
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
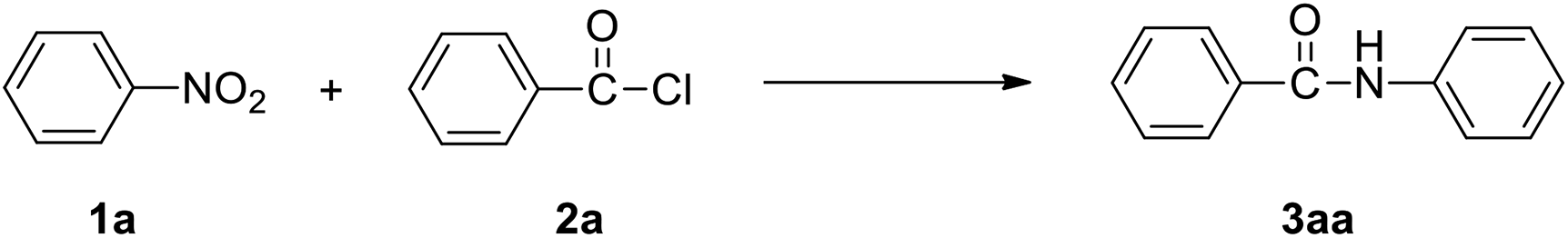
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe-mediated synthesis of N-aryl amides from nitroarenes and acyl chlorides†
Yundong Wua,
Lei Guo *a,
Yuxuan Liub,
Jiannan Xiangb and
Jun Jiang*c
*a,
Yuxuan Liub,
Jiannan Xiangb and
Jun Jiang*c
aSchool of Material and Chemical Engineering, Tongren University, Tongren 554300, China. E-mail: cqglei@163.com
bCollege of Chemistry and Chemical Engineering, Hunan University, Changsha 410082, China
cSchool of Chemistry and Chemical Engineering, Hunan University of Science and Technology, Xiangtan 411201, China. E-mail: jiangjunhust@163.com
First published on 23rd April 2021
Abstract
Amides are prevalent in nature and valuable functional compounds in agrochemical, pharmaceutical, and materials industries. In this work, we developed a selective and mild method for the synthesis of N-aryl amides. Starting from commercially available nitroarenes and acyl halides, N-aryl amides with good yields can be obtained in water. Especially in the process of transformation, Fe dust is the only reductant and additive, and the reaction can be easily performed on a large scale.
1. Introduction
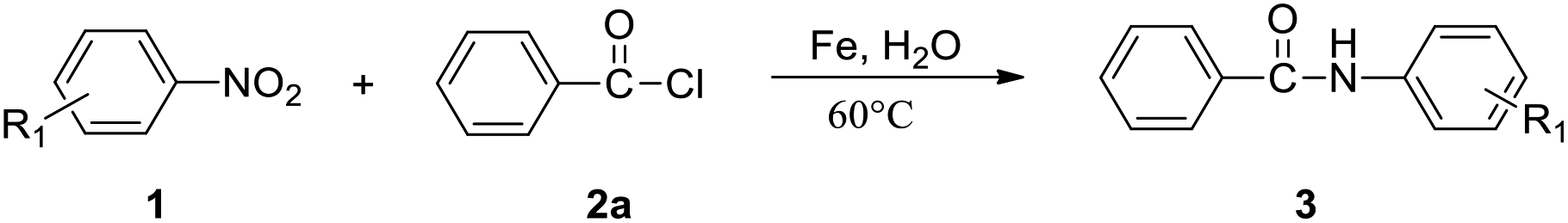
Amides are synthetically important intermediates and usually serve as useful building blocks for drugs, fine chemicals, and bioactive molecules such as peptides and proteins.1–8 The amidation reaction with anilines or amines as amino sources is used to construct amide motifs.9–13 It is more efficient to start directly from the nitrocompounds which are readily available industrial chemicals rather than to start from the corresponding amines or isocyanates.14,15 In general, the classical two-step approach should be often proposed when someone is asked to prepare an amide from a nitro compound, namely reduction to amine with subsequent acylation (Scheme 1a). More importantly, several methods for direct conversion of nitroarenes to acetanilides are carried out by Pt nanoparticle ZrO2 and acetic anhydride, molybdenum hexacarbonyl/acetic acid, and also anilides have been obtained either via nickel or rhodium or platinum catalyzed carbonylation of nitroarenes.16–18 However, 50–100 atm CO is required in each case. In 2018, Hu and co-workers described Mn-catalyzed reductive transamidation of tertiary amides with nitroarenes (Scheme 1b).19 Recently, the reductive amidation between esters and nitroarenes can be achieved by nickel catalysis by cleavage of acyl C–O bonds, wherein metallic zinc serves as efficient reductant (Scheme 1c).20–22 However, these methods have some obvious shortcomings such as the use of organic solvents and additives which are difficult to recover. To overcome the difficulties in the transformation of N-aryl amides, we need to find an efficient and improved synthesis route. Water, as a green solvent, is widely used in organic synthesis. In addition, Fe dust is a cheap and safe reducing agent and can be used in many organic reactions.23,24 We are interested in Fe-promoted organic reactions performed in water. Therefore, we developed a mild Fe-promoted reaction of nitroarenes with acyl chloride (fluoride) using water as solvent in this work. Using the Fe-mediated synthetic method, a series of N-aryl amides can be produced in moderate to good yields, showing its fascinating application prospects (Scheme 1d). | ||
| Scheme 1 Different approaches for amidation using nitroarenes as amino sources. | ||
2. Results and discussion
We used the reaction of nitrobenzene (1a) with benzoyl chloride (2a, 2 equiv.) as test reaction (Table 1). Using the conditions of benzoyl chloride (0.5 mmol) with nitrobenzenes (0.25 mmol), iron dust (4 equiv.) as reductant, and water (1 mL) as solvent, the transamidation was actually successful, giving the desired amide product N-phenyl benzamide (3aa), in 88% yield (Table 1, entry1). We optimized the Fe-mediated transamidation of 1a with 2a. Other metal reductants such as zinc, aluminium, and magnesium were not effective (Table 1, entries 2–4). By changing the temperature to 80 °C or 40 °C, the yield of 3aa was reduced (Table 1, entries 5 and 6). We believe that weak hydrolysis reaction may occur for acyl chlorides at elevated temperature, resulting in diminished yield of target product. The use of other solvent instead of water all led to a great reduction of yields (Table 1, entries 7–11). More loading of Fe (5 equiv.) or PhCOCl (2.5 equiv.) have no effect on yield, but lower loading of Fe (3 equiv.) or PhCOCl (1.5 equiv.) led to a modest reduction of yields (Table 1, entries 12–16).| Entry | Variations from standard conditions | Yieldb |
|---|---|---|
| a All reactions were performed with nitrobenzene (1a, 0.25 mmol), acyl chloride (2a, 0.5 mmol), reductant (1 mmol) and solvent (1 mL), 36 h.b Isolated yield. | ||
| 1 | No variation | 88 |
| 2 | Zn (4 equiv.) instead of Fe | 45 |
| 3 | Al (4 equiv.) instead of Fe | 17 |
| 4 | Mg (4 equiv.) instead of Fe | 18 |
| 5 | 80 °C instead of 60 °C | 68 |
| 6 | 40 °C instead of 60 °C | 78 |
| 7 | NMP instead of H2O | 30 |
| 8 | DMF instead of H2O | 22 |
| 9 | Toluene instead of H2O | 11 |
| 10 | PhCl instead of H2O | 14 |
| 11 | EtOH instead of H2O | 45 |
| 12 | Fe (5 equiv.) instead of (4 equiv.) | 88 |
| 13 | Fe (3 equiv.) instead of (4 equiv.) | 55 |
| 14 | Absence of Fe | Trace |
| 15 | PhCOCl (2.5 equiv.) instead of (2 equiv.) | 87 |
| 16 | PhCOCl (1.5 equiv.) instead of (2 equiv.) | 59 |
 |
||
The optimized reaction conditions were employed for the transamidation of nitroarenes (1a) with acyl chloride (2) (shown in Table 2). The reaction of nitrobenzene (1a) and benzoyl chloride (2a) efficiently offered 88% yield of 3aa in 36 h at 60 °C. Various acyl chlorides reacted smoothly with nitrobenzene to give the N-aryl amide products in medium to excellent yields. It was observed that benzoyl chlorides with electron donating substituents afforded the higher yields of corresponding products (3ab–3ad, 3ak). However, the yield of anilides with electron withdrawing substituents were reduced (3ae, 3ag–3aj). Notably, both isobutyryl chloride and cyclohexanecarbonyl chloride were proven to be effective to produce the desired products in medium yield (3af, 3al). However, when we used 4-nitrobenzoyl chloride (2n) or 2-nitrobenzoyl chloride (2m), no obvious reactions were observed. Moreover, benzoyl fluoride reacted with nitrobenzene to give 3aa in 56% yield (3aa).


Subsequently, various nitroarenes were reacted with benzoyl chloride (2a) as shown in Table 3. The reaction of 2a with 1-methyl-4-nitrobenzene, 1-methyl-3-nitrobenzene and 1-chloro-4-methyl-2-nitrobenzene gave the corresponding products, 3ba, 3ca and 3ga in good yields. The 1-chloro-4-nitrobenzene and 1-bromo-4-nitrobenzene also participated well in this reaction (3ea, 3fa). Meanwhile, this reaction is also tolerant of 1-chloro-4-methoxy-2-nitrobenzene and 1,4-dichloro-2-nitrobenzene, and the desired products 3ha and 3ia were produced in 78% and 64% yields. However, it was observed that nitromethane (1j) failed to produce desired N-methylbenzamide in practical yields.

To verify the practicality of the reaction, a scale–up reaction were carried out. Nitrobenzene (1a, 10 mmol) reacted with benzoyl chloride and Fe under standard conditions, and the expected product 3aa was obtained in 80% yield (Scheme 2).
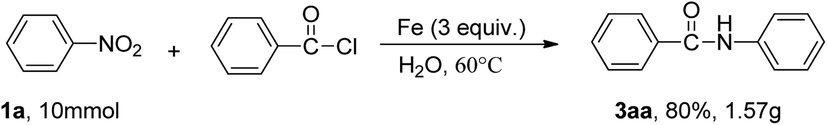 | ||
| Scheme 2 Gram scale synthesis. | ||
A series of control experiments were carried out to gain some insights into the possible mechanism. When radical trapping reagents (TEMPO) was added to the reaction mixture, the transformation was inhibited, implying that the transformation might proceed via radical process (Scheme 3a). Lacking of BzCl in the reaction, there was no PhNH2 generated (Scheme 3b). Then, we usedsd nitrosobenzene as the reactant under the standard conditions and the product 3aa was obtained (Scheme 3c), but N-phenyl-hydroxylamine or azobenzene or N,N′-diphenylhydrazine could not react with benzoyl chloride to generate 3aa (Scheme 3d) suggesting that nitrosobenzene might be involved as an intermediate in the transformation. Finally, when the reaction was conducted under a nitrogen atmosphere, we got the product 3aa in 86% yield (Scheme 3e). We observed that the color of the mixtures changed from colourless to pale brown and some brown solid was generated. We added 1 mL HCl (1 M) to the reaction mixtures and the precipitation was disappeared followed by color changing from pale brown to pale yellow. So we speculated that ferric hydroxide was generated in the transformation. Finally, the reaction of 1a with carboxylic acid anhydride Bz2O could not produce 3aa under the standard conditions (Scheme 3f).
 | ||
| Scheme 3 Control experiments. | ||
According to the aforementioned experimental results and literature reports,25 PhCOCl or PhCOF was activated by ferrous powder to produce a benzoyl radical (Scheme 4a). Then, the benzoyl radical reacted with nitrosoarene originating from the reduction of nitroarene by ferrous powder to form the N–C bond. Finally, the generated radical further transforms into the desired product 3 in the presence of Ferrous ion and water (Scheme 4b).
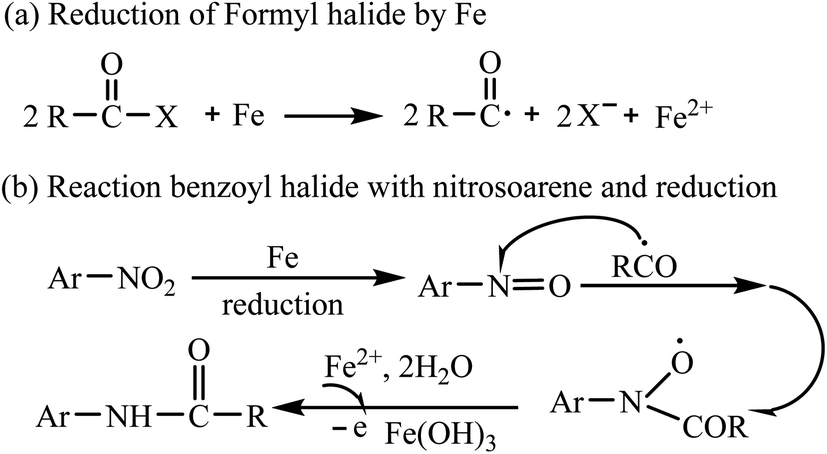 | ||
| Scheme 4 Proposed reaction mechanism. | ||
3. Conclusions
In conclusion, we have developed a step-economic amidation of PhCOX using nitroarenes as amino sources promoted by Fe. Broad scope and functional group compatibility have been demonstrated. The direct use of nitroarenes in place of anilines would provide potential advantages in cost and step economy.4. Experimental details
NMR spectroscopy was performed on a Bruker advanced spectrometer operating at 400 MHz (1H NMR) and 100 MHz (13C NMR). Melting point was measured with a XRC-1 melting point apparatus. FT infrared (IR) spectra was recorded using FD-5DX spectrometer. Mass spectrometric analysis was performed on MAT 95 XP (Thermo Finnigan).General procedure for synthesis of N-phenylacetamides (3)
A mixture of nitroarenes (0.25 mmol) and acyl chloride (fluoride) (0.5 mmol) and H2O (1 mL) was stirred at 60 °C for 36 h under air. After cooling to room temperature, water (10 mL) was added and the aqueous phase was extracted by EtOAc (3 × 10 mL). The combined organic phases were dried over Na2SO4, and concentrated in vacuum. The residue was purified by chromatography on silica gel with petroleum ether/ethyl acetate as eluent to afford the corresponding product.N-Phenylbenzamide (3aa)
Pale brown solid. Mp: 162–163 °C (lit12 164–165 °C). Yield 45 mg (88%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H), 8.01 (d, J = 7.2 Hz, 2H), 7.84 (d, J = 7.8 Hz, 2H), 7.62 (t, J = 7.2 Hz, 1H), 7.56 (t, J = 7.2 Hz, 2H), 7.39 (t, J = 7.2 Hz, 2H), 7.14 (t, J = 7.3 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 166.1, 139.7, 135.5, 132.4, 129.1, 128.9, 128.1, 124.1, 120.9.
4-Methyl-N-phenylbenzamide (3ab)
White solid. Mp: 142–143 °C (lit12 145–146 °C). Yield 39 mg (91%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.22 (s, 1H), 7.92 (d, J = 7.6 Hz, 2H), 7.83 (d, J = 7.6 Hz, 2H), 7.38 (t, J = 8.0 Hz, 4H), 7.12 (t, J = 7.0 Hz, 1H), 2.41 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 165.8, 142.0, 139.7, 132.6, 129.4, 129.0, 128.2, 124.0, 120.8, 21.5.
3-Methyl-N-phenylbenzamide (3ac)
White solid. Mp: 125–127 °C (lit12 125–126 °C). Yield 35 mg (67%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.28 (s, 1H), 7.86–7.78 (m, 4H), 7.44 (t, J = 7.2 Hz, 2H), 7.39 (t, J = 8.0 Hz, 2H), 7.13 (t, J = 7.2 Hz, 1H), 2.43 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 166.2, 139.7, 138.2, 135.5, 132.6, 129.1, 128.7, 128.6, 125.3, 124.1, 120.8, 21.4.
2-Methyl-N-phenylbenzamide (3ad)
White solid. Mp: 125–126 °C (lit12 125–126 °C). Yield 45 mg (86%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.34 (s, 1H), 7.78 (d, J = 8.0 Hz, 2H), 7.48 (d, J = 7.4 Hz, 1H), 7.42 (t, J = 7.2 Hz, 1H), 7.38–7.31 (m, 4H), 7.12 (t, J = 7.2 Hz, 1H), 2.41 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 168.3, 139.8, 137.7, 135.6, 131.0, 130.1, 129.2, 127.7, 126.1, 124.0, 120.1, 19.8.
3,5-Dichloro-N-phenylbenzamide (3ae)
White solid. Mp: 148–149 °C. Yield 47 mg (71%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.00 (s, 2H), 7.86 (s, 1H), 7.79 (d, J = 7.8 Hz, 2H), 7.39 (t, J = 7.2 Hz, 2H), 7.15 (t, J = 7.2 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 163.1, 139.1, 138.5, 134.7, 131.3, 129.1, 127.0, 124.6, 120.9.
HRMS (ESI) m/z calcd for C13H10Cl2NO [M + H]+: 266.0134, found 266.0135.
N-Phenylisobutyramide (3af)
Shallow brown solid. Mp: 101–102 °C (lit12 102–103 °C). Yield 25 mg (62%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 9.86 (s, 1H), 7.63 (d, J = 8.0 Hz, 2H), 7.30 (t, J = 7.4 Hz, 2H), 7.04 (t, J = 7.2 Hz, 1H), 2.65–2.58 (m, 1H), 1.12 (d, J = 6.8 Hz, 6H).
13C NMR (100 MHz, DMSO-d6) δ 175.7, 139.9, 129.1, 123.4, 119.5, 35.4, 20.0.
4-Fluoro-N-phenylbenzamide (3ag)
Pale brown solid. Mp: 182–184 °C (lit12 183–184 °C). Yield 33 mg (63%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.31 (s, 1H), 8.07 (t, J = 6.6 Hz, 2H), 7.80 (t, J = 8.0 Hz, 2H), 7.40 (t, J = 8.0 Hz, 4H), 7.13 (t, J = 7.2 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 164.9, 164.5 (d, JC–F = 247 Hz), 139.5, 131.9 (d, JC–F = 3 Hz), 130.9 (d, JC–F = 9 Hz), 129.1, 124.2, 120.9, 115.8 (d, JC–F = 22 Hz).
3-Bromo-N-phenylbenzamide (3ah)
White solid. Mp: 146–148 °C (lit12 145–147 °C). Yield 44 mg (64%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.39 (s, 1H), 8.18 (s, 1H), 7.99 (d, J = 7.8 Hz, 1H), 7.83–7.79 (m, 3H), 7.52 (t, J = 7.8 Hz, 1H), 7.39 (t, J = 7.8 Hz, 2H), 7.14 (t, J = 7.4 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 164.4, 139.4, 137.6, 134.7, 131.1, 130.7, 129.1, 127.3, 124.4, 122.2, 120.9.
4-Chloro-N-phenylbenzamide (3ai)
White solid. Mp: 197–198 °C (lit12 199–200 °C). Yield 41 mg (71%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.02 (d, J = 7.8 Hz, 2H), 7.81 (d, J = 7.8 Hz, 2H), 7.63 (t, J = 7.8 Hz, 2H), 7.38 (t, J = 7.2 Hz, 2H), 7.14 (t, J = 7.0 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 164.9, 139.4, 136.9, 134.1, 130.1, 129.1, 128.9, 124.3, 120.9.
N-Phenyl-4-(trifluoromethyl)benzamide (3aj)
Brown solid. Mp: 196–197 °C (lit12 196–197 °C). Yield 41 mg (73%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.58 (s, 1H), 8.00 (d, J = 6.8 Hz, 2H), 7.92 (d, J = 7.8 Hz, 2H), 7.25 (t, J = 7.2 Hz, 2H), 7.15 (t, J = 7.8 Hz, 2H), 7.05 (t, J = 7.0 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 160.0, 143.8, 137.6, 133.0 (q, JC–F = 26 Hz),129.7, 128.1, 126.9 (q, JC–F = 3 Hz), 125.0, 123.8 (q, JC–F = 271 Hz), 121.0.
N-Phenylfuran-2-carboxamide (3ak)
White solid. Mp: 178–179 °C. Yield 36 mg (78%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ = 10.17 (s, 1H), 7.93 (s, 1H), 7.75 (d, J = 7.6 Hz, 2H), 7.40–7.27 (m, 3H), 7.09 (t, J = 7.6 Hz, 1H), 6.70 (dd, J = 3.4, 1.8 Hz, 1H).
13C NMR (101 MHz, DMSO-d6) δ 156.7, 148.0, 146.1, 139.0, 129.1, 124.2, 120.8, 115.2, 112.6.
HRMS (ESI) m/z calcd for C11H10NO2 [M + H]+: 188.0712, found 188.0716.
N-Phenylcyclohexanecarboxamide (3al)
White solid. Mp: 133–135 °C. Yield 34 mg (67%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ = 9.78 (s, 1H), 7.61 (d, J = 8.0 Hz, 2H), 7.27 (t, J = 7.8 Hz, 2H), 7.00 (t, J = 7.5 Hz, 1H), 2.32 (t, J = 11.6 Hz, 1H), 1.78–1.73 (m, 4H), 1.67–1.63 (m, 1H), 1.45–1.38 (m, 2H), 1.27–1.23 (m, 3H).
13C NMR (101 MHz, DMSO-d6) δ 174.7, 140.0, 129.0, 123.3, 119.5, 45.3, 29.6, 25.7.
HRMS (ESI) m/z calcd for C13H18NO [M + H]+: 204.1388, found 204.1385.
N-(p-Tolyl)benzamide (3ba)
White solid. Mp: 158–159 °C (lit12 156–157 °C). Yield 50 mg (95%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ10.23 (s, 1H), 7.99 (d, J = 7.2 Hz, 2H), 7.72 (d, J = 8.0 Hz, 2H), 7.60 (d, J = 7.0 Hz, 1H), 7.55 (t, J = 7.4 Hz, 2H), 7.19 (d, J = 8 Hz, 2H), 2.31 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 165.8, 137.1, 135.5, 133.1, 131.9, 129.5, 128.8, 128.1, 120.9, 21.0.
N-(m-Tolyl)benzamide (3ca)
White solid. Mp: 125–126 °C (lit12 124–125 °C). Yield 45 mg (85%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.23 (s, 1H), 7.99 (d, J = 7.2 Hz, 2H), 7.68 (s, 1H), 7.61 (t, J = 7.4 Hz, 2H), 7.55 (t, J = 7.4 Hz, 2H), 7.29–7.23 (m, 1H), 6.95 (d, J = 7.2 Hz, 1H), 2.34 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 166.0, 139.6, 138.2, 135.5, 132.0, 128.9, 128.8, 128.1, 124.8, 121.4, 118.0, 21.7.
N-(o-Tolyl)benzamide (3da)
White solid. Mp: 145–146 °C (lit12 143–145 °C). Yield 42 mg (79%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.05 (d, J = 7.0 Hz, 2H), 7.64 (d, J = 6.8 Hz, 1H), 7.59 (d, J = 7.0 Hz, 2H), 7.41 (d, J = 6.8 Hz, 1H), 7.34 (t, J = 7.0 Hz, 1H), 7.28–7.23 (m, 2H), 2.30 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 165.8, 136.9, 135.0, 134.2, 132.0, 130.8, 128.9, 128.1, 127.1, 126.5, 18.4.
N-(4-Chlorophenyl)benzamide (3ea)
White solid. Mp: 195–196 °C (lit12 199–200 °C). Yield 42 mg (71%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.44 (s, 1H), 8.00 (d, J = 7.2 Hz, 2H), 7.88 (d, J = 8.2 Hz, 2H), 7.63–7.59 (m, 1H), 7.56 (t, J = 7.2 Hz, 2H), 7.44 (d, J = 8.0 Hz, 2H).
13C NMR (100 MHz, DMSO-d6) δ 166.1, 138.7, 135.2, 132.2, 129.0, 128.9, 128.2, 127.8, 122.3.
N-(4-Bromophenyl)benzamide (3fa)
White solid. Mp: 205–207 °C (lit12 205–206 °C). Yield 48 mg (70%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.03 (d, J = 7.2 Hz, 2H), 7.87 (d, J = 8.8 Hz, 2H), 7.65 (t, J = 7.2 Hz, 1H), 7.62–7.56 (m, 4H).
13C NMR (100 MHz, DMSO-d6) δ 166.1, 139.1, 135.2, 132.2, 131.9, 128.9, 128.2, 122.7, 115.8.
N-(2-Chloro-5-methylphenyl)benzamide (3ga)
White solid. Mp: 121–122 °C. Yield 49 mg (80%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.46 (s, 1H), 8.04 (d, J = 7.8 Hz, 2H), 7.67–7.62 (m, 1H), 7.57 (t, J = 6.8 Hz, 2H), 7.46 (t, J = 8.4 Hz, 2H), 7.14 (d, J = 8.2 Hz, 1H), 2.35 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 165.8, 137.5, 135.2, 134.5, 132.3, 129.6, 129.4, 129.0, 128.6, 128.1, 126.9, 20.9.
HRMS (ESI) m/z calcd for C14H13ClNO [M + H]+: 246.0680, found 246.0682.
N-(2-Chloro-5-methoxyphenyl)benzamide (3ha)
White solid. Mp: 124–125 °C. Yield 50 mg (78%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.03 (s, 1H), 8.03 (d, J = 7.8 Hz, 2H), 7.64 (t, J = 7.0 Hz, 1H), 7.57 (t, J = 7.2 Hz, 2H), 7.48 (d, J = 8.8 Hz, 1H), 7.29 (s, 1H), 6.92 (d, J = 8.8 Hz, 1H),3.81 (s, 3H).
13C NMR (100 MHz, DMSO-d6) δ 165.8, 158.7, 136.3, 134.4, 132.4, 130.3, 129.0, 128.2, 120.9, 113.9, 113.5, 56.1.
HRMS (ESI) m/z calcd for C14H13ClNO2 [M + H]+: 262.0629, found 262.0626.
N-(2,5-Dichlorophenyl)benzamide (3ia)
White solid. Mp: 122–123 °C (lit12 123–124 °C). Yield 42 mg (64%) at 0.25 mmol scale.1H NMR (400 MHz, DMSO-d6) δ 10.19 (s, 1H), 8.02 (d, J = 7.5 Hz, 2H), 7.80 (s, 1H), 7.67–7.62 (m, 2H), 7.58 (t, J = 7.2 Hz, 2H), 7.41 (d, J = 8.4 Hz, 1H).
13C NMR (100 MHz, DMSO-d6) δ 165.9, 136.9, 134.1, 132.6, 131.9, 131.4, 129.0, 128.4, 128.2, 128.1, 127.6.
Author contributions
Conceptualization: J. N. Xiang; data curation: Y. X. Liu and Y. D. Wu; formal analysis: Y. D. Wu. and J. Jiang; investigation: Y. D. Wu and L. Guo; methodology: Y. X. Liu and L. Guo; resources: J. N. Xiang; visualization: Y. D. Wu and Y. X. Liu; writing – original draft preparation: Y. D. Wu and L. Guo; writing – review and editing: Y. D. Wu, L. Guo, Y. X. Liu, J. N. Xiang; supervision: J. Jiang; project administration: L. Guo and J. N. Xiang; funding acquisition: L. Guo. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors have declared that no conflicting interests exist.Acknowledgements
This work was supported by the Guizhou Provincial Department of Education Foundation (QJHKY2020-067), the National Natural Science Foundation of China (21502049, 22062022), and the Foundation of the Department of Education of Guizhou Province (KY[2018]030).References
- C. W. Cheung, M. L. Ploeger and X. Hu, Nat. Commun., 2017, 8, 14878 CrossRef PubMed.
- R. M. de Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed.
- U. Manna and G. Das, Coord. Chem. Rev., 2021, 427, 213547 CrossRef CAS.
- J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed.
- J. Jiang, S. Zeng, D. Chen, C. Cheng, W. Deng and J. Xiang, Org. Biomol. Chem., 2018, 16, 5016–5020 RSC.
- C. W. Cheung, M. L. Ploeger and X. Hu, ACS Catal., 2017, 7, 7092–7096 CrossRef CAS.
- B. L. Korbad and S. H. Lee, Chem. Commun., 2014, 50, 8985–8988 RSC.
- C. L. Allen, B. N. Atkinson and J. M. Williams, Angew. Chem., Int. Ed. Engl., 2012, 51, 1383–1386 CrossRef CAS PubMed.
- H. Lundberg, F. Tinnis, N. Selander and H. Adolfsson, Chem. Soc. Rev., 2014, 43, 2714–2742 RSC.
- M. Sun, J. Jiang, J. Chen, Q. Yang and X. Yu, Tetrahedron, 2019, 75, 130456 CrossRef.
- C. L. Allen and J. M. Williams, Chem. Soc. Rev., 2011, 40, 3405–3415 RSC.
- R. Takise, K. Muto and J. Yamaguchi, Chem. Soc. Rev., 2017, 46, 5864–5888 RSC.
- A. S. Santos, A. M. S. Silva and M. M. B. Marques, Eur. J. Org. Chem., 2020, 2020, 2501–2516 CrossRef CAS.
- E. M. Nahmed and G. Jenner, Tetrahedron Lett., 1991, 32, 4917–4920 CrossRef CAS.
- B. N. Atkinson, A. R. Chhatwal, H. V. Lomax, J. W. Walton and J. M. Williams, Chem. Commun., 2012, 48, 11626–11628 RSC.
- M. L. Kantam, R. S. Reddy, K. Srinivas, R. Chakravarti, B. Sreedhar, F. Figueras and C. Venkat Reddy, J. Mol. Catal. A: Chem., 2012, 355, 96–101 CrossRef CAS.
- T. L. Ho, J. Org. Chem., 1977, 42, 3755 CrossRef CAS.
- Y. Watanabe, Y. Tsuji, T. Kondo and R. Takeuchi, J. Org. Chem., 1984, 49, 4451–4455 CrossRef CAS.
- C. W. Cheung, J. A. Ma and X. Hu, J. Am. Chem. Soc., 2018, 140, 6789–6792 CrossRef CAS PubMed.
- C. W. Cheung, M. Leendert Ploeger and X. Hu, Chem. Sci., 2018, 9, 655–659 RSC.
- L. Hie, N. F. Fine Nathel, T. K. Shah, E. L. Baker, X. Hong, Y. F. Yang, P. Liu, K. N. Houk and N. K. Garg, Nature, 2015, 524, 79–83 CrossRef CAS PubMed.
- C. W. Cheung, N. Shen, S.-P. Wang, A. Ullah, X. Hu and J.-A. Ma, Org. Chem. Front., 2019, 6, 756–761 RSC.
- L. N. de Paula, L. A. dos Reis Giusto, J. D. Ardisson and F. Magalhães, Environ. Chem. Lett., 2015, 13, 347–351 CrossRef.
- M. Zhang, J. Li, Q. Zeng and Q. Mou, Appl. Sci., 2019, 9, 3604 CrossRef CAS.
- C. W. Cheung and X. Hu, Nat. Commun., 2016, 7, 13776 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR spectra of new compounds. See DOI: 10.1039/d0ra10868e |
| This journal is © The Royal Society of Chemistry 2021 |