
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEfficient biodegradation of malachite green by an artificial enzyme designed in myoglobin†
Heng-Fang Xianga,
Jia-Kun Xu*b,
Jiao Liua,
Xin-Zhi Yangc,
Shu-Qin Gaoc,
Ge-Bo Wenc and
Ying-Wu Lin *ac
*ac
aSchool of Chemistry and Chemical Engineering, University of South China, Hengyang 421001, China. E-mail: ywlin@usc.edu.cn
bKey Lab of Sustainable Development of Polar Fisheries, Ministry of Agriculture and Rural Affairs, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Lab for Marine Drugs and Byproducts of Pilot National Lab for Marine Science and Technology, Qingdao 266071, China. E-mail: xujk@ysfri.ac.cn
cLaboratory of Protein Structure and Function, University of South China Medical School, Hengyang 421001, China
First published on 30th April 2021
Abstract
Synthetic dyes such as malachite green (MG) have a wide range of applications. Meanwhile, they bring great challenges for environmental security and cause potential damages to human health. Compared with traditional approaches, enzymatic catalysis is an emerging technique for wastewater treatment. As alternatives to natural enzymes, artificial enzymes have received much attention for potential applications. In previous studies, we have rationally designed artificial enzymes based on myoglobin (Mb), such as by introducing a distal histidine (F43H mutation) and creating a channel to the heme pocket (H64A mutation). We herein show that the artificial enzyme of F43H/H64A Mb can be successfully applied for efficient biodegradation of MG under weak acid conditions. The degradation efficiency is much higher than those of natural enzymes, such as dye-decolorizing peroxidase and laccase (13–18-fold). The interaction of MG and F43H/H64A Mb was investigated by using both experimental and molecular docking studies, and the biodegradation products of MG were also revealed by UPLC-ESI-MS analysis. Based on these results, we proposed a plausible biodegradation mechanism of MG. With the high-yield of overexpression in E. coli cells, this study suggests that the artificial enzyme has potential applications in the biodegradation of MG in fisheries and textile industries.
Introduction
Both natural and synthetic dyes are widely used in daily life, such as papermaking, food, leather and cosmetics.1 Compared with natural dyes, synthetic dyes have the advantages of being produced in large yields, with stable bright color that does not easily fade. Meanwhile, synthetic dyes also bring great challenges for environmental security. For example, in the dyeing process, more than a million tons of 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 types of dyes are released into waste water every year, including azo, anthraquinones, phthalocyanine and triarylmethane dyes.2
000 types of dyes are released into waste water every year, including azo, anthraquinones, phthalocyanine and triarylmethane dyes.2
Malachite green (MG) is a synthetic dye of N-methylated diaminotriphenylmethane (Fig. 1A), which has been extensively used in textile industries.3 Moreover, with availability at a low cost, MG was widely used as the most efficient antifungal agent in the fisheries for the treatment of bacterial and fungal infections. However, it is forbidden in the fisheries in many counties because of its potential toxicity for consumers.4 Therefore, it is vital to develop other approaches for the removal of MG from the environment and preventing potential damages to human health.
 | ||
| Fig. 1 (A) Chemical structure of malachite green and various degradation approaches; (B) rational design of artificial enzyme in the protein matrix of myoglobin (Mb) by introducing a distal histidine and creating a channel to the heme pocket. The X-ray structures of WT Mb (PDB code 1JP633), F43H Mb (PDB code 4PQC30), and F43H/H64A Mb (PDB code 5HLQ31) were shown for the heme active site, respectively. | ||
To date, there are various physical and chemical approaches have been developed to remove synthetic dyes from the environment.5–7 As an efficient and environment-friendly method, enzymatic catalysis has attracted much attention in the last decade. For example, Zhang et al. isolated and purified a copper-containing enzyme, laccase, from Bacillus vallismortis, and showed that it was able to degrade MG.8,9 Recently, Guan and colleagues expressed a mutant of CotA-laccase SF in E. coli cells and showed that it could decolorize MG under neutral and alkaline conditions.10 Enzyme immobilization was also used to enhance the stability of laccase in decolorization.11 Moreover, heme enzymes such as horseradish peroxidase and manganese peroxidase were also applied for the decolorization of MG.12,13 Meanwhile, the catalytic efficiency of these natural enzymes is not satisfied, and it is still needed to seek for more efficient enzymes for the biodegradation.
As alternatives to natural enzymes, artificial enzymes have received much attention for decades due to their potential applications, including in the environment.14–21 With the advantages of high-yield of overexpression in E. coli cells, myoglobin (Mb), an O2 carrier, has been favored for rational design of artificial enzymes with multiple functions.22 For example, Lu and colleagues rationally designed artificial heme-copper oxidases and nitric oxide reductases by creating CuB or FeB metal-binding site in Mb.23 Zhang and colleagues designed functional peroxidases by construction of Mn-reconstituted Mb.24 Our group also constructed multiple artificial enzymes using Mb as a model protein, such as dye-decolorizing peroxidases, dehaloperoxidases and artificial nucleases.25–28
Inspired by the finding from Watanabe and colleagues that the position of a distal histidine in Mb is crucial for H2O2 activation,29 we introduced a distal histidine (F43H mutation) and created a channel to the heme site (H64A mutation) of Mb, and solved the X-ray crystal structures of both F43H Mb and F43H/H64A Mb (Fig. 1B).30,31 The double mutant of F43H/H64A Mb was shown to exhibit not only enhanced peroxidase activity but also improved nitrite reductase activity compared with wild-type (WT) Mb.31,32
In this study, we further applied the artificial enzyme F43H/H64A Mb for the biodegradation of MG. We performed kinetic UV-Vis studies to determine the catalytic efficiency, and identified the degradation products by UPLC-ESI-MS analysis. Moreover, we performed a molecular modeling study for MG binding to the enzyme, and proposed a possible degradation mechanism based on the results.
Results and discussion
Molecular modeling of MG binding to F43H/H64A Mb
It is critical of substrate binding to the enzyme for efficient oxidation.34 To probe whether MG could bind to the protein scaffold of F43H/H64A Mb, we performed a molecular docking study based on the X-ray structure of F43H/H64A Mb. The results showed that ten most favorable complexes have the similar conformations with the similar binding energy (about −5.35 kcal mol−1, Table S1†). The overall structure of the MG–F43H/H64A Mb complexes were shown in Fig. 2A, which revealed that the molecule of MG, like the shape of a fan, binds to the cavity between helices E and F close to the heme group. The enlarged view provided more detailed information for MG–protein interactions (Fig. 2B), which showed that the benzene rings interact with the surrounding residues, including Phe138, Ile75 and Leu89, by hydrophobic interactions, such as van der Walls and π–π packing interactions. Moreover, an H-bond interaction may also occur between the methanaminium group and the backbone carbonyl group of Pro88 (∼3.6 Å), although it may not occur for Thr70 (∼4.7 Å). Therefore, these observations suggest that MG potentially binds to the protein of Mb, with a binding model favorable for oxidation.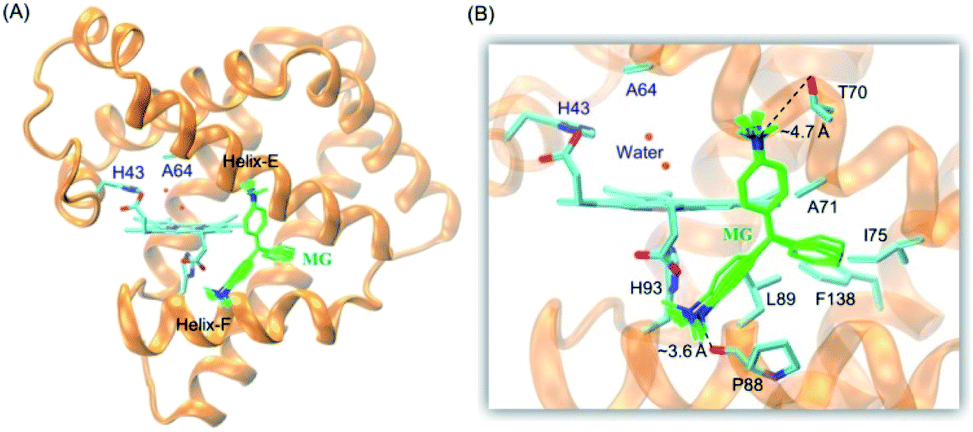 | ||
| Fig. 2 (A) Molecular modeling structure of the MG–F43H/H64A Mb complexes, and (B) detailed interactions between MG and the surrounding residues. | ||
Experimental studies of MG binding to F43H/H64A Mb
To provide experimental evidence for MG binding to F43H/H64A Mb, we first performed the ITC study (Fig. 3A). The results showed that the MG to F43H/H64A Mb titration caused decreases in the binding enthalpy (ΔH), indicating an exothermic nature of the binding. To evaluate the properties of binding thermodynamics, we analyzed the results by fitting the plot. It produced negative values for both binding enthalpy (ΔH) and binding entropy (ΔS), with a Ka value of 2.11 × 103 M−1, indicating a weak binding of MG to the protein. The negative binding free energy (ΔG) further suggests that MG binding is a spontaneous process.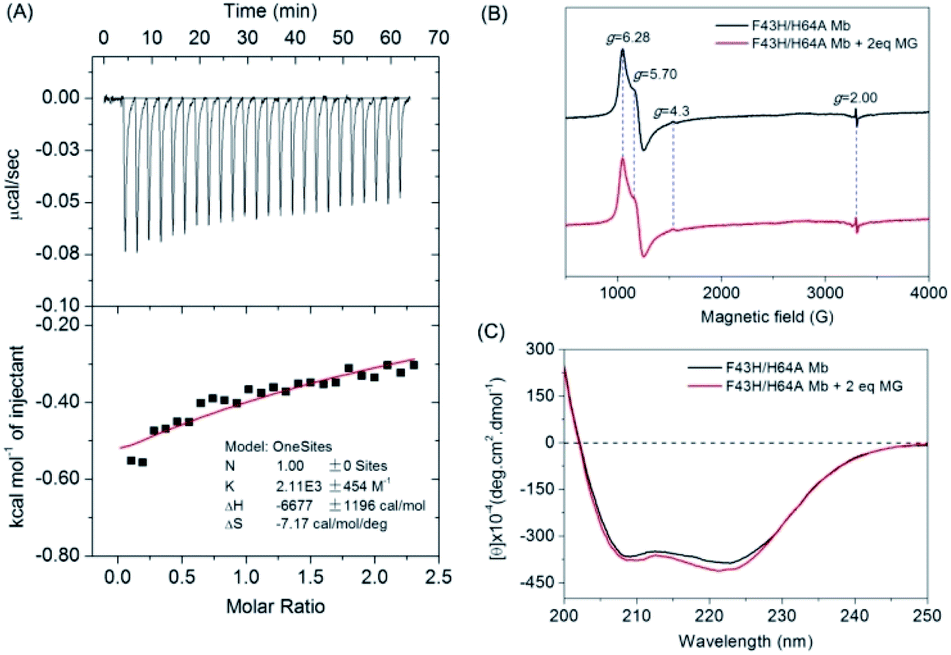 | ||
| Fig. 3 (A) ITC data for titration of F43H/H64A Mb (40 μM) with MG (0.5 mM) in KPi buffer (100 mM, pH 6.0) at 25 °C, showing the raw data (top) and the plot of integrated heats vs. MG/Mb ratio (bottom). (B) EPR spectra of ferric F43H/H64A Mb (0.5 mM) in the absence and presence of MG (1 mM) in KPi buffer (100 mM, pH 6.0), (C) CD spectra of ferric F43H/H64A Mb (1 μM) in the absence and presence of MG (1 μM) in KPi buffer (5 mM, pH 6.0). | ||
To probe whether MG binding affect the conformation of the heme active site, we performed EPR studies for ferric F43H/H64A Mb in the absence or presence of MG (Fig. 3B). The results showed that the ferric protein exhibited typical high-spin heme signals, with two g-values (5.70 and 6.28) close to g = 6 and the third value at g = 2, as observed previously for other Mbs by Reeder et al.35 Note that the tiny signal at g = 4.3 was originated from the rhombic ferric iron.36 These heme signals almost remained the same in the presence of MG, which suggests that the binding of MG does not affect the heme active site.
To further probe the effect of MG binding on the protein secondary structure, we collected the far UV-CD spectra of F43H/H64A Mb in the absence and presence of MG (Fig. 3C). The spectrum of F43H/H64A Mb exhibited two major absorptions at 209 nm and 223 nm, which indicates that the protein has more α-helix content than β-sheets. Upon the addition of MG, only slight decreases in ellipticity were observed for the characteristic regions of the secondary structure, suggesting that the binding of Mb almost has no effect on the secondary structure.
MG degradation catalyzed by F43H/H64A Mb
After confirming that MG does bind to the protein of F43H/H64A Mb, we tested whether the artificial enzyme could catalyze the oxidation of MG. We first observed the color of the MG solution with the enzyme by addition of H2O2 as an oxidant and increased its concentration from 0 to 1 mM (Fig. 4A). The digital photos showed that after reaction for 2 h, the typical green color of MG disappeared by an addition of 200 μM H2O2 (Fig. 4Ac), whereas the presence of higher concentrations of H2O2 such as 1 mM generated a light blue solution (Fig. 4Ah), indicating further reaction of the degradation products.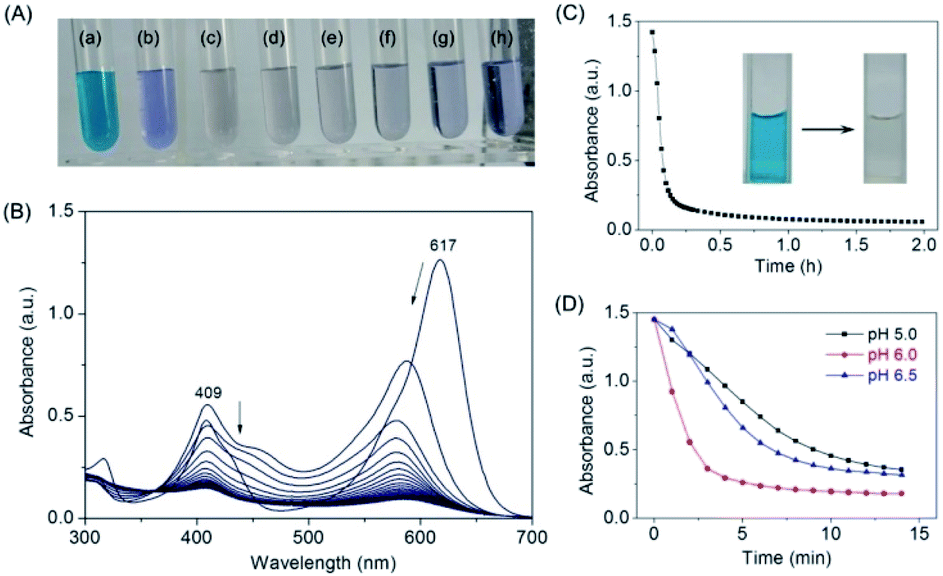 | ||
| Fig. 4 (A) Digital photos of MG (10 μM) oxidized by F43H/H64A Mb (2 μM) with different H2O2 concentrations ((a) 0 μM; (b) 100 μM; (c) 200 μM; (d) 300 μM; (e) 400 μM; (f) 600 μM; (g) 800 μM; (h) 1000 μM), in KPi buffer (100 mM, pH 6.0) at 25 °C for 2 h. (B) UV-Vis spectra of MG (10 μM) oxidized by F43H/H64A Mb (2 μM) with H2O2 (200 μM) under the same conditions. (C) Kinetic changes of absorption at 617 nm upon oxidation of MG catalyzed by F43H/H64A Mb. The digital photos of the solution before and after reaction were shown as insets. (D) Effect of pH values on the catalytic oxidation rate of MG by F43H/H64A Mb the similar conditions. | ||
We then monitored the UV-Vis spectra of MG oxidized by F43H/H64A Mb upon the addition of H2O2 (Fig. 4B). The results showed that the typical absorption of MG at 617 nm decreased and slightly blue-shifted over time. The kinetic changes of the absorption of MG at 617 nm were shown in Fig. 4C, which showed that MG was almost degraded within half an hour. The digital photos of the solution before and after reaction were shown in Fig. 4C as insets. Control studies with WT Mb and F43H Mb under the same conditions showed that F43H Mb was more reactive than that of WT Mb (Fig. S3†), and less reactive compared to the double mutant of F43H/H64A Mb, which indicate the critical roles of both His43 and the channel of Ala64.
Moreover, we tested the effect of pH values on the oxidation rates by monitoring the spectral changes at 617 nm. As shown in Fig. 4D, a maximal activity was observed at pH 6.0, which might facilitates the activation of H2O2 at this condition. Therefore, we selected as the optimal reaction condition (200 μM H2O2, pH 6.0) for the degradation of MG catalyzed by F43H/H64A Mb in following experiments.
Catalytic efficiency of F43H/H64A Mb toward MG
To obtain the kinetic parameters for degradation of MG by F43H/H64A Mb, we carried out kinetic UV-Vis studies using different concentrations of MG, and monitored the absorption changes at 617 nm in the UV-Vis spectra under the optimal reaction conditions. The values of kobs and the concentrations of MG were then plotted and fitted to the Michaelis–Menten equation (Fig. 5A). Table 1 listed the parameters of kcat and Km. The results showed that F43H/H64A Mb exhibited a kcat value of 0.0232 s−1, which is much higher than that of WT Mb (∼16-fold) and F43H Mb (∼6-fold), suggesting that not only the distal His43 but also the channel to heme pocket play crucial roles in the activity.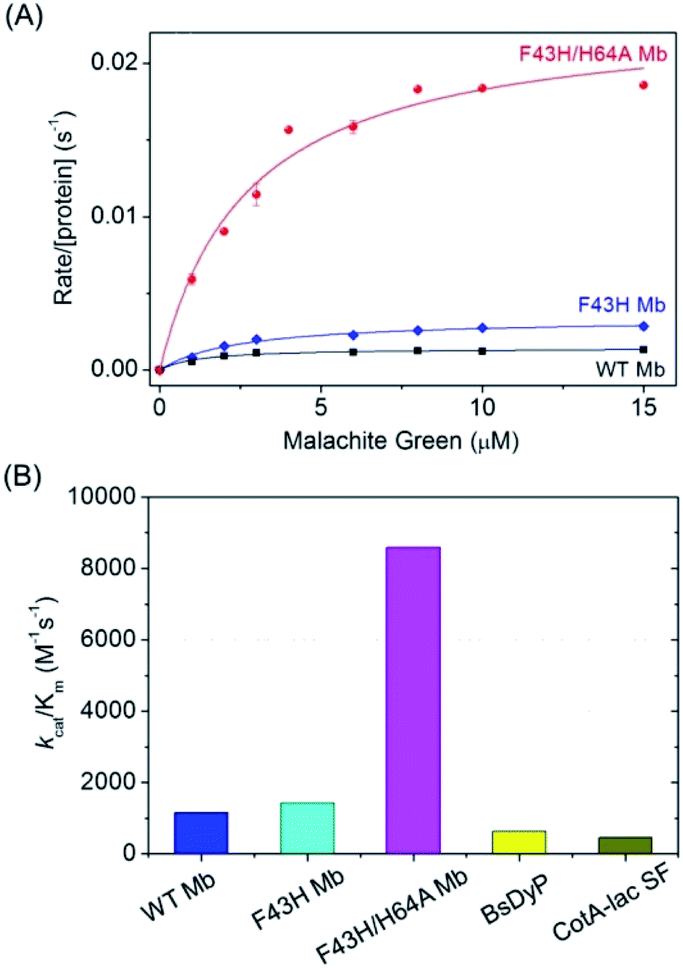 | ||
| Fig. 5 (A) Steady-state rates of oxidation as a function of MG concentrations (0–15 μM) catalyzed by WT Mb, F43H Mb and F43H/H64A Mb (2 μM) in 100 mM KPi buffer at pH 6.0, using H2O2 (0.2 mM) as an oxidant. (B) The comparison of the catalytic efficiency of WT Mb, F43H Mb, F43H/H64A Mb and the natural enzymes. | ||
On the other hand, both F43H Mb and F43H/H64A Mb exhibited showed a low Km value (2–3 μM), which is much less than those observed for natural enzymes (70 μM to 39.6 mM, Table 1). As a result, although the kcat value of F43H/H64A Mb is less than those of the natural enzymes, the catalytic efficiency (kcat/Km) was determined to be 8593 M−1 s−1, which is 13–18-fold higher than that reported for dye-decolorizing peroxidase from Bacillus subtilis (BsDyp)37 and the mutated CotA-laccase SF enzyme.10 Moreover, much less activity was reported previously for laccase from white-rot fungus Cerrenasp, with a catalytic efficiency as low as ∼12 M−1 s−1.38 These observations indicate that the protein matrix of Mb is more suitable for MG binding compared to that of natural enzymes, and the artificial enzyme designed in Mb is thus an alternative to natural enzymes for potential applications.
Product analysis of MG degradation by F43H/H64A Mb
To further identify the degradation products of MG catalyzed by F43H/H64A Mb, we performed UPLC-ESI-MS studies for the solution after reaction. As shown in Fig. 6A for the mass of MG (329 m/z), the peak disappeared after reaction and new peaks generated. These peaks included 315, 241, 226, 138 and 110 m/z, which corresponded the mass of N-methyl-N-(4-((4-(methylamino)phenyl)(phenyl)methylene)cyclohexa-2,5-dien-1-ylidene)methanaminium (Fig. 6B), N,N-dimethyl-4-((4-(methylamino)cyclohexa-2,5-dien-1-ylidene) methyl)aniline (Fig. 6C), (4-(dimethylamino)phenyl)(phenyl)methanone (Fig. 6D), 4-(dimethylamino)phenol (Fig. 6E), and 4-aminophenol (Fig. 6F), respectively. | ||
| Fig. 6 MS spectra of the degradation products of MG by F43H/H64A Mb. (A) Malachite green (m/z 329); (B) N-methyl-N-(4-((4-(methylamino)phenyl)(phenyl)methylene)cyclohexa-2,5-dien-1-ylidene)methanaminium (315 m/z); (C) N,N-dimethyl-4-((4-(methylamino)cyclohexa-2,5-dien-1-ylidene)methyl)aniline (241 m/z); (D) (4-(dimethylamino)phenyl)(phenyl)methanone (226 m/z); (E) 4-(dimethylamino)phenol (138 m/z); (F) 4-aminophenol (110 m/z). | ||
Based on these observations, we proposed a plausible biodegradation mechanism of MG catalyzed by F43H/H64A Mb. As shown in Scheme 1, the oxidation of MG at the tertiary carbon atom will generate two major products of (4-(dimethylamino)phenyl)(phenyl)methanone (Fig. 6D) and 4-(dimethylamino) phenol (Fig. 6E), as well as the product of N,N-dimethyl-4-((4-(methylamino)cyclohexa-2,5-dien-1-ylidene)methyl) aniline (Fig. 6C) by cleavage of the benzene group. Moreover, N-demethylation reaction occurred for MG, producing the product of N-methyl-N-(4-((4-(methylamino)phenyl)(phenyl)methylene)cyclohexa-2,5-dien-1-ylidene) methanaminium (Fig. 6B) that may also undergo oxidation, and the oxidation products further underwent N-demethylation as well. Note that both oxidation and N-demethylation reactions were reported for MG catalyzed by natural enzymes, such as laccase and heme enzymes.10,13,37,38 Therefore, the artificial enzyme of F43H/H64A Mb exhibited a general mechanism for the biodegradation of MG, which makes it an ideal alternative to that of natural enzymes.
 | ||
| Scheme 1 Proposed biodegradation mechanism for MG catalyzed by F43H/H64A Mb based on the MS analysis. | ||
Conclusions
In this study, we successfully achieved the degradation of MG using an artificial enzyme rationally designed in Mb, by constructing a distal histidine at position 43 and creating a channel to the heme active center via H64A mutation. Molecular modeling indicated that MG is favorable for binding to the protein scaffold of F43H/H64A Mb close to the heme group, which was further confirmed by ITC study. Both EPR and CD spectroscopy studies suggested that MG binding did not cause large effect on the heme active site and the protein secondary structure. Kinetic UV-Vis studies showed that the artificial enzyme F43H/H64A Mb was effective in catalyzing the degradation of MG, with the catalytic efficiency even exceeding those of natural enzymes such as BsDyP and CotA-laccase. The degradation products of MG were further identified by UPLC-ESI-MS analysis. Based on these results, we proposed a plausible biodegradation pathway of MG, involving both oxidation and N-demethylation mechanisms.Note that from the perspective of improving the catalytic activity of artificial enzymes, one should consider both to enhance the catalytic step (increase the kcat value) and to improve the substrate binding process (decrease the Km value). Because the protein scaffold of Mb already exhibits a low enough Km value, our next step will attempt to further increase the kcat value by rational modification of the heme center, which is currently in progress. Moreover, enzyme immobilization is also deserved to attempt for enhancing the stability and recycling of the artificial enzyme. Since F43H/H64A Mb is readily to obtain by overexpression in E. coli cells, and the oxidant H2O2 is relatively cheap, this artificial enzyme is expected to have practical applications in biodegradation of MG and other pollutions in fisheries and textile industries, by protecting environments toward a healthy future.
Materials and methods
Materials
Malachite green was purchased from Aladdin Industrial Corporation (Shanghai, China). Sperm whale WT Mb, F43H Mb and F43H/H64A Mb were expressed in BL21(DE3) cells and purified as previously reports,30–32 which were further confirmed by mass spectra (Fig. S1†). All other chemical reagents were commercial with analytical purity.Molecular docking study
The X-ray structure of F43H/H64A Mb, as reported in our previous study,32 was used as the initial structure for docking with MG using the Autodock 4.2.39 The heme iron was set to be the center, with a box size of ∼60 Å × 60 Å × 60 Å, which covered most of the protein surface. Amino acid residues Arg45, Lys63, Thr67 and Lys96 on the Mb were set as flexible residues. MG as a substrate for docking was generated using the Dundee Prodrg2 server.40 Docked conformations were ranked automatically by Autodock 4.2 using a binding-energy scoring function. The docking results with ten most favorable conformations after 2000 steps were then visualized and analyzed by using VMD 1.9.41UV-Vis spectroscopy
UV-Vis spectra were recorded on a Hewlett-Packard 8453 diode array spectrometer, which were recorded in a range of 200–700 nm with drop wise addition of MG to a final concentration of ∼0.1 mM in the absence or presence of H2O2. Protein concentration was determined with an extinction coefficient of ε409nm = 157 mM−1 cm−1, ε406nm = 155 mM−1 cm−1 and ε407nm = 157 mM−1 cm−1 for WT Mb, F43H Mb and F43H/H64A Mb in ferric state, respectively (Fig. S2†). To optimize the reaction conditions, the effects of pH values or H2O2 concentrations on the degradation activity of Mb were investigated on the same spectrometer by using MG as the substrate and H2O2 as the oxidant, respectively. The decrease in absorbance of MG at 617 nm (ε617nm = 148.9 mM−1 cm−1)42 in 100 mM potassium phosphate (KPi) buffer was monitored for the progress of the reaction.Kinetic UV-Vis study
The steady state experiments of MG oxidation were carried out on the Hewlett-Packard 8453 diode array. The reaction was carried out in 2 mL cuvette, in which WT Mb, F43H Mb or F43H/H64A Mb (2 μM in 100 mM KPi buffer, pH 6.0) was added by varying the concentration of MG (0–15 μM). The reaction was initiated by the addition of H2O2 (0.2 mM), and the decrease of absorption at 617 nm was monitored to determine the initial rate. The Michaelis–Menten equation, υ/[protein] = kcat[MG]/(Km + [MG]), was the used for data fitting to obtain the kinetic parameters.ITC study
Isothermal titration calorimetry (ITC) study of MG and F43H/H64A Mb was performed using Microcal VP-ITC (GE life sciences) at 25 °C. Both F43H/H64A Mb (40 μM in 100 mM KPi buffer, pH 6.0) and MG (0.5 mM in the same buffer) were degassed in the ThermoVal instrument (Microcal). For the titration experiment, 1.42 mL of F43H/H64A Mb solution was placed in the reaction cell, and the solution of MG was injected over 20 s with a total of 25 injections (2 μL for the first injection and 10 μL for the subsequent injections), with an interval of 150 s between each injection. The reaction cell was continuously stirred at a speed of 437 rpm, and the thermal change was recorded at 25 °C. The combined isotherm was fitted to a single point model in Origin 7.0 software (GE life sciences).EPR spectroscopy
Electron paramagnetic resonance (EPR) spectra of F43H/H64A Mb (0.5 mM, in 100 mM KPi buffer, pH 6.0) in the absence or presence of MG (1 mM) were recorded on a Bruker A300 spectrometer (X-band) equipped with Bruker ER4141VTM liquid nitrogen system. The sample was transferred into an EPR tube with a volume of 300 μL. The spectrum was measured at 100 K, with frequency of 9.43 GHz, center field 2200 G and sweep width 3600 G, microwave power 0.595 mW and modulation amplitude 3.0 G.CD spectroscopy
Circular dichroism (CD) spectroscopy was performed at Jasco 1500 spectropolarimeter, equipped with MCB-100 mini-circulating. The far-UV CD spectra were recorded in the range of 190–250 nm for F43H/H64A Mb (1 μM in 5 mM KPi buffer, pH 6.0) in the absence or presence of MG (2 μM) in 1.0 cm path quartz cuvette.UPLC-ESI-MS studies
The enzymatic oxidation products of MG were monitored by Ultra Performance Liquid Chromatography (UPLC) ESI-MS. The reaction mixtures (2 mL) contained 10 mM of MG dissolved in H2O, 500 μM H2O2, and 2 μM F43H/H64A Mb in KPi buffer (pH 6.0). After reaction for 2 h, aliquots were withdrawn and diluted in acetonitrile (1:1 ratio), and then analyzed in a Waters ACQUITY UPLC/Xevo G2 QTOF system, using a reverse-phase C-18 column (ACQUITY UPLC®BEH C18 1.7 μm, 2.1 mm × 50 mm) with a precolumn at 40 °C and a flow rate of 0.5 mL min−1. The column was further equilibrated with the mobile phase of 70% water (eluent A)/30% acetonitrile (eluent B) for 5 min. The mass spectrometer was operated in the ESI positive ion mode.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. S. G. Sligar and Prof. Y. Lu (University of Illinois at Urbana-Champaign) for providing the gene of sperm whale Mb. This study was supported by the National Natural Science Foundation of China (21977042), Special Scientific Research Funds for Central Nonprofit Institutes, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences (20603022016011), and Financial Fund of the Ministry of Agriculture and Rural Affairs, China (NFZX2018).References
- K. G. Pavithra, P. S. Kumar, V. Jaikumar and P. S. Rajan, J. Ind. Eng. Chem., 2019, 75, 1–19 CrossRef CAS.
- M. P. Shah, J. Biorem. Biodegrad., 2018, 9, 1000427 CrossRef.
- C.-J. Cha, D. R. Doerge and C. E. Cerniglia, Appl. Environ. Microbiol., 2001, 67, 4358–4360 CrossRef CAS PubMed.
- W. Andersen, C. Casey, T. Nickel, S. Young and S. Turnipseed, J. AOAC Int., 2018, 101, 1927–1939 CrossRef CAS PubMed.
- L. Yong, G. Zhanqi, J. Yuefei, H. Xiaobin, S. Cheng, Y. Shaogui, W. Lianhong, W. Qingeng and F. Die, J. Hazard. Mater., 2015, 285, 127–136 CrossRef CAS PubMed.
- Y. Y. Ling and F. B. Mohd Suah, J. Environ. Chem. Eng., 2017, 5, 785–794 CrossRef CAS.
- M. Lu, Y. Zhang, B. Li, X. Li, S. Xu, Z. Li and B. Yang, J. Mater. Sci.: Mater. Electron., 2018, 29, 12856–12870 CrossRef CAS.
- K.-Z. Xu, H.-R. Wang, Y.-J. Wang, J. Xia, H. Ma, Y.-J. Cai, X.-R. Liao and Z.-B. Guan, J. Biosci. Bioeng., 2020, 129, 405–411 CrossRef CAS PubMed.
- C. Zhang, S. Zhang, H. Diao, H. Zhao, X. Zhu, F. Lu and Z. Lu, J. Agric. Food Chem., 2013, 61, 5468–5473 CrossRef CAS PubMed.
- K.-Z. Xu, H. Ma, Y.-J. Wang, Y.-J. Cai, X.-R. Liao and Z.-B. Guan, Ecotoxicol. Environ. Saf., 2020, 193, 110335 CrossRef CAS PubMed.
- X. Zhang, S. Zhang, B. Pan, M. Hua and X. Zhao, Bioresour. Technol., 2012, 115, 16–20 CrossRef CAS PubMed.
- E. Kalaiarasan and T. Palvannan, Clean: Soil, Air, Water, 2015, 43, 846–856 CAS.
- X. Yang, J. Zheng, Y. Lu and R. Jia, Environ. Sci. Pollut. Res. Int., 2016, 23, 9585–9597 CrossRef CAS PubMed.
- E. N. Mirts, A. Bhagi-Damodaran and Y. Lu, Acc. Chem. Res., 2019, 52, 935–944 CrossRef CAS PubMed.
- Y. Yu, X. Liu and J. Wang, Acc. Chem. Res., 2019, 52, 557–565 CrossRef CAS PubMed.
- K. Oohora, A. Onoda and T. Hayashi, Acc. Chem. Res., 2019, 52, 945–954 CrossRef CAS PubMed.
- S. Ariyasu, J. K. Stanfield, Y. Aiba and O. Shoji, Curr. Opin. Chem. Biol., 2020, 59, 155–163 CrossRef CAS PubMed.
- Y.-W. Lin, Biotechnol. Appl. Biochem., 2020, 67, 484–494 CrossRef CAS PubMed.
- Y.-W. Lin, Coord. Chem. Rev., 2021, 434, 213774 CrossRef CAS.
- Y. Jiang, C. Wang, N. Ma, J. Chen, C. Liu, F. Wang, J. Xu and Z. Cong, Catal. Sci. Technol., 2020, 10, 1219–1223 RSC.
- S.-F. Chen, X.-C. Liu, J.-K. Xu, L. Li, J.-J. Lang, G.-B. Wen and Y.-W. Lin, Inorg. Chem., 2021, 60, 2839–2845 CrossRef CAS.
- Y. Lin, J. Wang and Y. Lu, Sci. China: Chem., 2014, 57, 346–355 CrossRef CAS.
- K. D. Miner, A. Mukherjee, Y. G. Gao, E. L. Null, I. D. Petrik, X. Zhao, N. Yeung, H. Robinson and Y. Lu, Angew. Chem., Int. Ed. Engl., 2012, 51, 5589–5592 CrossRef CAS PubMed.
- Y.-B. Cai, X.-H. Li, J. Jing and J.-L. Zhang, Metallomics, 2013, 5, 828–835 CrossRef CAS PubMed.
- L.-L. Li, H. Yuan, F. Liao, B. He, S.-Q. Gao, G.-B. Wen, X. Tan and Y.-W. Lin, Dalton Trans., 2017, 46, 11230–11238 RSC.
- L.-L. Yin, H. Yuan, C. Liu, B. He, S.-Q. Gao, G.-B. Wen, X. Tan and Y.-W. Lin, ACS Catal., 2018, 8, 9619–9624 CrossRef CAS.
- P. Zhang, J. Xu, X.-J. Wang, B. He, S.-Q. Gao and Y.-W. Lin, ACS Catal., 2019, 9, 7888–7893 CrossRef CAS.
- J. Luo, K.-J. Du, H. Yuan, C.-W. Wei, J.-J. Lang, G.-B. Wen, Y.-H. Wang and Y.-W. Lin, ACS Catal., 2020, 10, 14359–14365 CrossRef CAS.
- T. Matsui, S. Ozaki, E. Liong, G. N. Phillips Jr and Y. Watanabe, J. Biol. Chem., 1999, 274, 2838–2844 CrossRef CAS PubMed.
- J.-F. Du, W. Li, L. Li, G.-B. Wen, Y.-W. Lin and X. Tan, ChemistryOpen, 2015, 4, 97–101 CrossRef CAS PubMed.
- L.-B. Wu, H. Yuan, S.-Q. Gao, Y. You, C.-M. Nie, G.-B. Wen, Y.-W. Lin and X. Tan, Nitric Oxide, 2016, 57, 21–29 CrossRef CAS PubMed.
- L.-B. Wu, K.-J. Du, C.-M. Nie, S.-Q. Gao, G.-B. Wen, X. Tan and Y.-W. Lin, J. Mol. Catal. B: Enzym., 2016, 134, 367–371 CrossRef CAS.
- P. Urayama, G. N. Phillips Jr and S. M. Gruner, Structure, 2002, 10, 51–60 CrossRef CAS PubMed.
- P. Zhang, H. Yuan, J. Xu, X.-J. Wang, S.-Q. Gao, X. Tan and Y.-W. Lin, ACS Catal., 2020, 10, 891–896 CrossRef CAS.
- B. J. Reeder, D. A. Svistunenko, C. E. Cooper and M. T. Wilson, J. Am. Chem. Soc., 2012, 134, 7741–7749 CrossRef CAS PubMed.
- J. Peisach, W. E. Blumberg, S. Ogawa, E. A. Rachmilewitz and R. Oltzik, J. Biol. Chem., 1971, 246, 3342–3355 CrossRef CAS.
- P. Dhankhar, V. Dalal, J. K. Mahto, B. R. Gurjar, S. Tomar, A. K. Sharma and P. Kumar, Arch. Biochem. Biophys., 2020, 693, 108590 CrossRef CAS PubMed.
- J. Yang, X. Yang, Y. Lin, T. B. Ng, J. Lin and X. Ye, PLoS One, 2015, 10, e0127714 CrossRef PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- A. W. Schüttelkopf and D. M. F. v. Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- G. A. Kraus, I. Jeon, M. Nilsen-Hamilton, A. M. Awad, J. Banerjee and B. Parvin, Molecules, 2008, 13, 986–994 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI-MS, UV-Vis spectra, and molecular docking results. See DOI: 10.1039/d1ra02202d |
| This journal is © The Royal Society of Chemistry 2021 |