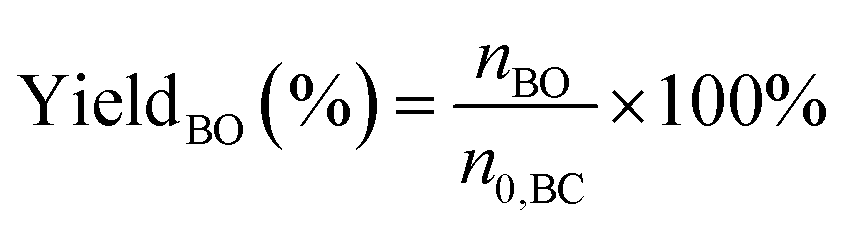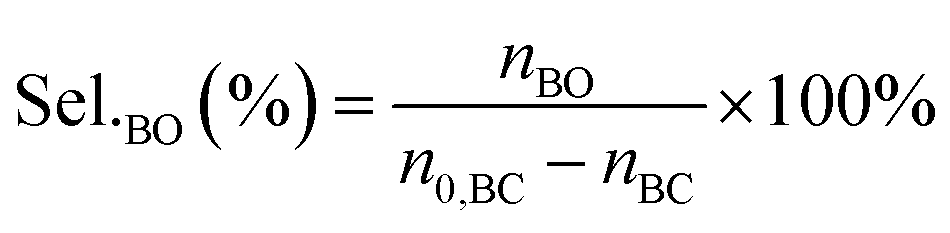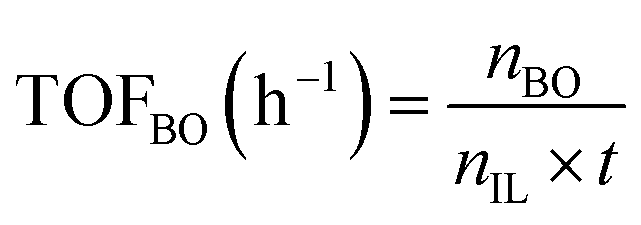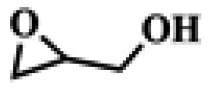DOI:
10.1039/D1RA01039E
(Paper)
RSC Adv., 2021,
11, 14193-14202
Tris-imidazolinium-based porous poly(ionic liquid)s as an efficient catalyst for decarboxylation of cyclic carbonate to epoxide†
Received
7th February 2021
, Accepted 3rd April 2021
First published on 15th April 2021
Abstract
A series of imidazolinium-based porous poly(ionic liquid)s (PILs) with different anions prepared by free-radical copolymerization of an arene-bridged tris-vinylimidazolium salt and divinylbenzene (DVB) were constructed. The as-prepared PILs were characterized by BET, SEM, TEM, TGA and Elemental Analysis (EA), and the results showed that they had plentiful ionic sites, and abundant and stable mesopores. In particular, the density of ionic sites and pore structure of PILs could be controlled by adjusting the content of DVB. Moreover, the PILs were used as efficient heterogeneous catalysts for the decarboxylation of cyclic carbonates to epoxides for the first time. Results showed that the catalytic activity of PILs was positively correlated with the nucleophilicity of the anions in PILs, and PDVB-[PhTVIM]Cl-1 with a chloride anion-enriched skeleton displayed the best catalytic performance. Without any solvent or cocatalyst, PDVB-[PhTVIM]Cl-1 achieved a TOF value of 108.1 h−1 and the yield of butylene oxide of 89.6%, which was even better than the homogeneous IL-based catalysts (TOF value: 8.7 h−1) that had been previously reported. Meanwhile, PDVB-[PhTVIM]Cl-1 also exhibited excellent recyclability and substrate compatibility.
1. Introduction
Epoxide as a kind of cyclic ether with a three-membered ring has attracted much attention owing to its broad applications in the fields of biomedicine and functional materials.1,2 The high stress of epoxy ring renders epoxides reactive intermediates, making it prone to ring-opening reaction with nucleophiles to form desired polymers.3,4 Typical epoxides such as ethylene oxide, propylene oxide and butylene oxide are often used as basic chemical raw materials to produce polyether4,5 and polyurethane.6,7 Among these epoxides, butylene oxide has relatively more methylene functional groups in its molecular structure. Therefore, polyether polyols and polyurethanes synthesized with butylene oxide as the copolymerization unit possess excellent hydrophobic properties and cold resistance properties. Meanwhile, as a crucial component of the polyether synthetic oil, the involvement of butylene oxide can significantly improve the oil solubility and increase the viscosity index of the lubricating oil.8
At present, the industrial production processes of epoxide are mainly chlorohydrin process, hydrogen peroxide combination process and hydroperoxide process. Chlorohydrin process is the earliest mature production process, which could be traced back to the 19th century.9 However, due to the inherent drawbacks such as environmental pollution and equipment corrosion, it has been gradually eliminated in recent years.10,11 While, hydrogen peroxide combination process uses hydrogen peroxide as an oxidant to convert olefins into epoxy compounds in one step.12,13 Although this process has advanced and green technology, the harsh operation conditions limit the efficiency of this method. As for the hydroperoxide process, alkylbenzenes are first oxidized to alkylbenzene hydroperoxides, which can further oxidize olefins to epoxides.13,14 This method suffers from inherent disadvantages, including high production cost and complex process. Recently, the use of cyclic carbonate as the raw material to produce epoxide through the direct decarboxylation process is worthy of attention from both economic and environmental perspectives.15,16 This is not only related to the environmentally friendly characteristics of cyclic carbonate as an chemical intermediate, but also because cyclic carbonate could be synthesized from polyols derived from a wide range of sources, such as coal-based17–20 and bio-based routes.21–23
In our previous work, a green process for the synthesis of butylene oxide through decarboxylation of butylene carbonate was reported.11 Using homogeneous ionic liquid (IL) catalyst, butylene oxide yield as high as 94% was obtained. However, the homogeneous and high viscosity properties of ILs accompany with problem of catalyst separation hindering their large-scale applications.24 Therefore, it is urgent to develop the green heterogeneous catalysts for the decarboxylation of cyclic carbonates to synthesize epoxides.
Poly(ionic liquid)s (PILs) are a subclass of polyelectrolytes constructed by incorporation of IL species into the polymeric backbone.25–29 Combining both advantages of ILs and polymers, such as facile adjustability and high mechanical stability, PILs have aroused considerable attention in the field of catalysts.30–34 In addition, PILs have the morphologies and properties which are inaccessible to ILs owing to the designability monomeric building blocks and the variability of polymerization method.35 Regulating their chemical compositions and porosities in principle can improve the catalytic activity and obtain excellent stability.36–39 In this context, porous PILs with abundant ionic sites are fabricated to accelerate the interfacial mass and energy transport.40–43 Therefore, reasonable expectation could be drawn that porous PILs could be used as an effective heterogeneous catalyst for the decarboxylation of cyclic carbonate.
Herein, a series of cross-linked tris-imidazolium-based PILs with different anions by copolymerization of DVB and ILs were constructed. The PILs were characterized by different techniques and evaluated as heterogeneous catalysts for the decarboxylation of cyclic carbonate to synthesize epoxide. Moreover, the reaction parameters, such as reaction temperature, reaction time and catalyst dosage on catalytic performance were optimized. In addition, the effects of anion and specific surface area on catalytic activity were investigated, and the corresponding catalytic decarboxylation mechanism was proposed.
2. Experimental
2.1 Materials
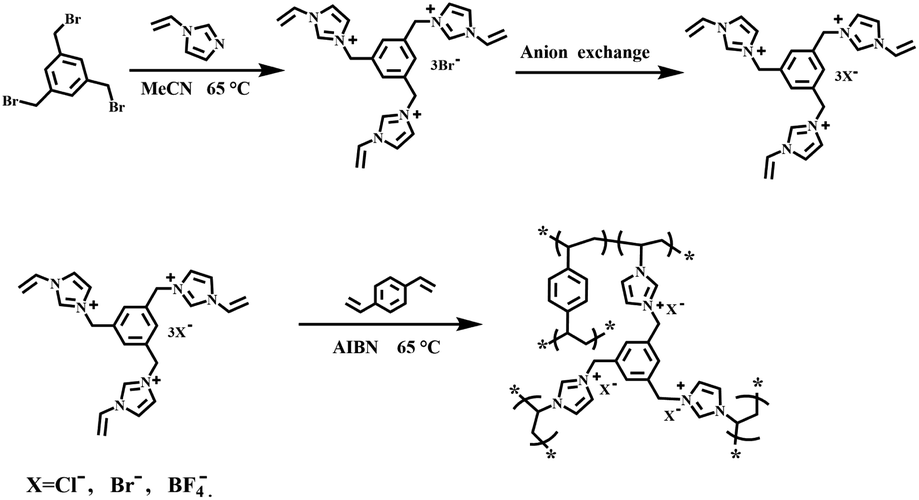
Acetonitrile (MeCN, ≥99.9%), divinylbenzene (DVB, ≥80%), 1,3,5-tris(bromomethyl)benzene (≥98%) and 1-vinylimidazole (99%) were purchased from Aladdin Industrial Corporation. 2,2′-Azobis(2-methylpropionitrile) (AIBN, 98%) was provided by Shanghai Macklin Biochemical Co., Ltd. Ethanol (AR), butylated hydroxytoluene (BHT, CP) were provided by Sinopharm Chemical Reagent Co., Ltd. Glycerol carbonate (90%) was purchased from ARK Pharm, Inc. 1,2-butylene carbonate (≥98%) was prepared through the method we have previously reported.11
2.2 Preparation of ionic liquid monomers-[PhTVIM]X
A modified method was explored to prepare the IL monomers with different anions.24,44 In a typical synthesis of [PhTVIM]Br, 1,3,5-tris(bromomethyl)benzene (6 g, 16.5 mmol), BHT (0.3 g), and a slight excess of 1-vinylimidazole (5.17 g, 54.4 mmol) were dissolved in MeCN (150 mL) in a 250 mL three-necked flask, and the mixture was kept at 65 °C with stirring and refluxing. The crude product was obtained by decantation, then thoroughly washed with MeCN. Thereafter, the white solid was dried under vacuum at 65 °C for 24 h, and the product was named as [PhTVIM]Br. Other IL monomers with different anions, [PhTVIM]Cl and [PhTVIM]BF4, were synthesized by the anion exchange of [PhTVIM]Br with AgCl, and AgBF4 in the aqueous solution, at room temperature, respectively. Meanwhile, the anion exchange rate of the sample [PhTVIM]BF4 was characterized by ICP based on the relative content of B and Br. And the anion exchange rate of [PhTVIM]Cl was characterized by ion chromatography based on the relative content of chloride anions and bromide anions. Table S1† showed that the anion exchange rate of all samples were higher than 99.0%.
2.2.1 [PhTVIM]Br. 1H NMR (600 MHz, D2O) δ 9.44 (s, 3H, –N–CH–N–), 8.05 (s, 3H, –N–CHCH–N–), 7.87 (s, 3H, –N–CHCH–N–), 7.86 (s, 3H, –Ph–(CH2)3–), 7.37 (m, 3H, –CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2–), 6.02 (dd, J = 15.6 Hz, 3H, 2.7 Hz, –CHCH2–), 5.77 (s, 6H, –Ph–(CH2)3–), 5.65–5.51 (dd, J = 8.7, 2.7 Hz, 3H, –CHCH2–). 13C NMR (151 MHz, D2O) δ 135.79, 135.11, 130.15, 128.55, 123.44, 120.31, 110.32, 52.75.
CH2–), 6.02 (dd, J = 15.6 Hz, 3H, 2.7 Hz, –CHCH2–), 5.77 (s, 6H, –Ph–(CH2)3–), 5.65–5.51 (dd, J = 8.7, 2.7 Hz, 3H, –CHCH2–). 13C NMR (151 MHz, D2O) δ 135.79, 135.11, 130.15, 128.55, 123.44, 120.31, 110.32, 52.75.
2.2.2 [PhTVIM]Cl. 1H NMR (600 MHz, D2O) δ 9.44 (s, 3H, –N–CH–N–), 8.05 (s, 3H, –N–CHCH–N–), 7.87 (s, 3H, –N–CHCH–N–), 7.86 (s, 3H, –Ph–(CH2)3–), 7.37 (m, 3H, –CHCH2–), 6.02 (dd, J = 15.6, 2.7 Hz, 3H, CHCH2–), 5.77 (s, 6H, –Ph–(CH2)3–), 5.65–5.51 (dd, J = 8.7, 2.7 Hz, 3H, –CHCH2–). 13C NMR (151 MHz, D2O) δ 135.74, 134.94, 129.50, 128.31, 123.09, 120.13, 110.08, 52.51.
2.2.3 [PhTVIM]BF4. 1H NMR (600 MHz, DMSO) δ 9.48 (s, 3H, –N–CH–N–), 8.24 (t, J = 1.8 Hz, 3H, –N–CHCH–N–), 7.83 (t, J = 1.7 Hz, 3H, –N–CHCH–N–), 7.50 (s, 3H, –Ph–(CH2)3–), 7.31 (dd, J = 15.6, 8.8 Hz, 3H, –CHCH2–), 5.97 (dd, J = 15.6, 2.5 Hz, 3H, –CHCH2–), 5.50–5.46 (m, 3H, –CHCH2–), 5.46 (s, 6H, –Ph–(CH2)3–). 13C NMR (151 MHz, DMSO) δ 136.20, 129.34, 123.81, 119.97, 109.55, 52.21.
2.3 Preparation of PILs
Imidazolinium-based PILs were prepared by free-radical copolymerization. In a typical procedure for the synthesis of PDVB-[PhTVIM]Br-1, [PhTVIM]Br (2.57 g, 4 mmol), DVB (0.52 g, 4 mmol) and AIBN (0.15 g, 5 wt%, based on the amount of both DVB and [PhTVIM]Br) were added into a 100 mL round bottom flask, which contained 6 mL deionized water and 26 mL ethanol as the mixed solvent, and then stirred at room temperature for 2 h under a nitrogen atmosphere for blending thoroughly. Thereafter, the resulting solution was heated to 65 °C and kept for 24 h with refluxing. The product was purified by decantation, and then washed with deionized water and ethanol three times, respectively. The white solid powder was obtained after drying in a vacuum at 65 °C for 24 h. The other parent poly(ionic liquid)s were obtained in a similar way, named as PDVB-[PhTVIM]X-n (X represents the anion species in the polymer framework, n is the initial mole ratio between DVB and the ionic liquid monomer). In addition, P[PhTVIM]Cl was synthesized through the self-polymerization of DVB and the IL monomer [PhTVIM]Cl under the similar conditions as above. Scheme 1 shows the preparation route of PDVB-[PhTVIM]X-n.
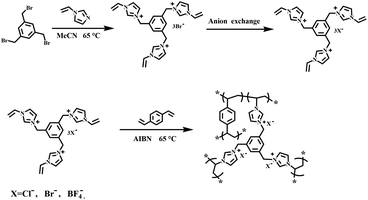 |
| | Scheme 1 The preparation route of PILs with different anions. | |
2.4 Decarboxylation of butylene carbonate
The performance of the catalysts for the decarboxylation of cyclic carbonate to epoxide was evaluated in the three-necked flask equipped with an oil bath. In a typical experiment, butylene carbonate and catalyst with a certain quality ratio were placed in a 100 mL three-necked flask. The reaction was carried out at a predetermined temperature under the slight negative pressure. The decarboxylation product was collected in the second round-bottomed flask, after condensed by the low temperature circulating cooler. After the reaction, the collection was determined by a gas chromatograph (GC-2010, Shimadzu), equipped with an FID detector and a capillary column (GSBP-1, 30 m × 0.25 mm). During investigation of the catalyst reusability, the catalyst was separated from the product by centrifugation, washed separately with deionized water and ethanol, thenceforth dried in a vacuum, and charged into the next cycle. Butylene oxide yield, butylene oxide selectivity and TOF value of catalysts were calculated according to the following formulas:| |
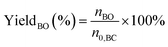 | (1) |
| |
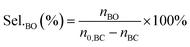 | (2) |
| |
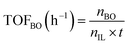 | (3) |
Where YieldBO and Sel.BO represent the yield and the selectivity of butylene oxide, respectively. TOFBO is turnover frequency of catalysts. nBO is the mole amount of pure butylene oxide produced after the decarboxylation reaction. While n0,BC and nBC are the mole amount of butylene carbonate added initially and remained after reaction, respectively, nIL is mole amount of ionic sites in PILs, t refers to reaction time, h.
2.5 Characterization methods
Fourier transform infrared (FT-IR) spectra were registered on a Nicolet IS 10 FT-IR instrument in the region of 400–4000 cm−1. 1H and 13C cross-polarization (CP)/magic-angle-spinning (MAS) nuclear magnetic resonance (NMR) spectra (spinning rate: 10 kHz) were collected on a Bruker AVANCE-III spectrometer. Brunauer–Emmett–Teller (BET) surface areas and N2 sorption isotherms were measured at 77 K by using an ASAP 2020 analyzer. The samples were pre-degassed at 373 K for 4 h under vacuum before analysis. The total pore volumes were calculated from the N2 isotherm at P/P0 = 0.99 and the pore size distribution curves were analyzed by BJH method. Thermo-gravimetric analysis (TGA) was performed on NETZSCH STA 449 F5/F3 Jupiter instrument at a heating rate of 10 K min−1 under a N2 atmosphere. CHN elemental analysis (EA) was carried out with Vario EL cube. The morphology of simples was studied by field emission scanning electron microscope (FESEM; SU8020, accelerated voltage: 5 kV). Transmission electron microscope (TEM) images were conducted on using a FEI Tecnai G2 F30 (300 kV) TEM instrument. The surface chemical composition was obtained by X-ray photoelectron spectroscopy (XPS) on a Thermo Escalab 250Xi X-ray photoelectron spectrometer. The anion exchange rate of ILs was determined using ion chromatography (IC, Thermo Dionex ICS-1100) and inductively coupled plasma-optical emission spectrometer (ICP-OES, Agilent ICPOES730).
3. Results and discussion
3.1 Catalysts characterization
TG analysis indicated that the initial decomposition temperature of all PILs were above 240 °C (Fig. S4†), reflecting relatively high thermal stability. The N2 adsorption analysis (Fig. 1(a)) exhibited a type IV isotherm with a H4 hysteresis loop in the region of 0.3 < P/P0 < 0.99, indicating a typical mesoporous structure of all PILs studied. Additionally, the N2 isotherms increased sharply at a high relative pressure (P/P0 > 0.90), demonstrating the existence of macropores. Specifically, compared with PILs with chloride or bromide anion-enriched skeleton, PDVB-[PhTVIM]BF4-1 displays more obvious mesoporous characteristics. The reason can be attributed to the different solvents used in the synthesis of PILs. Due to the hydrophobicity of IL monomer [PhTVIM]BF4, the solvent used for the synthesis of PDVB-[PhTVIM]BF4-1 is acetonitrile instead of a mixed solvent composed of ethanol and water. The above analysis verified the formation of hierarchical pore structure. Specifically, there was only a small amount of N2 adsorption for the isotherm of P[PhTVIM]Cl whether in the low P/P0 or high P/P0 region, indicating a poor pores structure of it. The textural properties of different PILs obtained by N2 adsorption analysis and elemental analysis were summarized in Table 1. It can be observed that with the value of “n” (the initial molar ratio of DVB to IL) increased from 0 to 5 (Table 1, entries 1–4), the surface area and pore volume of PDVB-[PhTVIM]Cl-n raised from 3 m2 g−1 and 0.01 cm3 g−1 to 394 m2 g−1 and 1.07 cm3 g−1, respectively. The reason can be ascribed to the high crosslinking degree of PILs enhanced by DVB.24 Meanwhile, Table 1 also showed that the IL content decreased with increasing surface area and pore volume, demonstrating that there was a trade-off between the surface area and the IL content in PILs.
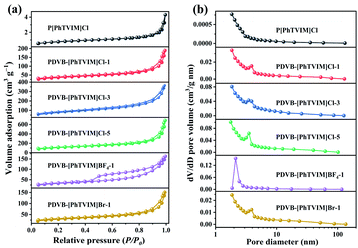 |
| | Fig. 1 (a) N2 sorption isotherms and (b) pore size distribution curves of different PILs. | |
Table 1 Textual properties of different PILs
| Entry |
Catalyst |
SBETa (m2 g−1) |
Davb (nm) |
VPc (cm3 g−1) |
Nd (%) |
IL contente (mmol g−1) |
| BET surface area. Average pore size. Total pore volume. Measured by elemental analysis. The IL content was calculated by N content. |
| 1 |
P[PhTVIM]Cl |
3 |
9.9 |
0.01 |
14.40 |
1.71 |
| 2 |
PDVB-[PhTVIM]Cl-1 |
125 |
9.9 |
0.29 |
11.47 |
1.19 |
| 3 |
PDVB-[PhTVIM]Cl-3 |
274 |
7.7 |
0.57 |
8.93 |
1.06 |
| 4 |
PDVB-[PhTVIM]Cl-5 |
394 |
11 |
1.07 |
6.63 |
0.79 |
| 5 |
PDVB-[PhTVIM]BF4-1 |
112 |
7.3 |
0.27 |
9.71 |
1.16 |
| 6 |
PDVB-[PhTVIM]Br-1 |
98 |
9.9 |
0.23 |
9.89 |
1.18 |
It was reported that the porous structure of the polymer could be benefit from the incorporation of the crosslinker DVB into the polymeric skeleton.45 In order to confirm the effect of DVB on the morphology of PILs, SEM and TEM analysis was applied. PDVB-[PhTVIM]Cl-1 was selected as a typical sample, and P[PhTVIM]Cl without incorporation of DVB was also studied as comparison. First of all, without incorporating DVB, the SEM image of P[PhTVIM]Cl in Fig. 2(a) showed structure of aggregated and highly fused large particles on the micrometer level. The granules of P[PhTVIM]Cl were tightly stacked and formed a highly dense structure, which was further confirmed by the TEM image (Fig. 2(c)). By contrast, after incorporating DVB, the nano-scale particles of PDVB-[PhTVIM]Cl-1 were interconnected with each other to form a cross-linked framework, and the SEM and TEM images of PDVB-[PhTVIM]Cl-1 in (Fig. 2(a) and (d)) showed a typically apparent spongy-like structure with abundant mesopores and macropores. This hierarchical pore structure could facilitate the mass transfer of the substrate and increases the chance of contact between the substrate and the ionic sites.46 The difference in the pore structures of the two PILs indicated that the incorporation of DVB could facilitate the forming of hierarchical pore structure of PILs by increasing the crosslinking density of PILs and reducing the size of the internal microspheres, which were in accordance with N2 adsorption analysis.
 |
| | Fig. 2 SEM images of (a) P[PhTVIM]Cl, (b) PDVB-[PhTVIM]Cl-1; TEM images of (c) P[PhTVIM]Cl, (d) PDVB-[PhTVIM]Cl-1. | |
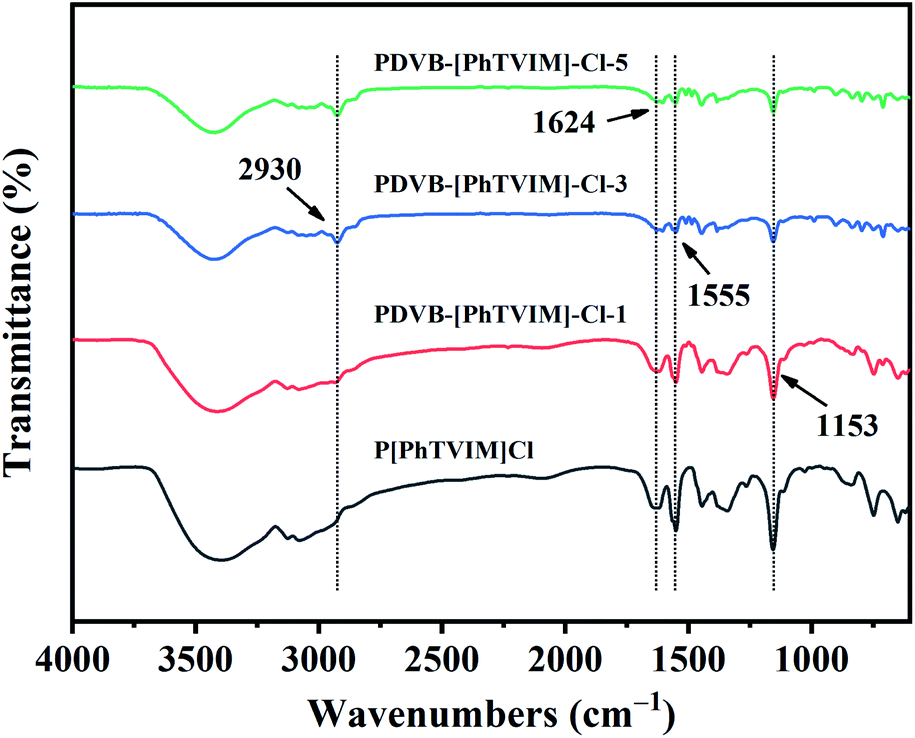
FT-IR spectra of different as-prepared PILs were shown in Fig. 3. All the PILs showed typical imidazolium ring stretching band at about 1153 cm−1 (C–N), 1555 cm−1 (CC), and 1624 cm−1 (CN). The C–H stretching at 2930 cm−1 which was originated from the methylene (–CH2–) used to link benzene ring and imidazole ring. Notably, with the content of DVB increased from 0 to 83 mol% (the initial molar ratio of DVB and IL (“n”) increased from 0 to 5), the intensity of the characteristic peak of the imidazolium ring gradually decreased, while the methylene (–CH2–) characteristic peak gradually increased, confirming the successful introduction of DVB in the skeleton. These results also proved the successful construction of the polymeric backbone.
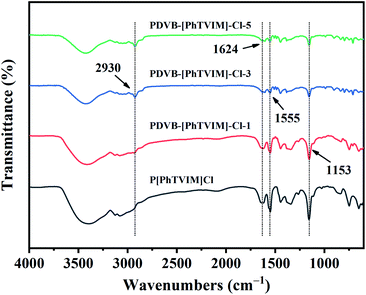 |
| | Fig. 3 FT-IR spectra of different PILs. | |
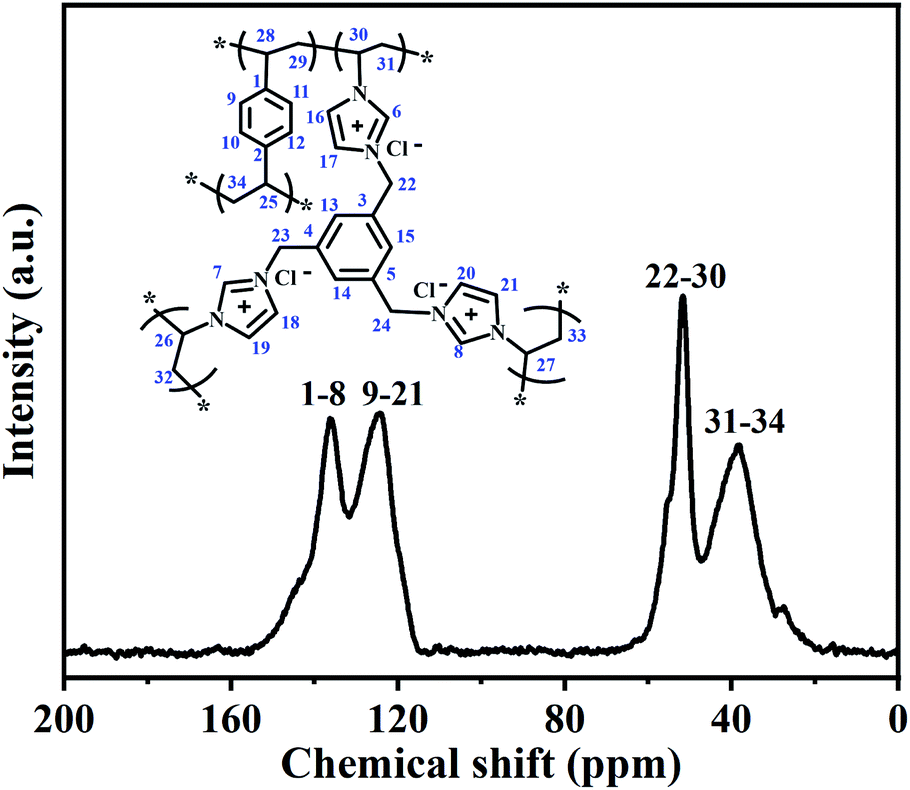
To further confirm the structure of the PILs, the selected typical PDVB-[PhTVIM]Cl-1 sample was also analysed by solid-state 13C CP/MAS NMR, and the results were shown in Fig. 4. It is seen from Fig. 4 that four distinct peaks at 39, 52, 124 and 136 ppm are observed, respectively. The signal at 136 ppm was ascribed to the substituted phenyl carbons and the imine-linked carbon (C2) in the imidazole ring, while the peak at 124 ppm corresponded to the non-substituted phenyl carbons and two adjacent unsaturated carbons (C4 and C5) in the imidazole ring. The signal at 51 ppm could be attributed to the methylene in the ILs units, and the chemical shift at 39 ppm was assigned to the methylene bridges formed after the free-radical copolymerization. These results are correspond to the literature reported.47–50
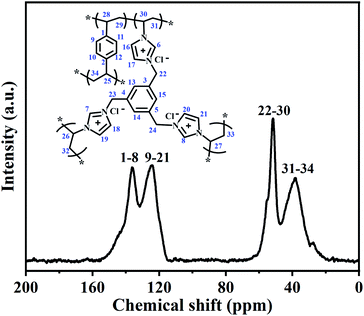 |
| | Fig. 4 13C CP/MAS NMR spectrum of PDVB-[PhTVIM]Cl-1. | |
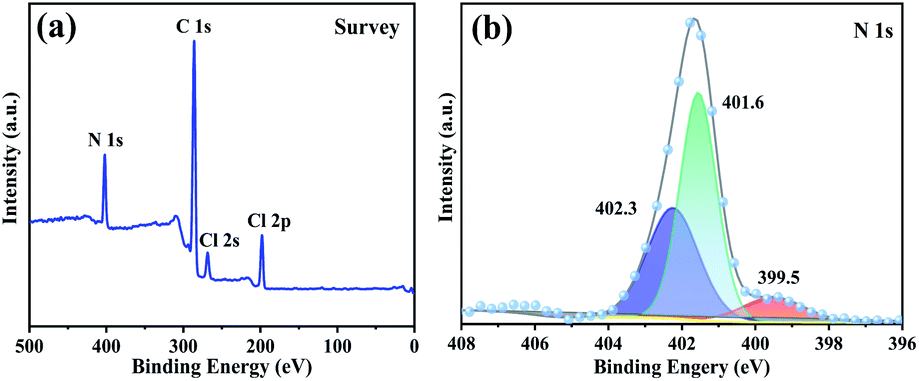
XPS analysis was also used to assess the structure of typical sample PDVB-[PhTVIM]Cl-1. The full survey spectra revealed the presence of four peaks, C 1s, N 1s, Cl 2s and Cl 2p (Fig. 5(a)), respectively. It is known that the imidazoline group has two nitrogen species, the imine-like (–N) and amine-like (–NH–).51 The N 1s spectral signal was deconvoluted into three peaks (Fig. 5(b)) representing the two nitrogen atoms with different chemical positions, which were ascribed to the imidazole moiety. Among them, two peaks at 401.6 eV and 402.3 eV were attributed to pyrrole nitrogen and the peak at 399.5 eV is related to pyridine nitrogen. Based on the above results, it could be concluded that the imidazolium-based IL moiety was proved to be successfully incorporated into the polymeric skeleton.
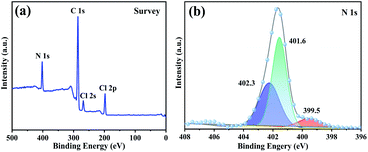 |
| | Fig. 5 (a) Survey scan and (b) N 1s XPS spectra of PDVB-[PhTVIM]Cl-1. | |
3.2 Catalytic performance of different PILs
Catalytic performance of PILs with different anions, as well as with different content of DVB incorporation was evaluated in the decarboxylation of butylene carbonate to butylene oxide. As could be seen in Table 2, PDVB-[PhTVIM]BF4-1 did not show any effect on the decarboxylation reaction (Table 2, entry 2); when using PDVB-[PhTVIM]Br-1 as catalyst in the decarboxylation, 6.0% butylene oxide yield and 14.9 h−1 TOF value was achieved (Table 2, entry 1). Whereas, PILs with Cl− anions exhibited much better catalytic activity than PILs with Br− and BF4− anions that, butylene oxide yield could increase to the range of 10.7–31.1%, with TOF values in the range of 38.9–68.6 h−1 (Table 2, entries 3–6). In brief, the activity of PILs decreased in the order of Cl− > Br− > BF4−. This trend is consistent with our previous studies that anions of imidazolium-based ILs play a key role in the ring-opening step of butylene carbonate via nucleophilic attack on the β-C atom of butylene carbonate by DFT calculation. It was reported52 that the pKa-value of three conjugate acids, HCl (hydrochloric acid), HBr (hydrobromic acid) and HBF4 (fluoroboric acid) were in the order of HCl (−0.4) > HBr (−4.9) > HBF4 (−10.3). Therefore, the nucleophilicity of anions decreased in the order of Cl− > Br− > BF4−, which was in the same order with the catalytic activity of PILs. The reason might be that the strong nucleophilicity of anion can facilitate the ring-opening of the butylene carbonate and thus accelerate the formation of butylene oxide.
Table 2 Evaluation of catalytic performance of catalystsa
| Entry |
Catalyst |
Yieldb % |
Sel.b % |
TOFc h−1 |
| Reaction conditions: butylene carbonate (20.0 g), catalyst dosage (0.2 g), reaction temperature (185 °C), reaction time (180 min), vacuum degree (5 kPa). The yield and selectivity of butylene oxide was determined by GC. TOF (h−1) = [mmol product]/([mmol ionic sites] × [h]). |
| 1 |
PDVB-[PhTVIM]Br-1 |
6.0 |
98.8 |
14.9 |
| 2 |
PDVB-[PhTVIM]BF4-1 |
0 |
0 |
0 |
| 3 |
PDVB-[PhTVIM]Cl-1 |
31.1 |
98.7 |
68.6 |
| 4 |
PDVB-[PhTVIM]Cl-3 |
19.2 |
98.8 |
52.1 |
| 5 |
PDVB-[PhTVIM]Cl-5 |
10.7 |
97.9 |
38.9 |
| 6 |
P[PhTVIM]Cl |
24.5 |
98.5 |
41.1 |
Also seen from Table 2, the content of DVB incorporation also played an important role in the catalytic activity of PILs. Butylene oxide yield of 24.5% and TOF value of 41.1 h−1 could be achieved under the P[PhTVIM]Cl without DVB incorporating (Table 2, entry 6). Whereas, with DVB incorporated into the PILs, butylene oxide yield and TOF value first increased and then decreased, and PDVB-[PhTVIM]Cl-1 revealed the best activity with the butylene oxide yield of 31.1%, TOF value of 68.6 h−1 (Table 2, entry 3). It was reported that both of the surface area and IL content would affect the catalyst activity.53,54 It is seen from Table 1 that, incorporation 50 mol% (n = 1) of DVB into P[PhTVIM]Cl leads to the largely increase of specific surface area from 3 m2 g−1 to 125 m2 g−1, with a decrease of IL content from 1.71 to 1.19 m2 g−1 (Table 1, entries 1–2). Hence the exposure of the active centers of PILs increased and facilitated the contact between the active center of PILs and the substrate, which finally resulted in the increase of the catalytic activity. However, when the content of DVB further increased to 83 mol% (n = 5), although specific surface area of PILs continue increased from 125 m2 g−1 of PDVB-[PhTVIM]Cl-1 to 394 m2 g−1 of PDVB-[PhTVIM]Cl-5 (Table 1, entries 2 and 4), the TOF value and butylene oxide yield gradually reduced to 38.9 h−1 and 10.7% (Table 2, entries 3–5). This might be due to the existence of the competitive relationship between the specific surface area and the IL content. An increase in the specific surface area will correspondingly decrease the IL content from 1.19 to 0.79 m2 g−1 and reduce the density of active sites. Therefore, the catalytic activity of PILs should be the synergetic effect between surface area and IL content. Therefore, PDVB-[PhTVIM]Cl-1 can be regarded as the most efficient catalyst for the decarboxylation of butylene carbonate with the SBET of 125 m2 g−1 and the IL content of 1.19 mmol g−1.
3.3 Effect of catalyst dosage
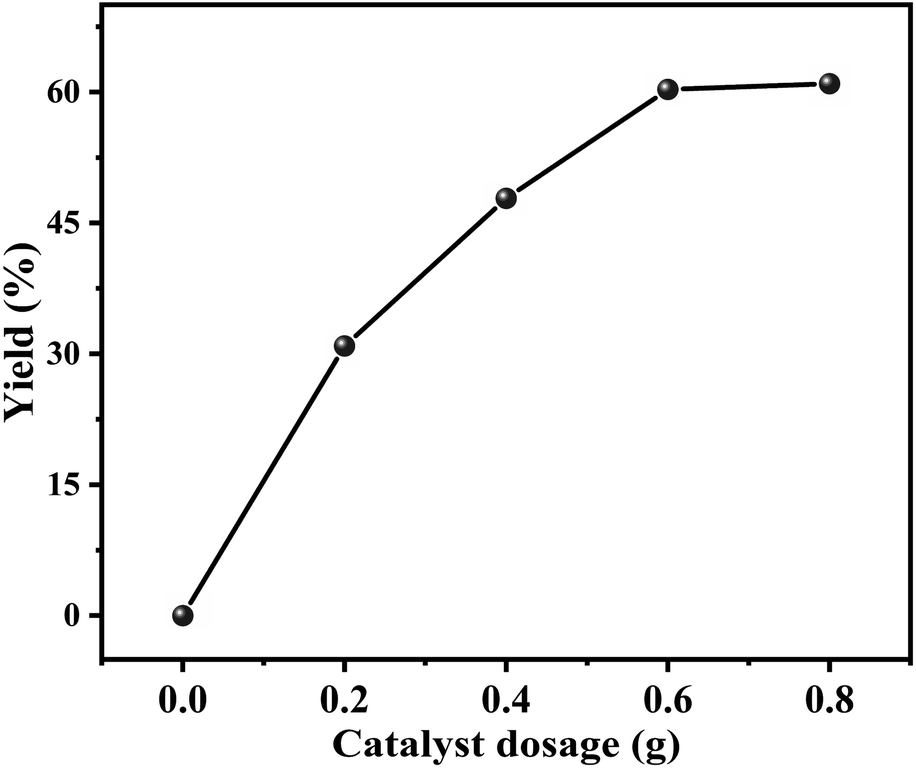
Effect of the catalyst dosage on the decarboxylation of butylene carbonate to produce butylene oxide was investigated in the range of 0–0.8 g, and the result was shown in Fig. 6. It could be observed that without catalyst, the decarboxylation reaction could not be carried out. As the catalyst dosage increased to 0.6 g, the yield of butylene oxide linearly increased to 60.3%. However, with further increasing the dosage of catalyst, the yield of butylene oxide hardly changed. The reason should be that the increasing amount of catalyst provided more active sites that facilitated its contact with the substrate, resulted in the increasing in butylene oxide yield.
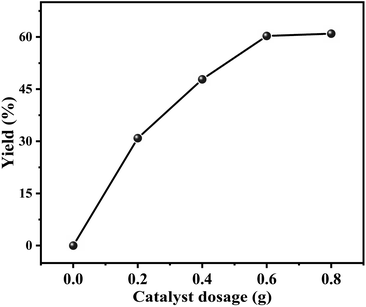 |
| | Fig. 6 Effect of catalyst dosage on the decarboxylation of butylene carbonate. Reaction conditions: butylene carbonate (20.0 g), reaction temperature (185 °C), vacuum degree (5 kPa), reaction time (180 min). | |
3.4 Effect of reaction temperature
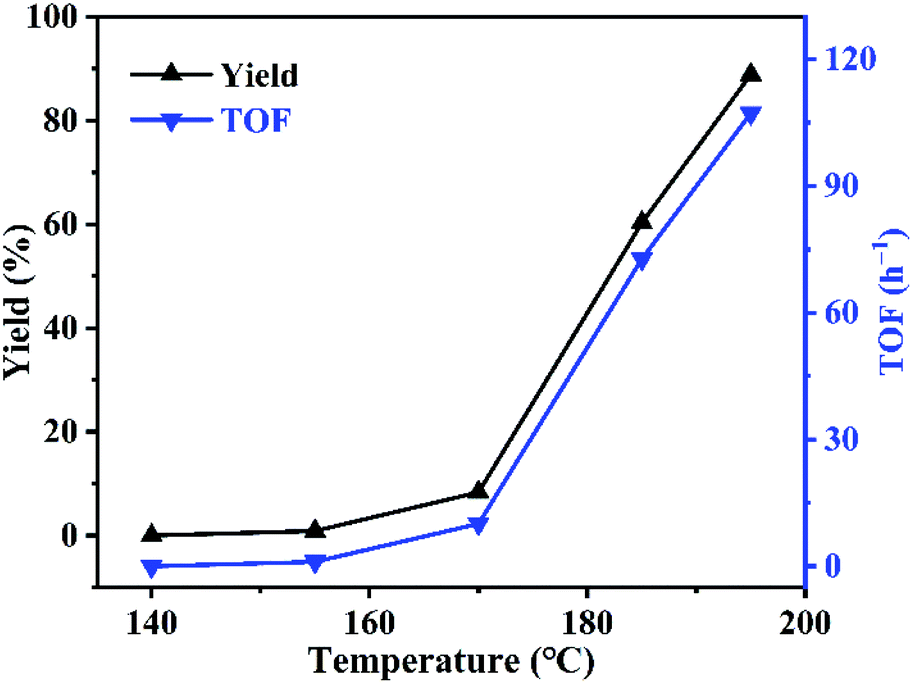
Effect of reaction temperature was investigated using PDVB-[PhTVIM]Cl-1 as catalyst. As can be seen in Fig. 7, the reaction rate is slow at first, and then turn fast. For example, at the temperature of 140 °C, the reaction did not carry out; with temperature increased from 140 to 170 °C, the yield of butylene oxide increased slightly from 0 to 8.4%. However, when temperature further increased to 195 °C, the yield of butylene oxide sharply increased to 88.9% with full butylene carbonate conversion. While, TOF value of PDVB-[PhTVIM]Cl-1 followed the same trend with butylene oxide yield. To our knowledge, the decarboxylation of cyclic carbonate is an endothermic process, hence, temperature increasing is beneficial to the reaction, and higher temperature is more favorable to this reaction.
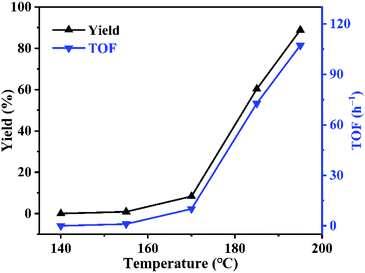 |
| | Fig. 7 Effect of reaction temperature on the decarboxylation of butylene carbonate. Reaction conditions: butylene carbonate (20.0 g), catalyst dosage (0.6 g), vacuum degree (5 kPa), reaction time (120 min). | |
3.5 Effect of reaction time
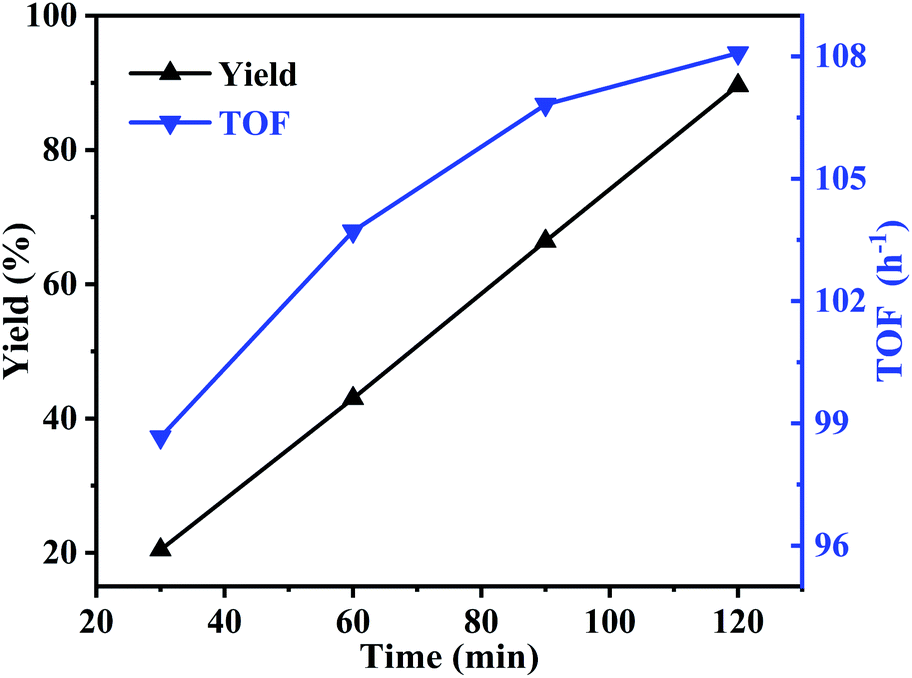
Meanwhile, the effect of reaction time was also studied at 195 °C. As can be seen in Fig. 8, with the reaction time increased from 30 to 120 min, the yield of butylene oxide increased linearly and rapidly from 20.5% to 89.6%, indicating high activity of the catalyst. While, TOF value showed similar trend with butylene oxide yield. When reaction time increased to 120 min, TOF value reached to 108.1 h−1, which was much higher than the homogeneous IL catalyst in our previous report (TOF value: 8.7 h−1).
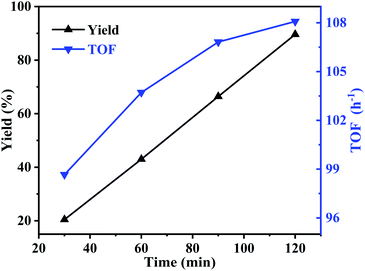 |
| | Fig. 8 Effect of reaction time on the decarboxylation of butylene carbonate. Reaction conditions: butylene carbonate (20.0 g), catalyst dosage (0.6 g), reaction temperature (195 °C), vacuum degree (5 kPa). | |
3.6 Reusability of the catalyst
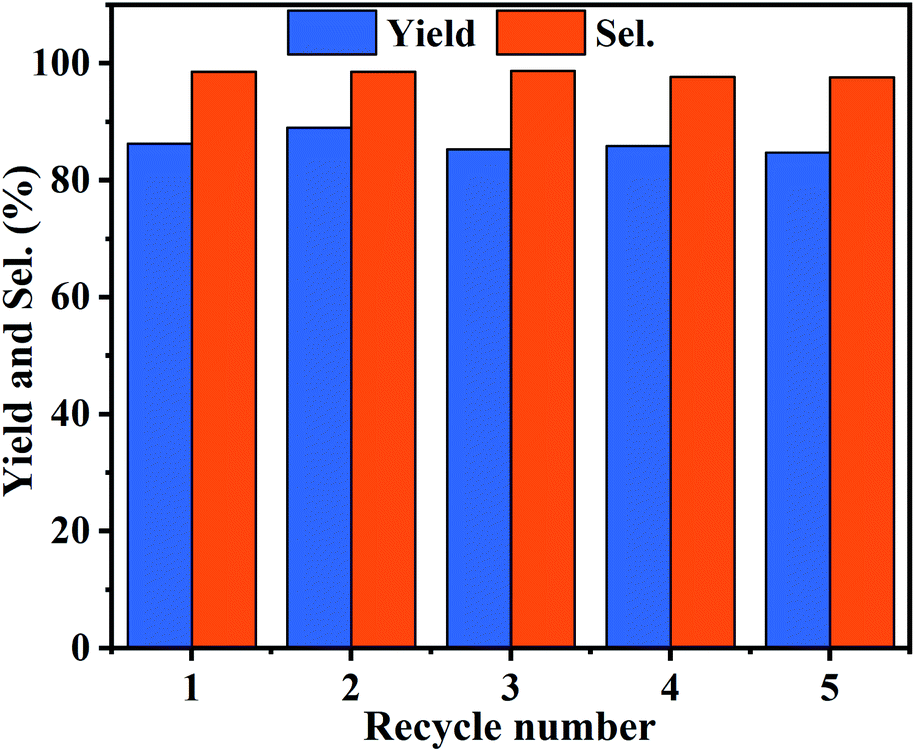
The reusability is significant for evaluating the industrial application of heterogeneous catalysts. Therefore, PDVB-[PhTVIM]Cl-1 was examined in a five-run recycling for decarboxylation of butylene carbonate to butylene oxide at the conditions of 0.6 g catalyst, 195 °C and 120 min. During 5 cycles, the yields and selectivity of butylene oxide have unnoticeable changes (Fig. 9). Moreover, the FT-IR spectrum (Fig. S5†) and SEM image (Fig. S6†) of the catalyst PDVB-[PhTVIM]Cl-1-Re recovered for 5 times showed that the typical imidazole rings stretching bands and morphology were still maintained compared with the fresh one. This further proved the excellent reusability of PDVB-[PhTVIM]Cl-1.
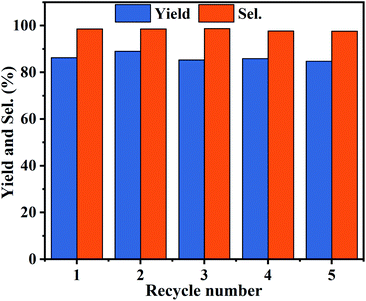 |
| | Fig. 9 Recycle performance of PDVB-[PhTVIM]Cl-1. Reaction conditions: butylene carbonate (20.0 g), catalyst dosage (0.6 g), reaction temperature (195 °C), vacuum degree (5 kPa), reaction time (120 min). | |
3.7 Substrate scope
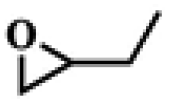
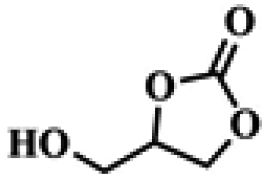
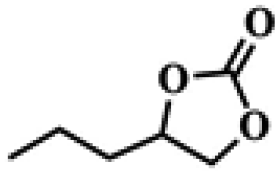
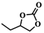
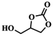
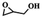
To investigate the versatility of PDVB-[PhTVIM]Cl-1 for the decarboxylation reaction of cyclic carbonates, a series of other cyclic carbonates, propylene carbonate, glycerol carbonate, pentenyl carbonate and hexenyl carbonate, were decomposed using PDVB-[PhTVIM]Cl-1 as the catalyst. As shown in Table 3, all substrates were effectively converted into corresponding epoxides with yields of 53.3–93.9% and TOF values of 51.8–126.0 h−1 under the further condensation and collection by liquid nitrogen. Notably, compared with pentenyl carbonate and hexenyl carbonate, substrates propylene carbonate and glycerol carbonate presented relatively higher reaction activity, the reason presumably attributed to the relatively shorter carbon-chain alkyl group can make substrates less sterically hindered and the ring-opening step easier to carry out.
Table 3 The decarboxylation with cyclic carbonates catalyzed by PDVB-[PhTVIM]Cl-1a
| Entry |
Substrate |
Product |
P/kPa |
Yieldb/% |
Sel.b/% |
TOFc/h−1 |
| Reaction conditions: substrate (20.0 g), PDVB-[PhTVIM]Cl-1 (0.6 g), reaction temperature (185 °C), reaction time (120 min). Determined by GC-MS and the yield referred to separation products. TOF (h−1) = [mmol product]/[mmol ionic sites] × [h]. |
| 1 |
 |
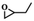 |
96 |
93.9 |
97.8 |
126.0 |
| 2 |
 |
 |
3 |
77.2 |
99.2 |
67.6 |
| 3 |
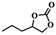 |
 |
96 |
57.4 |
98.9 |
61.8 |
| 4 |
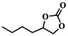 |
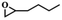 |
96 |
53.3 |
99.2 |
51.8 |
3.8 Proposed catalytic mechanism
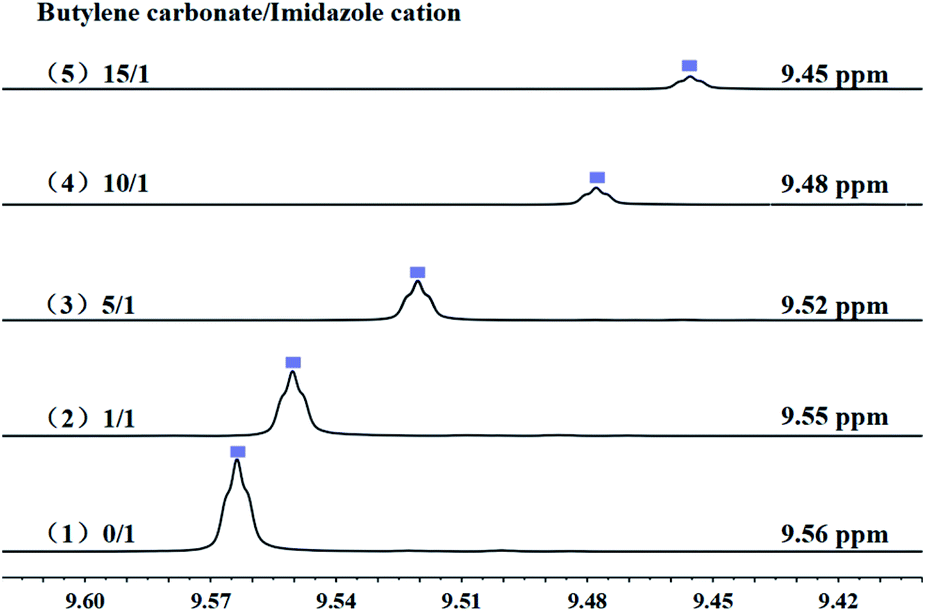
3.8.1 Interaction between catalyst and substrate. It was reported that, both cations and anions have impact on the substrate.55 Anions of imidazolium-based ILs play a key role in the ring-opening step of butylene carbonate via nucleophilic attack on the β-C atom of butylene carbonate, the results of which were verified in this paper and also by DFT calculation in our previous report.11While, in order to understand the role of cations of PDVB-[PhTVIM]Cl-1 in the decarboxylation of cyclic carbonates, the interaction between the imidazole cation of PDVB-[PhTVIM]Cl-1 and the butylene carbonate was determined by 1H NMR studies. [PhTVIM]Cl-1, the active units of PDVB-[PhTVIM]Cl-1, was selected as PIL fragment to provide imidazole cations. In a typical experiment, [PhTVIM]Cl and butylene carbonate were simultaneously dissolved in deuterated methanol (CD3OD) with the molar ratio of 0/1, 1/1, 5/1, 10/1, 15/1, respectively. As can be seen in Fig. 10, without butylene carbonate substrate, an obvious C2–H peak from imidazole cation at chemical shift of 9.56 ppm was detected; with adding butylene carbonate at the ratio of 1/1, the C2–H peak shifted to 9.55 ppm, and with gradually increasing the amount of butylene carbonate feeding from 1/1 to 15/1, the chemical shift of C2–H of imidazole cation also gradually shifted from 9.55 to 9.45 ppm, along with peak area decreasing. This result suggested that the strength of hydrogen-bond in this system tend to be weakened with the continuous addition of butylene carbonate. However, the widely accepted view56–59 is that as hydrogen-bonds are formed, the hydrogen atoms in the compound will move to the downfield in NMR analysis, the trend of which is opposite to the results in Fig. 10.
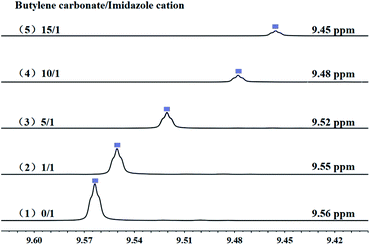 |
| | Fig. 10 The chemical shift of [PhTVIM]Cl in the 1H NMR spectra (CD3OD). The molar ratio of butylene carbonate to imidazole cations of [PhTVIM]Cl: (1) 0/1, (2) 1/1, (3) 5/1, (4) 10/1, (5) 15/1. | |
It is generally accepted that, both of Cl− of imidazolium-based ILs and carbonyl oxygen O1 of cyclic carbonate act as hydrogen-bond acceptors and had a hydrogen-bond interaction with the C2–H of the imidazole cations.60 However, compared with the hydrogen-bond formed between Cl− and C2–H in imidazolium, the hydrogen-bond formed by carbonyl oxygen O1 of butylene carbonate and C2–H is relatively weak. Therefore, it is speculated that the formation of the hydrogen-bond between the C2–H of imidazolium and carbonyl oxygen O1 of butylene carbonate lead to the high-field shift of the C2–H in imidazolium.
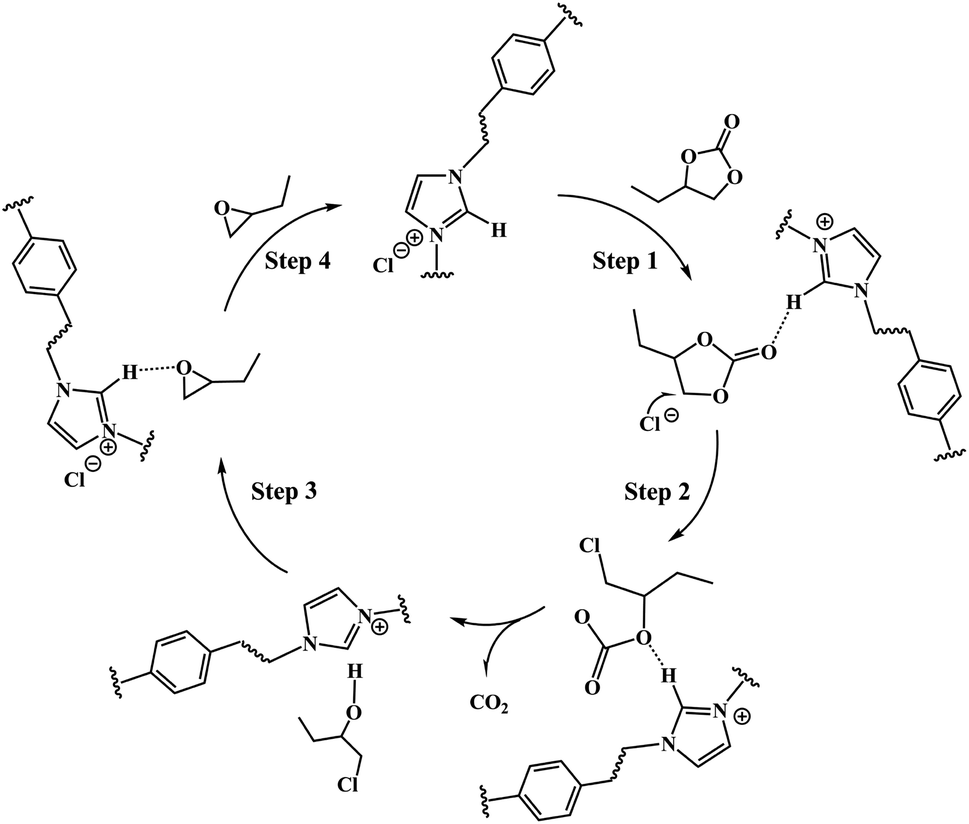
3.8.2 Possible mechanism. The above results proved the existence of the synergistic effect of anion and cation of the PILs catalyst on the substrate. The C2–H of the imidazole cations activate the carbonyl oxygen O1 of butylene carbonate by forming C–H⋯O hydrogen-bond, while the strong nucleophilic anions of PILs open the five-membered ring by attacking β-C of the cyclic carbonate.As a result, a plausible mechanism that contributed to the formation of butylene oxide through the decarboxylation of butylene carbonate catalyzed by PDVB-[PhTVIM]Cl-1 was proposed. As depicted in Scheme 2, firstly, the butylene carbonate was activated by the hydrogen-bond interaction formed between its own O atom and the H atom of the imidazole cation, thereby accelerating the ring opening of the cyclic carbonate (Step 1). At the same time, the Cl− anion nucleophilic attacked the less sterically hindered β-C on the butylene carbonate to form a ring-opening intermediate (Step 2). Subsequently, carbon dioxide dissociated from the molecular structure with the formation of chloroalkyl intermediates (Step 3). Finally, the epoxide was obtained by the intramolecular cyclization of the chloroalkyl intermediate and the departure of the chlorine process; and the catalyst was regenerated at the same time (Step 4).
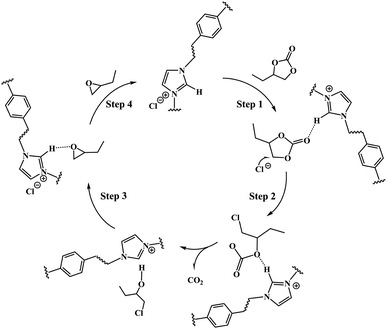 |
| | Scheme 2 Plausible mechanism of PDVB-[PhTVIM]Cl-1 catalyzed decarboxylation reaction of butylene carbonate to produce butylene oxide. | |
4. Conclusions
In summary, a series of DVB incorporated imidazolium-based porous PILs with different anions were successfully synthesized via free-radical copolymerization. FTIR, 13C CP/MAS NMR and XPS proved the successful incorporation of tris-imidazolium-based IL moiety into the polymer skeleton. N2 adsorption analysis showed the IL content and porosity of PILs were greatly affected by the content of DVB. This hierarchical pore structure which was also confirmed by SEM and TEM facilitated the mass transfer of the substrate, hence increase the catalytic activity of PILs. Performance of PILs with different anions indicated that, the activity of PILs was in the order of Cl− > Br− > BF4−, which was related to the nucleophilicity property of it. The polymeric imidazolium salt PDVB-[PhTVIM]Cl-1 simultaneously owned plentiful ionic sites and abundant mesopores, showing the best performance with butylene oxide yield of 89.6% and TOF value of 108.1 h−1. What's more, PDVB-[PhTVIM]Cl-1 could be easily reused with stable recycling performance. Meanwhile, PDVB-[PhTVIM]Cl-1 was also active in the decarboxylation reaction of various epoxides cyclic carbonates, suggesting its versatility for cyclic carbonates. At last, the cooperative catalytic mechanism between anions and cations of PILs was proposed. The ring opening step of cyclic carbonate was carried out through the hydrogen-bond interaction formed between the C2–H of the imidazole cations and the carbonyl oxygen O1 and the attack of anions on β-C of butylene carbonate, respectively. This work developed an effective and recyclable PILs catalyst for the green synthesis of epoxide through decarboxylation of cyclic carbonate.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financially supported by Science and Technology Service Network Initiative, Chinese Academy of Sciences (KFJ-STS-QYZD-138) and, “Transformational Technologies for Clean Energy and Demonstration”, Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA 21030600).
References
- Y. Zhu, C. Romain and C. K. Williams, Nature, 2016, 540, 354–362 CrossRef CAS PubMed.
- Z. F. Yan, J. X. Tian, K. Wang, K. D. P. Nigam and G. S. Luo, Chem. Eng. J., 2021, 229 Search PubMed.
- J. M. Longo, M. J. Sanford and G. W. Coates, Chem. Rev., 2016, 116, 15167–15197 CrossRef CAS PubMed.
- J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed.
- H. Misaka, E. Tamura, K. Makiguchi, K. Kamoshida, R. Sakai, T. Satoh and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 1941–1952 CrossRef CAS.
- J. Langanke, A. Wolf, J. Hofmann, K. Bohm, M. A. Subhani, T. E. Muller, W. Leitner and C. Gurtler, Green Chem., 2014, 16, 1865–1870 RSC.
- H. J. Zhang, C. Niu, Y. N. Zhang, X. Wang and B. Yang, Chim. Oggi, 2020, 298, 619–628 CAS.
- J. Seiwert, D. Leibig, U. Kemmer-Jonas, M. Bauer, I. Perevyazko, J. Preis and H. Frey, Macromolecules, 2016, 49, 38–47 CrossRef CAS.
- Z. F. Yan, J. X. Tian, K. Wang, K. D. P. Nigam and G. S. Luo, Chem. Eng. J., 2021, 229, 116071 CrossRef CAS.
- Q. L. Chen and E. J. Beckman, Green Chem., 2008, 10, 934–938 RSC.
- X. Niu, L. G. Wang, J. Y. Cao, Y. Cao, P. He, J. Y. Zhou and H. Q. Li, RSC Adv., 2019, 9, 10072–10080 RSC.
- F. Schmidt, M. Bernhard, H. Morell and M. Pascaly, Chim. Oggi, 2014, 32, 31–35 CAS.
- V. Russo, R. Tesser, E. Santacesaria and M. Di Serio, Ind. Eng. Chem. Res., 2013, 52, 1168–1178 CrossRef CAS.
- M. Lin, C. J. Xia, B. Zhu, H. Li and X. T. Shu, Chem. Eng. J., 2016, 295, 370–375 CrossRef CAS.
- C. L. Bolivar-Diaz, V. Calvino-Casilda, F. Rubio-Marcos, J. F. Fernandez and M. A. Banares, Appl. Catal., B, 2013, 129, 575–579 CrossRef CAS.
- Y. K. Endah, M. S. Kim, J. Choi, J. Jae, S. D. Lee and H. Lee, Catal. Today, 2017, 293, 136–141 CrossRef.
- R. P. Ye, L. Lin, C. C. Chen, J. X. Yang, F. Li, X. Zhang, D. J. Li, Y. Y. Qin, Z. F. Zhou and Y. G. Yao, ACS Catal., 2018, 8, 3382–3394 CrossRef CAS.
- Y. J. Zhao, H. H. Zhang, Y. X. Xu, S. N. Wang, Y. Xu, S. P. Wang and X. B. Ma, J. Energy Chem., 2020, 49, 248–256 CrossRef.
- D. W. Yao, Y. Wang, K. Hassan-Legault, A. T. Li, Y. J. Zhao, J. Lv, S. Y. Huang and X. B. Ma, ACS Catal., 2019, 9, 2969–2976 CrossRef CAS.
- J. Q. Feng, H. X. Guo, S. P. Wang, Y. J. Zhao and X. B. Ma, Chem. Eng. J., 2017, 321, 401–411 CrossRef CAS.
- A. Wang and T. Zhang, Acc. Chem. Res., 2013, 46, 1377–1386 CrossRef CAS PubMed.
- C. Li, X. Zhao, A. Wang, G. W. Huber and T. Zhang, Chem. Rev., 2015, 115, 11559–11624 CrossRef CAS PubMed.
- J. F. Pang, M. Y. Zheng, A. Q. Wang and T. Zhang, Ind. Eng. Chem. Res., 2011, 50, 6601–6608 CrossRef CAS.
- Y. Q. Xie, J. Liang, Y. W. Fu, M. T. Huang, X. Xu, H. T. Wang, S. Tu and J. Li, J. Mater. Chem. A, 2018, 6, 6660–6666 RSC.
- J. M. Lu, F. Yan and J. Texter, Prog. Polym. Sci., 2009, 34, 431–448 CrossRef CAS.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, M. Kar, S. Passerini, J. M. Pringle, H. Ohno, M. Watanabe, F. Yan, W. J. Zheng, S. G. Zhang and J. Zhang, Nat. Rev. Mater., 2016, 1, 15005 CrossRef CAS.
- W. Qian, J. Texter and F. Yan, Chem. Soc. Rev., 2017, 46, 1124–1159 RSC.
- J. Zhang, S. J. Zhang, J. X. Han, Y. H. Hu and R. Y. Yan, Chem. Eng. J., 2015, 271, 269–275 CrossRef CAS.
- D. Ager, V. Arjunan Vasantha, R. Crombez and J. Texter, ACS Nano, 2014, 8, 11191–11205 CrossRef CAS PubMed.
- Z. W. Wu, C. Chen, Q. R. Guo, B. X. Li, Y. G. Que, L. Wang, H. Wan and G. F. Guan, Fuel, 2016, 184, 128–135 CrossRef CAS.
- C. Gao, G. Chen, X. Wang, J. Li, Y. Zhou and J. Wang, Chem. Commun., 2015, 51, 4969–4972 RSC.
- X. Wang, Y. Zhou, Z. Guo, G. Chen, J. Li, Y. Shi, Y. Liu and J. Wang, Chem. Sci., 2015, 6, 6916–6924 RSC.
- S. Soll, P. F. Zhang, Q. Zhao, Y. Wang and J. Y. Yuan, Polym. Chem., 2013, 4, 5048–5051 RSC.
- F. Liu, L. Wang, Q. Sun, L. Zhu, X. Meng and F. S. Xiao, J. Am. Chem. Soc., 2012, 134, 16948–16950 CrossRef CAS PubMed.
- H. B. Song, Y. J. Wang, M. Xiao, L. Liu, Y. L. Liu, X. F. Liu and H. J. Gai, ACS Sustainable Chem. Eng., 2019, 7, 9489–9497 CrossRef CAS.
- Y. Zhou, W. L. Zhang, L. Ma, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9387–9398 CrossRef CAS.
- F. Liu, W. Li, Q. Sun, L. Zhu, X. Meng, Y. H. Guo and F. S. Xiao, ChemSusChem, 2011, 4, 1059–1062 CrossRef CAS PubMed.
- H. Zhou, Y. M. Zhou, L. Li, Y. H. Li, X. Q. Liu, P. Zhao and B. Gao, ACS Sustainable Chem. Eng., 2019, 7, 9281–9290 CrossRef CAS.
- K. Tauber, Q. Zhao, M. Antonietti and J. Y. Yuan, ACS Macro Lett., 2015, 4, 39–42 CrossRef.
- Q. Sun, Y. Jin, B. Aguila, X. Meng, S. Ma and F. S. Xiao, ChemSusChem, 2017, 10, 1160–1165 CrossRef CAS PubMed.
- Y. Y. Zhang, B. S. Wang, E. H. M. Elageed, L. Qjn, B. Ni, X. L. Liu and G. H. Gao, ACS Macro Lett., 2016, 5, 435–438 CrossRef CAS.
- T. Duan-Jian, L. F. Jian, W. Lin and J. L. Long, Appl. Catal., A, 2018, 564, 56–63 CrossRef.
- S. M. Morozova, A. S. Shaplov, E. I. Lozinskaya, D. Mecerreyes, H. Sardon, S. Zulfiqar, F. Suarez-Garcia and Y. S. Vygodskii, Macromolecules, 2017, 50, 2814–2824 CrossRef CAS.
- J. Y. Yuan, S. Prescher, K. Sakaushi and M. Antonietti, J. Mater. Chem. A, 2015, 3, 7229–7234 RSC.
- Q. Q. Liu, L. Wang and A. G. Xiao, Des. Monomers Polym., 2007, 10, 405–423 CrossRef CAS.
- L. Qin, B. Wang, Y. Zhang, L. Chen and G. Gao, Chem. Commun., 2017, 53, 3785–3788 RSC.
- Y. Zhou, W. L. Zhang, L. Ma, Y. Zhou and J. Wang, ACS Sustainable Chem. Eng., 2019, 7, 9026–9726 Search PubMed.
- Z. J. Guo, Q. W. Jiang, Y. M. Shi, J. Li, X. N. Yang, W. Hou, Y. Zhou and J. Wang, ACS Catal., 2017, 7, 6770–6780 CrossRef CAS.
- J. Li, D. G. Jia, Z. J. Guo, Y. Q. Liu, Y. N. Lyu, Y. Zhou and J. Wang, Green Chem., 2017, 19, 2675–2686 RSC.
- Z. J. Guo, X. C. Cai, J. Y. Xie, X. C. Wang, Y. Zhou and J. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 12812–12821 CrossRef CAS PubMed.
- D. G. Jia, L. Ma, Y. Wang, W. L. Zhang, J. Li, Y. Zhou and J. Wang, Chem. Eng. J., 2020, 390, 13 CrossRef.
- A. Kutt, T. Rodima, J. Saame, E. Raamat, V. Maemets, I. Kaljurand, I. A. Koppel, R. Y. Garlyauskayte, Y. L. Yagupolskii, L. M. Yagupolskii, E. Bernhardt, H. Willner and I. Leito, J. Org. Chem., 2011, 76, 391–395 CrossRef CAS PubMed.
- Y. Xie, Z. F. Zhang, T. Jiang, J. L. He, B. X. Han, T. B. Wu and K. L. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255–7258 CrossRef CAS PubMed.
- J. Biemol, I. M. Denekam, T. K. Slo, G. Rothenber and D. Eisenber, ChemSusChem, 2017, 10, 3862–3862 CrossRef.
- J. S. Choi, F. S. H. Simanjuntaka, J. Y. Oh, K. I. Lee, S. D. Lee, M. Cheong, H. S. Kim and H. Lee, J. Catal., 2013, 297, 248–255 CrossRef CAS.
- A. M. Hardman-Baldwin and A. E. Mattson, ChemSusChem, 2014, 7, 3275–3278 CrossRef CAS PubMed.
- J. Q. Wang and Y. G. Zhang, ACS Catal., 2016, 6, 4871–4876 CrossRef CAS.
- Z. F. Yang, J. Sun, W. G. Cheng, J. Q. Wang, Q. Li and S. J. Zhang, Catal. Commun., 2014, 44, 6–9 CrossRef CAS.
- S. Liang, H. Liu, T. Jiang, J. Song, G. Yang and B. Han, Chem. Commun., 2011, 47, 2131–2133 RSC.
- M. S. Liu, X. Wang, Y. C. Jiang, J. M. Sun and M. Arai, Catal. Commun., 2018, 61, 214–269 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01039e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *bcde,
Yan Caob,
Shuang Xub,
Peng Heb,
Huiquan Li
*bcde,
Yan Caob,
Shuang Xub,
Peng Heb,
Huiquan Li