
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIron-catalysed hydroboration of non-activated imines and nitriles: kinetic and mechanistic studies†
Adineh Rezaei Bazkiaei,
Michael Wiseman and
Michael Findlater *
*
Department of Chemistry & Biochemistry, Texas Tech University, Lubbock, Texas 79409, USA. E-mail: michael.findlater@ttu.edu
First published on 23rd April 2021
Abstract
Iron-catalysed hydroboration of imines and nitriles has been developed under low catalyst loading (1 mol%) in the presence of HBpin. A wide scope of substrate was found to smoothly undergo hydroboration, including electron releasing/withdrawing and halogen substitution patterns and cyclic substrates which all afforded the corresponding amines in good to excellent yields. Dihydroboration of nitriles was achieved conveniently under solvent free and additive free conditions. Promisingly, this catalytic system is also capable of the hydroboration of challenging ketimine substrates. Preliminary kinetic analysis of imine hydroboration reveals a first-order dependence on catalyst concentration. Both HBpin and 4-fluorophenyl-N-phenylmethanimine (1b) appear to exhibit saturation kinetics with first order dependence up to 0.5 mmol HBpin and 0.75 mmol imine, respectively. Temperature-dependent rate experiments for imine hydroboration have also been explored. Activation parameters for the hydroboration of FPhC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NPh (1b) were determined from the Eyring and Arrhenius plots with ΔS≠, ΔH≠, and Ea values of −28.69 (±0.3) e.u., 12.95 (±0.04) kcal mol−1, and 15.22 (±0.09) kcal mol−1, respectively.
NPh (1b) were determined from the Eyring and Arrhenius plots with ΔS≠, ΔH≠, and Ea values of −28.69 (±0.3) e.u., 12.95 (±0.04) kcal mol−1, and 15.22 (±0.09) kcal mol−1, respectively.
Introduction
Amines and their derivatives are found extensively in nature: proteins, nucleic acids and alkaloids contain the amine functionality. Amines are also key synthetic intermediates in the synthesis of bactericides, herbicides, rubber accelerators and many clinically applied drugs.1–5 A wide range of methods have been reported for the preparation of amines; the reduction of nitrogenous compounds using stoichiometric amounts of alkaline metal hydrides, LiAlH4 or NaBH4,6,7 hydrogenation8,9 catalysed by precious metals in the presence of either pressurized H2 or via hydrogen transfer10 and hydrosilylation.11 Hydroboration is an important transformation used to synthesize a wide variety of valuable products of industrial significance such as commodity and fine chemicals, agrochemicals and materials.12–15 Thus it is unsurprising that catalytic hydroboration of nitriles,16–20 carbodiimides21–25 and imines26–28 are common methods used to prepare amines. Many experimental protocols have been studied;29 catalysts employed in imine hydroboration range from main group elements,30–32 precious-metals33–36 and, rare earth metals.21,37The first report of imine hydroboration catalysed by a transition metal was by Baker and Westcott and used a bidentate phosphine ligated gold(I) complex (5 mol%) and catecholborane in 1995.38 In 2001, the Westcott group reported a rhodium catalysed hydroboration of allylamine in the presence of HBcat.33 Select examples of complexes capable of effecting catalytic imine hydroboration are shown in Fig. 1.
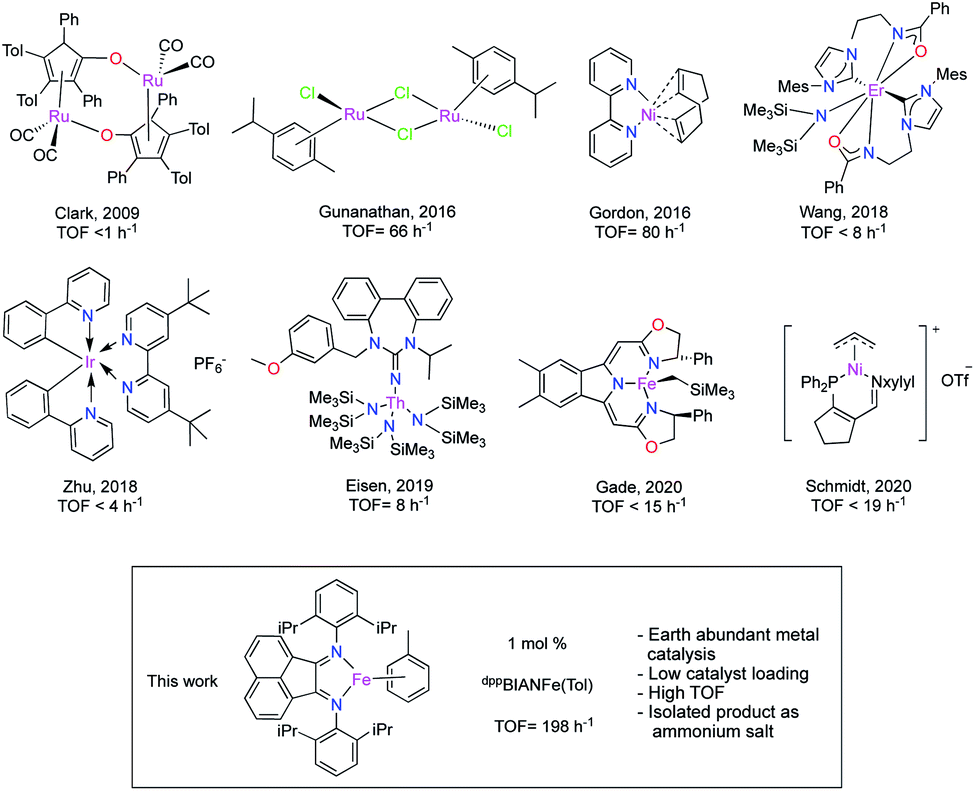 | ||
| Fig. 1 Select (pre)catalysts reported in the literature for imine hydroboration and this report. | ||
In 2009, Clark and co-workers reported a boron-substituted hydroxycyclopentadienyl ruthenium hydride catalyst with a limited substrate scope, three examples of aldimines, in hydroboration chemistry employing HBpin.34 Similarly, another ruthenium-based system capable of aldimine hydroboration was reported by Gunanathan in 2016 with a slightly broader substrate scope.35 A fascinating dual-catalytic system was recently reported by the Zhu group. In this protocol, an inverse imine hydroboration (where boron adds to the carbon of the CN double bond) was achieved through cooperative organocatalysis and photocatalysis.36 The Eisen group reported a thorium-catalysed hydroboration of aldimines with a broad substrate scope, however, this catalyst proved unsuccessful in ketimine reduction.37 While, there are several reports of catalytic imine hydroboration employing transition metals, first-row metal catalysis is still underdeveloped. In 2016, Gordon and co-workers demonstrated the catalytic hydroboration of imines with pinacolborane employing Ni(bpy)(cod) (bpy = 2,2-bipyridine, cod = 1,5-cyclooctadiene), as catalyst (in benzene) at room temperature with relatively low catalyst loadings. However, the substrate scope was very limited.39
Another base metal catalysed imine hydroboration was reported by the Zhang group in 2018, a Co(II) coordination polymer was employed as precatalyst, though this system was also effective with only a limited range of substrates.40 Very recently an iron catalyst was employed in enantioselective hydroboration of N-alkyl imines with a chiral bis(oxazolinylmethylidene)isoindoline pincer ligand by the Gade group. Asymmetric catalysis employing various acyclic N-alkyl imines afforded α-chiral amines in excellent yields.41 The Schmidt group recently reported a cationic [(iminophosphine)nickel(allyl)]+ complex in the hydroboration of N-allylimines (5 mol%). However, no successful hydroboration of ketimine is reported by this catalyst.42 Significantly, across all reports there have been limited examples of successful application of hydroboration strategies employing ketimine substrates; strongly indicating an ongoing need for more active/selective catalysts to be developed. The low abundance, high cost and toxicity of precious metal elements has prompted the catalytic community to focus heavily on an examination of the use of base metals in catalysis.43 Our group has a long standing interest in employing earth abundant metal catalysts in a variety of transformations such as aldehyde and ketone hydrosilylation,44 imine hydrosilylation,11 alkene and alkyne hydroboration,45 ester and amide reduction.46,47 Iron has emerged as a leading candidate in 1st-row metal catalysis as it is the most abundant transition metal and the fourth most abundant element in the Earth's crust, it is environmentally benign, cost effective and has long-term commercial availability.48 Given the scarce number of literature reports in iron catalysed imine hydroboration, coupled with a lack of success with ketimine substrates specifically, we decided to study the application of iron systems in imine hydroboration.
Results and discussion
Catalytic hydroboration of imines
Initially, we examined the catalytic hydroboration of imines using N-benzylideneaniline (1a) as our model substrate. dppBIANFe(Tol) is readily prepared following the reported literature procedure.44 Hydroboration of 1a proceeded smoothly, as judged by 1H NMR. As a control, the hydroboration reaction was repeated in the absence of dppBIANFe(Tol) and yield of 10% was observed (Table S1†). A range of different iron salts, from commercially available to synthesized complexes, was tested in catalysis and showed little to moderate activity (Table 1). In the interests of disclosure, use of Fe(OTf)3 revealed similar catalytic activity as dppBIANFe(Tol). However, our prior experience with dppBIANFe(Tol) and the synthetic modularity afforded by this ligand persuaded us to choose this precatalyst for further study. The use of an activator (NaOtBu) in conjunction with dppBIANFe(Tol) resulted in a modest improvement in yield. A number of reaction variables were screened in an effort to optimize product yield: catalyst loading, temperature, solvent and, activator. Ultimately, employing toluene and NaOtBu as the reaction solvent and activator, respectively, allowed us to maximize the yield of amine product (2a) to 97% (Tables S2–S4†). Finally, the reaction was found to be equally effective employing catecholborane (HBcat), the hydroboration product being isolated in 90% yield. However, a diminished yield (45%) was obtained when a sterically encumbered borane ((9-borabicyclo[3.3.1]nonane)) was used. With optimized conditions in hand, we screened a variety of aldimine and ketimine substrates, which were easily accessed using well established literature procedures.10 N-1-Diphenylethan-1-imine (1c) was cleanly converted to amine product (2c, 84%) at low catalyst loading (1 mol%) within 1 h.
|
||
|---|---|---|
| Entry | [Fe] | Yieldb (%) |
| a Reaction conditions: 1a (0.5 mmol), [Fe] (1 mol%), HBpin (1.5 equiv.), THF, 70 °C, 20 h.b As determined by 1H NMR employing mesitylene as internal standard.c Optimized conditions: 1a (0.5 mmol), dppBIANFe(Tol) (1 mol%), HBpin (1.5 equiv.), NaOtBu (2 mol%), toluene, 70 °C, 1 h. | ||
| 1 | dppBIANFeCl2 | 14 |
| 2 | Fe(CO)5 | 54 |
| 3 | dppBIANFe(Tol) | 87 |
| 4 | Fe(acac)3 | 65 |
| 5 | FeCl2-Bpy | <10 |
| 6 | Fe(OTf)3 | 84 |
| 7c | dppBIANFe(Tol) | 97 |

Encouraged by the successful conversion of a ketimine substrate employing dppBIANFe(Tol), we decided to examine more ketimine substrates. Substitution patterns ranging from electron-releasing, withdrawing, heteroatom and sterically hindered groups were studied. Excellent product yields of more than 90% (within ∼2 h) were obtained when employing substrates which feature electron withdrawing groups (2e, 2f, 2k). Similarly, a more electron-rich substrate (2d) also afforded product in very good yield (88%). A thiophene-based ketimine substrate (2g) was also found to perform well under optimized reaction conditions, achieving good conversion (88%) in only 1 h. Finally, substrates which feature an enlarged steric profile (2h, 2j) were also examined and revealed to be more recalcitrant substrates. Increased reaction times were required, and lower overall yields were observed. Moreover, to demonstrate the convenience of our approach, we have isolated reaction products as their ammonium salts following treatment with 1 M HCl in diethyl ether. Three examples are presented, 2a, 2b and 2g, affording isolated yields of 85, 65 and, 75%, respectively (Scheme 1). Moreover, a gram scale reaction of 1a was carried out to demonstrate the synthetic applicability of this protocol. An isolated yield of 90% was obtained as ammonium salt (Scheme 1c).
 | ||
| Scheme 1 Hydroboration of imines and their subsequent conversion to secondary amines. Reaction conditions: imine (0.5 mmol), HBpin (1.5 equiv.), dppBIANFe(Tol) (0.005 mmol), NaOtBu (0.01 mmol) at 70 °C in toluene; conversions were determined by 1H NMR employing mesitylene as internal standard. bIsolated yield as ammonium salt in parentheses. cScale-up reaction of catalytic hydroboration of N-benzylideneaniline; isolated yield as ammonium salt. | ||
Catalytic hydroboration of nitriles
We expanded our hydroboration studies to include nitriles, employing benzonitrile (3a) as a model substrate.16–20,37,49 Thus, exposure of 3a to dppBIANFe(Tol) (1 mol%) and 2.5 equiv. HBpin afforded diborylated amine (4a) in 98% conversion within 3.5 h. The reaction was carried out under solvent- and additive-free conditions under mild (RT) conditions. A small scope of substrate was explored, and the reaction was found to tolerate both electron-releasing and withdrawing substituents. Bromo- and chloro-substitution were tolerated and aliphatic (cyclic) substrates all afforded diborylated amines in good to excellent yields. Substrates with electron-withdrawing functional groups (–F, –Cl, –Br) on the backbone were well tolerated and yields ranging from good to excellent (81–95%) were obtained within 2 to 3 h at 70 °C (4b–4d). A nitrile substrate with an electron releasing group (–OMe) afforded product in 99% yield within 6 h at 70 °C (4e). Cyclohexanecarbonitrile also afforded the corresponding diborylated product (4f) in 83% yield in 6 h (Scheme 2). Following a similar procedure as employed in the isolation of imine hydroboration products, the treatment of nitrile hydroboration products with 1 M HCl (ether) facilitated easy isolation of the corresponding ammonium salts. Two substrates were chosen to demonstrate the utility of this approach, 4b and 4d were isolated in 84 and 77% yields, respectively. | ||
| Scheme 2 Catalytic hydroboration of nitriles to diboryl amines. aReaction conditions: nitriles (0.25 mmol), HBpin (2.5 equiv.), dppBIANFe(Tol) (0.0025 mmol) at 70 °C; yields were determined by 1H NMR employing mesitylene as internal standard. bReaction was carried out at room temperature. cIsolated yield as ammonium salt in parentheses. | ||
Preliminary mechanistic study of imine hydroboration
To gain insight into the mechanism of imine hydroboration including possible modes of activation of the precatalyst dppBIANFe(Tol), we conducted a series of experiments. Thus, stoichiometric reactions to generate plausible reaction intermediates, preliminary study of reactions kinetics including temperature-dependent reaction rate experiments were performed. Reactions between dppBIANFe(Tol) and model imine 1a (1 and 3 equiv. of the imine), were carried out at both room temperature and 70 °C; we concluded that dppBIANFe(Tol) by itself, does not appear to react directly with imine substrate even after prolonged exposure at elevated temperature (Fig. S8†). Moreover, stoichiometric reaction of dppBIANFe(Tol) with HBpin at either room temperature or 70 °C did not afford any detectable new product(s), as evidenced by in situ monitoring employing 11B NMR spectroscopy. However, upon addition of stoichiometric amounts of NaOtBu to the same reaction mixture we observed complete consumption of HBpin. The signal attributed to HBpin at δ 27 ppm in the 11B NMR spectrum disappeared, followed by the formation of a new signal at δ 20 ppm (Fig. S9†). This result can be interpreted as the requirement of precatalyst activation employing NaOtBu to form an active catalyst either via direct interaction with HBpin,50 then subsequent reaction with dppBIANFe(Tol) or a synergistic effect of all three species. Alas, our attempts to isolate organometallic species from stoichiometric reactions were unsuccessful. Preliminary kinetic analysis of the catalytic hydroboration reaction was performed by observing the disappearance of 4-fluorophenyl-N-phenylmethanimine (1b) which is conveniently monitored using 19F NMR spectroscopy (Fig. S1†). A plot of the initial rate of the disappearance of 1b exhibits a first-order dependence on the catalyst (Fig. 2). While varying the concentration of either HBpin or 1b appeared to reveal saturation kinetics, whereby the reaction is first order in both HBpin and imine (1b) only up to 0.5 and 0.75 mmol respectively (Fig. S2 and S4†). | ||
| Fig. 2 Plot of initial reaction rates (δp/δt) vs. dppBIANFe(Tol). | ||
The reaction profile of catalytic hydroboration of 4-fluorophenyl-N-phenylmethanimine at different temperatures is depicted in Fig. 3. Activation parameters for the hydroboration of FPhCNPh were determined from the Eyring and Arrhenius plots with ΔS≠, ΔH≠, and Ea values of −28.69 (±0.3) e.u., 12.95 (±0.04) kcal mol−1, and 15.22 (±0.09) kcal mol−1, respectively (Fig. S6 and S7†). The negative activation entropy indicates that the rate-determining step involves an associative process and ΔH≠ and ΔS≠ data are consistent with concerted bond-cleavage and bond-formation process with an organized four-centered transition state, respectively. Data obtained at 70 °C appears to show anomalously high reaction rates. Multiple kinetics runs at this same temperature showed the same data; we are still investigating the cause.
 | ||
| Fig. 3 Reaction profile of catalytic hydroboration of 4-fluorophenyl-N-phenylmethanimine with HBpin using dppBIANFe(Tol) as catalyst at different temperatures. | ||
On the basis of both our preliminary studies and previous reported literature, a plausible hydroboration mechanism is proposed for the reaction of dppBIANFe(Tol) and PhCNPh (1a) in the presence of HBpin (Scheme 3). The first step in the catalytic cycle is loss of toluene from dppBIANFe(Tol) concomitant with generation of a new HBpin complex. Exposure of this species to imine substrate produces an iron(boryl)(amine) after insertion of the CN bond into an iron-hydride. Reductive elimination of the aminoborane product followed by rebinding of the toluene-cap would close the catalytic cycle.
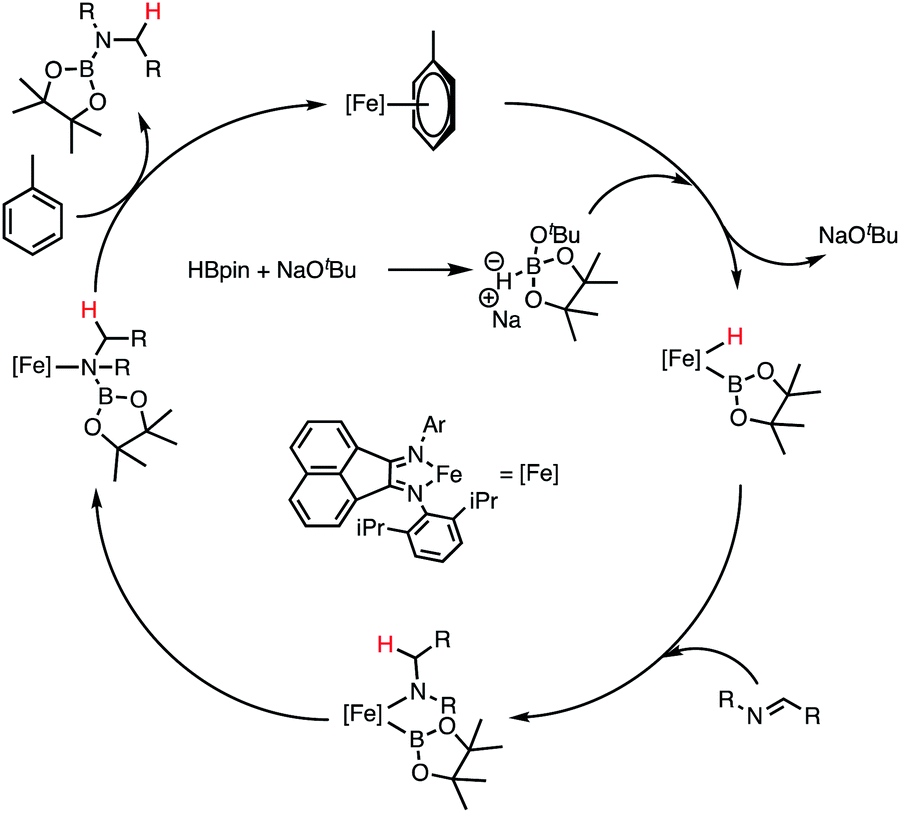 | ||
| Scheme 3 Plausible mechanism for imine hydroboration. | ||
Conclusions
In conclusion, the rapid hydroboration of imines to the corresponding secondary amines have been developed using dppBIANFe(Tol) as precatalyst in the presence of HBpin. Hydroboration products can be conveniently isolated as the corresponding ammonium salts via treatment with HCl as a 1 M solution in diethyl ether. To the best of our knowledge, this catalyst has the highest TOF (198 h−1) amongst previously reported transition and rare earth metal complexes to carry out the efficient hydroboration of imines. Moreover, this catalytic system proved capable of the reduction of ketimine substrates in good to excellent yields, which are considered more challenging substrates to reduce. In addition, dppBIANFe(Tol) was found to be an effective precatalyst in the hydroboration of nitriles under solvent free and additive free condition to form diboryl amines with excellent yields.Author contributions
ARB and MF designed and executed the project. ARB and MW carried out the experimental work. ARB and MF wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the National Science Foundation (CHE-1554906) for financial support of this work.Notes and references
- J. Verduyckt, R. Coeck and D. E. De Vos, ACS Sustainable Chem. Eng., 2017, 5, 3290–3295 CrossRef CAS.
- M. A. Blaskovich, J. Med. Chem., 2016, 59, 10807–10836 CrossRef CAS PubMed.
- R. Jastrząb, L. Łomozik and B. Tylkowski, Phys. Sci. Rev., 2016, 1, 69–106 Search PubMed.
- R. Mesnage, C. Benbrook and M. N. Antoniou, Food Chem. Toxicol., 2019, 128, 137–145 CrossRef CAS PubMed.
- B. Movassagh, H. Rooh and H. Bijanzadeh, Chem. Heterocycl. Compd., 2013, 48, 1719–1721 CrossRef CAS.
- J. T. Colyer, N. G. Andersen, J. S. Tedrow, T. S. Soukup and M. M. Faul, J. Org. Chem., 2006, 71, 6859–6862 CrossRef CAS PubMed.
- A. D. Dorsey, J. E. Barbarow and D. Trauner, Org. Lett., 2003, 5, 3237–3239 CrossRef CAS PubMed.
- S. F. Zhu, J. B. Xie, Y. Z. Zhang, S. Li and Q. L. Zhou, J. Am. Chem. Soc., 2006, 128, 12886–12891 CrossRef CAS PubMed.
- Y. Liu and H. Du, J. Am. Chem. Soc., 2013, 135, 6810–6813 CrossRef CAS PubMed.
- J. S. Samec and J. E. Bäckvall, Chem.–Eur. J., 2002, 8, 2955–2961 CrossRef CAS.
- A. Saini, C. R. Smith, F. S. Wekesa, A. K. Helms and M. Findlater, Org. Biomol. Chem., 2018, 16, 9368–9372 RSC.
- C. C. Chong, B. Rao and R. Kinjo, ACS Catal., 2017, 7, 5814–5819 CrossRef CAS.
- J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- S. R. Tamang, A. Singh, D. K. Unruh and M. Findlater, ACS Catal., 2018, 8, 6186–6191 CrossRef CAS.
- M. Haberberger and S. Enthaler, Chem.–Asian J., 2013, 8, 50–54 CrossRef CAS.
- T. Kitano, T. Komuro and H. Tobita, Organometallics, 2019, 38, 1417–1420 CrossRef CAS.
- H. Ben-Daat, C. L. Rock, M. Flores, T. L. Groy, A. C. Bowman and R. J. Trovitch, Chem. Commun., 2017, 53, 7333–7336 RSC.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed.
- M. Ito, M. Itazaki and H. Nakazawa, Inorg. Chem., 2017, 56, 13709–13714 CrossRef CAS PubMed.
- A. D. Ibrahim, S. W. Entsminger and A. R. Fout, ACS Catal., 2017, 7, 3730–3734 CrossRef CAS.
- Y. Yuan, X. Wang, Y. Li, L. Fan, X. Xu, Y. Chen, G. Li and W. Xia, Organometallics, 2011, 30, 4330–4341 CrossRef CAS.
- A. Ramos, A. Antiñolo, F. Carrillo-Hermosilla, R. Fernández-Galán and D. García-Vivó, Chem. Commun., 2019, 55, 3073–3076 RSC.
- Y. Ding, X. Ma, Y. Liu, W. Liu, Z. Yang and H. W. Roesky, Organometallics, 2019, 38, 3092–3097 CrossRef CAS.
- M. Rauch, S. Ruccolo and G. Parkin, J. Am. Chem. Soc., 2017, 139, 13264–13267 CrossRef CAS PubMed.
- Q. Shen, X. Ma, W. Li, W. Liu, Y. Ding, Z. Yang and H. W. Roesky, Chem.–Eur. J., 2019, 25, 11918–11923 CrossRef CAS PubMed.
- Q. Yin, Y. Soltani, R. L. Melen and M. Oestreich, Organometallics, 2017, 36, 2381–2384 CrossRef CAS.
- M. Arrowsmith, M. S. Hill and G. Kociok-Köhn, Chem.–Eur. J., 2013, 19, 2776–2783 CrossRef CAS PubMed.
- D. Bedi, A. Brar and M. Findlater, Green Chem., 2020, 22, 1125–1128 RSC.
- D. Hayrapetyan and A. Y. Khalimon, Chem.–Asian J., 2020, 15, 2575–2587 CrossRef CAS PubMed.
- M. R. Adams, C. H. Tien, B. S. Huchenski, M. J. Ferguson and A. W. Speed, Angew. Chem., 2017, 56, 6268–6271 CrossRef CAS PubMed.
- M. K. Bisai, S. Pahar, T. Das, K. Vanka and S. S. Sen, Dalton Trans., 2017, 46, 2420–2424 RSC.
- C. H. Tien, M. R. Adams, M. J. Ferguson, E. R. Johnson and A. W. Speed, Org. Lett., 2017, 19, 5565–5568 CrossRef CAS PubMed.
- C. M. Vogels, P. E. O'Connor, T. E. Phillips, K. J. Watson, M. P. Shaver, P. G. Hayes and S. A. Westcott, Can. J. Chem., 2001, 79, 1898–1905 CrossRef CAS.
- L. Koren-Selfridge, H. N. Londino, J. K. Vellucci, B. J. Simmons, C. P. Casey and T. B. Clark, Organometallics, 2009, 28, 2085–2090 CrossRef CAS.
- A. Kaithal, B. Chatterjee and C. Gunanathan, J. Org. Chem., 2016, 81, 11153–11161 CrossRef CAS PubMed.
- N. Zhou, X. A. Yuan, Y. Zhao, J. Xie and C. Zhu, Angew. Chem., 2018, 130, 4054–4058 CrossRef.
- S. Saha and M. S. Eisen, ACS Catal., 2019, 9, 5947–5956 CrossRef CAS.
- R. T. Baker, J. C. Calabrese and S. A. Westcott, J. Organomet. Chem., 1995, 498, 109–117 CrossRef CAS.
- A. E. King, S. C. E. Stieber, N. J. Henson, S. A. Kozimor, B. L. Scott, N. C. Smythe, A. D. Sutton and J. C. Gordon, Eur. J. Inorg. Chem., 2016, 1635–1640 CrossRef CAS.
- J. Wu, H. Zeng, J. Cheng, S. Zheng, J. A. Golen, D. R. Manke and G. Zhang, J. Org. Chem., 2018, 83, 9442–9448 CrossRef CAS PubMed.
- C. K. Blasius, N. F. Heinrich, V. Vasilenko and L. H. Gade, Angew. Chem., 2020, 132, 16108–16111 CrossRef.
- I. Hossain and J. A. R. Schmidt, Eur. J. Inorg. Chem., 2020, 1877–1884 CrossRef CAS.
- S. R. Tamang and M. Findlater, Molecules, 2019, 24, 3194 CrossRef CAS PubMed.
- F. S. Wekesa, R. Arias-Ugarte, L. Kong, Z. Sumner, G. P. McGovern and M. Findlater, Organometallics, 2015, 34, 5051–5056 CrossRef CAS.
- A. Singh, S. Shafiei-Haghighi, C. R. Smith, D. K. Unruh and M. Findlater, Asian J. Org. Chem., 2020, 9, 416–420 CrossRef CAS.
- S. R. Tamang, A. F. Cozzolino and M. Findlater, Org. Biomol. Chem., 2019, 17, 1834–1838 RSC.
- S. R. Tamang, A. Singh, D. Bedi, A. R. Bazkiaei, A. A. Warner, K. Glogau, C. McDonald, D. K. Unruh and M. Findlater, Nat. Catal., 2020, 3, 154–162 CrossRef CAS.
- M. D. Greenhalgh, A. S. Jones and S. P. Thomas, ChemCatChem, 2015, 7, 190–222 CrossRef CAS.
- Z. Huang, S. Wang, X. Zhu, Q. Yuan, Y. Wei, S. Zhou and X. Mu, Inorg. Chem., 2018, 57, 15069–15078 CrossRef CAS PubMed.
- J. H. Docherty, J. Peng, A. P. Dominey and S. P. Thomas, Nat. Chem., 2017, 9, 595 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental details and NMR spectra. See DOI: 10.1039/d1ra02001c |
| This journal is © The Royal Society of Chemistry 2021 |