
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxidative cross-dehydrogenative coupling (CDC) via C(sp2)–H bond functionalization: tert-butyl peroxybenzoate (TBPB)-promoted regioselective direct C-3 acylation/benzoylation of 2H-indazoles with aldehydes/benzyl alcohols/styrenes†
Richa Sharma,
Lalit Yadav,
Ravi Kant Yadav and
Sandeep Chaudhary *
*
Laboratory of Organic and Medicinal Chemistry (OMC Lab), Department of Chemistry, Malaviya National Institute of Technology, Jawaharlal Nehru Marg, Jaipur-302017, India. E-mail: schaudhary.chy@mnit.ac.in; Fax: +911412529029; Tel: +911412713319
First published on 15th April 2021
Abstract
An efficient, cost-effective, transition-metal-free, oxidative C(sp2)–H/C(sp2)–H cross-dehydrogenative coupling via a C(sp2)–H bond functionalization protocol for the regioselective direct C-3 acylation/benzoylation of substituted 2H-Indazoles 1a–m with substituted aldehydes 2a–q/benzyl alcohols 5a–e/styrenes 6a–e is reported. The operationally simple protocol proceeds in the presence of tert-butyl peroxybenzoate (TBPB) as an oxidant in chlorobenzene (PhCl) as a solvent at 110 °C for 24 h under an inert atmosphere, which furnished a diverse variety of substituted 3-(acyl/benzoyl)-2H-indazoles 3a–q/4a–l in up to 87% yields. The reaction involves a free-radical mechanism and proceeds via the addition of an in situ generated acyl radical (from aldehydes/benzyl alcohols/styrenes) on 2H-indazoles. The functional group tolerance, broad substrate scope, control/competitive experiments and gram-scale synthesis and its application to the synthesis of anti-inflammatory agent 11 and novel indazole-fused diazepine 13 further signify the versatile nature of the developed methodology.
Introduction
During the last two decades, oxidative cross-dehydrogenative coupling, often abbreviated to “oxidative CDC reaction”, between two C–H bonds has been recognized as a greener fundamental synthetic approach in C–C bond forming reactions due to its association with several advantages such as being metal-free, high yields, cost effectiveness, operational simplicity, high selectivity, step-/atom-economy, product selectivity, no pre-functionalization of starting materials, reduced waste, energy and resource competence, etc.1 Indazoles are renowned bioactive heterocyclic scaffolds which are found abundantly in several pharmaceutically privileged bioactive natural products/therapeutics/drugs molecules2 and are bestowed with a wide array of biological activities such as antimicrobial, anticancer, antitumor, HIV-protease inhibition, antiplatelet, anti-depressant, and anti-inflammatory actions.3Indazole functionalization has achieved emerging demand in the fields of organic and medicinal chemistry as the functionalization of indazoles can be rendered into advantageous structural motifs for various medications.4 Therefore, the development of a new synthetic pathway to introduce various functional groups on 2H-indazoles leading to an increase in the molecular abundance and the formation of new bioactive molecules, will always be of the utmost importance to medicinal chemistry and drug discovery.
In particular, acylated 2H-indazoles have received tremendous attention as a pharmaceutically important class of structural motifs in a large number of bioactive skeletons/therapeutic molecules. Noticeably, C3-acylated-(2H)-indazoles I–V are endowed with several biological activities, such as antiemetic and anti-inflammatory properties (Fig. 1).5 Therefore, the development of a direct synthetic strategy via an oxidative cross-dehydrogenative coupling pathway for the C-3 acylation of 2H-indazoles is a highly desirable and challenging area of investigation. It becomes more appealing if it proceeds through a transition-metal-free approach. A few transition-metal-catalyzed (Ag, Ni) synthetic approaches via C(sp2)–H bond activation/functionalization have been reported for the direct, regioselective, C-3 acylation of (2H)-indazoles with either Ag-catalyzed decarboxylative cross-coupling of α-keto acids6 or with an Ni-catalyzed reaction on substituted aldehydes7 and with an Ag-catalyzed reaction with substituted Hantzsch esters as acyl radical sources8 (Scheme 1). However, these strategies are associated with several drawbacks such as the use of costly metals, the use of additives/ligands, and limited substrate scope. In addition, the separate synthesis of an acylation/benzoylation source further amplifies its limitations and increases the tediousness of these methodologies. However, there has been no report of the direct C-3 acylation of 2-aryl-2H-indazoles with metal-free and ligand-/additive-free catalysis.
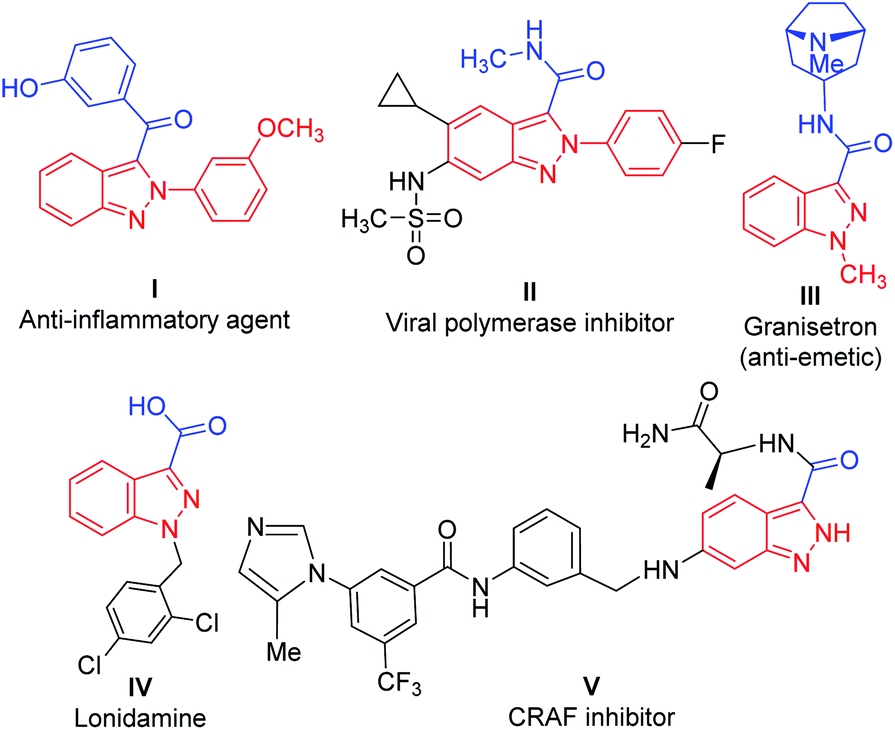 | ||
| Fig. 1 Representative examples of various bioactive molecules/drugs/therapeutics with a 2H-indazole moiety. | ||
 | ||
| Scheme 1 Previous methodologies and present work: TBPB-promoted approach to regioselective direct C-3 acylation/benzoylation of 2-aryl-2H-indazoles with substituted aldehydes/benzyl alcohols/styrenes. | ||
Herein, we report a cost-effective, highly efficient, tert-butyl peroxybenzoate (TBPB)-promoted, regioselective, direct C-3 acylation/benzoylation of 2-aryl-2H-indazoles 1a–m with different aldehydes 2a–q/benzyl alcohols 5a–e/styrenes 6a–e via C(sp2)–H/C(sp2)–H cross-dehydrogenative coupling at 110 °C for 24 h under N2 atmosphere which furnished 3-(acylated/benzoylated)-2-aryl-2H–indazoles 3a–q/4a–l in excellent (87%) yields, with a broad range of functional group tolerances and varied substrate compatibilities [Scheme 1]. Succinctly, this is the first detailed investigation of an oxidant-promoted C(sp2)–H/C(sp2)–H cross-dehydrogenative coupling method for the regioselective direct C-3 acylation/benzoylation of 2-aryl-2H-indazoles.
Results and discussion
We initiated our optimization study by investigating direct C(sp2)–C(sp2) coupling by taking 2-(4-methoxyphenyl)-2H-indazole 1a and 4-methylbenzaldehyde 2a as the starting substrate (Table 1).
|
|||||
|---|---|---|---|---|---|
| S. no. | Oxidant (2.5 equiv.) | Catalyst (20 mol%) | Solvent | Temp. (°C) | Yield (%) |
| a Reaction conditions: 2-(4-methoxyphenyl)-2H-indazole 1a (0.13 mmol), 4-methyl benzaldehyde 2a (0.27 mmol), oxidant (0.33 mmol), catalyst (20 mol%), N2 atm, 110 °C, 24 h.b For details, see ref. 9f.c TBHP (0.53 mmol) and NCS (30 mol%) were used.d TFA (0.6 equiv.) was used.e 2a (1 equiv.) was used.f Reaction was allowed to run for 16 h. | |||||
| 1 | TBHPb | NCS | DCE | 110 | NRc |
| 2 | TBHP | NCS | DCE | 110 | 30 |
| 3 | TBHP | TFA | DCE | 110 | 40d |
| 4 | TBHP | TBAI | DCE | 110 | NR |
| 5 | TBHP | NaI | — | 110 | NR |
| 6 | TBHP | KI | DCE | 110 | NR |
| 7 | TBHP | I2 | DCE | 110 | NR |
| 8 | TBHP | — | DCE | 110 | 65 |
| 9 | TBHP | — | DCE | 110 | 10e |
| 10 | TBHP | — | PhCl | 110 | 68 |
| 11 | DTBP | — | PhCl | 110 | 70 |
| 12 | TBPB | — | PhCl | 110 | 82 |
| 13 | DCP | — | PhCl | 110 | 51 |
| 14 | Lauroyl peroxide | — | PhCl | 110 | 18 |
| 15 | H2O2 | — | PhCl | 110 | NR |
| 16 | Cumene hydroperoxide | — | PhCl | 110 | Trace |
| 17 | K2S2O8 | — | PhCl | 110 | NR |
| 18 | Oxone | — | PhCl | 110 | NR |
| 19 | (NH4)2S2O8 | — | PhCl | 110 | NR |
| 20 | PIDA | — | PhCl | 110 | Trace |
| 21 | O2 | — | PhCl | 110 | NR |
| 22 | TBPB | — | ACN | 110 | 71 |
| 23 | TBPB | — | Toluene | 110 | 70 |
| 24 | TBPB | — | DMSO | 110 | 18 |
| 25 | TBPB | — | Dioxane | 110 | 15 |
| 26 | TBPB | — | DMF | 110 | Trace |
| 27 | TBPB | — | THF | 110 | Trace |
| 28 | TBPB | — | H2O | 110 | Trace |
| 29 | TBPB | — | AcOH | 110 | Trace |
| 30 | TBHP | — | TFA | 110 | NR |
| 31 | TBPB | — | PhCl | 80 | 72 |
| 32 | TBPB | — | PhCl | 140 | 68 |
| 33 | TBPB | — | PhCl | 110 | 71f |

It has been noted that TBHP, either alone or in combination, has been utilized in several oxidative cross-dehydrogenative coupling reactions;9 we had chosen tert-butyl hydroperoxide (TBHP) as an oxidant and N-chlorosuccinimide (NCS) as a catalyst for the beginning of our study. Therefore, using the procedure in the literature,9f we carried out a reaction of 1a with 2a in the presence of TBHP (0.53 mmol, 4 equiv.) and NCS (30 mol%) in dichloroethane (DCE) at 110 °C for 24 h under an inert atmosphere. However, the desired product 3a was not found at all and several spots appeared on TLC (Table 1, entry 1). Then, keeping all other reaction parameters the same, we carried out the same reaction as shown in entry no. 1 with reduced equivalents of both TBHP and NCS catalyst. Intriguingly, 3a was obtained, albeit in low (30%) yield (Table 1, entry 2). Subsequently, we examined the screening of some other well-known reagents which had previously been utilized extensively in CDC reactions (Table 1, entries 3–7). While the reaction performed in TFA furnished 3a in 40% yield, iodine-based catalysts were found ineffective in improving the yield of the reaction. Furthermore, keeping all the reaction conditions the same, the reaction was performed without a catalyst, which furnished 3a in 65% yield (Table 1, entry 8). This observation instructed us to stop further use of any additives as a catalyst. However, the reaction performed with less equivalents of 2a (1 equiv.) drastically reduced the yield of 3a (Table 1, entry 9). It has been reported in the literature that chlorobenzene has been utilized as an effective solvent for cross-dehydrogenative coupling reactions.10 Therefore, we conducted the same reaction in chlorobenzene instead of DCE and 3a was obtained in 68% yield (Table 1, entry 10).
Then, we carried out the screening of several organic as well as inorganic oxidants, such as di-tert-butyl peroxide (DTBP), tert-butyl peroxybenzoate (TBPB), dicumyl peroxide (DCP), lauroyl peroxide, H2O2, cumene hydroperoxide, K2S2O8, oxone, (NH4)2S2O8, (diacetoxyiodo)benzene (PIDA), and oxygen gas, while keeping all the other reaction parameters the same (Table 1, entries 11–21). It was found that while DTBP, TBPB and DCP furnished 3a in 70%, 82% and 51% yields, respectively; the reaction performed using lauroyl peroxide gave 3a in only 18% yield (Table 1, entries 11–14). The rest of the oxidants either did not furnish 3a at all or afforded 3a in only trace amounts (Table 1, entries 15–21).
Sequentially, the screening of several polar/non-polar solvents was also carried out (Table 1, entries 22–30). It should be noted that none of the solvents were found to be effective except for chlorobenzene. Afterwards, keeping all reaction parameters the same, the effect of variation in temperature and time was studied. It was observed that increasing or decreasing the temperature and time did not have a beneficial effect on the yield of the reaction (Table 1, entries 31–33). Thus, overall, 2 equivalents of substituted aldehydes, 2.5 equivalents of tert-butyl peroxybenzoate (TBPB) dissolved in chlorobenzene as solvent at 110 °C for 24 h under N2 atmosphere were found to be the best optimization reaction conditions for the direct C-3 acylation/benzoylation of 2-aryl-2H-indazoles via a C(sp2)–H/C(sp2)–H cross-dehydrogenative coupling methodology.
Taking 2-(4-methoxyphenyl)-2H-indazole 1a as a starting substrate, several substituted aromatic/aliphatic/hetero-aromatic aldehydes 2a–q were reacted under the optimized reaction conditions, which furnished substituted 3-(acylated/benzoylated)-2-(4-methoxyphenyl)-2H-indazoles 3a–q in 57–86% yield (Scheme 2). Like 3a, 1a was reacted with benzaldehyde 2b under the optimized reaction conditions and furnished 3b in 80% yield. It has been noted that aromatic aldehydes with electron-donating groups (EDGs) at the p-position were found to be well-tolerated and afforded the corresponding C-3 benzoylated-2H-indazoles 3a and 3c in 82% and 86% yields, respectively. However, aromatic aldehyde 2d with two EDG (i.e., OCH3) groups was subjected to reaction with 1a under the optimized conditions; 3d was afforded in slightly lower (78%) yield compared to 3a and 3c. This could be due to the steric hindrance created by the OCH3 group at the o-position to the aldehydic functionality. In the case of aromatic aldehydes containing a halo (X = F, Cl, Br) group at the para-position, the reaction of 1a with 2e–g under optimized conditions furnished 3e–g in 81–84% yield. However, keeping all the reaction conditions the same, a decrease in the reactivity of aromatic aldehydes containing halo groups at the meta-/ortho-positions, was observed and 3h and 3i were obtained in 67% and 58% yields, respectively. In addition, the aromatic aldehyde containing an electron-withdrawing group (EWG) 2p was found to be totally reluctant to undergo the optimized reaction conditions. Similarly, phenyl acetaldehyde 2q was also found to be unreactive. Furthermore, in order to check the versatility of the methodology, a few aliphatic aldehydes 2j–k were reacted with 1a under the optimized reaction conditions and afforded 3j and 3k in 68% and 61% yields, respectively. Comparing aromatic aldehydes with aliphatic aldehydes, the latter were found to be less reactive than the former. A different observation was noticed in the current protocol when the number of carbon atoms increased to four (unbranched and/or branched) in the aldehydes.7 Compounds 2l–m on reaction with 1a under the optimized reaction conditions did not furnish C-3 acylated 2H-indazoles but incorporated the corresponding alkyl group of the 2l and 2m via decarbonylation and furnished 3l and 3m in 60% and 71% yields, respectively. We could speculate on the stability of the corresponding generated free-radicals on treatment with TBPB based on the greater +I effect of the propyl group (generated after decarbonylation of n-butyraldehyde) compared to the +I effect of the ethyl group (generated after decarbonylation of n-propionaldehyde). This could lead to the formation of unprecedented 3l from 2l in 60% yield. Furthermore, the instability of the acyl radical formed from 2m on treatment with TBPB can be understood by the formation of a more stable secondary free-radical of isobutyraldehyde (generated after decarbonylation of n-isobutyraldehyde).6,7 Later on, the generated alkyl free-radical attacks at the C-3 position of 2-aryl-2H-indazole, subsequently leading to the formation of C-3-alkylated-2-aryl-2H-indazoles.6,7 Our protocol was also found to be feasible with hetero-aromatic aldehydes. Significantly, good yields were observed when pyrrole-2-carboxaldehyde 2n, furan-2-carboxaldehyde 2o, 5-methyl-furan-2-carboxaldehyde 2p and 5-bromothiophene-2-carboxaldehyde 2q were reacted under the optimized reaction conditions to furnish 3n–q.
 | ||
| Scheme 2 Substrate scope: reaction of 2-(4-methoxyphenyl)-2H-indazole 1a with various aldehydes 2a–q for the synthesis of substituted 3-acylated 2-(4-methoxyphenyl)-2H-indazoles 3a–qa,b. aReaction conditions: 2-(4-methoxyphenyl)-2H-indazole 1a (0.5 mmol), substituted aldehydes 2a–q (1.0 mmol), oxidant (1.25 mmol), PhCl, N2 atm, 110 °C, 24 h. bIsolated yield. | ||
Overall, the reactivity order for different types of aldehydes has been depicted as:
| Aromatic > aliphatic > hetero-aromatic |
On the other hand, electron-donating groups (EDGs) containing aldehydes were found to be more favorable to the optimized reaction conditions compared to electron-withdrawing groups (EWGs) containing aldehydes. The synthetic utility was also demonstrated by performing a gram-scale synthesis of 3a by the reaction of 1a with 2a under our optimized reaction conditions, which furnished 3a in 65% isolated yield (Scheme 2).
Sequentially, a diverse variety of substituted 2H-indazoles 1b–m were reacted with substituted benzaldehyde 2a–b under the optimized reaction conditions, which furnished the 3-benzyolated-2H-indazoles 4a–l in 54–80% yield (Scheme 3). 2H-Indazoles with EDGs, i.e., 1b (R1 = –OCH2O–), 1d (R1 = –OCH3), 1e (R1 = CH3) and 1g (R1 = –OCH2O–), were reacted with 2a under the optimized reaction conditions, which furnished 4a, 4c, 4d and 4f in 55%, 65%, 68% and 58% moderate yields, respectively. However, 2H-indazoles with a halo group (R1 = F, Cl) 1c and 1f, when subjected to CDC reaction with 2a under the optimized reaction conditions afforded 4b and 4e in 71% and 78% yields, respectively. 2H-Indazoles 1g–i (R2 = p-OCH3, CH3, Cl) and 1m (R2 = H) on coupling with benzaldehyde 2a and 2b afforded C-3 benzoylated product 4f–h in 58–85% yields and 4l in 80% yield, respectively. Similarly, heteroaryl 2H-indazoles 1j–l were reacted with 4-methylbenzaldehyde 2a, which furnished 3-benzyolated-(heteroaryl)-2H-indazoles 4i–k in 72%, 71% and 82% yields, respectively. The substrates, 2-cyclohexyl-2H-indazole 1n and 1H-indazole 1o were found to be unreactive under the optimized reaction conditions. This clearly illustrates that the 2-aryl substitution in 2H-indazole plays a dynamic role in stabilizing the intermediate for the coupling of aldehydes. This transition-metal-free, regioselective, direct C-3 benzoylation of 2-aryl-2H-indazoles via a C(sp2)–H/C(sp2)–H cross-dehydrogenative coupling also works effectively with several substituted benzyl alcohols 5a–e and styrenes 6a–e on reaction with 1a, which furnished C-3-benzoylated-2-aryl-2H-indazole products 3a–c and 3e–f in good yields (Schemes 4 and 5).
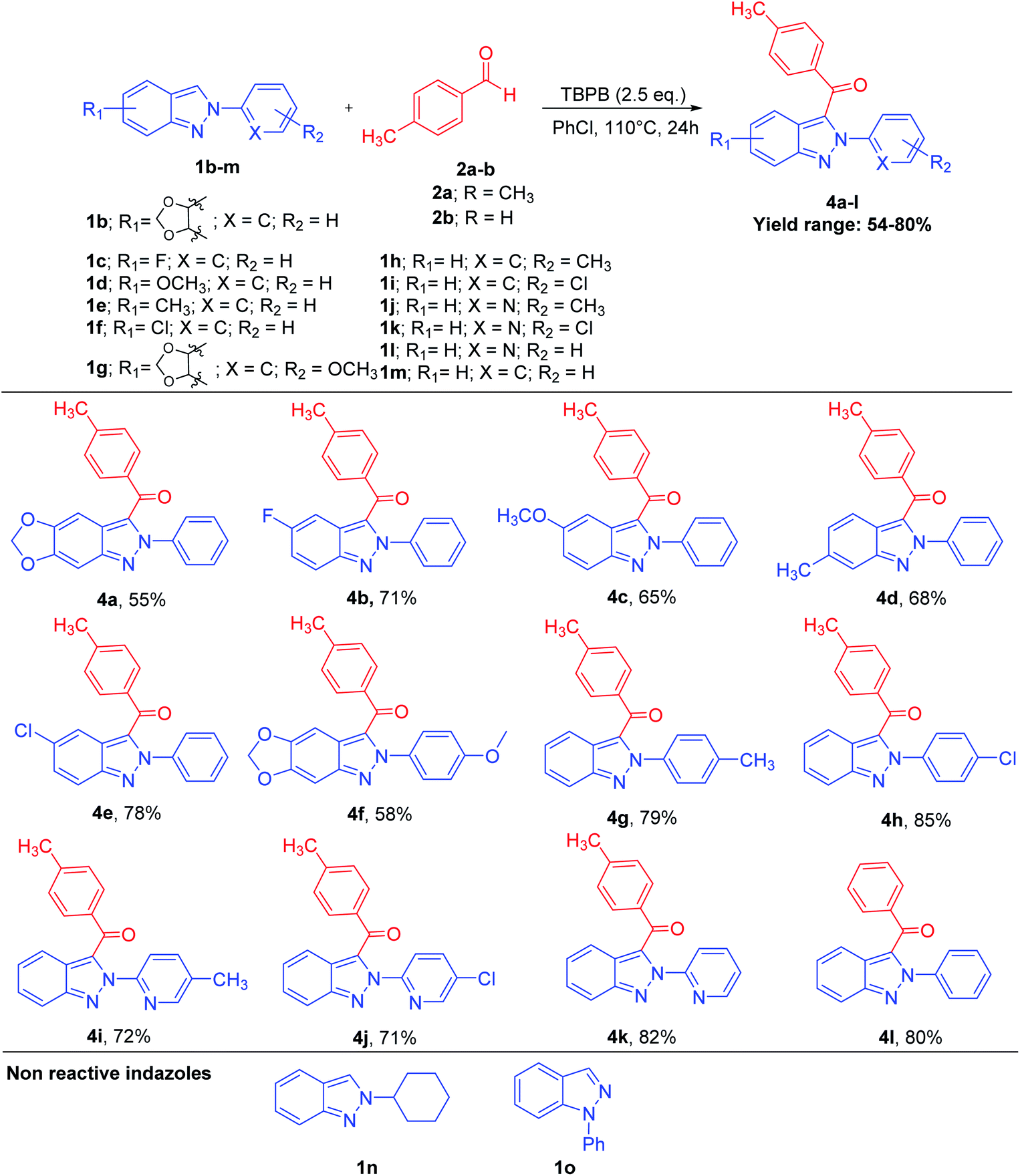 | ||
| Scheme 3 Substrate scope: reaction of different substituted 2H-indazoles 1b–m with benzaldehyde 2a–b for the synthesis of 4a–la,b. aReaction conditions: substituted 2H-indazoles 1b–m (0.5 mmol), substituted aldehydes 2a–b (1.0 mmol), oxidant (1.25 mmol), PhCl, N2 atm, 110 °C, 24 h. bIsolated yield. | ||
 | ||
| Scheme 4 Reaction of oxidative coupling of different substituted benzyl alcohol 5a–e with 2H-indazole 1aa,b. aReaction conditions: 2-(4-methoxyphenyl)-2H-indazole 1a (0.5 mmol), substituted benzyl alcohols 5a–e (1.0 mmol), TBPB (1.25 mmol), PhCl, N2 atm, 110 °C, 24 h. bIsolated yield. | ||
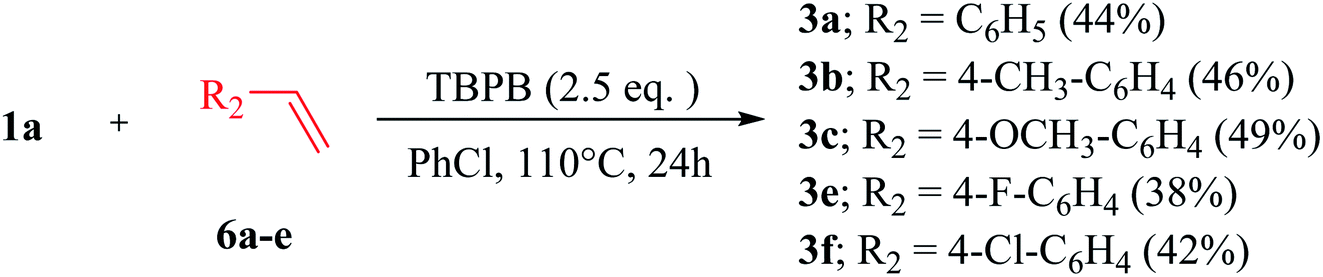 | ||
| Scheme 5 Benzoylation reaction of different substituted styrenes 6a–e with 2H-indazole 1aa,b. aReaction conditions: 2-(4-methoxyphenyl)-2H-indazole 1a (0.5 mmol), substituted styrenes 6a–e (1.0 mmol), TBPB (1.25 mmol), PhCl, N2 atm, 110 °C, 24 h. bIsolated yield. | ||
In order to understand the mechanism of this unique protocol, control experiments were conducted [Scheme 6, eqn (i)–(iv)]. It has already been exemplified in the literature that tert-butyl peroxybenzoate (TBPB) acts as a radical initiator.11 Keeping all the reaction conditions the same, 1a was reacted with 2a under the optimized reaction conditions in the presence of TEMPO (1 equiv.), which furnished 3a in 51% yield [Scheme 6, eqn (i)]. However, 3a was not formed and the reaction was completely aborted when TEMPO (2.5 equiv.) was added under the standard reaction conditions. We were successful in isolating the TEMPO-trapped acyl adduct 7 in 71% yield [Scheme 6, eqn (ii)]. The isolation of adduct 7 confirmed that the synthetic pathway towards the regioselective synthesis of 3-(acyl/benzoyl)-2-aryl-2H-indazoles proceed via the free-radical pathway. This underlines the importance of the oxidant in this free-radical-catalyzed reaction. In order to interpret the reactivity order among EDGs and EWGs while keeping all the reaction conditions the same, a competitive experiment was executed in which substrate 1a was reacted with an equimolar amount of 2c and 2e, which afforded the C-3 benzoylated products 3c and 3e in 56% and 30% yields, respectively [Scheme 6, eqn (iii)]. Therefore, it has been concluded that 2c with an EDG showed higher reactivity than 2e with an EWG. Another competitive experiment was carried out to analyse the reactivity order of this CDC reaction between aliphatic and aromatic aldehydes. Thus, substrate 1a was reacted with an equimolar amount of 2a and 2j under optimized reaction conditions, which furnished 3-(4-methylbenzyolated)-2-aryl-2H-indazole 3a and 3-(acetyl)-2-aryl-2H-indazole 3j in 55% and 29% yields, respectively [Scheme 6, eqn (iv)]. This reaction demonstrated that the aromatic aldehydes were found to be more reactive than aliphatic aldehydes.
 | ||
| Scheme 6 Control experiments. | ||
A plausible free-radical mechanism for this regioselective benzoylation method has been depicted in Fig. 2. Initially, thermal cleavage of TBPB generates a tBuO free-radical and a carboxyl (PhCOO) free-radical. Then, the tBuO free-radical through hydrogen radical abstraction (HRA) from 4-methylbenzaldehyde 2a generates a benzoyl (acyl) free-radical 8 [note: the radical species 8 was trapped with TEMPO to give adduct 7]. The free-radical 8 can also be derived from benzyl alcohol 5a as well as styrene 6a.10a,b This benzoyl free-radical species 8 was regioselectively added to the C-3 position of 1a. Subsequently, the carboxyl free-radical abstracted the hydrogen radical from the intermediate radical species 9 to afford the desired product 3a.
 | ||
| Fig. 2 Plausible mechanism. | ||
To illustrate the synthetic application of the developed CDC methodology, using our optimized reaction conditions, we synthesized the indazole-based anti-inflammatory agent 11 (Scheme 7). The substrate 1p on reaction with m-acetoxybenzaldehyde 2r under the optimized reaction conditions furnished 10, which on deacetylation furnished anti-inflammatory agent 11 in 51% yield. It has been noted that compound 11 was earlier synthesized in 7 steps;12 in contrast, we were successful in synthesizing 11 in 51% in two steps. Progressively, the (2-bromophenyl)(2-(4-methoxyphenyl)-2H-indazol-3-yl)methanone 12 was synthesized by the reaction of 1a with o-bromobenzaldehyde using our optimized reaction conditions which on further subjection to Pd-catalyzed biaryl coupling leads to the formation of a novel class of heterocycles, i.e., 3-methoxy-9H-dibenzo[4,5:6,7]azepino[1,2-b]indazol-9-one 13 in 75% yield, which can be utilized for medicinal chemistry applications.13 In addition, the synthesized benzoylated product 3a on subjection to NaBH4 reduction in methanol furnished the reduced hydroxylated product, i.e., (2-(4-methoxyphenyl)-2H-indazol-3-yl)(p-tolyl)methanol 14, in 90% yield, which can be utilized further for derivatization/functionalization and other organic transformations.
 | ||
| Scheme 7 Practical synthetic applications. | ||
Conclusions
In summary, we have developed an efficient, cost-effective, transition-metal-free, regioselective, TBPB-promoted, oxidative C(sp2)–H/C(sp2)–H cross-dehydrogenative coupling protocol for the direct C-3 acylation/benzoylation of 2-substituted-2H-indazoles 3a–q/4a–l via the reaction of various 2-substituted-2H-indazoles 1a–m with different substituted aldehydes 2a–q/benzyl alcohols 5a–e/styrenes 6a–e in up to 87% yields. The operationally simple, oxidant-promoted protocol exhibits a variety of functional group tolerances and wide substrate compatibilities. The reaction involves a free-radical mechanism and proceeds via the addition of an in situ generated acyl radical (from aldehydes/benzyl alcohols/styrenes) on 2H-indazoles. The gram-scale synthesis, and facile synthesis of anti-inflammatory agent 11, 3-methoxy-9H-dibenzo[4,5:6,7]azepino[1,2-b]indazol-9-one 13 and hydroxylated product 14 further highlights the versatile nature of the developed methodology. We believe that our acylation/benzoylation regioselective CDC protocol will accomplish several kinds of utilization in constructing a ubiquitous class of bioactive azaheterocycles.Experimental
General
Oven-dried laboratory glassware was used for carrying out all the synthetic procedures. Melting points were taken in open capillaries on Sisco melting point apparatus and are presented uncorrected. 1H NMR and 13C NMR spectra were recorded on a JEOL ECS-400 spectrometer (2-channel console with a flexible broadband RF performance), which was operating at 400 MHz for 1H and 100 MHz for 13C NMR and utilized CDCl3 and DMSO-d6 as solvents for sample preparation. Tetramethylsilane (δ 0.00 ppm) and CDCl3 both served as internal standards in 1H NMR (δ 7.246 ppm) and 13C (δ 77.0 ppm) NMR. Patterns of chemical shifts are reported in parts per million. Peak splitting patterns are described as singlet (s), broad singlet (brs), doublet (d), double doublet (dd), triplet (t), and multiplet (m). Coupling constants (J) are reported in Hertz (Hz). High-Resolution Electron Impact Mass Spectra (HR-EIMS) were obtained on Xevo G2-S Q-Tof (Waters, USA) compatible with ACQUITY UPLC® and nano ACQUITY UPLC® systems. Column chromatography was performed over normal (particle size: 60–120 mesh, 100–200 mesh) and flash (particle size: 230–400 mesh) silica gel, which was procured from Qualigens™ (India), Rankem (India), and Spectrochem (India). TLC plates coated with silica gel (Kiesel 60-F254, Merck (India)) were used for monitoring the progress of the reactions. The visualizing agents used for TLC was UV light. A BUCHI Rotavapor R-210 was used for all drying and concentration procedures. All the analytical grade supplied solvents such as MeOH, EtOH were used without further purification. All the AR grade chemicals and reagents obtained from Sigma Aldrich (USA), Merck (India), TCI (India) and/or Spectrochem (India) etc. were used without further purification.Synthesis of starting material 2H-indazoles 1a–o (Scheme 8)14
A solution of 2-bromobenzaldehyde or substituted-2-bromobenzaldehyde (1.5 mmol) and aromatic/hetero-aromatic/aliphatic/cyclic amine (2.4 mmol) in methanol (20 mL) was refluxed for 6–7 h at 80 °C. After the completion of the reaction, the solvent was evaporated under reduced pressure to get the corresponding imine product, which was further used in the next step without prior purification.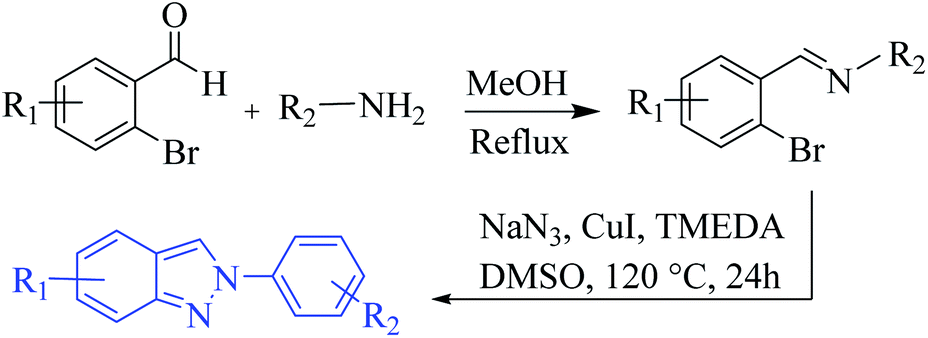 | ||
| Scheme 8 General procedure for the synthesis of 2H-indazole 1a–o. | ||
The imine product was dissolved in anhydrous DMSO (10.0 mL) and CuI (38 mg, 0.20 mmol), NaN3 (261 mg, 4.0 mmol) and TMEDA (22 mg, 0.20 mmol) were added. The reaction mixture was heated at 120 °C for 12 h. After cooling the reaction mixture, it was poured into chloroform (70.0 mL) and sequentially washed with water (3 × 30 mL) and brine (3 × 30 mL), then dried over anhyd. Na2SO4. Then, after evaporation of the solvent under reduced pressure, the crude product was further purified by column chromatography (hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :EtOAc = 99:1 to 90:10) to produce the corresponding 2H-indazole 1a–m.
:EtOAc = 99:1 to 90:10) to produce the corresponding 2H-indazole 1a–m.
Synthetic procedure for the C-3 benzoylation of 2H-indazoles 3a–q/4a–l (Schemes 2 and 3)
For the C-3 benzoylation of 2H-indazoles 3a–q/4a–l, a mixture of 2H-indazole 1a–m (0.5 mmol), aromatic/hetero-aromatic/aliphatic aldehydes 2a–q (1 mmol) and TBPB (1.25 mmol) dissolved in PhCl were taken in a sealed reaction tube under nitrogen atmosphere. Then the reaction mixture was stirred at 110 °C for 24 h. After completion of the reaction, the reaction mixture was cooled and the work-up was done with a saturated solution of NaHCO3 (10 mL) and ethyl acetate (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to get the crude residue which was further purified through column chromatography on silica gel (100–200 mesh) using hexane:ethyl acetate (95:5 ratio) as an eluent to afford the corresponding desired product.
General procedure for the gram-scale synthesis of compound 3a
A mixture of 2-(4-methoxyphenyl)-2H-indazole 1a (1.0 g, 4.46 mmol), 4-methyl benzaldehyde 2a (1.05 mL, 8.92 mmol) and TBPB (2.12 mL, 11.15 mmol) was taken in a 50 mL round-bottom flask under nitrogen atmosphere. Then, the reaction mixture was stirred at 110 °C for 24 h. After completion of the reaction, the reaction mixture was cooled and the work-up was done with a saturated solution of NaHCO3 (10 mL) and ethyl acetate (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to get the crude residue which was further purified through column chromatography on silica gel (100–200 mesh) using hexane:ethyl acetate (95:5 ratio) as an eluent to afford compound 3a as a yellow solid in 65% yield (0.97 g).
Characterization of C-3 acylated/benzoylated 2H-indazoles
:EtOAc = 95:05) as a yellow solid in 82% yield (140 mg, 0.40 mmol); mp: 153–155 °C; Rf (hexane:EtOAc = 85:15): 0.48; 1H NMR (400 MHz, chloroform-d) δ 7.86 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.49–7.44 (m, 2H), 7.38–7.33 (m, 2H), 7.27 (d, J = 8.4 Hz, 2H), 7.17–7.13 (m, 1H), 6.93 (d, J = 8.8 Hz, 2H), 3.82 (s, 3H), 2.44 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.87, 159.94, 148.41, 144.80, 135.37, 133.75, 132.41, 130.29, 129.47, 126.89, 126.70, 124.69, 123.86, 120.68, 118.44, 114.33, 55.63, 21.89; HRMS (ESI/QTOF) m/z: calcd for C22H18N2O2, 343.1441 [M + H]+; found, 343.1446.:EtOAc = 95:05) as a yellow solid in 80% yield (131 mg, 0.39 mmol); mp: 93–95 °C; Rf (hexane:EtOAc = 85:15): 0.45; 1H NMR (400 MHz, chloroform-d) δ 7.88–7.84 (m, 3H), 7.59 (t, J = 7.2 Hz, 1H), 7.47–7.43 (m, 4H), 7.38–7.32 (m, 2H), 7.17–7.13 (m, 1H), 6.91 (d, J = 8.8 Hz, 2H), 3.81 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 186.15, 159.99, 148.46, 137.99, 133.74, 133.63, 132.20, 130.01, 128.73, 126.95, 126.78, 124.95, 124.05, 120.63, 118.51, 114.33, 55.63; HRMS (ESI/QTOF) m/z: calcd for C21H16N2O2, 329.1285 [M + H]+; found, 329.1282.:EtOAc = 95:05) as a yellow solid in 86% yield (154 mg, 0.43 mmol); mp: 123–125 °C; Rf (hexane:EtOAc = 85:15): 0.38; 1H NMR (400 MHz, chloroform-d) δ 7.89–7.83 (m, 3H), 7.47–7.44 (m, 2H), 7.39–7.33 (m, 2H), 7.16–7.12 (m, 1H), 6.94–6.91 (m, 4H), 3.87 (s, 3H), 3.81 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 184.89, 164.25, 159.91, 148.39, 133.73, 132.63, 132.51, 130.67, 126.90, 126.63, 124.56, 123.71, 120.65, 118.40, 114.35, 114.07, 55.70, 55.64; HRMS (ESI/QTOF) m/z: calcd for C22H18N2O3, 359.1390 [M + H]+; found, 359.1392.:EtOAc = 95:05) as a brown oil in 78% yield (152 mg, 0.39 mmol); Rf (hexane:EtOAc = 85:15): 0.30; 1H NMR (400 MHz, chloroform-d) δ 7.84 (d, J = 8.8 Hz, 1H), 7.44 (d, J = 9.2 Hz, 2H), 7.35–7.33 (m, 2H), 7.17–7.14 (m, 1H), 7.04 (d, J = 3.2 Hz, 1H), 6.99–6.96 (m, 1H), 6.88 (d, J = 9.2 Hz, 2H), 6.74 (d, J = 9.2 Hz, 1H), 3.80 (s, 3H), 3.76 (s, 3H), 3.48 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 184.23, 159.38, 153.19, 151.66, 147.87, 133.21, 133.15, 128.89, 126.53, 126.34, 124.61, 123.37, 120.08, 118.91, 117.86, 113.95, 113.37, 112.36, 55.70, 55.49, 55.13. HRMS (ESI/QTOF) m/z: calcd for C23H20N2O4, 389.1496 [M + H]+; found, 389.1498.:EtOAc = 95:05) as a yellow solid in 83% yield (143 mg, 0.41 mmol); mp: 135–137 °C; Rf (hexane:EtOAc = 85:15): 0.42; 1H NMR (400 MHz, chloroform-d) δ 7.92–7.87 (m, 3H), 7.46–7.44 (m, 2H), 7.40–7.36 (m, 2H), 7.22–7.18 (m, 1H), 7.14 (t, J = 8.8 Hz, 2H), 6.94 (d, J = 9.2 Hz, 2H), 3.83 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 184.52, 165.94 (JC-F = 254.7 Hz), 159.96, 148.37, 134.18 (JC-F = 2.9 Hz), 133.52, 132.60 (JC-F = 9.4 Hz), 131.81, 126.95, 126.65, 125.03, 123.90, 120.30, 118.52, 115.91 (JC-F = 22.0 Hz), 114.28, 55.57; HRMS (ESI/QTOF) m/z: calcd for C21H15FN2O2, 347.1191 [M + H]+; found, 347.1194.:EtOAc = 95:05) as a yellow solid in 81% yield (147 mg, 0.41 mmol); mp: 142–144 °C; Rf (hexane:EtOAc = 85:15): 0.45; 1H NMR (400 MHz, chloroform-d) δ 7.86 (d, J = 8.8 Hz, 1H), 7.78 (d, J = 8.8 Hz, 2H), 7.44–7.40 (m, 4H), 7.37–7.33 (m, 2H), 7.19–7.16 (m, 1H), 6.91 (d, J = 8.8 Hz, 2H), 3.81 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 184.77, 160.07, 148.45, 140.09, 136.28, 133.57, 131.75, 131.36, 129.09, 127.05, 126.76, 125.23, 124.01, 120.38, 118.63, 114.37, 55.65; HRMS (ESI/QTOF) m/z: calcd for C21H15ClN2O2, 363.0895 [M + H]+; found, 363.0897.:EtOAc = 95:05) as a yellow solid in 84% yield (170 mg, 0.42 mmol); mp: 141–143 °C; Rf (hexane:EtOAc = 85:15): 0.48; 1H NMR (400 MHz, chloroform-d) δ 7.89–7.87 (m, 1H), 7.73 (d, J = 8.4 Hz, 2H), 7.61 (d, J = 8.4 Hz, 2H), 7.44 (d, J = 9.2 Hz, 2H), 7.45–7.35 (m, 2H), 7.22–7.18 (m, 1H), 6.94 (d, J = 8.8 Hz, 2H), 3.84 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 184.93, 160.08, 148.46, 136.71, 133.56, 132.07, 131.70, 131.43, 128.83, 127.05, 126.76, 125.26, 124.00, 120.38, 118.64, 114.38, 55.66; HRMS (ESI/QTOF) m/z: calcd for C21H15BrN2O2, 407.0390 [M + H]+; found, 407.0392.:EtOAc = 95:05) as a yellow solid in 67% yield (122 mg, 0.34 mmol); mp: 155–157 °C; Rf (hexane:EtOAc = 85:15): 0.44; 1H NMR (400 MHz, chloroform-d) δ 7.88 (d, J = 8.8 Hz, 1H), 7.78 (s, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.54 (d, J = 8.0 Hz, 1H), 7.43–7.37 (m, 5H), 7.23–7.19 (m, 1H), 6.92 (d, J = 8.8 Hz, 2H), 3.81 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 184.63, 160.07, 148.51, 139.52, 134.98, 133.57, 133.37, 131.66, 130.01, 129.76, 128.03, 127.11, 126.81, 125.45, 124.20, 120.37, 118.66, 114.37, 55.66; HRMS (ESI/QTOF) m/z: calcd for C21H15ClN2O2, 363.0895 [M + H]+; found, 363.0892.:EtOAc = 95:05) as a yellow solid in 58% yield (105 mg, 0.29 mmol); mp: 90–92 °C; Rf (hexane:EtOAc = 85:15): 0.44; 1H NMR (400 MHz, chloroform-d) δ 7.8–7.78 (m, 1H), 7.39 (d, J = 8.8 Hz, 2H), 7.36–7.24 (m, 5H), 7.14–7.09 (m, 1H), 7.06–7.04 (m, 1H), 6.84 (d, J = 8.8 Hz, 2H), 3.75 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 183.82, 160.11, 148.53, 138.80, 133.59, 132.21, 132.16, 131.94, 130.37, 129.90, 127.20, 127.09, 126.08, 124.52, 120.20, 118.77, 114.09, 55.68; HRMS (ESI/QTOF) m/z: calcd for C21H15ClN2O2, 363.0895 [M + H]+; found, 363.0898.:EtOAc = 95:05) as a yellow oil in 68% yield (90 mg, 0.34 mmol); Rf (hexane:EtOAc = 85:15): 0.36; 1H NMR (400 MHz, chloroform-d) δ 7.70–7.68 (m, 1H), 7.60 (d, J = 8.4 Hz, 1H), 7.45 (d, J = 8.8 Hz, 2H), 7.32–7.28 (m, 1H), 7.06–7.00 (m, 3H), 3.87 (s, 3H), 2.60 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 188.07, 159.75, 148.49, 133.02, 132.07, 127.53, 127.06, 126.70, 120.86, 120.02, 117.52, 114.37, 55.69, 11.12; HRMS (ESI/QTOF) m/z: calcd for C16H14N2O2, 267.1128 [M + H]+; found, 267.1131.:EtOAc = 95:05) as a yellow oil in 61% yield (86 mg, 0.31 mmol); Rf (hexane:EtOAc = 85:15): 0.31; 1H NMR (400 MHz, chloroform-d) δ 7.86–7.59 (m, 2H), 7.47–7.40 (m, 2H), 7.31–7.28 (m, 1H), 7.07–7.00 (m, 3H), 3.87 (s, 3H), 3.02 (q, J = 7.6 Hz, 1H), 2.60 (s, 1H), 1.27–1.23 (m, 3H); 13C NMR (100 MHz, chloroform-d) δ 188.08, 159.92, 159.76, 148.55, 138.10, 133.09, 127.36, 127.06, 126.59, 120.86, 120.84, 120.15, 117.61, 114.36, 55.68, 18.91, 14.15; HRMS (ESI/QTOF) m/z: calcd for C17H16N2O2, 281.1285 [M + H]+; found, 281.1287.:EtOAc = 95:05) as a yellow oil in 60% yield (88 mg, 0.30 mmol); Rf (hexane:EtOAc = 85:15): 0.32; 1H NMR (400 MHz, chloroform-d) δ 7.70–7.64 (m, 2H), 7.45–7.40 (m, 2H), 7.31–7.27 (m, 1H), 7.06–7.00 (m, 3H), 3.87 (s, 3H), 2.96 (t, J = 7.6 Hz, 2H), 1.66 (q, J = 7.6 Hz, 2H), 0.88 (t, J = 7.2 Hz, 3H); 13C NMR (100 MHz, chloroform-d) δ 159.89, 148.49, 136.89, 133.16, 127.49, 127.06, 126.56, 120.84, 120.29, 117.58, 114.37, 114.32, 55.68, 27.36, 22.87, 14.08; HRMS (ESI/QTOF) m/z: calcd for C17H18N2O, 267.1492 [M + H]+; found, 267.1495.:EtOAc = 95:05) as a yellow solid in 71% yield (104 mg, 0.35 mmol); mp: 98–100 °C; Rf (hexane:EtOAc = 85:15): 0.34; 1H NMR (400 MHz, chloroform-d) δ 7.81 (dd, J = 8.4 Hz, 0.8 Hz, 1H), 7.69 (d, J = 8.8 Hz, 1H), 7.39–7.37 (m, 2H), 7.30–7.26 (m, 1H), 7.03–7.00 (m, 3H), 3.87 (s, 3H), 3.36–3.29 (m, 1H), 1.46 (d, J = 7.2 Hz, 6H); 13C NMR (100 MHz, chloroform-d) δ 160.01, 148.77, 142.10, 133.18, 127.70, 126.28, 120.97, 120.56, 119.06, 117.86, 114.29, 55.69, 27.20, 22.51; HRMS (ESI/QTOF) m/z: calcd for C17H18N2O, 267.1492 [M + H]+; found, 267.1498.:EtOAc = 95:05) as a brown solid in 58% yield (92 mg, 0.29 mmol); mp: 134–136 °C; Rf (hexane:EtOAc = 85:15): 0.29; 1H NMR (400 MHz, chloroform-d) δ 10.35 (b, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.74 (dd, J = 8.4 Hz, 0.8 Hz, 1H), 7.51–7.49 (m, 2H), 7.38–7.34 (m, 1H), 7.21–7.17 (m, 1H), 7.03 (s, 1H), 6.96–6.94 (m, 3H), 6.33–6.31 (m, 1H), 3.83 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 174.34, 159.87, 148.50, 133.85, 132.45, 132.25, 127.24, 126.95, 126.66, 124.31, 123.14, 121.36, 120.80, 118.21, 114.29, 111.60, 55.65; HRMS (ESI/QTOF) m/z: calcd for C19H15N3O2, 318.1237 [M + H]+; found, 318.1233.:EtOAc = 95:05) as a brown solid in 57% yield (91 mg, 0.28 mmol); mp: 70–72 °C; Rf (hexane:EtOAc = 85:15): 0.29; 1H NMR (400 MHz, chloroform-d) δ 7.85 (d, J = 9.6 Hz, 1H), 7.68 (d, J = 8.4 Hz, 1H), 7.60 (b, 1H), 7.46 (d, J = 8.8 Hz, 2H), 7.40–7.36 (m, 1H), 7.24–7.20 (m, 2H), 6.93 (d, J = 8.8 Hz, 2H), 6.56–6.54 (m, 1H), 3.81 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 178.93, 172.80, 159.93, 152.39, 148.49, 147.78, 133.70, 127.06, 126.55, 124.94, 123.70, 121.20, 120.42, 118.45, 114.35, 112.79, 55.64; HRMS (ESI/QTOF) m/z: calcd for C19H14N2O3, 319.1077 [M + H]+; found, 319.1073.:EtOAc = 95:05) as a brown solid in 61% yield (101 mg, 0.30 mmol); mp: 76–78 °C; Rf (hexane:EtOAc = 85:15): 0.24; 1H NMR (400 MHz, chloroform-d) δ 7.83 (d, J = 9.2 Hz, 1H), 7.72 (d, J = 8.8 Hz, 1H), 7.48 (d, J = 8.8 Hz, 2H), 7.39–7.35 (m, 1H), 7.24–7.19 (m, 1H), 7.12 (dd, J = 3.6, 0.7 Hz, 1H), 6.93 (d, J = 8.8 Hz, 2H), 6.17–6.16 (m, 1H), 3.82 (s, 3H), 2.33 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 172.24, 159.88, 159.60, 151.18, 148.50, 133.84, 131.85, 127.02, 126.44, 124.64, 123.63, 120.52, 118.32, 114.31, 109.71, 55.66, 14.18; HRMS (ESI/QTOF) m/z: calcd for C20H16N2O3, 333.1234 [M + H]+; found, 333.1235.:EtOAc = 95:05) as a yellow solid in 59% yield (122 mg, 0.29 mmol); mp: 70–72 °C; Rf (hexane:EtOAc = 85:15): 0.34; 1H NMR (400 MHz, chloroform-d) δ 7.78 (d, J = 8.8 Hz, 1H), 7.56 (d, J = 8.8 Hz, 1H), 7.39 (d, J = 9.2 Hz, 2H), 7.33–7.29 (m, 2H), 7.17–7.13 (m, 1H), 7.02–7.03 (m, 1H), 6.88 (d, J = 8.8 Hz, 2H), 3.76 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 176.80, 160.09, 148.49, 145.70, 135.74, 133.39, 131.53, 131.17, 127.12, 126.59, 125.03, 124.85, 123.46, 120.22, 118.55, 114.45, 55.67; HRMS (ESI/QTOF) m/z: calcd for C19H13BrN2O2S, 412.9954 [M + H]+; found, 412.9957.:EtOAc = 95:05) as a yellow oil in 55% yield (91 mg, 0.26 mmol); Rf (hexane:EtOAc = 85:15): 0.40; 1H NMR (400 MHz, chloroform-d) δ 7.75 (d, J = 8.4 Hz, 2H), 7.47–7.45 (m, 2H), 7.39–7.35 (m, 4H), 7.23 (s, 1H), 7.08 (s, 1H), 6.58 (s, 1H), 5.98 (s, 2H), 2.41 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.80, 149.62, 147.86, 146.11, 144.59, 140.49, 135.13, 132.16, 130.03, 129.39, 129.00, 128.34, 125.08, 120.78, 101.40, 95.69, 94.54, 21.79; HRMS (ESI/QTOF) m/z: calcd for C22H16N2O3, 357.1234 [M + H]+; found, 357.1239.:EtOAc = 95:05) as an off-white solid in 71% yield (101 mg, 0.31 mmol); mp: 65–67 °C; Rf (hexane:EtOAc = 85:15): 0.51; 1H NMR (400 MHz, chloroform-d) δ 7.88–7.84 (m, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.54–7.51 (m, 2H), 7.44–7.42 (m, 3H), 7.28 (d, J = 7.6 Hz, 2H), 7.18 (td, J = 9.2, 2.4 Hz, 1H), 6.97–6.94 (m, 1H), 2.45 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.31, 160.09 (JC-F = 243.3 Hz), 145.89, 144.94, 140.39, 134.92, 130.05, 129.51, 129.14, 129.02, 125.35, 123.78 (JC-F = 11.9 Hz), 120.77 (JC-F = 9.9 Hz), 118.67 (JC-F = 29.0 Hz), 103.63 (JC-F = 25.5 Hz), 97.22, 21.83.3; HRMS (ESI/QTOF) m/z: calcd for C21H15FN2O, 331.1241 [M + H]+; found, 331.1244.:EtOAc = 95:05) as a brown solid in 65% yield (111 mg, 0.32 mmol); mp: 140–142 °C; Rf (hexane:EtOAc = 85:15): 0.48; 1H NMR (400 MHz, chloroform-d) δ 7.75–7.72 (m, 3H), 7.48–7.46 (m, 2H), 7.36–7.32 (m, 3H), 7.20 (d, J = 7.2 Hz, 2H), 7.07–7.04 (m, 1H), 6.67 (s, 1H), 3.68 (s, 3H), 2.39 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.93, 157.54, 145.56, 144.41, 140.72, 135.32, 131.84, 130.07, 129.29, 129.08, 128.61, 125.39, 125.01, 122.14, 119.92, 97.13, 55.44, 21.84; HRMS (ESI/QTOF) m/z: calcd for C22H18N2O2, 343.1441 [M + H]+; found, 343.1445.:EtOAc = 95:05) as a yellow solid in 68% yield (111 mg, 0.34 mmol); mp: 102–104 °C; Rf (hexane:EtOAc = 85:15): 0.44; 1H NMR (400 MHz, chloroform-d) δ 7.80 (d, J = 8.4 Hz, 2H), 7.61 (s, 1H), 7.53–7.38 (m, 6H), 7.27–7.25 (m, 2H), 7.00 (d, J = 8.8 Hz, 1H), 2.47 (s, 3H), 2.44 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.70, 149.13, 144.67, 140.56, 136.99, 135.24, 132.28, 130.18, 129.35, 129.04, 128.71, 127.75, 125.43, 122.38, 120.14, 116.69, 22.08, 21.80; HRMS (ESI/QTOF) m/z: calcd for C22H18N2O, 327.1492 [M + H]+; found, 327.1494.:EtOAc = 95:05) as a yellow solid in 78% yield (134 mg, 0.39 mmol); mp: 158–160 °C; Rf (hexane:EtOAc = 85:15): 0.50; 1H NMR (400 MHz, chloroform-d) δ 7.81 (dd, J = 9.2 Hz, 0.8 Hz, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.52–7.50 (m, 2H), 7.42–7.37 (m, 4H), 7.32–7.26 (m, 3H), 2.26 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.29, 146.83, 145.14, 140.22, 134.78, 132.19, 130.71, 130.11, 129.55, 129.16, 129.09, 128.54, 125.33, 124.19, 120.00, 119.36, 21.85; HRMS (ESI/QTOF) m/z: calcd for C21H15ClN2O, 347.0946 [M + H]+; found, 347.0947.:EtOAc = 95:05) as a yellow oil in 58% yield (112 mg, 0.29 mmol); Rf (hexane:EtOAc = 85:15): 0.40; 1H NMR (400 MHz, chloroform-d) δ 7.75 (d, J = 8.4 Hz, 2H), 7.39 (dd, J = 6.8 Hz, 2.0 Hz, 2H), 7.26 (s, 2H), 7.09 (s, 1H), 6.90 (dd, J = 6.8 Hz, 2.0 Hz, 2H), 6.56 (s, 1H), 5.99 (s, 2H), 3.81 (s, 3H), 2.44 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.62, 159.25, 149.25, 147.49, 145.60, 144.32, 135.00, 133.54, 131.87, 129.82, 129.17, 126.08, 120.30, 113.95, 101.14, 95.52, 94.31, 55.31, 21.58; HRMS (ESI/QTOF) m/z: calcd for C23H18N2O4, 387.1340 [M + H]+; found, 387.1346.:EtOAc = 95:05) as a yellow solid in 79% yield (129 mg, 0.40 mmol); mp: 167–169 °C; Rf (hexane:EtOAc = 85:15): 0.48; 1H NMR (400 MHz, chloroform-d) δ 7.87 (dd, J = 8.4 Hz, 0.8 Hz, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 7.35 (t, J = 8.4 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 7.22 (d, J = 8.4 Hz, 2H), 7.16–7.13 (m, 1H), 2.44 (s, 3H), 2.38 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.71, 148.38, 144.72, 138.94, 138.08, 135.22, 132.33, 130.21, 129.67, 129.37, 126.81, 125.17, 124.62, 123.79, 120.59, 118.41, 21.80, 21.19; HRMS (ESI/QTOF) m/z: calcd for C22H18N2O, 327.1492 [M + H]+; found, 327.1495.:EtOAc = 95:05) as a yellow semi-solid in 85% yield (142 mg, 0.41 mmol); Rf (hexane:EtOAc = 85:15): 0.48; 1H NMR (400 MHz, chloroform-d) δ 7.86 (dd, J = 8.8 Hz, 0.8 Hz, 1H), 7.81 (d, J = 8.0 Hz, 2H), 7.52–7.48 (m, 2H), 7.41 (d, J = 8.8 Hz, 2H), 7.38–7.33 (m, 2H), 7.30 (d, J = 8.0 Hz, 2H), 7.19–7.15 (m, 1H), 2.46 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 185.59, 148.70, 145.14, 139.10, 135.13, 134.90, 132.54, 130.30, 129.58, 129.35, 127.29, 126.74, 125.12, 123.97, 120.72, 118.55, 21.93; HRMS (ESI/QTOF) m/z: calcd for C21H15ClN2O, 347.0946 [M + H]+; found, 347.0942.:EtOAc = 95:05) as a yellow solid in 72% yield (118 mg, 0.36 mmol); mp: 130–132 °C; Rf (hexane:EtOAc = 85:15): 0.34; 1H NMR (400 MHz, chloroform-d) δ 8.08 (s, 1H), 7.91 (d, J = 8.0 Hz, 1H), 7.83 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 8.0 Hz, 2H), 7.69–7.66 (m, 1H), 7.43 (d, J = 8.8 Hz, 1H), 7.38–7.34 (m, 1H), 7.24 (d, J = 7.6 Hz, 2H), 7.15–7.11 (m, 1H), 2.42 (s, 3H), 2.32 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 186.88, 149.95, 148.92, 148.30, 144.37, 139.19, 135.40, 133.35, 132.60, 129.95, 129.39, 127.60, 124.43, 123.49, 120.67, 118.40, 116.99, 21.86, 18.10; HRMS (ESI/QTOF) m/z: calcd for C21H17N3O, 328.1445 [M + H]+; found, 328.1449.:EtOAc = 95:05) as a white solid in 71% yield (124 mg, 0.35 mmol); mp: 104–106 °C; Rf (hexane:EtOAc = 85:15): 0.38; 1H NMR (400 MHz, chloroform-d) δ 8.18–8.17 (m, 1H), 8.03–8.00 (m, 1H), 7.85–7.75 (m, 4H), 7.42–7.34 (m, 2H), 7.24 (d, J = 8.8 Hz, 2H), 7.14–7.10 (m, 1H), 2.41 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 186.65, 150.10, 149.12, 146.77, 144.62, 138.41, 135.09, 132.81, 131.34, 129.85, 129.42, 127.98, 124.70, 123.56, 120.59, 118.33, 118.07, 21.80; HRMS (ESI/QTOF) m/z: calcd for C20H14ClN3O, 348.0898 [M + H]+; found, 348.0895.:EtOAc = 95:05) as a yellow solid in 82% yield (131 mg, 0.42 mmol); mp: 98–100 °C; Rf (hexane:EtOAc = 85:15): 0.33; 1H NMR (400 MHz, chloroform-d) δ 8.25–8.23 (m, 1H), 8.06–8.04 (m, 1H), 7.89–7.82 (m, 2H), 7.77 (d, J = 8.4 Hz, 2H), 7.46 (d, J = 8.8 Hz, 1H), 7.40–7.35 (m, 1H), 7.23–7.21 (m 3H), 7.16–7.12 (m, 1H), 2.40 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 186.81, 151.87, 149.01, 148.04, 144.29, 138.64, 135.30, 132.73, 129.78, 129.30, 127.74, 124.47, 123.50, 123.28, 120.62, 118.35, 117.33, 21.76; HRMS (ESI/QTOF) m/z: calcd for C20H15N3O, 31.1288 [M + H]+; found, 314.1286.:EtOAc = 95:05) as a yellow solid in 80% yield (119 mg, 0.40 mmol); mp: 125–127 °C; Rf (hexane:EtOAc = 85:15): 0.42; 1H NMR (400 MHz, chloroform-d) δ 7.89–7.85 (m, 3H), 7.62–7.57 (m, 1H), 7.55–7.52 (m, 2H), 7.47–7.35 (m, 7H), 7.19–7.15 (m, 1H); 13C NMR (100 MHz, chloroform-d) δ 186.07, 148.66, 140.58, 137.90, 133.67, 132.35, 130.00, 129.18, 129.03, 128.74, 127.12, 125.63, 125.12, 124.16, 120.68, 118.63; HRMS (ESI/QTOF) m/z: calcd for C20H14N2O, 299.1179 [M + H]+; found, 299.1176.General procedure for the synthesis of 11
The starting substrate 1p was synthesized using the reported procedure15 and was reacted with m-acetoxybenzaldehyde 2r under the optimized reaction conditions to afford 10. In the reaction vessel, 2-(3-methoxyphenyl)-2H-indazole (1p, 112 mg, 0.5 mmol) was charged with m-acetoxybenzaldehyde (2r, 164 mg, 1 mmol) and TBPB (238 μL, 1.25 mmol) dissolved in chlorobenzene. The reaction mixture was then stirred under N2 atmosphere at 110 °C for 24 h. After completion of the reaction, the reaction mixture was cooled and the work-up was done with a saturated solution of NaHCO3 (10 mL) and ethyl acetate (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to get the crude residue which was further purified through column chromatography on silica gel (100–200 mesh) using hexane:ethyl acetate (95:5 ratio) as an eluent to afford the corresponding product 10 which upon deprotection under basic conditions furnished the desire product 11 (anti-inflammatory agent).
:EtOAc = 95:05) as a white solid in 51% yield (87 mg, 0.25 mmol); Rf (hexane:EtOAc = 85:15): 0.20; 1H NMR (400 MHz, DMSO-d6) δ 9.84 (s, 1H), 7.84 (d, J = 8.8 Hz, 1H), 7.40–7.17 (m, 6H), 7.13 (t, J = 2.4 Hz, 1H), 7.11–7.09 (m, 1H), 7.04–6.96 (m, 3H), 3.72 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 186.09, 160.00, 158.10, 148.30, 141.66, 139.09, 132.77, 130.49, 127.70, 125.58, 123.85, 121.48, 121.08, 120.75, 118.74, 118.22, 116.01, 115.27, 111.66, 56.02; HRMS (ESI/QTOF) m/z: calcd for C21H16N2O3, 345.1234 [M + H]+; found, 345.1236.General procedure for the synthesis of 13
The starting material 12 was synthesized by utilizing our general procedure which on subjection to Pd-catalyzed direct biaryl coupling using the reported procedure16 afforded a new class of bio-azaheterocycles, i.e., 3-methoxy-9H-dibenzo[4,5:6,7]azepino[1,2-b]indazol-9-one (indazole-fused diazepine).:DCM = 90:10) as a yellow solid in 75% yield (123 mg, 0.37 mmol); mp: 108–110 °C; Rf (hexane:DCM = 50:50): 0.5; 1H NMR (400 MHz, chloroform-d) δ 8.14–8.09 (m, 2H), 8.05–8.03 (m, 1H), 7.82 (d, J = 8.8 Hz, 1H), 7.77 (d, J = 7.6 Hz, 1H), 7.66–7.62 (m, 1H), 7.55–7.51 (m, 1H), 7.41–7.37 (m, 1H), 7.31–7.27 (m, 1H), 7.19 (d, J = 2.8 Hz, 1H), 7.09 (dd, J = 9.2, 2.8 Hz, 1H), 3.89 (s, 3H); 13C NMR (100 MHz, chloroform-d) δ 183.27, 159.12, 148.62, 139.52, 138.11, 134.80, 132.75, 131.61, 130.70, 130.17, 129.16, 128.72, 127.93, 127.60, 125.89, 123.69, 121.09, 118.05, 116.36, 115.47, 55.84; HRMS (ESI/QTOF) m/z: calcd for C21H14N2O2, 327.1128 [M + H]+; found, 327.1126.General procedure for the synthesis of 14
To a solution of 3a (0.29 mmol; 1.0 equiv.) in methanol (10–15 mL) was added NaBH4 (0.58 mmol; 2.0 equiv.) portionwise at room temperature and the reaction was stirred at the same temperature for 1 h. After completion of the reaction monitored by TLC, the solvent was evaporated under reduced pressure and the crude solid product was extracted with ethyl acetate (3 × 20 mL) and water (10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to get the crude residue which was further purified through column chromatography on silica gel (100–200 mesh) using hexane:ethyl acetate (80:20 ratio) as an eluent to afford the corresponding product 14 as a white solid in 90% yield.
:EtOH = 80:20) as a white solid in 90% yield (90 mg, 0.26 mmol); mp: 106–108 °C; Rf (hexane:EtOH = 70:30): 0.3; 1H NMR (400 MHz, chloroform-d) δ 7.51 (d, J = 9.2 Hz, 1H), 7.39 (d, J = 9.2 Hz, 1H), 7.15–7.09 (m, 3H), 7.04 (d, J = 8.4 Hz, 2H), 6.98 (d, J = 8.0 Hz, 2H), 6.87–6.83 (m, 1H), 6.69 (d, J = 8.8 Hz, 2H), 5.93 (s, 1H), 3.69 (s, 3H), 2.22 (s, 3H); 13C NMR (101 MHz, chloroform-d) δ 159.98, 148.56, 138.37, 137.65, 137.54, 132.49, 129.26, 127.48, 126.69, 126.47, 121.84, 121.40, 120.39, 117.40, 114.16, 68.75, 55.62, 21.25; HRMS (ESI/QTOF) m/z: calcd for C22H20N2O2, 345.1598 [M + H]+; found, 345.1596.General procedure for the competition reaction
A mixture of 2-(4-methoxyphenyl)-2H-indazole 1a (112 mg, 0.5 mmol), 4-methoxybenzaldehyde 2c (61 μL, 0.5 mmol) and 4-fluorobenzaldehyde 2e (53 μL, 0.5 mmol) and TBPB (238 μL, 1.25 mmol) was taken in a sealed reaction tube under nitrogen atmosphere. Then, the reaction mixture was stirred at 110 °C for 24 h. After completion of the reaction, the reaction mixture was cooled and the work-up was done with a saturated solution of NaHCO3 (10 mL) and ethyl acetate (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to get the crude residue which was further purified through column chromatography on silica gel (100–200 mesh) using hexane:ethyl acetate (95:5 ratio) as an eluent to afford the corresponding product 3c in 56% (101 mg, 0.28 mmol as a yellow solid) and 3e in 30% (52 mg, 0.15 mmol as a yellow solid) yields, respectively. Similarly, a mixture of 2-(4-methoxyphenyl)-2H-indazole 1a (112 mg, 0.5 mmol), 4-methylbenzaldehyde 2a (59 μL, 0.5 mmol) and acetaldehyde 2j (28 μL, 0.5 mmol) and TBPB (238 μL, 1.25 mmol) was taken in a sealed reaction tube under nitrogen atmosphere. Then the reaction mixture was stirred at 110 °C for 24 h. After work-up, the crude residue was further purified through column chromatography on silica gel (100–200 mesh) using hexane:ethyl acetate (95:5 ratio) as an eluent to afford the corresponding product 3a in 55% (94 mg, 0.27 mmol as a yellow solid) and 3j in 29% (39 mg, 0.14 mmol as a yellow solid) yields, respectively.
Synthetic procedure for performing the control experiment
A mixture of 2-(4-methoxyphenyl)-2H-indazole 1a (112 mg, 0.5 mmol), 4-methylbenzaldehyde 2a (118 μL, 1 mmol) and TBPB (238 μL, 1.25 mmol) and TEMPO (195 mg, 1.25 mmol) was taken in a sealed reaction tube under nitrogen atmosphere. Then, the reaction mixture was stirred at 110 °C for 24 h. After completion of the reaction, the reaction mixture was cooled and the work-up was done with a saturated solution of NaHCO3 (10 mL) and ethyl acetate (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to get the crude residue which was further purified through column chromatography on silica gel (100–200 mesh) using hexane:ethyl acetate (95:5 ratio) as an eluent to afford the TEMPO trapped acyl adduct (7) as a viscous liquid. TLC observation showed that there was no formation of 3a in the TEMPO-assisted reaction. The intermediate, i.e. benzoyl free radical, was trapped with TEMPO to afford the TEMPO-acyl adduct 7.
:ethyl acetate = 85:15) 0.5; 1H NMR (400 MHz, chloroform-d) δ 7.93 (d, J = 8.4 Hz, 2H), 7.21 (d, J = 8.0 Hz, 2H), 2.37 (s, 3H), 1.77–1.63 (m, 3H), 1.55–1.52 (m, 2H), 1.43–1.40 (m, 1H), 1.23 (s, 6H), 1.08 (s, 6H); 13C NMR (100 MHz, chloroform-d) δ 166.50, 143.56, 129.66, 129.22, 128.52, 126.98, 60.40, 39.11, 32.03, 25.43, 21.72, 20.91, 17.08; HRMS (ESI/QTOF) m/z: calcd for C17H25NO2, 276.1958 [M + H]+; found, 276.1955.Conflicts of interest
The author declares no competing financial interest.Acknowledgements
S. C. acknowledges DST-NRF Indo-South Africa Joint research project [DST/INT/South Africa/P-19/2016]. R. S. acknowledges MNIT Jaipur for providing an institute fellowship. L. Y. and R. K. Y. thanks DST, New Delhi and UGC, New Delhi for providing financial assistance in the form of a senior research fellowship. Materials Research Centre (MRC), MNIT Jaipur is gratefully acknowledged for providing analytical facilities.Notes and references
- (a) C.-Y. Huang, H. Kang, J. Li and C.-J. Li, J. Org. Chem., 2019, 84, 12705–12721 CrossRef CAS PubMed; (b) S. Girard, T. Knauber and C. -J. Li, Angew. Chem., Int. Ed., 2014, 53, 74–100 CrossRef CAS PubMed; (c) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed.
- (a) C. N. Sternberg, I. D. Davis, J. Mardiak, C. Szczylik, E. Lee, J. Wagstaff, C. H. Barrios, P. Salman, O. A. Gladkov, A. Kavina, J. J. Zarbá, M. Chen, L. McCann, L. Pandite, D. F. Roychowdhury and R. E. Hawkins, J. Clin. Oncol., 2010, 28, 1061–1068 CrossRef CAS PubMed; (b) P. Jones, S. Altamura, J. Boueres, F. Ferrigno, M. Fonsi, C. Giomini, S. Lamartina, E. Monteagudo, J. M. Ontoria, M. V. Orsale, M. C. Palumbi, S. Pesci, G. Roscilli, R. Scarpelli, C. Schultz- Fademrecht, C. Toniatti and M. Rowley, J. Med. Chem., 2009, 52, 7170–7185 CrossRef CAS PubMed; (c) A. Floridi, M. G. Paggi, M. L. Marcante, B. Silvestrini, A. Caputo and C. D. Martino, J. Natl. Cancer Inst., 1981, 66, 497 CAS; (d) H. Shen, S. Gou, J. Shen, Y. Zhu, Y. Zhang and X. Chen, Bioorg. Med. Chem. Lett., 2010, 20, 2115 CrossRef CAS PubMed; (e) L. A. Clutterbuck, C. G. Posada, C. Visintin, D. R. Riddal, B. Lancaster, P. J. Gane, J. Garthwaite and D. L. Selwood, J. Med. Chem., 2009, 52, 2694 CrossRef CAS PubMed; (f) D. Szymon, B. Marta, P. Arkadiusz, P. Michał, F. Michał, W. Maria, D. Anna, S. Sebastian, T. Adam and K. Marcin, Int. J. Cardiol., 2018, 269, 283 CrossRef PubMed; (g) M. C. Vega, M. Rolón, A. Montero-Torres, C. Fonseca-Berzal, J. A. Escario, A. Gómez-Barrio, J. Gálvez, Y. Marrero-Ponce and V. J. Arán, Eur. J. Med. Chem., 2012, 58, 214 CrossRef CAS PubMed; (h) A. Tanitame, Y. Oyamada, K. Ofuji, Y. Kyoya, K. Suzuki, H. Ito, M. Kawasaki, K. Nagai, M. Wachi and J. Yamagishi, Bioorg. Med. Chem. Lett., 2004, 14, 2857 CrossRef CAS PubMed; (i) W. Han, J. C. Pelletier and C. N. Hodge, Bioorg. Med. Chem. Lett., 1998, 8, 3615 CrossRef CAS PubMed.
- (a) A. Thangadurai, M. Minu, S. Wakode, S. Agrawal and B. Narasimhan, Med. Chem. Res., 2012, 21, 1509 CrossRef CAS; (b) I. Denya, S. F. Malan and J. Joubert, Expert Opin. Ther. Pat., 2018, 28, 441 CrossRef CAS PubMed; (c) H. Cerecetto, A. Gerpe, M. Gonzalez, V. Aran and C. de Ocariz, Mini-Rev. Med. Chem., 2005, 5, 869 CrossRef CAS PubMed; (d) J. Dong, Q. Zhang, Z. Wang, G. Huang and S. Li, ChemMedChem, 2018, 13, 1490 CrossRef CAS PubMed; (e) F.-Y. Lee, J.-C. Lien, L.-J. Huang, T.-M. Huang, S.-C. Tsai, C.-M. Teng, C.-C. Wu, F.-C. Cheng and S.-C. Kuo, J. Med. Chem., 2001, 44, 3746 CrossRef CAS PubMed; (f) Y. Ikeda, N. Takano, H. Matsushita, Y. Shiraki, T. Koide, R. Nagashima, Y. Fujimura, M. Shindo, S. Suzuki and T. Iwasaki, Arzneim. Forsch., 1979, 29, 511 CAS; (g) S.-G. Zhang, C.-G. Liang and W.-H. Zhang, Molecules, 2018, 23, 2783 CrossRef PubMed.
- (a) L. Yadav and S. Chaudhary, Org. Biomol. Chem., 2020, 18, 5927–5936 RSC; (b) S. Ghosh, S. Mondal and A. Hajra, Adv. Synth. Catal., 2020, 362, 3768–3794 CrossRef CAS.
- H. Oh, S. Han, A. K. Pandey, S. H. Han, N. K. Mishra, S. Kim, R. Chun, H. S. Kim, J. Park and I. S. Kim, J. Org. Chem., 2018, 83, 4070–4077 CrossRef CAS PubMed.
- G. Bogonda, H. Y. Kim and K. Oh, Org. Lett., 2018, 9, 2711–2715 CrossRef PubMed.
- Y.-L. Liu, Y.-L. Pan, G.-J. Li, H.-F. Xu and J.-Z. Chen, Org. Biomol. Chem., 2019, 17, 8749–8755 RSC.
- L. Liu, P. Jiang, Y. Liu, H. Du and J. Tan, Org. Chem. Front., 2020, 7, 2278–2283 RSC.
- (a) R. Chen, J. Chen, J. Zhang and X. Wan, Chem. Rec., 2018, 18, 1292 CrossRef CAS PubMed; (b) S. Rajamanickam, G. Majji, S. K. Santra and B. K. Patel, Org. Lett., 2015, 17, 5586 CrossRef CAS PubMed; (c) G. Majji, S. Rajamanickam, N. Khatun, S. K. Santra and B. K. Patel, J. Org. Chem., 2015, 80, 3440 CrossRef CAS PubMed; (d) A. Gogoi, A. Modi, S. Guin, S. K. Rout, D. Das and B. K. Patel, Chem. Commun., 2014, 50, 10445 RSC; (e) X.-F. Wu, J.-L. Gonga and X. Qia, Org. Biomol. Chem., 2014, 12, 5807 RSC; (f) Y. Siddaraju and K. R. Prabhu, Tetrahedron, 2016, 72, 959–967 CrossRef CAS.
- (a) N. Khatun, A. Banerjee, S. K. Santra, A. Behera and B. K. Patel, RSC Adv., 2014, 4, 54532 RSC; (b) S. K. Rout, S. Guin, A. Gogoi, G. Majji and B. K. Patel, Org. Lett., 2014, 16, 1614 CrossRef CAS PubMed.
- (a) J.-J. Cao, T.-H. Zhu, S.-Y. Wang, Z.-Y. Gu, X. Wang and S.-J. Ji, Chem. Commun., 2014, 50, 6439–6442 RSC; (b) S.-L. Zhou, L.-N. Guo, H. Wang and X.-H. Duan, Chem.–Eur. J., 2013, 19, 12970–12973 CrossRef CAS PubMed; (c) T. He, H. Li, P. Li and L. Wang, Chem. Commun., 2011, 47, 8946–8948 RSC; (d) H. Zhang, Z. Gu, P. Xu, H. Hu, Y. Chenga and C. Zhu, Chem. Commun., 2016, 52, 477–480 RSC.
- R. J. Steffan, E. M. Matelan, [(1H-Indazol-3-ly)Methyl]Phenols and (Hydroxyphenyl), (1H-Indazol-3-yl)Methanones, WO Pat., 2006050006, 2006.
- A. H. Shinde, S. Vidyacharan and D. S. Sharada, Org. Biomol. Chem., 2016, 14, 3207–3211 RSC.
- M. R. Kumar, A. Park, N. Park and S. Lee, Org. Lett., 2011, 13, 3542–3545 CrossRef CAS PubMed.
- N. E. Genung, L. Wei and G. E. Aspnes, Org. Lett., 2014, 16, 3114–3117 CrossRef CAS PubMed.
- A. P. Kozikowski and D. Ma, Tetrahedron Lett., 1991, 32, 3317–3320 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data, 1H NMR and 13C NMR spectra. See DOI: 10.1039/d1ra02225c |
| This journal is © The Royal Society of Chemistry 2021 |