
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
CuI nanoparticles supported on a novel polymer-layered double hydroxide nanocomposite: an efficient heterogeneous nanocatalyst for the synthesis of bis-N-arylsulfonamides†
Jamshid Babamoradi,
Ramin Ghorbani-Vaghei * and
Sedigheh Alavinia
* and
Sedigheh Alavinia
Department of Chemistry, Bu-Ali Sina University, Zip Code 65174, Hamedan, Iran. E-mail: rgvaghei@yahoo.com; Tel: +98-81-38380647
First published on 26th May 2021
Abstract
A new type of polymer-layered double hydroxide nanocomposite bearing thiazole moieties was used to support CuI nanoparticles (NPs) as a heterogeneous catalyst for the synthesis of bis-N-arylsulfonamides. The prepared nanostructured catalyst (LDH@MPS-GMA-TZ-CuI) showed high catalytic activity, as well as excellent recyclability for the preparation of bis-N-arylsulfonamides via the chemoselective reaction of 1,3-disulfonyl chloride and nitroarenes. The superior catalytic activity of the LDH@MPS-GMA-TZ-CuI is related to the high loading of CuI NPs and favorable surface properties.
1. Introduction
Sulfonamide is a wonderful compound that has a clear biological effect, as it is used in the treatment of various diseases. Several studies in the field of medical chemistry have proven that sulfonamide derivatives possess a range of beneficial biological activities, being antimicrobial, antiviral, antitubercular, and anticancer agents.1–3 Although the amination of arylsulfonyl chlorides is still a valuable method for the synthesis of N-arylsulfonamides, the mentioned reactions typically require the use of strong bases that can negatively effect the recovery of the catalyst.4 Recently, transition metal-catalyzed carbon–nitrogen bond forming via cross-coupling reactions has represented a powerful means for the preparation of diverse sulfonamide compounds, which have high utilities in pharmaceutical, materials and chemical science.5 However, some of them require harsh reaction conditions and expensive noble and precious metal-based catalyst systems, which can increase the production costs due to difficult separation of the desired value-added products. Thus, it is of great significance to develop a new catalyst system capable of providing functionalized N-aryl sulfonamides with excellent yields under mild conditions. In this regard, the reaction of nitroarenes and sulfonyl chloride has been proven to be a practical method in the synthesis of N-arylsulfonamides. Hence, some research has been recently reported to synthesis of N-aryl sulfonamide derivatives from nitroarenes, such as combination of FeCl2 and trans-N,N′-dimethyl-1,2-diaminocyclohexane (DMDACH),6 Fe dust as a reducing agent,7 iron powder,8 iron-based metal–organic framework,9 insertion of sulfur dioxide,10 and electrochemical synthesis.11 However, despite recent progress, it is still a big challenge to design new heterogeneous support, focusing on reducing the use of expensive or homogeneous catalysts to achieve mild reaction conditions.In this sense, layered double hydroxides (LDHs) with the formula [M2+ (1 − x) Mx3+(OH)2] (An−) x/n zH2O] have been widely investigated as heterogeneous support because of their low cost, ease of accessibility, large surface area, non-toxicity, recyclability and high stability.12 However, low loading problem has a negative effect on the catalytic activity of heterogeneous supports. In order to overcome this limitation, the functionalization of LDH; for example, post-synthesis, grafting and polymerization are particularly important methods to affect the catalytic activity of LDHs.13 Herein, we can refer to some specific methods applied to improve the catalytic performance of Cu–Zn–Al LDH support: (1) radical polymerization of glycidyl methacrylate (GMA) linkage, (2) presenting thiazole moieties in order to decrease the aggregation of CuI NPs, (3) and immobilization of CuI NPs.
Following our recent aim of designing multifunctional catalyst;14 in this article, we synthesized a LDH@MPS-GMA-TZ as a novel support with a thiazole moieties. After immobilization of CuI NPs, we aimed at analyzing the catalytic performance of LDH@MPS-GMA-TZ-CuI (Scheme 1) for the generation of bis-N-aryl sulfonamides from the reaction of 1,3-disulfonylchloride and nitroarenes with high yields (Scheme 2). The prepared nanocomposite revealed excellent catalytic function in the synthesis of different bis-N-aryl sulfonamides.
 | ||
| Scheme 1 Schematic representation for preparation of LDH@MPS-GMA-TZ-CuI nanocomposite. | ||
 | ||
| Scheme 2 Catalytic efficiency of LDH@MPS-GMA-TZ-CuI in the synthesis of bis-N-arylsulfonamides. | ||
2. Experimental
2.1. Synthesis of LDH
Cu–Zn–Al-LDH was synthesized through the coprecipitation method as per the previously reported procedure.15 In a typical procedure, solution A was prepared by dissolving Zn(NO3)2·6H2O, Al(NO3)2·9H2O and Cu(NO3)3·6H2O in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in deionized water (100 mL). Solution B was also prepared by dissolving Na2CO3 and NaOH in 100 mL of deionized water in order to form a mixed base solution. Then, the solution B was slowly added to the solution A at a pH value of 12. Then, the reaction – without being stirred – was set at 80 °C for 6 h. The solid compound obtained was separated by filtration then washed with distilled water and dried under vacuum conditions at 80 °C for 12 h. The synthesized Cu–Zn–Al-LDH was calcined in static air at 600 °C for 4 h (Scheme 1).
:1:1 in deionized water (100 mL). Solution B was also prepared by dissolving Na2CO3 and NaOH in 100 mL of deionized water in order to form a mixed base solution. Then, the solution B was slowly added to the solution A at a pH value of 12. Then, the reaction – without being stirred – was set at 80 °C for 6 h. The solid compound obtained was separated by filtration then washed with distilled water and dried under vacuum conditions at 80 °C for 12 h. The synthesized Cu–Zn–Al-LDH was calcined in static air at 600 °C for 4 h (Scheme 1).
2.2. Preparation of silica coated LDH (LDH@MPS)
At first, Cu–Zn–Al-LDH (0.2 g) was dispersed in of DMF (10 mL) for 5 min in an ultrasonic cleaning apparatus. After sonication, ammonia solution (2 mL of 25 wt% solution) and 3-(trimethoxysilyl) propyl methacrylate (2 mL) were added carefully in to a round bottom flask. The resulting suspension was stirred at 50 °C for 24 h. The resultant suspension was filtered out and washed with ethanol (20 mL) and water (40 mL) and, then, dried at 50 °C (Scheme 1).162.3. Preparation of LDH coated with glycidyl methacrylate (LDH@MPS-GMA)
The LDH@MPS-GMA were synthesized through free radical polymerization method. To synthesize the LDH@MPS-GMA, the obtained LDH@MPS (0.2 g) was dispersed in 30 mL of toluene for 30 min, and then glycidyl methacrylate (2 mL) and azobisisobutyronitrile (0.25 g) were added to the above mixture and refluxed for 24 h. The functionalized LDH@MPS-GMA were filtered off and washed with toluene (20 mL), and dried under vacuum conditions at room temperature for 12 h (Scheme 1).162.4. Synthesis of LDH@MPS-GMA-TZ
For synthesis of LDH@MPS-GMA-TZ, the prepared LDH@MPS-GMA (0.2 g) was sonicated in DMF (50 mL) for 15 min. Afterwards, to the reaction solution, 2-aminobenzothiazole (0.6 g) was slowly added and, then, it was refluxed for 24 h. Subsequently, the LDH@MPS-GMA-TZ precipitate were collected, washed with DMF, and dried under vacuum (Scheme 1).2.5. Procedure for the preparation of LDH@MPS-GMA-TZ-CuI
The CuI NPs were synthesized through our previously work.14c The prepared support (0.1 g) and CuI NPs (0.05 g) were dispersed in 100 mL of water and then stirred for 5 h at room temperature. Subsequently, the LDH@MPS-GMA-TZ-CuI nanocomposite was separated through centrifusion, washed with water, and dried under vacuum. The ICP report indicated that the amount of copper in the final catalyst is 20.3%.2.6. General procedure for the synthesis of bis-N-aryl sulfonamide derivatives (3a-o)
At first, the catalyst (50 mg) was dispersed in H2O:C2H4Cl2 (1:1) (2 mL) by sonication for 10 min and then nitroarene (0.5 mmol), 1,3-disulfonyl chloride (0.25 mmol), pyridine (0.5 mmol) and NaBH4 (1.0 mmol) was added to the mixture and stirred untill completion of the reaction monitored by TLC (n-hexane/ethyl acetate, 8:2). After the completion of the reaction, the reaction mixture was diluted with EtOAc (5 mL) and the LDH@MPS-GMA-TZ-CuI was separated by centrifuging and washed with ethanol (20 mL). The pure product was obtained by extraction with ethyl acetate (3 × 5 mL). The organic solution was dried by anhydrous sodium sulfate and, the solvent evaporated. Finally, the product was washed three times with HCl solution (10 mL, 0.1 M) and dried. In some products, the solid formed was filtered, dried and recrystallized from ethanol (10 mL) as yellow crystals.
2.7. Spectral data of compounds
3. Results and discussions
The synthesized LDH@MPS@GMA-TZ-CuI catalyst was fully characterized by FT-IR, TGA, ICP-MS, and FESEM-EDX mapping techniques. The FT-IR spectrum of Cu–Zn–Al LDH, LDH@MPS, LDH@MPS-GMA, LDH@MPS-GMA-TZ and LDH@MPS-GMA-TZ-CuI is displayed in Fig. 1. The results obtained from FT-IR spectrum show that: (I) the presence of LDHs coated by silica (LDH@MPS), (II) the presence of glycidyl methacrylate linker (LDH@MPS-GMA), (III) the presence of thiazole moieties (IV), and the interaction of CuI NPs with the prepared support. FT-IR spectrum of Cu–Zn–Al LDH displayed vibration bands at 500 and 786 cm−1 (due to stretching of M–O and O–M–O bonds). The broad peaks at 3396 cm−1 (due to hydroxy group) can also be seen in the LDH structure (Fig. 1A). The FT-IR spectrum of LDH@MPS displayed new bands at 1722 cm−1 are related to ester carbonyl groups.17 The presence of this peak confirmed successful grafting of MPS on the surface of LDH. In Fig. 1C, the presence of the new sharp band at 1722 cm−1 (due to the ester carbonyl groups) was also confirmed the successful polymerization of glycidyl methacrylate.18 After grafting of 2-aminothiazole, the next spectrum indicated the new peak at 1646 cm−1 corresponds to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N group (Fig. 1D). In addition, the ester carbonyl peaks shifted from 1722 cm−1 to 1684 cm−1. In case of LDH@MPS-GMA-TZ-CuI (Fig. 1 E), after interaction of CuI NPs with prepared support, the band at 3392 cm−1 (due to the NH2 stretching) shifted to lower wave number (3428 and 3130 to 3375 and 3108 cm−1). In addition, the CN peaks shifted from 1665 to 1646 cm−1 (Fig. 1E).
N group (Fig. 1D). In addition, the ester carbonyl peaks shifted from 1722 cm−1 to 1684 cm−1. In case of LDH@MPS-GMA-TZ-CuI (Fig. 1 E), after interaction of CuI NPs with prepared support, the band at 3392 cm−1 (due to the NH2 stretching) shifted to lower wave number (3428 and 3130 to 3375 and 3108 cm−1). In addition, the CN peaks shifted from 1665 to 1646 cm−1 (Fig. 1E).
 | ||
| Fig. 1 FT-IR spectra of LDH (A), LDH@MPS (B), LDH@MPS-GMA (C), LDH@MPS-GMA-TZ (D) and LDH@MPS-GMA-TZ-CuI (E). | ||
The FE-SEM images of Cu–Zn–Al LDH (Fig. 2a and b), LDH@MPS (Fig. 2c and d), LDH@MPS-GMA-TZ (Fig. 2e and f), CuI NPs (Fig. 2g) and the final synthesized catalyst (LDH@MPS-GMA-TZ-CuI) (Fig. 2h) are shown in Fig. 2. The FE-SEM images of LDHs indicated that Cu–Zn–Al LDH was grown in the form of sheet (Fig. 2a and b). FE-SEM image of LDH@MPS confirmed the attachment of 3-(trimethoxysilyl) propyl methacrylate to the LDHs surface (Fig. 2c and d). FE-SEM image of LDH@MPS-GMA-TZ clearly indicated the successful grafting of organic groups on the surface of LDHs (Fig. 2e and f). The FE-SEM micrograph of CuI NPs showed spherical CuI NPs were successfully fabricated in nanometric size (Fig. 2g). FE-SEM image of the LDH@MPS-GMA-TZ-CuI (Fig. 2h) showed that the CuI nanoparticles were successfully immobilized on the surface of LDHs@MPS-GMA-TZ support.
 | ||
| Fig. 2 SEM image of (a and b) Cu–Zn–Al LDHs, (c and d) LDH@MPS, (e and f) LDH@MPS-GMA-TZ, (g) CuI NPs, and (h) LDH@MPS-GMA-TZ-CuI. | ||
The EDX technique was applied for studying the elemental analysis of the prepared final catalyst (Fig. 3). The presence of all elements (C, N, Cu, Zn, S, Al, I and O) in the catalyst was proved by this technique. Elemental mapping exhibited the uniform distribution of all the elements, as shown in Fig. 4.
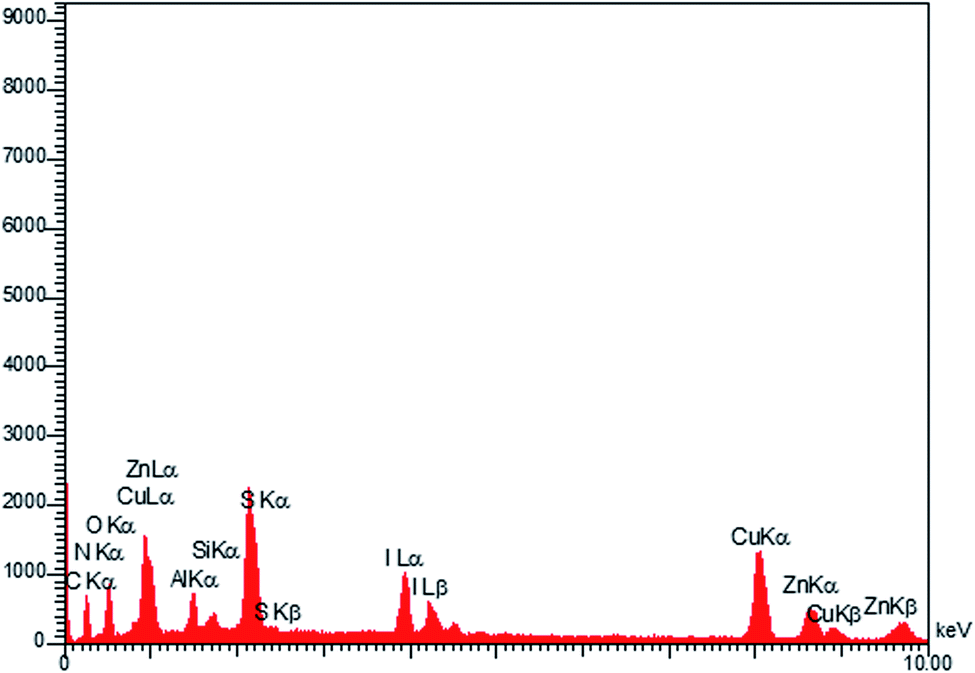 | ||
| Fig. 3 EDX analysis of LDH@MPS-GMA-TZ-CuI. | ||
 | ||
| Fig. 4 Elemental mapping of the A (Zn), B (S), C (O), D (N), E (I), F (Cu), G (C), H (Al) atoms achieved from SEM micrographs. | ||
Fig. 5 shows the TGA curve of LDH@MPS-GMA-TZ-CuI in which a small loss weight from 30 to 100 °C was witnessed, clearly connected to the physically absorbed water. After this, the weight decrease shown in the range of 220–450 °C. This weight loss clearly indicated the degradation of organic ligand and linker that immobilized on the surface of LDHs.
 | ||
| Fig. 5 Thermogravimetric diagram of LDH@MPS-GMA-TZ-CuI nanocomposite. | ||
The crystallinity and phases structure of the LDH@MPS-GMA-TZ-CuI nanocomposite was ascertained by XRD and presented in Fig. 6. The sharp peaks at 2θ = 25.64°, 42.36° was attributed to the Cu–Zn–Al LDHs,19 the diffraction peaks appeared at 2θ = 29°, 35° and 61.30° can be attributed to copper iodide nanoparticles.20 Additionally, the diffraction peaks at 2θ = 67.52° and 77.32° (due to the presence of GMA)21 and the diffraction peaks at 2θ = 38.95° (due to the presence of MPS)22 confirms the successful coating and polymerization of LDH material. The sharp peak at 2θ = 50.12° is a strong reason for the presence of thiazole groups. The location and intensity of the peaks indicate the correct synthesis and high crystallinity of the prepared composite. Using the Scherer equation, the size of the crystals is calculated to be 14.1 nm. Also, the absence of additional impurities related to impurities indicates the efficiency of the proposed method in synthesis.
 | ||
| Fig. 6 XRD pattern of LDH@MPS-GMA-TZ-CuI nanocomposite. | ||
The N2 adsorption–desorption isotherms of the LDH@MPS-GMA-TZ (Fig. 7A) and LDH@MPS-GMA-TZ-CuI (Fig. 7B) were measured in order to determine the textural properties. It can be seen from Fig. 7 that both LDH@MPS-GMA-TZ and LDH@MPS-GMA-TZ-CuI indicate a typical type IV isotherm with type H3 hysteresis (defined by IUPAC),23 which are identified as mesoporous materials. The BJH method is used to obtain the pore size distribution. As can be seen, although the maximum frequency of pores with a radius of 2.31 nm is micro pore and mesopore , and there is a heterogeneous distribution of pores in the structure. The average pore size is 1.21 nm, which is in the mesopore range. The changes associated to the textural properties of the final catalyst (LDH@MPS-GMA-TZ-CuI) can be due to the fact that copper nanoparticles which were distributed inside the LDH@MPS-GMA-TZ cavities (Table 1).
 | ||
| Fig. 7 The N2 adsorption–desorption isotherm and BJH pore size distribution for the (A) LDH@MPS-GMA-TZ, (B) LDH@MPS-GMA-TZ-CuI. | ||
| Parameter | LDH@MPS-GMA-TZ-CuI | LDH@MPS-GMA-TZ |
|---|---|---|
| as (m2 g−1) | 86.614 | 153.51 |
| Vm (cm3(STP) g−1) | 19.9 | 35.27 |
| Vp (cm3 g−1) | 0.0072 | 0.0087 |
| ap (m2 g−1) | 3.11 | 4.30 |
After the fully characterization of prepared LDH@MPS-GMA-TZ-CuI catalyst, we aimed at analyzing the optimization of reaction parameters, i.e. temperature, catalyst loading and solvent over LDH@MPS-GMA-TZ-CuI for the model reaction (1,3-disulfonyl chloride and nitrobenzene). The results are presented in Table 2. As per catalytic results in different solvents (entries 1–8), a mixture of dichloroethane (DCE) with water (1:1 in volume) gave the best results (entry 8), and changing the ratio of solvents led to a decrease in the efficiency of the reaction. As the amount of catalyst increased (entries 8–11), the yield of the product increased rapidly in 0.05 g of the prepared catalyst (entry 8). By investigation of the effect of bases on the reaction progress, the reaction reached 92% yield after 0.5 h with pyridine at room temperature. Other inorganic bases also showed good yields (entries 13–17). However, a low yield was obtained in the absence of base (entry 18). In optimization of the temperature, increasing the temperature (up to 50 °C) negatively affected the progress of the model reaction (entry 19). The model reaction was then performed in the different amounts of sodium borohydride (0.5, 1, and 2 mmol). It was found that 0.5 mmol of NaBH4 provided the desired product with excellent yield (entries 20–21). Under this condition, the catalytic performance of the LDH@MPS@GMA-TZ-CuI was further examined and compared to LDH and different amount of CuI NPs (entries 22–26). The results showed that the grafting of organic groups and immobilization of the metal increased its catalytic activity, shortened the reaction time and improved the product efficiency.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Reductant (mmol) | Cat. (mg) | Solvent | Base | Time (h) | Yieldb (%) |
| a Reaction conditions: nitroarene (0.5 mmol), LDH@MPS-GMA-TZ-CuI (5 mg), 1,3-disulfonylchloride (0.25 mmol), pyridine (0.5 mmol), NaBH4 (1.0 mmol) and H2O:C2H4Cl2 (1:1, 2 mL), at room temperature.b Isolated yield.c The reaction was investigated at 50 °C.d The reaction was investigated in the presence of Cu–Zn–Al LDH.e The reaction was investigated in the presence of CuI NPs (50 mg).f The reaction was investigated in the presence of CuI NPs (40 mg).g The reaction was investigated in the presence of CuI NPs (70 mg).h The reaction was investigated in the presence of CuI NPs (100 mg). |
||||||
| 1 | NaBH4 (1) | 50 | EtOH | Pyridine | 2 | Trace |
| 2 | NaBH4 (1) | 50 | CH3CN | Pyridine | 2 | 45 |
| 3 | NaBH4 (1) | 50 | Ethyl acetate | Pyridine | 2 | 15 |
| 4 | NaBH4 (1) | 50 | H2O | Pyridine | 2 | Trace |
| 5 | NaBH4 (1) | 50 | DMF | Pyridine | 2 | 7 |
| 6 | NaBH4 (1) | 50 | CH2Cl2 | Pyridine | 2 | 55 |
| 7 | NaBH4 (1) | 50 | C2H4Cl2 | Pyridine | 2 | 78 |
| 8 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
Pyridine | 0.5 | 92 |
| 9 | NaBH4 (1) | 40 | H2O:C2H4Cl2 |
Pyridine | 0.5 | 88 |
| 10 | NaBH4 (1) | 20 | H2O:C2H4Cl2 |
Pyridine | 1 | 60 |
| 11 | NaBH4 (1) | 10 | H2O:C2H4Cl2 |
Pyridine | 1 | 40 |
| 12 | NaBH4 (1) | 60 | H2O:C2H4Cl2 |
Pyridine | 0.5 | 92 |
| 13 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
Et3N | 2 | 41 |
| 14 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
K2CO3 | 2 | 56 |
| 15 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
Na2CO3 | 2 | 50 |
| 16 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
KOH | 2 | 70 |
| 17 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
NaHCO3 | 2 | 60 |
| 18 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
— | 24 | 40 |
| 19 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
Pyridine | 0.5 | 88c |
| 20 | NaBH4 (2) | 50 | H2O:C2H4Cl2 |
Pyridine | 0.5 | 92 |
| 21 | NaBH4 (0.5) | 50 | H2O:C2H4Cl2 |
Pyridine | 2 | 60 |
| 22 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
Pyridine | 12 | Traced |
| 23 | NaBH4 (1) | 50 | H2O:C2H4Cl2 |
Pyridine | 12 | 50e |
| 24 | NaBH4 (1) | 40 | H2O:C2H4Cl2 |
Pyridine | 12 | 39f |
| 25 | NaBH4 (1) | 70 | H2O:C2H4Cl2 |
Pyridine | 12 | 59g |
| 26 | NaBH4 (1) | 100 | H2O:C2H4Cl2 |
Pyridine | 12 | 72h |

Under the optimal conditions, we evaluated its catalytic efficiency for synthesis of bis-N-aryl sulfonamides from various substituted nitroarenes (Table 3). We found that a variety of functionalized bis-N-aryl sulfonamides are accessible in good to excellent yields using this method. With regard to the scope of nitroarenes, electron-donating nitrobenzenes such as 2,4-dimethoxy, 2,4-dimethyl, 2,6-dimethyl, 2,3-dimethyl, o-CH3 (3b–h), electron-withdrawing substituted nitrobenzenes such as p-Br, p-Cl, p-I, p-F, m-NO2, (3i–m), heterocyl nitroarene like 2-nitropyridine (3n), and 1-nitronaphthalene (3o) are all good substrates. Compared with electron-withdrawing nitroarenes, nitroarenes with electron-donating substituents gave the desired products in a better yields. In addition, the reaction was performed with nitrobenzenes containing substituents at the ortho position (3d–g), and the efficiency of forming the desired product was good.
| Entry | Substrate | Productb | Time (h) | Yieldc (%) |
|---|---|---|---|---|
| a Reaction condition: nitroarene (0.5 mmol), LDH@MPS-GMA-TZ-CuI (5 mg), 1,3-disulfonylchloride (0.25 mmol), pyridine (0.5 mmol), NaBH4 (1.0 mmol) and H2O:C2H4Cl2 (1:1, 2 mL), at room temperature.b All the products were characterized by HNMR, CNMR, mass and FT-IR.c Isolated yields. |
||||
| 1 | Nitrobenzene | 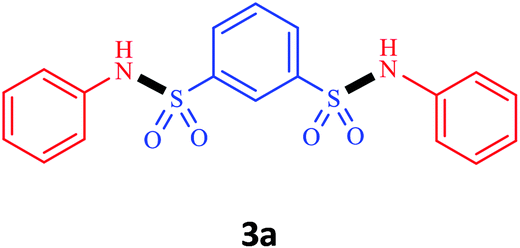 |
0.5 | 92 |
| 2 | 1-Methyl-4-nitrobenzene |  |
0.5 | 98 |
| 3 | 1-Methoxyl-4-nitrobenzene |  |
1 | 91 |
| 4 | 2,4-Dimethoxy-1-nitrobenzene |  |
1 | 85 |
| 5 | 2,4-Dimethyl-1-nitrobenzene |  |
1 | 88 |
| 6 | 1,3-Dimethyl-2-nitrobenzene |  |
1 | 78 |
| 7 | 1,2-Dimethyl-3-nitrobenzene |  |
1.5 | 88 |
| 8 | 1-Methyl-2-nitrobenzene |  |
1.5 | 92 |
| 9 | 1-Bromo-4-nitrobenzene |  |
1.5 | 88 |
| 10 | 1-Chloro-4-nitrobenzene |  |
1.5 | 90 |
| 11 | 1-Iodo-4-nitrobenzene |  |
2 | 78 |
| 12 | 1-Fluoro-4-nitrobenzene |  |
1 | 80 |
| 13 | 1-Bromo-3-nitrobenzene |  |
2.5 | 88 |
| 14 | 2-Nitropyridine |  |
1.5 | 90 |
| 15 | 1-Nitronaphthalene |  |
4 | 75 |
3.1. Catalyst recycling
Fig. 8 represents the regeneration of LDH@MPS-GMA-TZ-CuI up to six consecutive recycles. After each recycle, very little and insignificant change in the product yield has been noticed (92, 92, 90, 89, 87, 85) (Fig. 8). | ||
| Fig. 8 Reusability of catalyst for the model reaction (1,3-disulfonyl chloride and nitrobenzene). | ||
The efficiency of LDH@MPS@GMA@TZ-CuI for this method was compared to some other related reports (Table 4). In the present work, using LDH@MPS-GMA-TZ-CuI as a recyclable catalyst can remarkably enhance the product yield and shorten the reaction time.
| Entry | Conditions | Yield (%) [ref] |
|---|---|---|
| 1 | Sodium arylsulfinates (0.75 mmol), nitrobenzene (0.5 mmol), NaHSO3 (1.5 mmol), FeCl2 (10 mol%), DMDACH (20 mol%), DMSO (2 mL), 12 h, Ar | 90 [6] |
| 2 | Nitrobenzene (0.25 mmol), benzene sulfonyl chlorides (0.5 mmol), Fe dust (1.0 mmol), H2O (1 mL), 60 °C, 36 h | 85 [7] |
| 3 | Nitrobenzene (0.3 mmol), benzene sulfonyl chlorides (0.6 mmol), iron powder (1.15 mmol), water (1.5 mL), 60 °C, 40 h. | 85 [8] |
| 4 | Nitrobenzene (0.5 mmol), sodium arylsulfinates (1 mmol), NaHSO3 (1 mmol), MIL-101(Fe) (10 mg, 8 mol% of Fe), H2O (2 mL), 60 °C, 20 h | 91 [9] |
| 5 | A solution (80 mL) of water (phosphate buffer, pH = 3.0, c = 0.2 M)/ethanol mixture (20/80, v/v), arylsulfinic acids sodium salt (2 mmol), p-nitroaniline (1 mmol) | 60 [11] |
| 6 | Nitrobenzene (0.5 mmol), LDH@MPS-GMA-TZ-CuI (5 mg), 1,3-disulfonylchloride (0.25 mmol), pyridine (0.5 mmol), NaBH4 (1.0 mmol) and H2O:C2H4Cl2 (1:1, 2 mL), room temperature, 0.5 h |
92 [This work] |
A plausible mechanism for the synthesis of bis-N-aryl sulfonamides from the reaction of 1,3-disulfonyl chloride and nitroarenes in the presence of LDH@MPS-GMA-TZ-CuI catalyst has been proposed in Scheme 3. According to the figure, it is believed that effective electronic interactions between copper and heteroatoms can create an appropriate substrate for this type of reaction.24 These close electronic interactions of heteroatoms with dissociated hydrogen atoms from sodium borohydride, onto the surface of the Cu NPs cause nitrobenzene derivatives to be adsorbed and converted to anilines during successive dehydration processes. In fact, sodium borohydride is considered as a substantial H-supporter for the reduction process and can effectively interact with Cu NPs surfaces from its nitrogen sites.
 | ||
| Scheme 3 The proposed mechanism for the synthesis of bis-N-arylsulfonamides in the presence of LDH@MPS-GMA-TZ-CuI catalyst. | ||
First of all, the reaction of sodium borohydride and copper can lead to forming copper hydride species. Subsequently, the hydride group transfers to nitroarene species. In mechanistic pathway, the intermediates nitrosobenzene (A) and phenyl hydroxyl amine (B) are generated, followed by water elimination and hydride transfer process to intermediate (B) and generated the aniline intermediate (Scheme 3). Finally, the bis-N-aryl sulfonamides were obtained from the amination of 1,3-disulfonyl chlorides in the presence of pyridine base.25
4. Conclusions
The aim of the present research was to synthesize a new type of polymer–LDH nanocomposite with thiazole for immobilization of CuI NPs. The catalytic activity of LDH@MPS-GMA-TZ-CuI was explored for effective synthesis of bis-N-aryl sulfonamides from the reaction of 1,3-disulfonyl chloride and nitroarenes. The results illustrated that the high metal loading results in the high performance of the catalyst for the synthesis of bis-N-aryl sulfonamides.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors wish to thank Bu-Ali Sina University, Center of Excellence Developmental of Environmentally Friendly Methods for Chemical Synthesis (CEDEFMCS) and Iran National Science Foundation (INSF) for financial support to carry out this research.References
- N. Sun, X. Ma, K. Zhou, C. Zhu, Z. Cao, Y. Wang, J. Xu and W. Fu, Eur. J. Med. Chem., 2020, 187, 111984–112000 CrossRef CAS PubMed.
- Y. Liu, Y. Wu, l. Sun, Y. Gu and L. Hu, Eur. J. Med. Chem., 2020, 191, 112181–112198 CrossRef CAS PubMed.
- P. Gholamzadeh, G. Mohammadi Ziarani, F. Zandi, A. Abolhasani Soorki, A. Badiei and F. Yazdian, C. R. Chim., 2017, 20, 833–840 CrossRef CAS.
- M. Harmata, P. Zheng, C. F. Huang, M. G. Gomes, W. Ying, K. Ranyanil, G. Balan and N. L. Calkins, J. Org. Chem., 2007, 72, 683–685 CrossRef CAS PubMed.
- M. Negwer, in Organic Chemical Drugs and their Synonyms: An International Survey, 7th edn, Akademie Verlag, Berlin, 1994 Search PubMed.
- W. Zhang, J. Xie, B. Rao and M. Luo, J. Org. Chem., 2015, 80, 3504 CrossRef CAS PubMed.
- J. Jiang, S. Zeng, D. Chen, C. Cheng, W. Deng and J. Xiang, Org. Biomol. Chem., 2018, 16, 5016–5020 RSC.
- Y. Huilan, B. Pengli, W. Leilei, L. Xiaoxia, Y. Daoshan, W. Hua and W. Wei, Chin. J. Org. Chem., 2019, 39, 463 CrossRef.
- X. Li, F. Chen and G.-P. Lu, Tetrahedron Lett., 2018, 59, 4226–4230 CrossRef CAS.
- X. Wang, M. Yang, Y. Kuang, J.-B. Liu, X. Fan and J. Wu, Chem. Commun., 2020, 56, 3437–3440 RSC.
- B. Mokhtari, D. Nematollahi and H. Salehzadeh, Green Chem., 2018, 20, 1499–1505 RSC.
- P. M. Pardeshi, A. K. Mungray and A. A. Mungray, Desalination, 2017, 421, 149–159 CrossRef CAS.
- (a) S. Rohani, G. Mohammadi-Ziarani, A. Badiei, A. Ziarati and R. Luque, Catal. Lett., 2019, 149, 2984–2993 CrossRef CAS; (b) R. Ghorbani-Vaghei and N. Sarmast, Res. Chem. Intermed., 2018, 44, 4483–4501 CrossRef CAS; (c) Z. Karamshahi and R. Ghorbani-Vaghei, Appl. Organomet. Chem., 2019, 34, e5347 Search PubMed.
- (a) F. Hamidi Dastjerdi, R. Ghorbani-Vaghei and S. Alavinia, Catal. Lett., 2020, 150, 3514–3522 CrossRef CAS; (b) R. Ghorbani-Vaghei, S. Alavinia, Z. Merati and V. Izadkhah, Appl. Organomet. Chem., 2018, 32, e4127 CrossRef; (c) S. Alavinia, R. Ghorbani-Vaghei, J. Rakhtshah, J. Yousefi Seyf and I. Ali Arabian, Appl. Organomet. Chem., 2020, 34, e5449 CrossRef CAS; (d) A. Fatehi, R. Ghorbani-Vaghei, S. Alavinia and J. Mahmoodi, ChemistrySelect, 2020, 5, 944 CrossRef CAS; (e) S. Alavinia and R. Ghorbani-Vaghei, New J. Chem., 2020, 44, 13062–13073 RSC; (f) S. Alavinia and R. Ghorbani-Vaghei, J. Phys. Chem. Solids, 2020, 146, 109573 CrossRef CAS; (g) M. Malaekeh poor, K. Derakhshandeh, R. Haddadi, A. R. Nouriand and R. Ghorbani-Vaghei, Polym. Chem., 2020, 11, 2408–2417 RSC.
- S. Zhang, Q. Liu and G. Fan, Catal. Lett., 2012, 142, 1121–1127 CrossRef CAS.
- Y. Wei, T. Wang, C. Liu, Q. Zhang, L. Wang, G. Tang and J. Kang, Chin. J. Chem., 2013, 31, 715–720 CrossRef CAS.
- A. Pourjavadi, S. Hassan Hosseini and S. S. Amin, Chem. Eng. J., 2014, 247, 85–92 CrossRef CAS.
- A. Bukowska, W. Bukowski, K. Hus, J. Depciuch and M. Parlińska-Wojtan, eXPRESS Polym. Lett., 2017, 11, 2–13 CrossRef CAS.
- S. Chakraborty, I. Sarkar, A. Ashok, I. Sengupta, S. K. Pal and S. Chakraborty, Appl. Therm. Eng., 2018, 141, 339–351 CrossRef CAS.
- N. Raman and R. Mahalakshmi, Inorg. Chem. Commun., 2014, 40, 157–163 CrossRef CAS.
- X. Cheng, R. Cheng, S. Ou and Y. Li, Carbohydr. Polym., 2013, 96, 320–325 CrossRef CAS PubMed.
- C. H. Campos, B. F. Urbano and B. L. Rivas, Composites, Part B, 2014, 57, 1–7 CrossRef CAS.
- S. W. Sing and W. Kenneth, Adsorpt. Sci. Technol., 2004, 22, 773–782 CrossRef.
- M. B. Gawande, H. Guo, A. K. Rathi, P. S. Branco, Y. Chen, R. S. Varma and D. L. Peng, RSC Adv., 2013, 3, 1050–1054 RSC.
- O. Mazaheria and R. Javad Kalbasi, RSC Adv., 2015, 5, 34398–34414 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedure and FT-IR and MS of all compounds. See DOI: 10.1039/d1ra02086b |
| This journal is © The Royal Society of Chemistry 2021 |