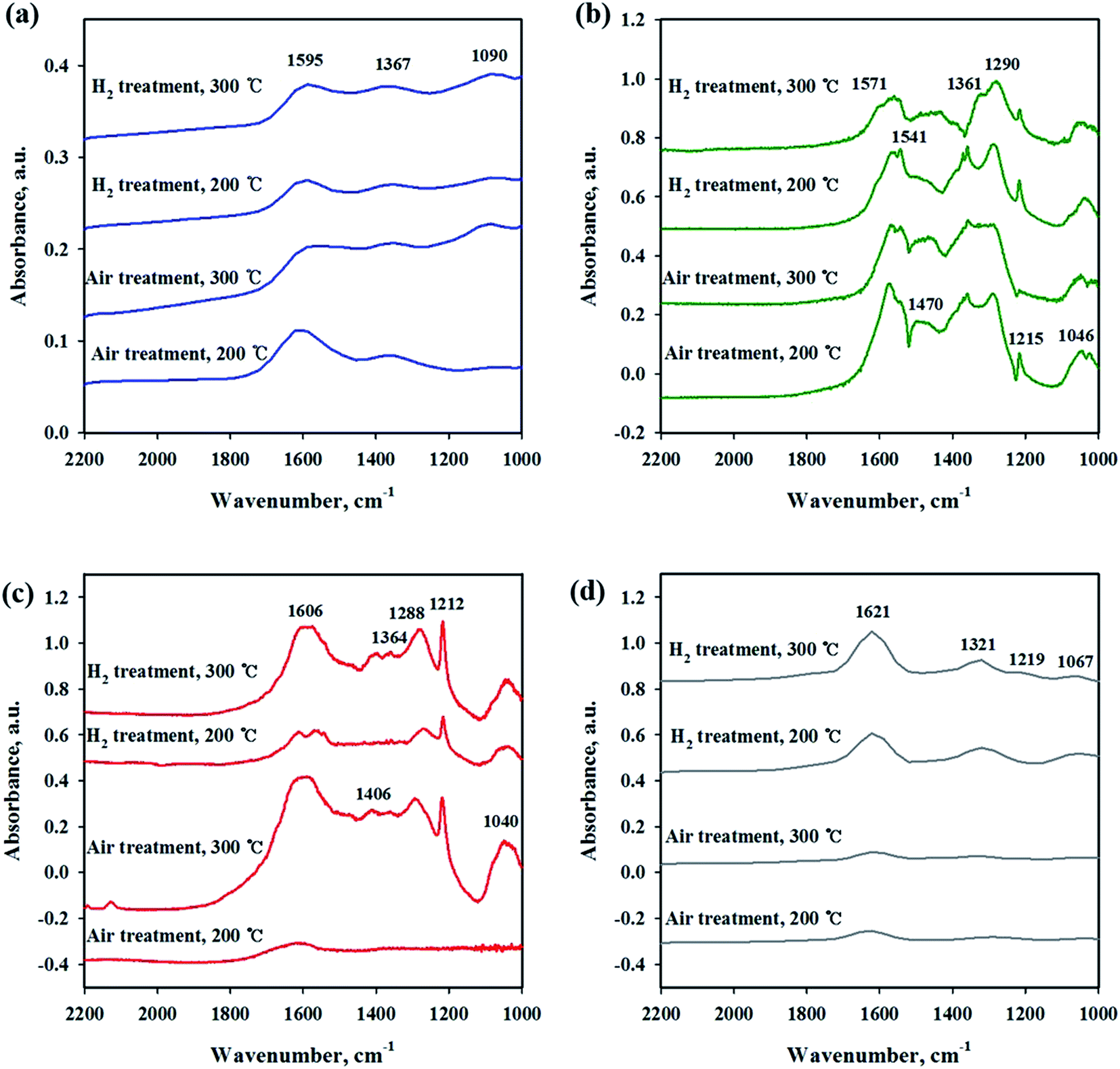
To investigate the interaction of CO2 with catalysts, in situ DRIFT studies were performed on the reduced and unreduced catalysts at 200 and 300 °C as shown in Fig. 6. Fig. 6(a) shows the CO2 adsorption profile on the Ni/CeO2 catalyst surface. The assignment of CO2 adsorption bands for the Ni/Ce0.5Zr0.5O2 catalyst was performed by reported in the previous literature.41,42 The spectra of CO2 adsorption on Ni/CeO2 are similar to those of the Ni/Ce0.5Zr0.5O2 catalyst. The band observed at 1595 cm−1 can be assigned to formate species and the bands centered at 1367 cm−1 was assigned to monodentate carbonate. These two main bands were still maintained at 300 °C, implying that the formation of formate and monodentate carbonate species are favored at high temperature, as the main intermediate during direct hydrogenation of CO2.41,43–46 The reduced and unreduced Ni/CeO2 catalysts exhibited similar CO2 adsorption bands, regardless of calcination or reduction treatments, indicating that the CO2 adsorption properties on Ni/CeO2 catalyst may depend on the CeO2 support, not NiO and metallic Ni species. CO2 adsorption bands were compared for Ni/Y2O3 and Ni/Al2O3 in Fig. 6(b) and (c). The main adsorption bands were detected at 1541, 1290, 1215 and 1046 cm−1 (formate), and 1571 cm−1 (bidentate carbonate) for Ni/Y2O3 catalyst and at 1606, 1406 and 1364 cm−1 (formate), and 1364 cm−1 (bidentate carbonate) for Ni/Al2O3. The formate bands were maintained at 300 °C for Ni/Al2O3 and Ni/Y2O3 catalysts. The intensity of main peaks increased with higher temperature (300 °C) for Ni/Al2O3 catalyst. In Fig. 6(d), the CO2 adsorption bands were observed for the Ni/TiO2 catalyst. The bands centered at 1219 and 1067 cm−1 were attributed to monodentate species and the bands centered at 1620 and 1219 cm−1 were assigned to hydrogen carbonate peaks. The intensity of CO2 absorbed peaks decreased at calcined catalyst while these peaks increased with H2 reduction treatment, indicating that a small part of catalytic sites become active by surface OH groups, and CO2 adsorption peaks are increased. Fig. 7 shows the CO2 adsorption profiles of all reduced and unreduced catalysts in the CO2–4H2 atmosphere as a function of different temperatures. The two main bands attributed to formate and monodentate carbonate were still maintained at 200 and 300 °C for the calcined Ni/CeO2 catalyst when introducing CO2 and H2 (Fig. 7(a)). These peaks were maintained at the reaction temperature of 200 °C for reduced Ni/CeO2 catalyst but the intensity of peaks decreased obviously at reaction temperature of 300 °C and the two bands were detected at 1900 and 2038 cm−1 which was assigned bridged CO and linear CO bands adsorbed on Ni, respectively.47 This result indicated that CO2 was readily dissociated to CO and it does participate in the CO2 methanation reaction at high temperature for Ni/CeO2 catalyst. Aldana et al.44 reported that methanation proceeds through formate species originated from the hydrogenation of carbonates and CO was formed by a redox cycle on reduced ceria support. At 300 °C, only monodentate carbonate bands well remained. This result indicated that the monodentate carbonates are not easily hydrogenated; this result was in good agreement with Pan et al.41 As shown in Fig. 7(b), although hydrogen is injected, the main formate and bidentate carbonate bands were still maintained at 200 and 300 °C for the calcined Ni/Y2O3 catalyst. The small and broad formate bands observed at 300 °C for the reduced Ni/Y2O3 catalyst and the two small bands were detected at 1900 and 2038 cm−1 which was assigned bridged CO and linear CO bands adsorbed on Ni, respectively, it form a similar pattern with reduced Ni/CeO2 catalyst. This formation of active species as formate leads to higher catalytic activity at high temperature. It is expected that sufficient reduction pretreatment of reducible supports such as CeO2 and Y2O3 supported catalysts efforts on CO2 decomposition into CO which is an crucial step in CO2 conversion into CH4 at high temperature and thus catalytic activity using Ni/CeO2 and Ni/Y2O3 catalysts is enhanced.13 In Fig. 7(c), the CO2 absorption bands were still maintained at 200 °C for the calcined Ni/Al2O3 catalyst, it forms a similar pattern with CO2 adsorption pattern as shown in Fig. 6(c). CO2 methanation is not progressed in this low temperature for Ni/Al2O3 catalyst. The similar bands were detected at 300 °C for the calcined Ni/Al2O3 catalyst, but the small band observed at 2027 cm−1 which was assigned linear CO bands adsorbed on Ni. It is known from the H2 TPR results that the release of bonding oxygen atoms within Ni–O–Al by H2 reduction at 300 °C, CO2 was readily dissociated to CO on reduction active site for the calcined Ni/Al2O3 catalyst. The spectra for the reduced Ni/Al2O3 catalyst exhibited small overlapped bands of formate species and the intensity of bridged CO and linear CO peaks centered at 1870 and 2027 cm−1 increased. This result indicated that CO2 was readily dissociated to CO and it does participate in the CO2 methanation reaction at high temperature for reduced Ni/Al2O3 catalyst. The Ni/TiO2 catalyst showed a different adsorption bands than other catalyst as shown in Fig. 7(d). The intensity of broad hydrogen carbonate peak centered at 1000–1300 cm−1 increased for calcination Ni/TiO2 catalyst and the two small bands were observed at 1336 and 1077 cm−1 which was assigned monodentate peaks regardless of reaction temperature. The amount of CO2 adsorbed onto Ni/TiO2 was much larger than that onto other catalyst, but CO2 methanation reaction cannot occur regardless of reaction temperature. Due to the low Ni dispersion, it can be inferred that adsorbed monodentate and hydrogen carbonate species are difficult to react with adsorbed hydrogen atoms on active metallic Ni site to form methane so that CO2 methanation reaction does not take place at all temperatures. Numerous studies about reaction mechanism of CO2 methanation have been investigated. The reaction mechanisms are normally classified into two reaction pathways. One involves CO2 conversion to CO as intermediate, which then follows the same mechanism as CO methanation.48 The other one involves formate and carbonate as main intermediate, which directly hydrogenate without forming CO.44 From the results in this study, it was assumed that the methanation reaction of CO2 will follow the first mechanism for Ni/Al2O3 catalyst and formate and carbonate as main intermediate mechanism at low temperature (200 °C) for Ni/Y2O3 and Ni/CeO2 catalysts. However, the methanation reaction of CO2 will follow the first mechanism at high temperature (300 °C) for Ni/Y2O3 and Ni/CeO2 catalysts. These different mechanisms might be one of the reasons why the role of supports. The Ni supported irreducible Al2O3 catalyst favors the CO2 conversion to CO and then follows the same mechanism as CO methanation. While carbonate and formate species were found to be the main intermediate on surface oxygen vacancy site such as Ce3+ sites, which could enhance the catalytic activity at low temperature compare to Ni/Al2O3 catalyst.3 The carbonate and formate species were mainly present as an adsorption intermediate on surface oxygen vacancy site (Ce3+ sites), while the CO species were found to be the main adsorption intermediate for the irreducible Al2O3 supported Ni catalyst. According to TPR and DRIFT results, the interaction between Ni and Ce or Y may facilitate the formation of the OH groups on the Ni–OH or Y–OH, Ce–OH bonds. The dissociated hydrogen atoms on the Ni metal spillover onto the ceria or yttria support surface, and undergo surface diffusion at low temperature by introduction of hydrogen. Superior reducibility of
both Ni-rich surface species and CeO2 support by strong interaction between Ni and CeO2 or Y2O3 supports are important factor and then weakly adsorbed CO2 species such as formate and carbonate on the surface oxygen vacancy site easily reacted with hydrogen dissociation on the metal followed by spillover at low temperature.
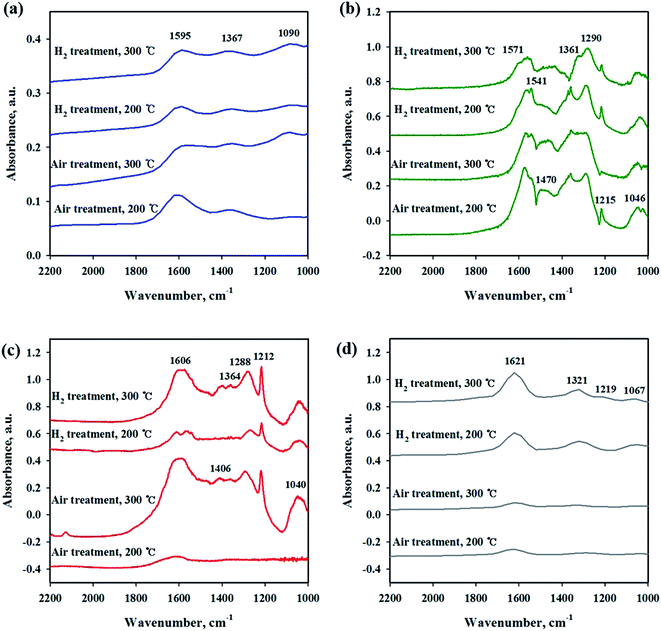 |
| | Fig. 6 In situ DRIFTS studies of CO2 adsorption on Ni/metal oxide catalysts with different pretreatment and temperature conditions. (a) Ni/CeO2, (b) Ni/Y2O3, (c) Ni/Al2O3 and (d) Ni/TiO2. | |
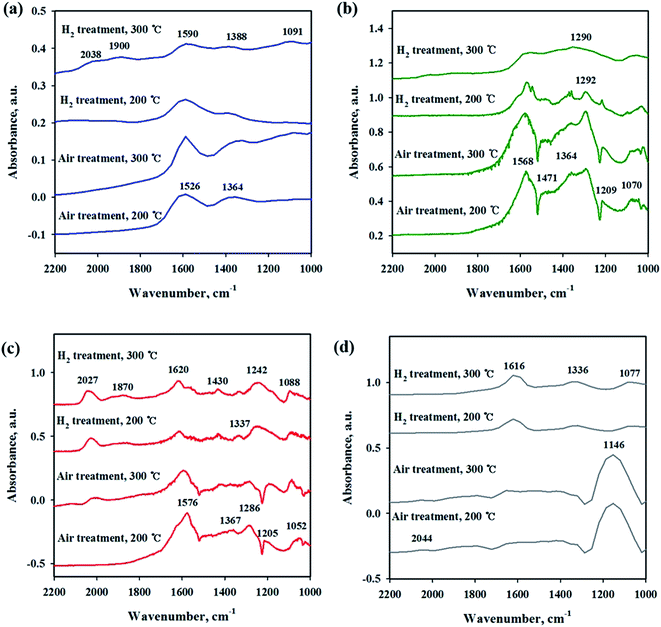 |
| | Fig. 7 In situ DRIFTS studies of CO2 methanation (CO2 + H2) on Ni/metal oxide catalysts with different pretreatment and temperature conditions. (a) Ni/CeO2, (b) Ni/Y2O3, (c) Ni/Al2O3 and (d) Ni/TiO2. | |
4 Conclusions
The Ni/CeO2 and Ni/Y2O3 catalysts show highly enhanced catalytic activity for low-temperature CO2 methanation, as compared with the Ni/Al2O3 and Ni/TiO2 catalysts. The increase in the low-temperature CO2 methanation activity can be directly correlated with an enhancement in the reducibility and small Ni particle size of the Ni/CeO2 and Ni/Y2O3 catalyst, which is caused by a strong interaction between reduced CeO2 or Y2O3 oxide support and Ni. When Ni species are dispersed on “reducible” oxides, such as CeO2 and Y2O3, carbonate and formate species were mainly present as an adsorption intermediate on the surface oxygen vacancy site such as Y(3−x)+ or Ce3+ sites at low temperature, which plays a key role in enhancing the low catalytic activity. Due to the low Ni dispersion of Ni/TiO2 catalyst, it can be concluded that adsorbed CO2 species are difficult to react with hydrogen atoms on active metallic Ni site to form methane. When the Ni species were dispersed on “irreducible” oxides, species such as Al2O3 and CO were found to be the main adsorption intermediates. Overall, CeO2 and Y2O3 supports are promising support for the Ni-based catalyst and further improvement in low-temperature CO2 methanation activity can be made by modification of the mixed CeO2 and Y2O3 support with small Ni particle size and oxygen vacancies.
Author contributions
Ye Hwan Lee: conceptualization, writing – original draft. Jeong Yoon Ahn: experiment and evaluation. Dinh Duc Nguyen: data curation, validation, formal analysis. Soon Woong Chang: writing – review & editing. Sung Su Kim: writing – review & editing. Sang Moon Lee: supervision, project administration.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1D1A1B03036192).
Notes and references
- F. Ocampo, B. Louis, L. Kiwi-Minsker and A. C. Roger, Appl. Catal., A, 2011, 392, 36–44 CrossRef CAS.
- K. Ghaib, K. Nitz and F.-Z. Ben-Fares, ChemBioEng Rev., 2016, 3, 266–275 CrossRef.
- M. A. A. Aziz, A. A. Jalil, S. Triwahyono and A. Ahmad, Green Chem., 2015, 17, 2647–2663 RSC.
- Q. Pan, J. Peng, T. Sun, D. Gao and S. Wang, Fuel Process. Technol., 2014, 123, 166–171 CrossRef CAS.
- W. J. Shen, M. Okumura, Y. Matsumura and M. Haruta, Appl. Catal., A, 2001, 213, 225–232 CrossRef CAS.
- V. M. Shinde and G. Madras, AIChE J., 2014, 60, 1027–1035 CrossRef CAS.
- M. Yamasaki, H. Habazaki, T. Yoshida, E. Akiyama, A. Kawashima, K. Asami, K. Hashimoto, M. Komori and K. Shimamura, Appl. Catal., A, 1997, 163, 187–197 CrossRef CAS.
- G. D. Weatherbee and C. H. Bartholomew, J. Catal., 1981, 68, 67–76 CrossRef CAS.
- M. Yamasaki, H. Habazaki, K. Asami, K. Izumiya and K. Hashimoto, Catal. Commun., 2006, 7, 24–28 CrossRef CAS.
- A. E. Aksoylu and Z. I. Önsan, Appl. Catal., A, 1997, 164, 1–11 CrossRef.
- F. Ocampo, B. Louis and A. C. Roger, Appl. Catal., A, 2009, 369, 90–96 CrossRef CAS.
- X. Zhang, W. J. Sun and W. Chu, J. Fuel Chem. Technol., 2013, 41, 96–101 CrossRef.
- S. Tada, T. Shimizu, H. Kameyama, T. Haneda and R. Kikuchi, Int. J. Hydrogen Energy, 2012, 37, 5527–5531 CrossRef CAS.
- N. Takezawa, H. Terunuma, M. Shimokawabe and H. Kobayashib, Appl. Catal., 1986, 23, 291–298 CrossRef CAS.
- K. Zhao, W. Wang and Z. Li, J. CO2 Util., 2016, 16, 236–244 CrossRef CAS.
- H. Muroyama, Y. Tsuda, T. Asakoshi, H. Masitah, T. Okanishi, T. Matsui and K. Eguchi, J. Catal., 2016, 343, 178–184 CrossRef CAS.
- G. Zhou, H. Liu, K. Cui, A. Jia, G. Hu, Z. Jiao, Y. Liu and X. Zhang, Appl. Surf. Sci., 2016, 383, 248–252 CrossRef CAS.
- J. Liu, C. Li, F. Wang, S. He, H. Chen, Y. Zhao, M. Wei, D. G. Evans and X. Duan, Catal. Sci. Technol., 2013, 3, 2627–2633 RSC.
- E. Jwa, S. B. Lee, H. W. Lee and Y. S. Mok, Fuel Process. Technol., 2013, 108, 89–93 CrossRef CAS.
- J. Ashok, M. L. Ang and S. Kawi, Catal. Today, 2017, 281, 304–311 CrossRef CAS.
- S. Abate, C. Mebrahtu, E. Giglio, F. Deorsola, S. Bensaid, S. Perathoner, R. Pirone and G. Centi, Ind. Eng. Chem. Res., 2016, 55, 4451–4460 CrossRef CAS.
- S. Damyanova, B. Pawelec, R. Palcheva, Y. Karakirova, M. C. C. Sanchez, G. Tyuliev, E. Gaigneaux and J. L. G. Fierro, Appl. Catal., B, 2018, 225, 340–353 CrossRef CAS.
- P. Frontera, A. Macario, M. Ferraro and P. L. Antonucci, Catalysts, 2017, 7, 1–28 CrossRef.
- T. A. Le, M. S. Kim, S. H. Lee, T. W. Kim and E. D. Park, Catal. Today, 2017, 293–294, 89–96 CrossRef CAS.
- S. Abelló, C. Berrueco and D. Montané, Fuel, 2013, 113, 598–609 CrossRef.
- C. Vogt, E. Groeneveld, G. Kamsma, M. Nachtegaal, L. Lu, C. J. Kiely, P. H. Berben, F. Meirer and B. M. Weckhuysen, Unravelling structure sensitivity in CO2 hydrogenation over nickel, Nat. Cat., 2018, 1, 127–134 CrossRef CAS.
- Y. Yan, Y. Dai, H. He, Y. Yu and Y. Yang, Appl. Catal., B, 2016, 196, 108–116 CrossRef CAS.
- D. Wierzbicki, R. Baran, R. Dębek, M. Motak, M. E. Gálvez, T. Grzybek, P. Da Costa and P. Glatzel, Appl. Catal., B, 2018, 232, 409–419 CrossRef CAS.
- K. Ray and G. Deo, Appl. Catal., B, 2017, 218, 525–537 CrossRef CAS.
- W. Cai, Q. Zhong and Y. Zhao, Catal. Commun., 2013, 39, 30–34 CrossRef CAS.
- M. Guo and G. Lu, Catal. Commun., 2014, 54, 55–60 CrossRef CAS.
- A. Trovarelli, Catalysis by ceria and related materials, World Scientific Publishing Co., Singapore, 2002 Search PubMed.
- F. T. Zangeneh, S. Sahebdelfar and M. T. Ravanchi, J. Nat. Gas Chem., 2011, 20, 219–231 CrossRef CAS.
- H. Zhang, Y. Dong, W. Fang and Y. Lian, Chin. J. Catal., 2013, 34, 330–335 CrossRef CAS.
- H. Singh, A. Rai, R. Yadav and A. K. Sinha, Mol. Catal., 2018, 451, 186–191 CrossRef CAS.
- S. Hwang, J. Lee, U. G. Hong, J. H. Baik, D. J. Koh, H. Lim and I. K. Song, J. Ind. Eng. Chem., 2013, 19, 698–703 CrossRef CAS.
- J. Ren, X. Qin, J. Z. Yang, Z. F. Qin, H. L. Guo, J. Y. Lin and Z. Li, Fuel Process. Technol., 2015, 137, 204–211 CrossRef CAS.
- B. Mutz, H. W. P. Carvalho, S. Mangold, W. Kleist and J. D. Grunwaldt, J. Catal., 2015, 327, 48–53 CrossRef CAS.
- Y. Zeng, H. Ma, H. Zhang, W. Ying and D. Fang, Fuel, 2015, 162, 16–22 CrossRef CAS.
- M. Ding, J. Tu, Q. Zhang, M. Wang, N. Tsubaki, T. Wang and L. Ma, Biomass Bioenergy, 2016, 85, 12–17 CrossRef CAS.
- Q. Pan, J. Peng, S. Wang and S. Wang, Catal. Sci. Technol., 2014, 4, 502–509 RSC.
- S. Tada, O. J. Ochieng, R. Kikuchi, T. Haneda and H. Kameyama, Int. J. Hydrogen Energy, 2014, 39, 10090–10100 CrossRef CAS.
- S. Sharma, Z. Hu, P. Zhang, E. W. Mcfarland and H. Metiu, J. Catal., 2011, 278, 297–309 CrossRef CAS.
- P. A. U. Aldana, F. Ocampo, K. Kobl, B. Louis, F. Thibault-Starzyk, M. Daturi, P. Bazin, S. Thomas and A. C. Roger, Catal. Today, 2013, 215, 201–207 CrossRef CAS.
- Q. Pan, J. Peng, T. Sun, S. Wang and S. Wang, Catal. Commun., 2014, 45, 74–78 CrossRef CAS.
- C. Schild, A. Wokaun and A. Baiker, J. Mol. Catal., 1990, 63, 223–242 CrossRef CAS.
- S. ichiro Fujita, M. Nakamura, T. Doi and N. Takezawa, Appl. Catal., A, 1993, 104, 87–100 CrossRef.
- A. Beuls, C. Swalus, M. Jacquemin, G. Heyen, A. Karelovic and P. Ruiz, Appl. Catal., B, 2012, 113–114, 2–10 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  b,
Soon Woong Changb,
Sung Su Kim
b,
Soon Woong Changb,
Sung Su Kim