
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConstruction of cyclopentane-fused coumarins via DBU-catalyzed [3+2] cycloaddition of 3-homoacyl coumarins with cyclic 1-azadienes†
Huawei Lina,
Huimin Yanga,
Qi Gonga,
Shan Luoa,
Jing Gua,
Xiaoqun Caoa,
Biming Mao*b,
Yanqing Ge *a and
Chunhao Yuan*a
*a and
Chunhao Yuan*a
aSchool of Chemistry and Pharmaceutical Engineering, Shandong First Medical University, Shandong Academy of Medical Sciences, Taian 271016, Shandong, P. R. China. E-mail: yuanchunhao2017@163.com; geyanqing2016@126.com
bInstitute of Materia Medica, Shandong First Medical University, Shandong Academy of Medical Sciences, Jinan 250117, Shandong, P. R. China. E-mail: maobiming@sdfmu.edu.cn
First published on 4th June 2021
Abstract
The metal-free DBU catalyzed [3+2] cycloaddition of 3-homoacyl coumarins with cyclic 1-azadienes proceeded smoothly to furnish the corresponding highly functionalized cyclopentane-fused coumarins with excellent diastereoselectivity and complete chemoselectivity and in good yields under mild conditions.
Coumarins1 and cyclopentane scaffolds2 are widely distributed in natural products and display a wide range of biological and pharmacological activities. When combining coumarin skeletons with cyclopentane moieties, the cyclopentane-fused coumarins show interesting biological activities. For example, aflatoxins, which occur naturally, exhibit acute toxicity, teratogenicity, mutagenicity and carcinogenicity (Fig. 1).3 Herbertenolide, which belongs to the family of sesquiterpenoids, was first isolated from the leafy liverwort Herberta adunca, the extract of which showed significant inhibition against the growth of certain plant pathogenic fungi (Fig. 1).4 Not surprisingly, the strategies for synthesis of cyclopentane-fused coumarins have attracted much attention.5
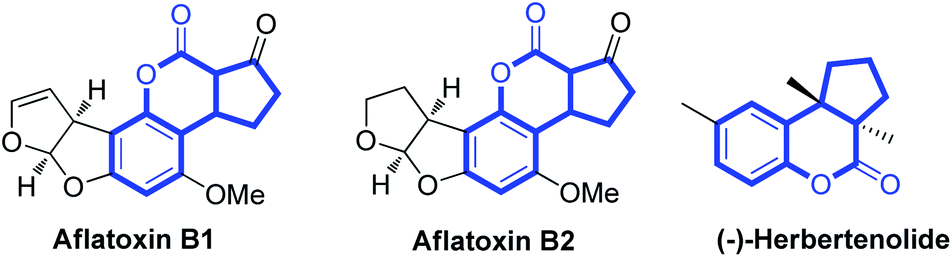 | ||
| Fig. 1 Bioactive molecule bearing cyclopentane-fused coumarin. | ||
Recently, the group of Lin developed a 1,3-dipolar precursor 3-homoacyl coumarin, which is an efficient synthon for the construction of cyclopentane-fused coumarins under the catalysis of bases (Scheme 1a and b).6 However, the partners reacted with 3-homoacyl coumarins were focus on α,β-unsaturated carbonyl compounds and conjugated dienes. The other dipolarophiles, such as aza-dienes, might also be potential candidates for the [3+n] cycloadditions with 3-homoacyl coumarins but never been developed.
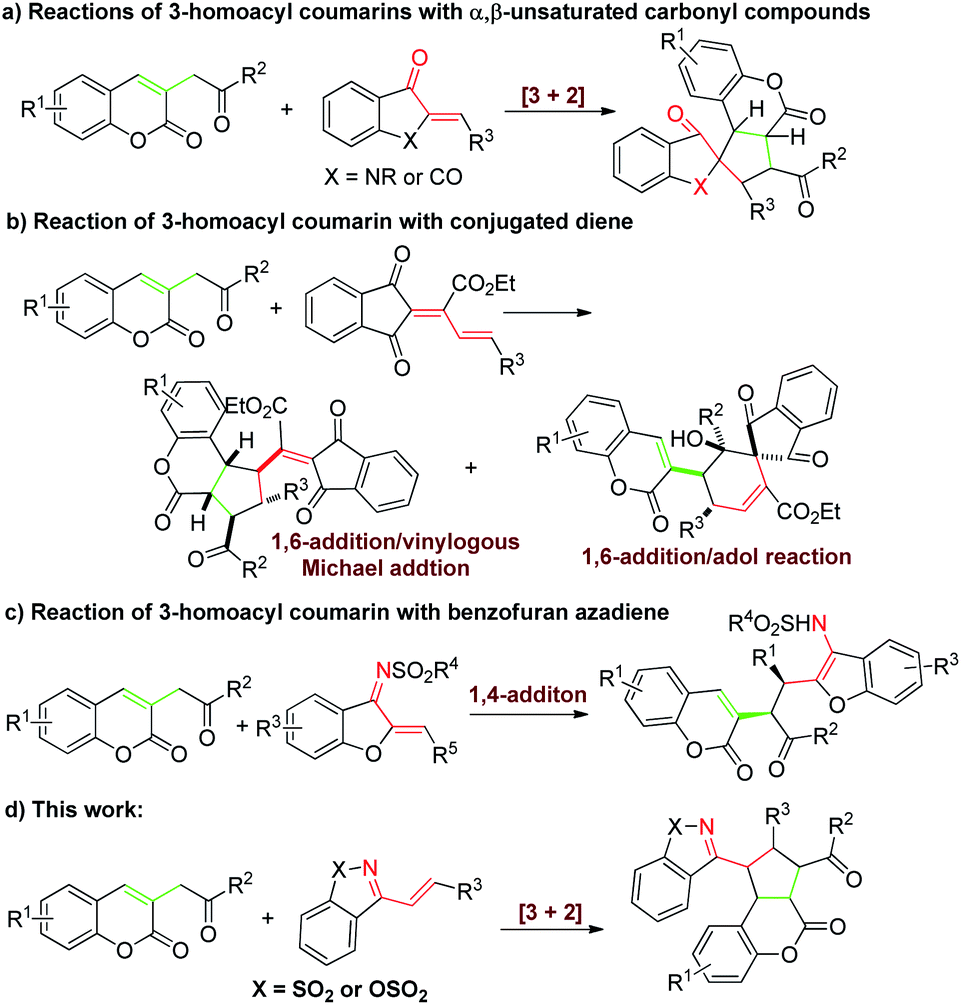 | ||
| Scheme 1 The reaction of 3-homoacyl coumarins with dipolarophiles catalyzed by Brønsted base. | ||
The cyclic 1-azadienes are extensive used dipolarophiles and have been widely involved in a series of cyclization reactions as two-,7 three-8 or four9 member synthons. While the organocatalytic [3+2] cycloaddition of cyclic 1-azadiene as two synthons has rarely been investigated.7b,c In 2016, Chen's7b and Guo's7c group respectively developed a asymmetric [3+2] annulation reaction of Morita–Baylis–Hillman carbonates with cyclic 1-azadienes catalyzed by Lewis base. Encouraged by these works above and as our continuing efforts on cycloadditions,10 herein we expected to achieve the first [3+2] cycloaddition reaction of 3-homoacyl coumarins with cyclic 1-azadienes catalyzed by Brønsted base for synthesis of various functionalized cyclopentane-fused coumarins derivatives efficiently (Scheme 1d). However, Huang's group reported a enantioselective 1,4-addition reaction of benzofuran azadiene with 3-homoacyl coumarin, instead of cycloaddition (Scheme 1c).11 To achieve our assumption in high chemoselectivity would be a challenging work.
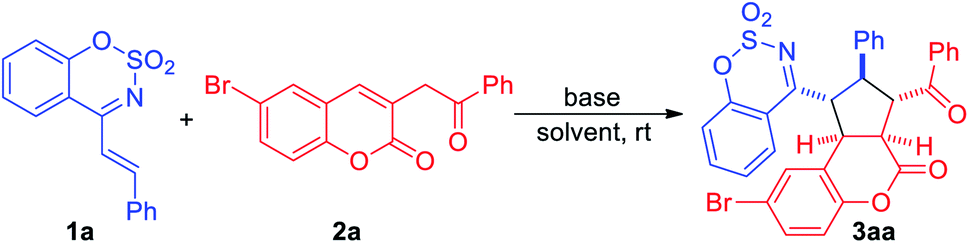
In an initial experiment, cyclic 1-azadiene 1a and 6-bromo-3-(2-oxo-2-phenylethyl)-2H-chromen-2-one 2a were employed as the model substrates to carry out the reaction in CH2Cl2 at room temperature in the presence of DABCO. To our delight, the desired [3+2] cycloadduct 3aa was obtained in 56% yield (Table 1, entry 1). Subsequently, several bases were screened and when the use of stronger base (Table 1, entries 2–4), DBU, the reaction gave a higher yield in 12 h, and no 1,4-addition product was observed (Table 1, entry 3). Further screening of several representative solvents, such as THF, toluene, DCE and CH3CN, revealed that the reaction proceeded better in THF with 86% yield (Table 1, entry 5). Therefore, the best reaction conditions were determined as below: DBU, THF and room temperature (Table 1, entry 5).
|
|||||
|---|---|---|---|---|---|
| Entry | Base | Solvent | Time (h) | Yieldb (%) | drc |
| a Reactions were carried out with 1a (0.1 mmol), 2a (0.12 mmol), and base (20 mol%) in 2 mL of solvent at rt.b Isolated yields.c Determined by 1H NMR. | |||||
| 1 | DABCO | CH2Cl2 | 24 | 56 | >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
| 2 | DMAP | CH2Cl2 | 24 | 60 | >20:1 |
| 3 | DBU | CH2Cl2 | 12 | 78 | >20:1 |
| 4 | Et3N | CH2Cl2 | 24 | 67 | >20:1 |
| 5 | DBU | THF | 12 | 86 | >20:1 |
| 6 | DBU | Toluene | 12 | 31 | >20:1 |
| 7 | DBU | DCE | 12 | 76 | >20:1 |
| 8 | DBU | CH3CN | 12 | 73 | >20:1 |
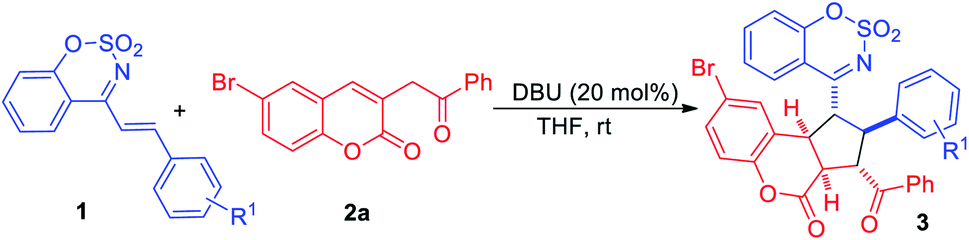
Under optimal reaction conditions, the substrate scope of the cyclic 1-azadienes 1 was investigated and the results were summarized in Table 2. As expected, the desired [3+2] cycloadducts 3ba–3qa were obtained in moderate to good yields. Both electron-withdrawing (entries 1–8) and electron-donating substituents (entries 9–14) on the benzene ring were tolerated and the yields of the former were slightly higher than the latter. And either para-, meta- or ortho-substituted phenyl cyclic 1-azadienes 1 could serve as suitable reaction partners, while 1m bearing ortho-methoxyphenyl gave moderate yield (67% yield) due to the steric hindrance (entry 12). Moreover, 2-naphthyl and 2-thienyl substituted substrates 1p and 1q exhibited good reactivities, delivering the desired products 3pa in 72% yield and 3qa in 74% yield, respectively (entries 15–16).
|
||||
|---|---|---|---|---|
| Entry | R1 in 1 | 3 | Yieldb (%) | drc |
| a Reactions were carried out with 1 (0.1 mmol), 2a (0.12 mmol), and DBU (20 mol%) in 2 mL of THF at rt for 12–48 h.b Isolated yields.c Determined by 1H NMR. | ||||
| 1 | 2-FC6H4 (1b) | 3ba | 85 | >20:1 |
| 2 | 3-FC6H4 (1c) | 3ca | 90 | >20:1 |
| 3 | 4-FC6H4 (1d) | 3da | 85 | >20:1 |
| 4 | 3-ClC6H4 (1e) | 3ea | 86 | >20:1 |
| 5 | 4-ClC6H4 (1f) | 3fa | 80 | >20:1 |
| 6 | 3-BrC6H4 (1g) | 3ga | 79 | >20:1 |
| 7 | 4-BrC6H4 (1h) | 3ha | 80 | >20:1 |
| 8 | 4-CNC6H4 (1i) | 3ia | 75 | >20:1 |
| 9 | 2-MeC6H4 (1j) | 3ja | 79 | >20:1 |
| 10 | 3-MeC6H4 (1k) | 3ka | 76 | >20:1 |
| 11 | 4-MeC6H4 (1l) | 3la | 73 | >20:1 |
| 12 | 2-OMeC6H4 (1m) | 3ma | 67 | >20:1 |
| 13 | 3-OMeC6H4 (1n) | 3na | 75 | >20:1 |
| 14 | 4-OMeC6H4 (1o) | 3oa | 77 | >20:1 |
| 15 | 2-Naphthyl (1p) | 3pa | 72 | >20:1 |
| 16 | 2-Thienyl (1q) | 3qa | 74 | >20:1 |
Subsequently, we performed the application of cyclic 1-azadiene 1a in DBU-catalyzed [3+2] cycloaddition with a variety of 3-homoacyl coumarins 2 under the optimal conditions (Table 3). And substrates 2 with electron-withdrawing (F, Cl, Br, Table 2, entries 1–5) or electron-donating (Me, MeO, entries 6–9) substituents at 6 or 7 position were all suitable for the cycloaddition, affording the cycloadducts 3aa–3ai in good to excellent yields of 78–94%. Replacing the R1 group with H, the desired product 3aj was obtained in 85% yield (entry 10). Notably, when the R2 were para-substituted phenyl groups, the cycloaddition reactions underwent smoothly to deliver the products 3ak–3am in up to 92% yield, and para-methyl substituted 2m gave a lower yield compared to para-electron-withdrawing substituted 2k and 2l (entries 11–13). The structure of product 3aj was confirmed by its X-ray crystallographic data.12
|
||||
|---|---|---|---|---|
| Entry | R1/R2 | 3 | Yieldb (%) | drc |
| a Reactions were carried out with 1a (0.1 mmol), 2 (0.12 mmol), and DBU (20 mol%) in 2 mL of THF at rt for 12–48 h.b Isolated yields.c Determined by 1H NMR. | ||||
| 1 | 6-Br/C6H5 (2a) | 3aa | 86 | >20:1 |
| 2 | 6-F/C6H5 (2b) | 3ab | 88 | >20:1 |
| 3 | 6-Cl/C6H5 (2c) | 3ac | 78 | >20:1 |
| 4 | 7-Cl/C6H5 (2d) | 3ad | 84 | >20:1 |
| 5 | 7-Br/C6H5 (2e) | 3ae | 86 | >20:1 |
| 6 | 6-Me/C6H5 (2f) | 3af | 87 | >20:1 |
| 7 | 6-OMe/C6H5 (2g) | 3ag | 94 | >20:1 |
| 8 | 7-Me/C6H5 (2h) | 3ah | 86 | >20:1 |
| 9 | 7-OMe/C6H5 (2i) | 3ai | 83 | >20:1 |
| 10 | H/C6H5 (2j) | 3aj | 83 | >20:1 |
| 11 | H/4-FC6H4 (2k) | 3ak | 90 | >20:1 |
| 12 | H/4-BrC6H4 (2l) | 3al | 92 | >20:1 |
| 13 | H/4-MeC6H4 (2m) | 3am | 80 | >20:1 |
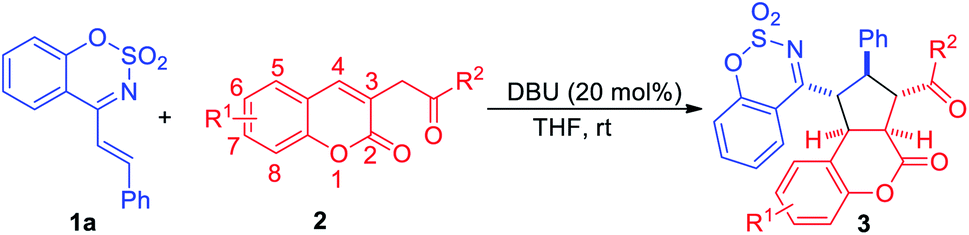
To explore the asymmetric variant of this [3+2] cycloaddition reaction of 1a and 2j, a series of commercially available chiral amines were screened, and unfortunately, this reaction did not proceeded in CH2Cl2 and THF. However, when CH3CN was employed as a solvent, this reaction could be catalysed by a few of chiral amines, giving poor enantioselectivities and low to moderate yields (see ESI Table S1†). As shown in Table 3, cinchona catalyst C1 catalyzed the reaction to afford the 3aj in 46% yield with the highest 27.3% ee, and the reaction could be catalyzed by diimidazole catalyst C16 to give the highest 73% yield but poor 11% ee. The subsequent attempts to find the optimal asymmetric reaction conditions failed (Table 4).
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Yieldb (%) | drc | eed (%) |
| a Reactions were carried out with 1a (0.1 mmol), 2j (0.12 mmol), and 20 mol% catalyst in 2 mL of CH3CN at rt.b Isolated yields.c Determined by 1H NMR.d Determined by HPLC analysis. | |||||
| 1 | C1 | 120 | 46 | >20:1 |
27.3 |
| 2 | C2 | 120 | 52.4 | >20:1 |
9.5 |
| 3 | C3 | 120 | 60 | >20:1 |
8 |
| 4 | C9 | 120 | 31 | >20:1 |
4.5 |
| 5 | C16 | 120 | 73 | >20:1 |
11 |
| 6 | C18 | 120 | 36 | >20:1 |
11 |
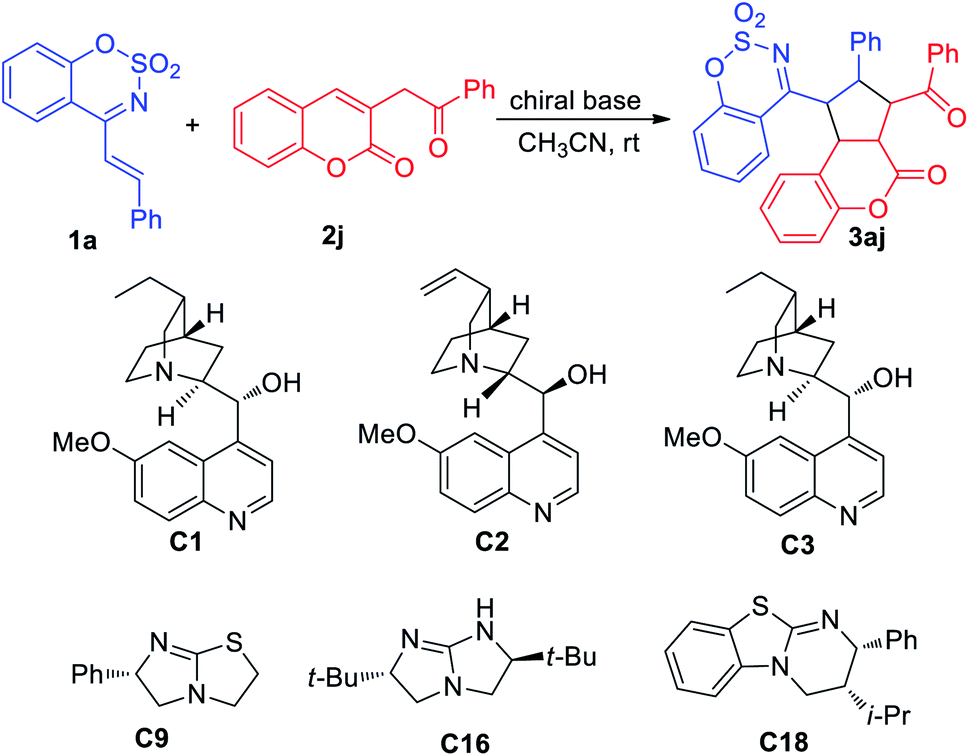
On the basis of the results and previous literature,6 herein we proposed a plausible mechanism for the [3+2] cycloaddition reaction (Scheme 2), which proceeded via stepwise mechanism with zwitterion.13 Firstly, 1a is deprotonated to deliver the dienolate intermediate A under basic conditions. Subsequently, the α-carbanion of A attracks the olefinic bond of 2a to form the anion B. Then through cyclization and protonation, the final [3+2] cycloaddition product 3aa is given.
 | ||
| Scheme 2 Plausible reaction mechanism. | ||
As shown in Scheme 3, the saccharin-derived cyclic 1-azadiene 4 was tested under the optimized reaction conditions. Delightfully, the [3+2] cycloadduct 5 could also be easily prepared in 85% yield and >20:1 dr. To explore the synthetic utility of this cycloaddition, a gram scale reaction was carried out to obtain the desired cycloadduct 3aa without any loss of yield and diastereoselectivity. The lactone of 3aa was opened under basic condition to give the multisubstituted cyclopentane 6 in 79% yield and >20:1 dr (Scheme 3).
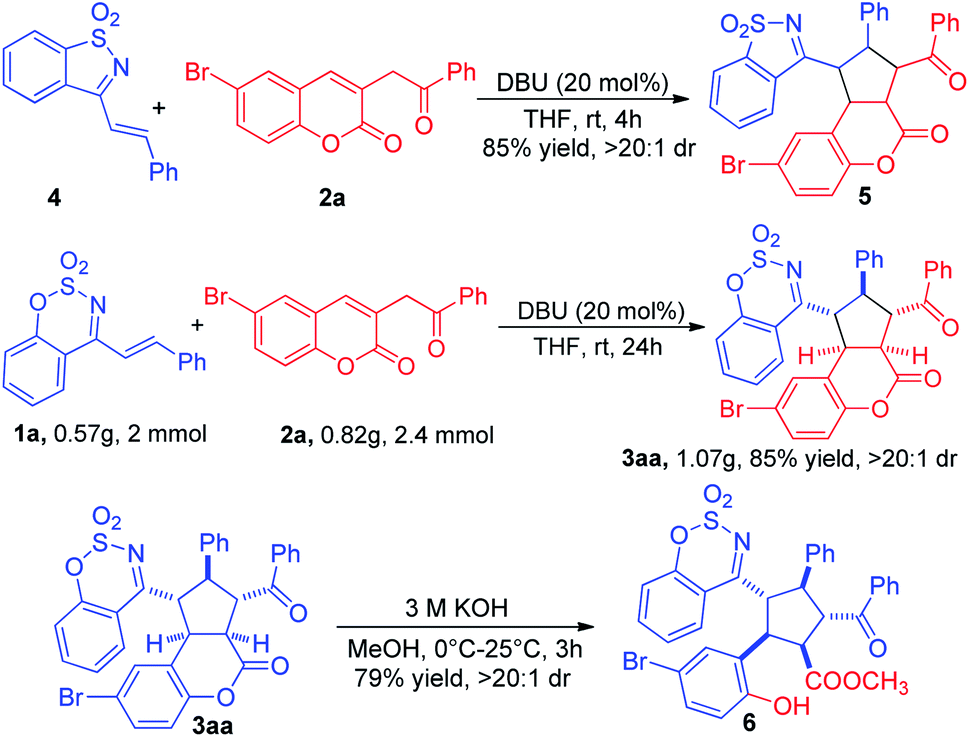 | ||
| Scheme 3 [3+2] cycloaddition of 4 and 2a, gram-scale reaction and further transformation. | ||
Conclusions
In summary, we have successfully developed a DBU catalyzed [3+2] cycloaddition reaction of 3-homoacyl coumarins with cyclic 1-azadienes. The present protocol offers an efficient methodology to synthesize cyclopentane-fused coumarin derivatives with complete chemoselectivity and excellent diastereoselectivity in good yields. Efforts on further investigations of this protocol are underway in our group.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21801180), the Natural Science Foundation of Shandong Province (ZR2018LB010, ZR2019BB054), Doctoral Scientific Research Foundation of Shandong First Medical University, the Innovative Research Programs of Higher Education of Shandong Province (2019KJC009), Academic promotion programme of Shandong First Medical University (2019QL008).Notes and references
- (a) G. S. Clark, Perfum. Flavor., 1995, 20, 23–34 CAS; (b) D. Yu, M. Suzuki, L. Xie, S. L. Morris-Natschke and K.-H. Lee, Med. Res. Rev., 2003, 23, 322–345 CrossRef CAS PubMed; (c) F. Borges, F. Roleira, N. Milhazes, L. Santana and E. Uriarte, Curr. Med. Chem., 2005, 12, 887–916 CrossRef CAS PubMed; (d) A. M. Breul, M. D. Hager and U. S. Schubert, Chem. Soc. Rev., 2013, 42, 5366–5407 RSC; (e) F. G. Medina, J. G. Marrero, M. Macías-Alonso, M. C. González, I. Córdova-Guerrero, A. G. T. García and S. Osegueda-Robles, Nat. Prod. Rep., 2015, 32, 1472–1507 RSC.
- (a) E. B. Melian and K. L. Goa, Drugs, 2002, 62, 107–133 CrossRef CAS PubMed; (b) J. L. Reino, R. Durán-Patrón, I. Segura, R. Hernández-Galán, H. H. Riese and I. G. Collado, J. Nat. Prod., 2003, 66, 344–349 CrossRef CAS PubMed; (c) B. M. Trost and T. M. Lam, J. Am. Chem. Soc., 2012, 134, 11319–11321 CrossRef CAS PubMed; (d) A. Goto, S. Yoshimura, Y. Nakao, M. Inai, T. Asakawa, M. Egi, Y. Hamashima and M. Kondo, Org. Lett., 2017, 19, 3358–3361 CrossRef CAS PubMed; (e) H. P. A. Khan, D. Das and T. K. Chakraborty, J. Org. Chem., 2018, 83, 6086–6092 CrossRef CAS PubMed.
- M. McLean and M. F. Dutton, Pharmacol. Ther., 1995, 65, 163–192 CrossRef CAS PubMed.
- (a) A. Matsuo, S. Yuki and M. Nakayama, J. Chem. Soc., Perkin Trans. 1, 1986, 701–710 RSC; (b) D. Ng, Z. Yang and M. A. Garcia-Garibay, Org. Lett., 2004, 6, 645–647 CrossRef CAS PubMed; (c) A. Srikrishna and B. Vasantha Lakshmi, Tetrahedron Lett., 2005, 46, 4879–4881 CrossRef CAS; (d) P. Kamat, S. G. Tilve, V. P. Kamat and J. K. Kirtany, Org. Prep. Proced. Int., 2015, 47, 1–79 CrossRef; (e) M. Michalak, K. Michalak and J. Wicha, Nat. Prod. Rep., 2017, 34, 361–410 RSC; (f) M. I. L. Soares, C. S. B. Gomes, S. C. C. Nunes, A. A. C. C. Pais and T. M. V. D. P. e. Melo, Eur. J. Org. Chem., 2019, 32, 5441–5451 CrossRef.
- (a) Y. Fukuyama, H. Yuasa, Y. Tonoi, K. Harada, M. Wada, Y. Asakawa and T. Hashimoto, Tetrahedron, 2001, 57, 9299–9307 CrossRef CAS; (b) C. E. Henry and O. Kwon, Org. Lett., 2007, 9, 3069–3072 CrossRef CAS PubMed; (c) A. Marinetti, M. Neel, J. Gouin and A. Voituriez, Synthesis, 2011, 12, 2003–2009 CrossRef; (d) A. Bhunia, A. Patra, V. G. Puranik and A. T. Biju, Org. Lett., 2013, 15, 1756–1759 CrossRef CAS PubMed; (e) Q. Liu, X.-Y. Chen, R. Puttreddy, K. Rissanen and D. Enders, Angew. Chem., Int. Ed., 2018, 57, 17100–17103 CrossRef CAS PubMed.
- (a) Y.-R. Chen, M. R. Ganapuram, K.-H. Hsieh, K.-H. Chen, P. Karanam, S. S. Vagh, Y.-C. Liou and W. Lin, Chem. Commun., 2018, 54, 12702–12705 RSC; (b) S. S. Vagh, P. Karanam, C.-C. Liao, T.-H. Lin, Y.-C. Liou, A. Edukondalu, Y.-R. Chen and W. Lin, Adv. Synth. Catal., 2020, 362, 1679–1685 CrossRef CAS; (c) M. Wang, P. Y. Tseng, W. J. Chi, S. Suresh, A. Edukondalu, Y. R. Chen and W. Lin, Adv. Synth. Catal., 2020, 362, 3407–3415 CrossRef CAS.
- (a) C. Ma, J. Gu, B. Teng, Q. Zhou, R. Li and Y.-C. Chen, Org. Lett., 2013, 15, 6206–6209 CrossRef CAS PubMed; (b) K.-K. Wang, T. Jin, X. Huang, Q. Ouyang, W. Du and Y.-C. Chen, Org. Lett., 2016, 18, 872–875 CrossRef CAS PubMed; (c) Y. Wu, Y. Liu, W. Yang, H. Liu, L. Zhou, Z. Sun and H. Guo, Adv. Synth. Catal., 2016, 358, 3517–3521 CrossRef CAS; (d) L. Yu, Y. Cheng, F. Qi, R. Li and P. Li, Org. Chem. Front., 2017, 4, 1336–1340 RSC; (e) Y.-H. Chen, D.-H. Li and Y.-K. Liu, ACS Omega, 2018, 3, 16615–16625 CrossRef CAS PubMed; (f) Y. Lin, Q. Wang, Y. Wu, C. Wang, H. Jia, C. Zhang, J. Huang and H. Guo, RSC Adv., 2018, 8, 40798–40803 RSC; (g) Q. Zhou, B. Chen, X. Huang, Y. Zeng, W. Chu, L. He and Q. Liu, Org. Chem. Front., 2019, 6, 1891–1894 RSC.
- (a) X. Yin, Y. Zheng, X. Feng, K. Jiang, X.-Z. Wei, N. Gao and Y.-C. Chen, Angew. Chem., Int. Ed., 2014, 53, 6245–6248 CrossRef CAS PubMed; (b) X. Chen, J.-Q. Zhang, S.-J. Yin, H.-Y. Li, W.-Q. Zhou and X.-W. Wang, Org. Lett., 2015, 17, 4188–4191 CrossRef CAS PubMed; (c) X.-L. He, Y.-C. Xiao, W. Du and Y.-C. Chen, Chem.–Eur. J., 2015, 21, 3443–3448 CrossRef CAS PubMed.
- (a) X. Feng, Z. Zhou, C. Ma, X. Yin, R. Li, L. Dong and Y.-C. Chen, Angew. Chem., Int. Ed., 2013, 52, 14173–14176 CrossRef CAS PubMed; (b) J. Gu, C. Ma, Q.-Z. Li, W. Du and Y.-C. Chen, Org. Lett., 2014, 16, 3986–3989 CrossRef CAS PubMed; (c) Q. An, J. Shen, N. Butt, D. Liu, Y. Liu and W. Zhang, Adv. Synth. Catal., 2015, 357, 3627–3638 CrossRef CAS; (d) J. Izquierdo and M. A. Pericàs, ACS Catal., 2016, 6, 348–356 CrossRef CAS; (e) Z.-Q. Liang, D.-L. Wang, C.-L. Zhang and S. Ye, Org. Biomol. Chem., 2016, 14, 6422–6425 RSC; (f) Z. Wang, H. Xu, Q. Su, P. Hu, P.-L. Shao, Y. He and Y. Lu, Org. Lett., 2017, 19, 3111–3114 CrossRef CAS PubMed; (g) Z. Zhou, Z.-X. Wang, Q. Ouyang, W. Xiao, W. Du and Y.-C. Chen, Chem.–Eur. J., 2017, 23, 2945–2949 CrossRef CAS PubMed; (h) X. Ren, J. Lin, X. Hu and P. Xu, Org. Chem. Front., 2019, 6, 2280–2283 RSC.
- (a) C. Yuan, H. Liu, Z. Gao, L. Zhou, Y. Feng, Y. Xiao and H. Guo, Org. Lett., 2015, 17, 26–29 CrossRef CAS PubMed; (b) C. Yuan, L. Zhou, Z. Sun and H. Guo, RSC Adv., 2016, 6, 77931–77936 RSC; (c) C. Yuan, L. Zhou, M. Xia, Z. Sun, D. Wang and H. Guo, Org. Lett., 2016, 18, 5644–5647 CrossRef CAS PubMed; (d) C. Yuan, Y. Wu, D. Wang, Z. Zhang, C. Wang, L. Zhou, C. Zhang, B. Song and H. Guo, Adv. Synth. Catal., 2018, 360, 652–658 CrossRef CAS.
- J. Yan, X. Li, Y. Chen, Y. Li, W. Chen, R. Zhan and H. Huang, J. Org. Chem., 2020, 85, 12175–12186 CrossRef PubMed.
- Crystallographic data for 3aj have been deposited with the Cambridge Crystallographic Data Centre as deposition number CCDC 2073841.†.
- R. Jasiński and E. Dresler, Organics, 2020, 1, 49–69 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental conditions and spectroscopic data of all new compounds. CCDC 2073841. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra03387e |
| This journal is © The Royal Society of Chemistry 2021 |