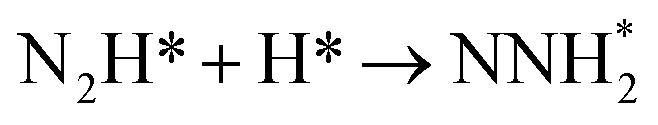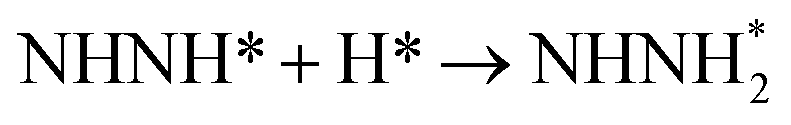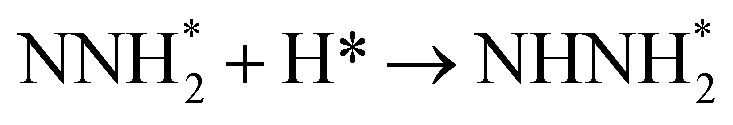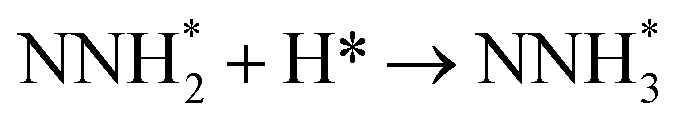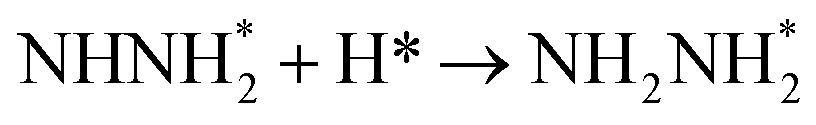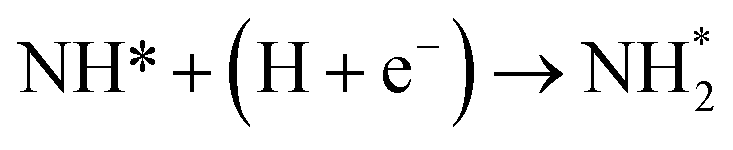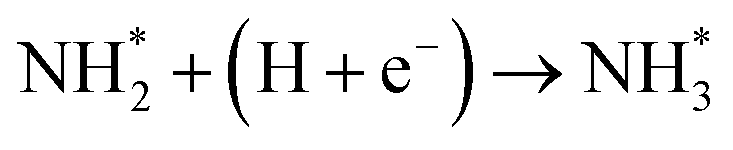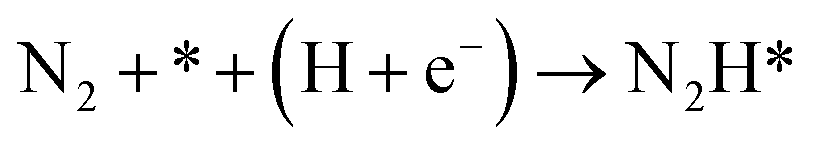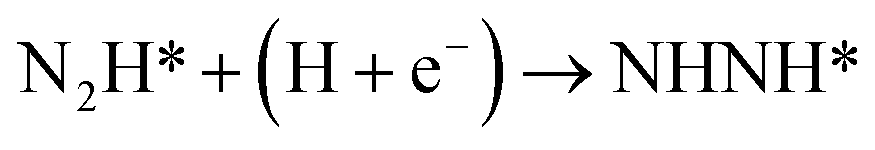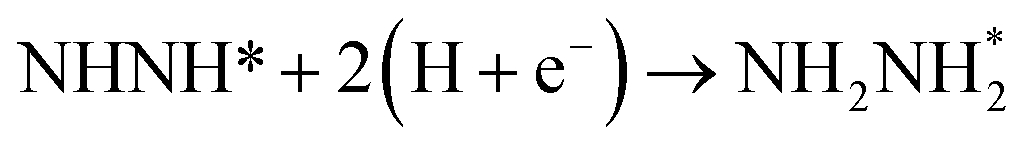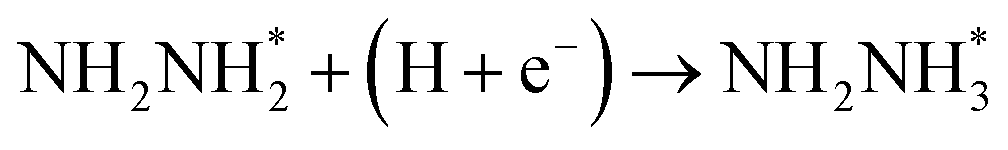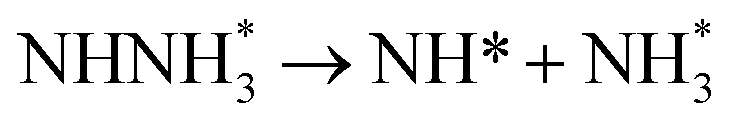DOI:
10.1039/D1RA01978C
(Paper)
RSC Adv., 2021,
11, 17828-17839
Theoretical insights into the electroreduction mechanism of N2 to NH3 from an improved Au(111)/H2O interface model†
Received
12th March 2021
, Accepted 7th May 2021
First published on 17th May 2021
Abstract
An improved H coverage-dependent Au(111)/H2O electrochemical interface model is proposed in this paper, which is firstly used to study electroreduction mechanisms of N2 into NH3 at the thermodynamical equilibrium potential in cooperation with electronic structure analysis. The results show that the associative mechanism is more favorable on Au(111) and therein alternating and distal pathways may be able to parallelly occur in gas phase and the present simulated electrochemical interface. The initial N2 reduction into the N2H intermediate is the rate determining step, which may be able to be regarded as the origin of the observed experimentally high overpotential during N2 electroreduction. The presence of an electrochemical environment can significantly change the N2 reduction pathway and decrease the barrier of the rate determining step, which can be ascribed to the significant electron accumulation and interaction between N2 molecules and H2O clusters. The theoretical results display excellent consistency with the available experimental data, confirming the rationality of the present proposed electrochemical model. The comparison of the barrier between the hydrogen evolution reaction and rate determining step well explains why the activity of Au electrodes is usually unsatisfactory. Accordingly, a single descriptor can be proposed, in which an ideal electrocatalyst should be able to reduce the barrier for initial N2 electroreduction into N2H. In this way, N2 electroreduction pathways can be facilitated and the yield of NH3 can be enhanced. We believe that the present study can represent progress to study N2 electroreduction mechanisms from an improved electrochemical model.
1. Introduction
NH3 as a promising carbon-free alternative energy carrier can be employed in fertilizers, production of important chemicals, NH3 fuel cells and indirectly hydrogen fuel cells.1–3 At present, NH3 is synthesized primarily by reaction of N2 with H2 on an Fe/Ru-based catalyst through the industrial Haber–Bosch (HB) process under harsh conditions such as high temperatures and pressures,4,5 which is an energy intensive chemical process and consumes ca. 2% of annual global energy because of the stability and chemical inertness of N2 molecule.6–8 By contrast, the electrochemical reduction of N2 into NH3 with proton and electron transfer in an aqueous environment is more energy efficient and attracts extensive interest in recent years since it can be operated at ambient conditions and powered by renewable electricity.9–14 Unfortunately, the required large overpotential and competing hydrogen evolution reaction lead to low faradaic efficiency and poor selectivity for N2 electroreduction into NH3 on most electrocatalysts, thus impeding its practical applications.15–18 The poor activity of the cathode electrocatalyst may put a major limitation on production of NH3 product in significant yields at ambient conditions. To achieve the rational design of more selective and active electrocatalysts, the system understanding on electroreduction mechanism of N2 into NH3 is extremely urgent and essential.
Although tremendous efforts in recent years, N2 electroreduction mechanism remains elusive. The present most studies on N2 electroreduction reaction primarily focus on the synthesis of electrocatalysts including metals such as Pt, Ru, Fe, Au, Pd, Rh, Fe, Ni, Mo and Bi,19–30 alloys such as Pt–Ru,31 metal nitrides and sulfides,32,33 and carbon-based materials.34–38 However, these reported electrocatalysts suffer from low NH3 faradaic efficiency and N2 electroreduction reaction is still plagued. Based on these previous studies, the development of more active and selective electrocatalysts is highly desired but remains challenging. Study of N2 electroreduction reaction mechanism will help design electrocatalysts with high activity and selectivity. In the most recent reviews from Shao et al. and Zhang et al.,39,40 N2 electroreduction mechanisms and reaction intermediates that obtained in experiments by various spectroscopy techniques are summarized. Currently, it is generally accepted that there are two main reaction mechanisms for N2 electroreduction into NH3, namely, dissociative pathways and associative pathways. N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond in N2 molecule is broken to form N atoms on electrocatalyst surface before hydrogenation in dissociative mechanism, whereas the hydrogenation of N2 molecule occurs before NN bond is broken in associative mechanism. It is currently believed that the dissociative mechanism is dominant in the HB process of N2 reduction into NH3.15,41 An associative distal or alternating mechanism may be followed during N2 electroreduction into NH3 through experimental identification of some intermediates. For example, using surface-enhanced infrared absorption spectroscopy, Shao et al., studied for the first time N2 electrochemical reduction reaction mechanism on Au thin film, in which the adsorbed N2Hy (1 ≤ y ≤ 4) species was detected at potentials below 0 V (vs. RHE),42 thus indicating that N2 electroreduction may follow the associative alternating and distal mechanisms on Au surface. Combing surface-enhanced infrared absorption spectroscopy with electrochemical measurements, the adsorbed N2Hx (0 ≤ x ≤ 2) species was detected at potentials below 0.2 V (vs. RHE) on Ru thin film in the subsequent study from Shao et al., and notably increased coverage of N2Hx was observed as the potential decreasing from 0.2 to −0.4 V, thus the associative distal mechanism may be able be concluded.20 By performing isotope-labelled experiments, Yin et al., found only a trace amount of N2H4 intermediate during N2 electroreduction on bismuth surface, suggesting that associative distal mechanism may be more favorable.30 Despite of understanding of N2 electroreduction reaction mechanism is of importance for rationally designing more efficient electrocatalysts, the experimental studies are very rare at present in this area since low selectivity makes experimental determination of mechanism rather difficult.
N bond in N2 molecule is broken to form N atoms on electrocatalyst surface before hydrogenation in dissociative mechanism, whereas the hydrogenation of N2 molecule occurs before NN bond is broken in associative mechanism. It is currently believed that the dissociative mechanism is dominant in the HB process of N2 reduction into NH3.15,41 An associative distal or alternating mechanism may be followed during N2 electroreduction into NH3 through experimental identification of some intermediates. For example, using surface-enhanced infrared absorption spectroscopy, Shao et al., studied for the first time N2 electrochemical reduction reaction mechanism on Au thin film, in which the adsorbed N2Hy (1 ≤ y ≤ 4) species was detected at potentials below 0 V (vs. RHE),42 thus indicating that N2 electroreduction may follow the associative alternating and distal mechanisms on Au surface. Combing surface-enhanced infrared absorption spectroscopy with electrochemical measurements, the adsorbed N2Hx (0 ≤ x ≤ 2) species was detected at potentials below 0.2 V (vs. RHE) on Ru thin film in the subsequent study from Shao et al., and notably increased coverage of N2Hx was observed as the potential decreasing from 0.2 to −0.4 V, thus the associative distal mechanism may be able be concluded.20 By performing isotope-labelled experiments, Yin et al., found only a trace amount of N2H4 intermediate during N2 electroreduction on bismuth surface, suggesting that associative distal mechanism may be more favorable.30 Despite of understanding of N2 electroreduction reaction mechanism is of importance for rationally designing more efficient electrocatalysts, the experimental studies are very rare at present in this area since low selectivity makes experimental determination of mechanism rather difficult.
Because of the experimental limitations to identify intermediates and complexity of N2 electroreduction process involving 6 elementary reaction steps, theoretical calculations have become a powerful tool for studying electrocatalytic reactions by employing density functional theory (DFT).43,44 DFT calculations can give mechanistic information that is not accessible based on experiments alone and identify favored reaction intermediates. Theoretical work by Skúlason et al. and Montoya et al. thermodynamically indicated that the associative distal mechanism would be more favorable since more positive reaction free energies are expected for the associative alternating mechanism by combining DFT calculations with the computational hydrogen electrode model.15,45 Their findings had shown that the potential limiting step is N2 reductive protonation to the adsorbed N2H species on the transition metal surface with relatively weak N adsorption such as Pt, Pd, Ag, Au, Ni, Co and Ru during N2 electroreduction into NH3, whereas the potential limiting step is determined by the protonation of the adsorbed NH into NH2 species for more reactive transition metals, such as Mo. On the basis of DFT-based computational hydrogen electrode model, the electrocatalytic activity of various binary transition metals are systematically examined in recent theoretical work from Zhao et al.,46 and the binary FeRh catalyst was thought to have the optimal catalytic performance due to its lowest limiting potential and best suppressing effect on hydrogen evolution reaction during N2 electroreduction reaction. Furthermore, their study indicated that N2 reduction reaction prefers to proceed through associative distal mechanisms than alternating pathways on the FeRh catalyst. These above theoretical studies assumed that activation barriers of reaction pathways is related with reaction free energies on different transition metal surfaces and only considered various elementary step thermodynamics, whereas the potential-dependent kinetic barriers that predicting catalytic activity were not further calculated. Most recently, Janik et al. calculated explicitly the potential-dependent activation barriers for elementary electroreduction reactions included in associative distal and alternating pathways during N2 electroreduction using their previously developed method,47–49 and concluded that the alternating mechanism by direct surface hydrogenation may be more favorable on late transition metals due to smaller barriers at 0 V (vs. RHE),50,51 in which N2 electroreduction into N2H species is rate determining step of overall reaction. However, despite various theoretical efforts, N2 electroreduction mechanisms are still not systematically understood and mechanistic inconsistencies remain exist. Furthermore, the key factor such as solvent effect was not included in previous theoretical studies on N2 electroreduction mechanisms. Thus, the modeling of electrocatalytic reaction systems occurred at the complex electrode/aqueous interfaces remains a subject of ongoing discussion.
In the present paper, Au electrocatalyst is selected due to its excellent durability, relatively high faradaic efficiency and low hydrogen evolution reaction activity in N2 electroreduction. Furthermore, electrocatalytic N2 reduction reaction on the Au surface is indeed possible under ambient conditions.25,52 Our previous validated explicit solvation model with two relaxed H2O bilayer structure is employed to simulate solvent effect,53–55 which allows us to better model the interactions among adsorbates, surface and solvents and determine the kinetic barriers for various elementary reaction steps. Thus, an improve H coverage-dependent Au(111)/H2O electrochemical interface model is proposed, by which the electroreduction mechanisms of N2 into NH3 can be identified. Simultaneously, solvation effect on N2 reduction mechanisms is also considered in this work. Our present used model is differentiated from previous theoretical work on N2 electroreduction into NH3. The available experimental results on Au electrodes will be used to examine whether the currently employed computational model is enough accurate by comparing with our present theoretical study.
2. H coverage-dependent Au(111)/H2O interface model
2.1 Determination method of equilibrium potentials
Surface and solvation model, and computational parameters included in computational details have been represented in detail in ESI (see Fig. S1).† It is known that the hydrogen evolution reaction and proton-coupled electron transfer usually occur during N2 electroreduction on the Au electrodes, thereby the surface adsorbed H atoms as the intermediate may be involved.24,25,53 Moreover, the difference of electrostatic potential in electric double layer can be controlled and adjusted by changing number of the surface adsorbed H atoms.56–58 Thus, the H coverage-dependent equilibrium potentials can be determined and potential-dependent N2 electroreduction mechanisms can be speculated. The surface adsorbed H atom can be formed during N2 electroreduction by following eqn (1):
At different H coverage conditions, the Gibbs free energy of eqn (1), ΔG(θ) can be calculated by eqn (2) on the basis of the methodology proposed by Nørskov et al., Chen et al. and Strasser et al. for oxygen reduction, hydrogen evolution and CO2 electrochemical reduction reactions,56,59–63 in which ΔE(θ), ΔS(θ), ΔZPE and kBT![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ln(θ/1 − θ) represent the differential adsorption energy of surface adsorbed H atoms, entropy change, zero-point energy change and the contributions of configuration entropy to ΔG(θ), respectively. Herein, coverage θ = n/N, in which n is the number of surface adsorbed H atoms and N is the total number of surface Au atoms. Thus, the H coverage-dependent equilibrium potential U (vs. RHE) can be determined at Au(111)/H2O interface when the Gibbs free energy, ΔG(θ) is equal with zero.
ln(θ/1 − θ) represent the differential adsorption energy of surface adsorbed H atoms, entropy change, zero-point energy change and the contributions of configuration entropy to ΔG(θ), respectively. Herein, coverage θ = n/N, in which n is the number of surface adsorbed H atoms and N is the total number of surface Au atoms. Thus, the H coverage-dependent equilibrium potential U (vs. RHE) can be determined at Au(111)/H2O interface when the Gibbs free energy, ΔG(θ) is equal with zero.
| | |
ΔG(θ) = ΔE(θ) + eU − TΔS(θ) + ΔZPE + kBTln(θ/1 − θ)
| (2) |
According to
eqn (3), the differential adsorption energy of surface adsorbed H atoms, Δ
E(
θ) can be calculated, where
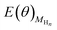
is the total energy of the Au surface with different coverage of adsorbed H atoms.
| |
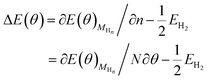 | (3) |
The contributions from ZPE and entropy changes together to ΔG(θ) are estimated to be ca. 0.24 eV at standard temperature (298 K) based on the available data from previous literature.64 Therefore, combining eqn (2) with eqn (3), the eqn (4) can be obtained. The values of 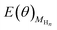 and
and 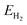 is directly available via DFT calculations. A series of values of
is directly available via DFT calculations. A series of values of 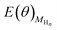 can be calculated by changing the coverage of surface adsorbed H atoms on Au(111). At the different coverages, the values of ΔE(θ) can be obtained by differentiating the plots of
can be calculated by changing the coverage of surface adsorbed H atoms on Au(111). At the different coverages, the values of ΔE(θ) can be obtained by differentiating the plots of 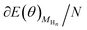 against θ on the basis of eqn (3).
against θ on the basis of eqn (3).
| |
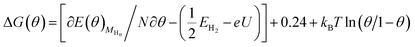 | (4) |
2.2 Relationship between H coverage and equilibrium potentials
In the present study, we consider various possible surface adsorption configurations of H atoms and coverage dependence. It is observed that H atoms prefer to adsorb at 3-fold face-centered cubic hollow (fcc) sites on Au(111) so that they can stay away from each other in order to minimize the repulsive reactions when H coverage (θH) is below and equal with 1 monolayer (ML) (see Fig. S2†). The coverages of H atoms above 1 ML are not further analyzed on Au(111) because of the observed spontaneous formations of hydrate proton by adsorbed H atoms with adjacent H2O molecules caused by strong repulsive interactions among adsorbed H atoms. Fig. 1(a) exhibits a reasonable polynomial relationship between the differential adsorption energy of surface adsorbed H atoms, ΔE(θ) and θH, indicating that the Langmuir adsorption isotherms may be followed on Au(111). Thereby, ΔE(θ) can be calculated at any θH through polynomial fitting of ΔE(θ) ∼ θH data. We can calculated the equilibrium potentials (U) based on the eqn (4) at any θH and then the polynomial relationships between θH and U can be obtained on Au(111). It is found that the more negative equilibrium potentials can be obtained with the increasing H coverage, as shown in Fig. 1(b). In fact, the previous theoretical study from Skúlason et al. also showed that most surfaces will be fully covered with the adsorbed H atoms at more negative electrode potentials,56 confirming the reasonability of our present proposed H coverage-dependent Au(111)/H2O electrochemical interface model to some degree. The equilibrium potential is calculated as ca. −0.22 V (vs. RHE) when θH is equal with zero based on the polynomial relationship between θH and U, which is the closest to the thermodynamical value of ca. −0.06 V (vs. RHE) for N2 electroreduction under ambient conditions,20 thus being regarded as the thermodynamical equilibrium potential. Our present study will focus on the electroreduction mechanisms of N2 into NH3 at the thermodynamical required equilibrium potential with the aim of applying the present proposed H coverage-dependent Au(111)/H2O interface model. The mechanistic understanding on N2 electroreduction can provide scientific guideline for rational design of efficient electrocatalysts. The potential-dependent N2 electroreduction mechanisms will be further studied in our future work.
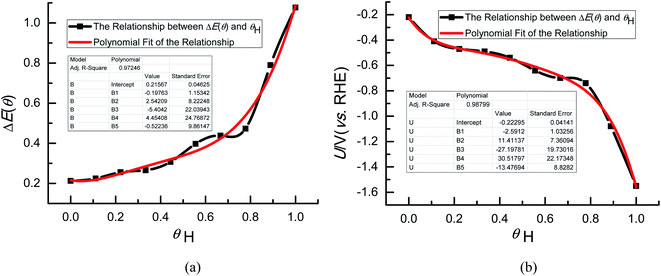 |
| | Fig. 1 (a) The relationship between the differential adsorption energy of H atoms and H coverage (θH), ΔE(θ); (b) the relationship between the calculated equilibrium potentials (U) and H coverage (θH). | |
3. Results and discussion
3.1 N2 reduction mechanism on Au(111) in gas phase
For comparison and consideration of solvation effect on N2 reduction mechanisms, we firstly present calculated results of N2 reduction to NH3 via dissociative and associative mechanisms on Au(111) in gas phase. In dissociative mechanism, N2 is initially dissociated to form the adsorbed N atoms on Au(111) with an extremely high activation barrier of ca. 6.02 eV, as can be seen in Fig. 2. Subsequently, the adsorbed N atoms are further reduced to form NH3 molecules via serial surface hydrogenation steps with corresponding barriers of ca. 0.40, 0.47 and 0.57 eV, respectively. By comprehensively scrutinizing the overall energy pathway diagram of N2 reduction into NH3 in gas phase, N2 dissociation pathway is rate determining step of overall reaction in dissociative mechanism on Au(111).
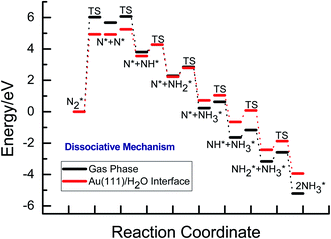 |
| | Fig. 2 Overall energy pathway diagram of N2 reduction and electroreduction into NH3 via dissociative mechanism on Au(111) in gas phase and the present simulated Au(111)/H2O electrochemical interface. | |
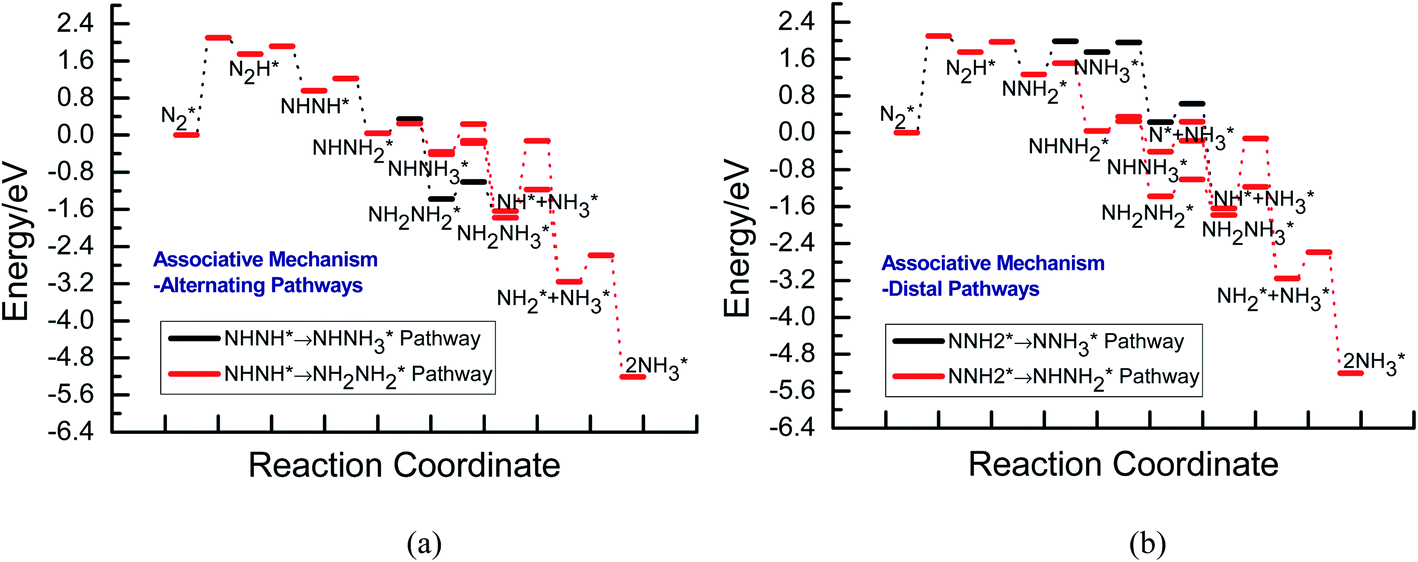
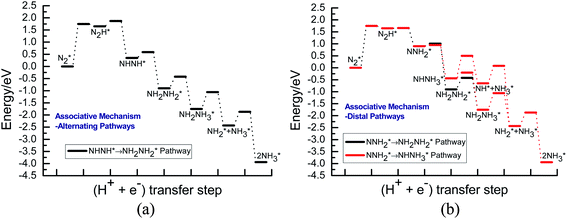
Fig. 3 shows the overall energy pathway diagram of N2 reduction into NH3 via associative alternating and distal mechanisms in gas phase. In these both mechanisms, the activation barrier is calculated as ca. 2.10 eV for the first hydrogenation step of N2 molecule into the adsorbed N2H species. Beginning with the further reduction of N2H, there may be two possibilities to occur. One is N2H hydrogenation to form the surface adsorbed NHNH species, which is defined alternating pathways; another is N2H hydrogenation to form the surface adsorbed NNH2 species, being defined as distal pathways. The required barrier for the formation of NHNH species is ca. 0.17 eV in alternating pathways. NHNH species can further be reduced to form the adsorbed NHNH2 species with an activation barrier of ca. 0.26 eV. Two possibilities are considered for NHNH2 subsequent further reduction, the adsorbed NH2NH2 and NHNH3 species may be formed via surface hydrogenation. The corresponding barrier is 0.31 and 0.21 eV, respectively, which is extremely low and surmountable at room temperature, indicating that NHNH2 further reduction into NH2NH2 and NHNH3 species may be parallel pathways in alternating pathways. NH2NH2 further reduction to form the first NH3 molecule may be able to be separated into two elementary reaction steps, namely, NH2NH2 species surface hydrogenation into the adsorbed NH2NH3 species, and subsequent formation of the adsorbed NH2 species and NH3 product via N–N bond cleavage. The calculated barrier is ca. 0.37 and 1.66 eV, respectively, as shown in Fig. 3(a). Surface hydrogenation of NH2 intermediate can finally leads to production of the second NH3 molecule.
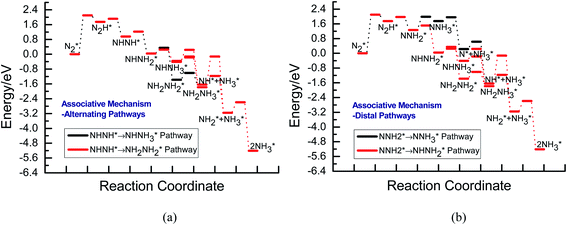 |
| | Fig. 3 Overall energy pathway diagram of N2 reduction into NH3 via associative mechanism on Au(111) in gas phase: (a) alternating pathways; (b) distal pathways. | |
In distal pathways, surface hydrogenation of NNH2 intermediate is possible to form the adsorbed NNH3 and NHNH2 species. However, the significant higher barrier for formation of NNH3 than NHNH2 is observed (0.72 eV vs. 0.24 eV), indicating that the NNH3 formation is kinetically inhibited, as shown in Fig. 3(b). Starting with NHNH2 further reduction, the first NH3 molecule may be produced via two elementary reaction steps involving hydrogenation into the adsorbed NHNH3 species and its subsequent N–N bond scission with the formations of the adsorbed NH species and NH3 product, and the corresponding barrier is calculated as ca. 0.21 and 0.65 eV. The adsorbed NH2NH2 intermediate is also possible to be formed through NHNH2 surface hydrogenation with the surmountable barrier of ca. 0.31 eV at room temperature in distal pathways. As above elaborated, NH2NH2 species can further be reduced into the adsorbed NH2 species and the first NH3 molecule via surface hydrogenation and N–N bond scission. The adsorbed NH and NH2 intermediates can finally lead to the second NH3 molecule via surface hydrogenation in distal pathways. However, we note that the extremely high barrier is required for N–N bond cleavage in NH2NH3 species to form NH3 product (ca. 1.66 eV) in associative alternating and distal mechanisms, thereby it can be concluded that NH2NH3 species is only a spectator during N2 reduction in gas phase due to its easy formation via NHNH3 and NH2NH2 hydrogenation. The corresponding energetics of for the possible elementary reaction steps involved in N2 reduction pathways on Au(111) in gas phase are summarized in Table 1.
Table 1 Activation barriers (ΔEact, eV) and reaction energies (ΔEreac, eV) for the possible elementary reaction steps involved in N2 reduction pathways on Au(111) in gas phase
| Elementary reaction stepsa |
ΔEact, eV |
ΔEreac, eV |
| The asterisk (*) indicates that the species is adsorbed on the Au(111) surface. |
| N2 + * → N* + N* |
6.02 |
5.67 |
| N* + H* → NH* |
0.40 |
−1.87 |
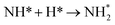 |
0.47 |
−1.52 |
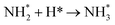 |
0.57 |
−2.05 |
| N2 + H* → N2H* |
2.10 |
1.75 |
| N2H* + H* → NHNH* |
0.17 |
−0.79 |
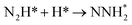 |
0.22 |
−0.48 |
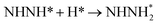 |
0.26 |
−0.92 |
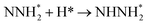 |
0.24 |
−1.23 |
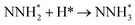 |
0.72 |
0.48 |
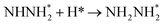 |
0.31 |
−1.42 |
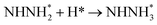 |
0.21 |
−0.45 |
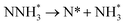 |
0.21 |
−1.52 |
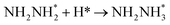 |
0.37 |
−0.40 |
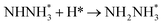 |
0.24 |
−1.37 |
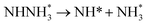 |
0.65 |
−1.23 |
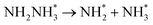 |
1.66 |
−1.38 |
By scrutinizing the overall energy pathway diagram of associative alternating and distal mechanisms (see Fig. 3), N2 hydrogenation into N2H species is rate determining step with the barrier of ca. 2.10 eV, suggesting that both alternating and distal pathways may be parallel and operable on Au(111) in gas phase. It is found that the barrier of rate determining step for the associative mechanism is notably lower than that of dissociative mechanism by comparing the barriers between these both mechanisms (ca. 2.10 eV vs. 6.02 eV), suggesting that the associative mechanism including the adsorbed NHNH, NNH2, NH2NH2, NHNH3 and NH2NH3 intermediates is more favorable. The optimal associative mechanisms including alternating and distal pathways on Au(111) in gas phase are summarized in Fig. 4. Images of reactants, products and transition states for N2 reduction into NH3 via the optimal associative mechanisms are included in ESI, as shown in Fig. S3–S13.† However, we also note that the required barrier for initial N2 hydrogenation is extremely high, which may be able to ascribed to weakly bonded N2 molecule on Au(111) with the adsorption energy of ca. −0.03 eV. Furthermore, NN bond length of N2 on Au(111) is almost identical with that of isolated N2 molecule, ca. 1.11 Å.
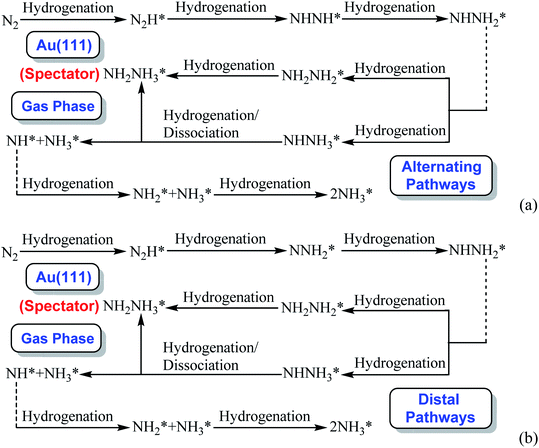 |
| | Fig. 4 The optimal associative mechanisms on Au(111) in gas phase: (a) alternating pathways via NHNH species; (b) distal pathways via NNH2 species (* represents surface adsorption). | |
3.2 N2 electroreduction mechanism at Au(111)/H2O interface
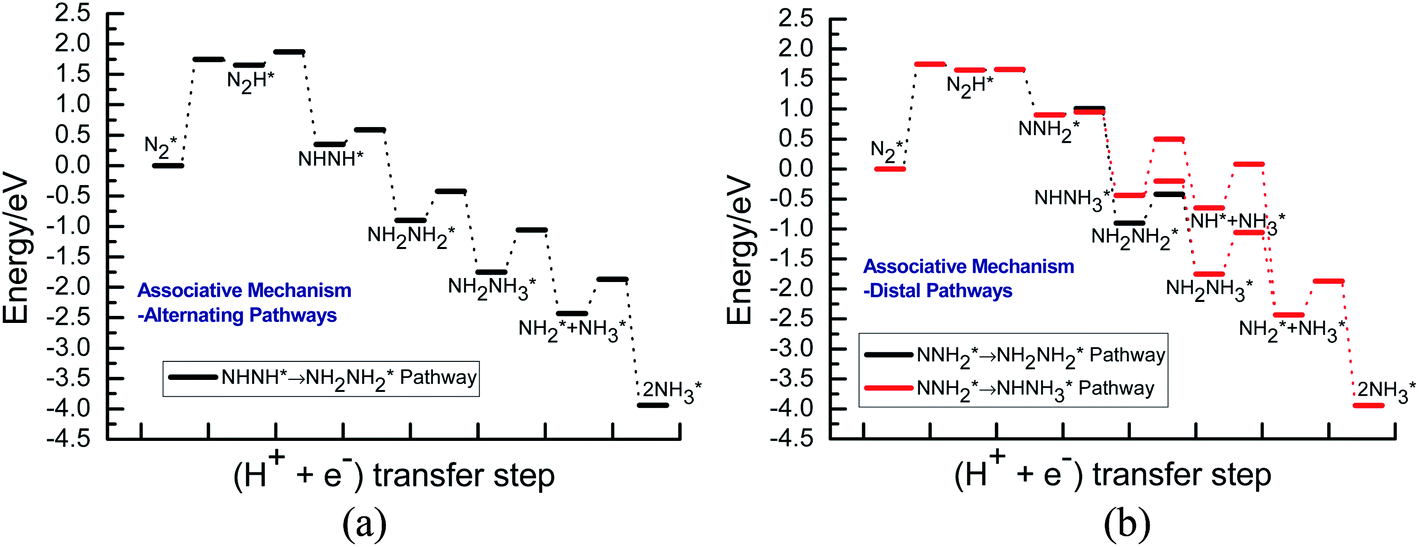
Our present proposed H coverage-dependent Au(111)/H2O electrochemical interface model is utilized to simulate N2 electroreduction pathways, including dissociative and associative mechanisms. The present calculated thermodynamically required equilibrium potential of ca. −0.22 V (vs. RHE) when θH is equal with zero is focused. Fig. 1 shows the overall energy pathway diagram of N2 electroreduction into NH3 through dissociative mechanism. The largest barrier for this mechanism is initial NN bond scission to form the adsorbed N atoms with an activation barrier of ca. 4.90 eV at −0.22 V (vs. RHE), thus being rate determining step of overall reaction. Although the barrier for N2 dissociation is significantly decreased at Au(111)/H2O electrochemical interface by comparing with that in gas phase (4.90 eV vs. 6.02 eV), it is still extremely high and difficult to be overcome. An activation barrier of ca. 1.75 eV is required for initial N2 electroreduction into N2H species at the present simulated Au(111)/H2O interface in associative mechanisms, as can be seen in Fig. 5. The barrier for N2H further electroreduction into the surface adsorbed NHNH and NNH2 species in alternating and distal pathways is calculated as ca. 0.22 and 0.01 eV, respectively, suggesting that both NHNH and NNH2 species are possible key intermediates during N2 electroreduction on Au(111) because of extremely low formation barriers.
 |
| | Fig. 5 Overall energy pathway diagram of N2 electroreduction into NH3 via associative mechanism at the present proposed Au(111)/H2O interface: (a) alternating pathways; (b) distal pathways. | |
Beginning with NHNH further electroreduction in alternating pathways, we find that the adsorbed NHNH2 species is unstable at Au(111)/H2O interface, which can be spontaneously electrochemically reduced to form the surface adsorbed NH2NH2 species by proton-coupled electron transfer with the surmountable barrier of ca. 0.24 eV at room temperature, as shown in Fig. 5(a). The first NH3 molecule can be produced by further electroreduction of NH2NH2 species through two elementary reaction steps including NH2NH3 formation and its subsequent N–N bond scission. The corresponding barrier is calculated as ca. 0.48 and 0.69 eV, respectively. It is noted that the barrier for NH2NH3 further electroduction into the adsorbed NH2 species and NH3 product is significantly decreased at Au(111)/H2O interface compared with that in gas phase (0.69 eV vs. 1.66 eV), suggesting that the presence of electrochemical interface may be able to alter N2 reduction mechanisms. The formed NH2 species can finally lead to production of the second NH3 molecule with an activation barrier of ca. 0.56 eV. Starting with NNH2 intermediate formed in distal pathways, it is found that the adsorbed NHNH2 and NNH3 species observed in gas phase are unstable at Au(111)/H2O interface, which can be also spontaneously electrochemically reduced to form the surface adsorbed NH2NH2 and NHNH3 intermediates by proton-coupled electron transfer process. The required barrier for NNH2 electroreduction into NH2NH2 and NHNH3 is calculated as only ca. 0.11 and 0.05 eV, respectively, as shown in Fig. 5(b), suggesting that NH2NH2 and NHNH3 species is possible intermediate in distal pathways. Similarly, NH2NH2 intermediate can be further electrochemically reduced to form the adsorbed NH2 species and the first NH3 molecule via abovementioned two elementary reaction steps. Two possibilities are also considered for further electroreduction of NHNH3 intermediate at Au(111)/H2O interface, one is NH2NH3 formation, and another is formation of the adsorbed NH species and production of the first NH3 molecule via N–N bond scission. It is found that the required barrier for the former is notably lower than that of the latter (0.24 eV vs. 0.94 eV), indicating that NH2NH3 formation is more favorable. Thus, we can concluded that the first NH3 molecule is possible to be produced by N–N bond scission of NH2NH3 species at Au(111)/H2O interface, rather than NHNH3 species as observed in gas phase, again suggesting that the influence of electrochemical interface containing solvation effect on N2 reduction pathways. The corresponding energetics of for various possible reaction steps involved during N2 electroreduction at Au(111)/H2O interface are summarized in Table 2.
Table 2 Activation barriers (ΔEact, eV) and reaction energies (ΔEreac, eV) for various possible reaction steps involved in N2 electroreduction pathways at Au(111)/H2O interface
| Reaction stepsa |
ΔEact, eV |
ΔEreac, eV |
| The asterisk (*) indicates that the species is adsorbed on the Au(111) surface. |
| N2 + * → N* + N* |
4.93 |
4.92 |
| N* + (H + e−) → NH* |
0.33 |
−1.37 |
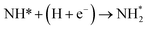 |
0.73 |
−1.78 |
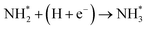 |
0.56 |
−1.51 |
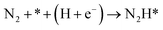 |
1.75 |
1.65 |
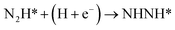 |
0.22 |
−1.30 |
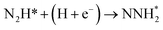 |
0.01 |
−0.75 |
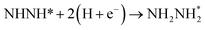 |
0.24 |
−1.25 |
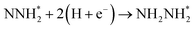 |
0.11 |
−1.80 |
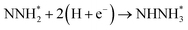 |
0.05 |
−1.34 |
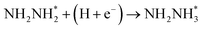 |
0.48 |
−0.85 |
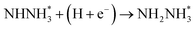 |
0.24 |
−1.31 |
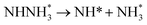 |
0.94 |
−0.21 |
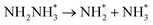 |
0.69 |
−0.68 |
By scrutinizing the overall energy pathway diagram, our present simulation results reveal that the rate determining step for N2 electroreduction into NH3 via associative alternating and distal mechanisms at Au(111)/H2O interface is N2 electroreduction into form the adsorbed N2H species, suggesting that these both mechanisms may be able to parallelly occur. The corresponding barrier is significantly lower than that of rate determining step in the dissociative mechanism (1.75 eV vs. 4.90 eV). Thus, it can be concluded that the associative mechanisms are more facile to occur at the present simulated Au(111)/H2O interface. The optimal associative alternating and distal mechanisms are summarized in Fig. 6. Images of reactants, products and transition states for N2 electroreduction into NH3 via the optimal associative mechanisms at the present simulated Au(111)/H2O interface are included in ESI, as shown in Fig. S14–S23.†
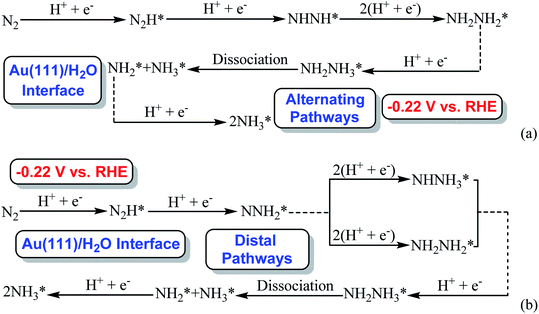 |
| | Fig. 6 The optimal associative mechanisms at the present simulated Au(111)/H2O interface: (a) alternating pathways via NHNH species; (b) distal pathways via NNH2 species (* represents surface adsorption). | |
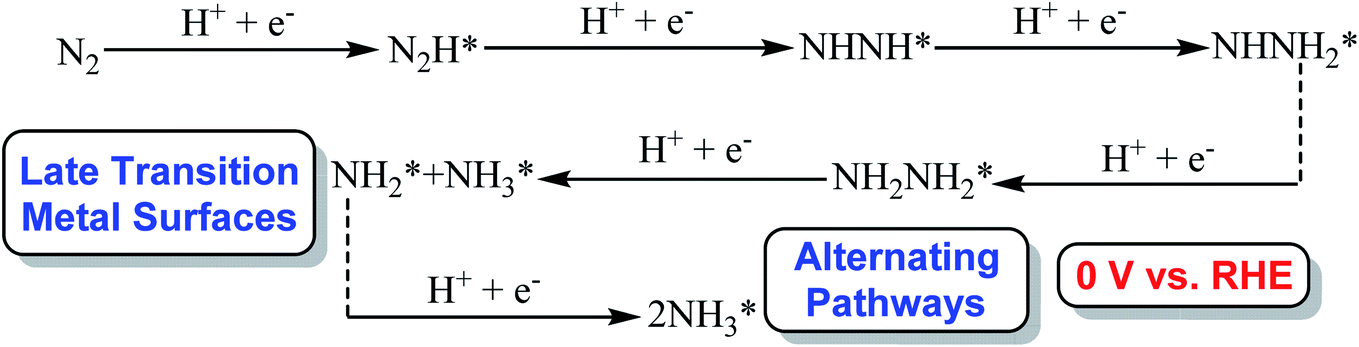
Our present calculated N2 electroreduction mechanisms at Au(111)/H2O electrochemical interface are partially inconsistent with the previous theoretical study from Janik et al., in which only alternating pathway via NHNH species is favorable on late transition metals at 0 V (vs. RHE) with rate determining step of N2 electroreduction into N2H species by calculating explicitly the potential-dependent barriers for elementary electroreduction reactions (see Fig. 7).51 Furthermore, the predicted NHNH2 species on late transition metals at 0 V (vs. RHE) is found to may be unstable at Au(111)/H2O interface, which can be spontaneously reduced to form adsorbed NH2NH2 species by proton-coupled electron transfer with the surmountable barrier at room temperature. The difference of interface model may lead to partially inconsistent N2 electroreduction mechanisms, in which only a H2O molecule is employed to simulate solvent effect in previous theoretical work from Janik et al., being insufficient to model interactions among solvent, adsorbates and surface. However, our present theoretical results can be confirmed by the most recent experimental study from Shao et al., in which the adsorbed intermediates such as N2H, NHNH, NNH2, NH2NH2, NH2NH3 and NH2 may be able to be formed during N2 electroreduction on the Au electrodes at potentials of ca. −0.10 V (vs. RHE) or lower due to detected N2Hy (1 ≤ y ≤ 4) reaction species using the surface-enhanced infrared absorption spectroscopy technique,42 further validating the rationality of our present employed Au(111)/H2O interface model. Simultaneously, we also note that the presence of electrochemical interface makes the barrier of rate determining step in the associative mechanisms decrease compared with that in gas phase (1.75 eV vs. 2.10 eV), indicating that the presence of solvent effect could help stabilize the adsorbed N2H species and lower the corresponding barrier value to ca. 1.75 eV. Even so, barrier of 1.75 eV is still high for N2 electroreduction, making this process be still challenging, which may be able to be regarded as origin of experimentally observed high overpotential. The almost not changed NN bond length of ca. 1.11 Å at Au(111)/H2O interface again shows weakly adsorbed N2 molecule compared with that of isolated N2 molecule.
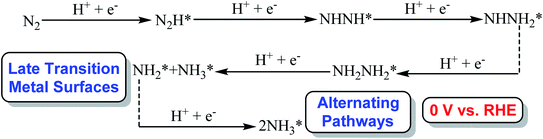 |
| | Fig. 7 The optimal associative alternating pathways via NHNH species on late transition metal surfaces at 0 V (vs. RHE) from previous work conducted by Janik et al. (* represents surface adsorption).51 | |
The potential of the hydrogen evolution reaction is very approximate to the thermodynamically required potential for N2 electroreduction under ambient conditions. Therefore, overcoming the undesirable hydrogen evolution reaction competition may become the critical challenge during N2 electroreduction at present, which can greatly reduce the faradaic efficiency of reaction. Herein, the barrier of hydrogen evolution reaction is evaluated by using our present proposed H coverage-dependent Au(111)/H2O interface model at −0.22 V (vs. RHE) and compared with that of rate determining step for the optimal associative mechanisms during N2 electroreduction into NH3 to quantify the challenge in designing the high efficient electrocatalysts. As can be seen in Fig. 8, the calculated activation barrier of ca. 0.24 eV is remarkably lower than that of initial N2 electroreduction into N2H intermediate (ca. 1.75 eV). Furthermore, we also observe that the formed N2H intermediate may be unstable, which can be facile to back to N2 molecule with extremely low barrier of ca. 0.10 eV. The present comparison and analysis can well explain why the catalytic activity of Au electrodes is usually unsatisfactory although the relatively high faradaic efficiency can be achieved experimentally during N2 electroreduction.24,25,42 Accordingly, the single descriptor may be able to be proposed to scale catalytic activity of electrocatalysts for N2 electroreduction, in which an ideal electrocatalyst should be able to reduce barrier for initial N2 electroreduction into N2H intermediate. In this way, N2 electroreduction pathways can be facilitated and the yield of NH3 can be enhanced.
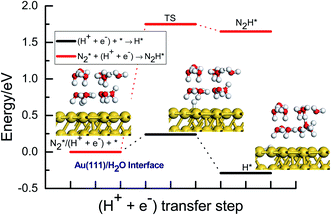 |
| | Fig. 8 Energy pathway diagram of competitive hydrogen evolution reaction and initial N2 electroreduction into N2H species at the present proposed Au(111)/H2O electrochemical interface. | |
3.3 Origin of solvation effect on N2 reduction mechanisms
As above discussed, the presence of electrochemical interface including solvation effect could change N2 reduction mechanism, especially it can reduce the barrier of rate determining step. To ascertain the origin of difference of N2 reduction mechanisms at gas phase and the present simulated electrochemical interface on Au(111), the charge density difference analyses are carried out in our present work taking example for N2 adsorption on Au(111). It is observed that there is almost not significant electron accumulation and interaction between N2 molecule and surface Au atoms under gas- and aqueous-phase environment, as shown in Fig. 9, confirming the above-mentioned weakly binding N2 molecule on Au(111) and almost identical NN bond length compared with isolated N2 molecule, thus explaining why high barrier is required for initial N2 reduction. However, the significant electron interactions are found between N2 and H2O cluster at the present simulated electrochemical interface. The existence of H bonds and interactions of N2 with H2O molecules at the Au(111)/H2O interface may lead to easier initial N2 electroreduction, as observed decreased barrier value.
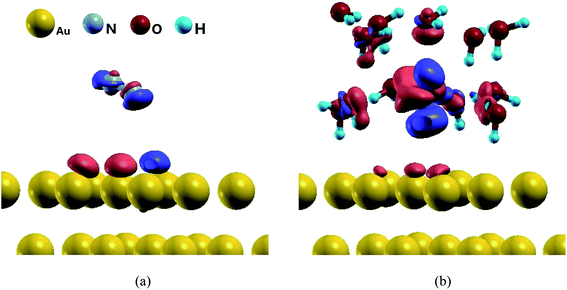 |
| | Fig. 9 The charge density difference maps for N2 (a) in gas-phase and (b) at the Au(111)/H2O interface. | |
Based on the above conclusions, we can conclude that the decreased barrier for rate determining step may be able to be ascribed to electronic interactions between N2 and H2O cluster. Therefore, the quantitative analysis of electronic structures will facilitate well understanding the origin of solvation effect on N2 electroreduction mechanisms. According to projected electron densities of states, the Löwdin charge (the number of valence electron) of N2 can be obtained from Löwdin population analyses on Au(111) in gas phase and at the present simulated electrochemical interface. Table 3 gives the electron gains (Δq) for N2 molecule, respectively, which could be obtained by subtracting the Löwdin charge of isolated N2 molecule from that in the optimized structure. Simultaneously, Δq of H2O cluster at the present simulated electrochemical interfaces are also given in Table 3 compared with that of free N2 adsorbed Au(111)/H2O interface. A positive value of Δq will imply a gain of electron by the component. It can be found that only slight electron transfer occurs between N2 molecule and Au(111) surface in gas phase due to little electron gains of total, s and p orbitals of N2, as found weakly bonded N2 molecule on Au(111) with the adsorption energy of only ca. −0.03 eV, confirming the above observed no significant electron accumulation and interaction based on the charge density difference analyses. However, we notice that the nature of electronic interactions at the Au(111)/H2O interface is practically not the same as that observed in gas phase. The significant electron transfer occurs between N2 molecule and H2O cluster, namely, the total net electrons of H2O cluster are positive because s orbital can gain more electrons although p orbital loses electrons, whereas the total net electrons of N2 is negative, in which s orbital loses electrons and p orbital gains electrons. Thus, it can be concluded that the notable different electron interactions on Au(111) in gas phase and at the electrochemical interface may result in the difference of N2 reduction mechanisms.
Table 3 The electron gains (Δq) of total, s and p orbitals of N2 molecule on Au(111) and at the present simulated Au(111)/H2O interface; Δq of total, s and p orbitals of H2O cluster in N2 adsorbed Au(111)/H2O interface
| |
Difference of electron (Δq) |
| Total |
s |
p |
| Au(111) |
N2 |
−0.0284 |
−0.0271 |
−0.0013 |
| Au(111)/H2O interface |
N2 |
−0.0147 |
−0.0385 |
+0.0238 |
| H2O cluster |
+0.0188 |
+0.0384 |
−0.0195 |
4. Conclusions
In the present paper, an improved H coverage-dependent Au(111)/H2O electrochemical interface model is proposed, which is firstly used to study electroreduction mechanisms of N2 to NH3 at the thermodynamical equilibrium potential cooperated with electronic structure analysis. The calculated results show that the associative mechanism is more favorable on Au(111) and therein alternating and distal pathways may be able to parallelly occur in gas phase and the present simulated electrochemical interface. The initial N2 reduction into N2H intermediate is rate determining step, which may be able to be regarded as the origin of observed experimentally high overpotential during N2 electroreduction. However, the presence of electrochemical environment can significantly change N2 reduction pathway and decrease the barrier of rate determining step, in which NHNH2 and NNH3 species observed in gas phase are unstable at Au(111)/H2O interface, which can be spontaneously electrochemically reduced to form adsorbed NH2NH2 and NHNH3 intermediates by proton-coupled electron transfer process. The significant electron accumulation and interaction between N2 molecule and H2O cluster may result in different N2 electroreduction pathways and the decreased barrier of rate determining step. The theoretical results display excellent consistency with the available experimental data, confirming the rationality of the present used Au(111)/H2O interface model. The comparison of barrier between hydrogen evolution reaction and rate determining step well explains why the catalytic activity of Au electrodes is usually unsatisfactory although the relatively high faradaic efficiency can be experimentally achieved. Accordingly, the single descriptor may be able to be proposed, in which an ideal electrocatalyst should be able to reduce barrier for initial N2 electroreduction into N2H intermediate. In this way, N2 electroreduction pathways can be facilitated and the yield of NH3 can be enhanced. We believe that the present study represents a progress to systematically study N2 electroreduction mechanisms based on an improved electrochemical interface model.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Key Program of Education Department of Hunan Province (Grant No. 19A337); Hunan Provincial Natural Science Foundation of China (Grant No. 2018JJ2273); Key Program of Hunan University of Arts and Science (19ZD06) and National Natural Science Foundation of China (Grant No. 21303048).
References
- V. Smil, Detonator of the Population Explosion, Nature, 1999, 400, 415 CrossRef CAS.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Beyond Fossil Fuel-Driven Nitrogen Transformations, Science, 2018, 360, eaar6611 CrossRef PubMed.
- A. Klerke, C. H. Christensen, J. K. Nørskov and T. Vegge, Ammonia for Hydrogen Storage: Challenges and Opportunities, J. Mater. Chem., 2008, 18, 2304–2310 RSC.
- J. A. Pool, E. Lobkovsky and P. J. Chirik, Hydrogenation and Cleavage of Dinitrogen to Ammonia with a Zirconium Complex, Nature, 2004, 427, 527–530 CrossRef CAS PubMed.
- G. Ertl, Reactions at Surfaces: From Atoms to Complexity, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS PubMed.
- H. K. Lee, C. S. L. Koh, Y. H. Lee, C. Liu, I. Y. Phang, X. Han, C. K. Tsung and X. Y. Ling, Favoring the Unfavored: Selective Electrochemical Nitrogen Fixation Using a Reticular Chemistry Approach, Sci. Adv., 2018, 4, eaar3208 CrossRef PubMed.
- M. Kitano, Y. Inoue, Y. Yamazaki, F. Hayashi, S. Kanbara, S. Matsuishi, T. Yokoyama, S. W. Kim, M. Hara and H. Hosono, Ammonia Synthesis Using a Stable Electride as an Electron Donor and Reversible Hydrogen Store, Nat. Chem., 2012, 4, 934–940 CrossRef CAS PubMed.
- J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont and W. Winiwarter, How a Century of Ammonia Synthesis Changed the World, Nat. Geosci., 2008, 1, 636–639 CrossRef CAS.
- C. J. M. van der Ham, M. T. M. Koper and D. G. H. Hetterscheid, Challenges in Reduction of Dinitrogen by Proton and Electron Transfer, Chem. Soc. Rev., 2014, 43, 5183–5191 RSC.
- V. Kyriakou, I. Garagounis, E. Vasileiou, A. Vourros and M. Stoukides, Progress in the Electrochemical Synthesis of Ammonia, Catal. Today, 2017, 286, 2–13 CrossRef CAS.
- M. A. Shipman and M. Symes, Recent Progress towards the Electrosynthesis of Ammonia from Sustainable Resources, Catal. Today, 2017, 286, 57–68 CrossRef CAS.
- C. X. Guo, J. R. Ran, A. Vasileff and S. Z. Qiao, Rational Design of Electrocatalysts and Photo (Electro) Catalysts for Nitrogen Reduction to Ammonia (NH3) under Ambient Conditions, Energy Environ. Sci., 2018, 11, 45–56 RSC.
- N. Cao and G. F. Zheng, Aqueous Electrocatalytic N2 Reduction under Ambient Conditions, Nano Res., 2018, 11, 2992–3008 CrossRef CAS.
- J. Deng, J. A. Iñiguez and C. Liu, Electrocatalytic Nitrogen Reduction at Low Temperature, Joule, 2018, 2, 846–856 CrossRef CAS.
- J. H. Montoya, C. Tsai, A. Vojvodic and J. K. Nørskov, The Challenge of Electrochemical Ammonia Synthesis: A New Perspective on the Role of Nitrogen Scaling Relations, ChemSusChem, 2015, 8, 2180–2186 CrossRef CAS PubMed.
- R. Ran, J. T. S. Irvine and S. Tao, Synthesis of Ammonia Directly from Air and Water at Ambient Temperature and Pressure, Sci. Rep., 2013, 3, 1145 CrossRef PubMed.
- S. Licht, B. Cui, B. Wang, F. F. Li, J. Lau and S. Liu, Ammonia Synthesis by N2 and Steam Electrolysis in Molten Hydroxide Suspensions of Nanoscale Fe2O3, Science, 2014, 345, 637–640 CrossRef CAS PubMed.
- G. Qing, R. Kikuchi, S. Kishira, A. Takagaki, T. Sugawara and S. T. Oyama, Ammonia Synthesis by N2 and Steam Electrolysis in Solid-State Cells at 220 °C and Atmospheric Pressure, J. Electrochem. Soc., 2016, 163, E282–E287 CrossRef CAS.
- V. Kordali, G. Kyriacou and C. Lambrou, Electrochemical Synthesis of Ammonia at Atmospheric Pressure and Low Temperature in a Solid Polymer Electrolyte Cell, Chem. Commun., 2000, 17, 1673–1674 RSC.
- Y. Yao, H. J. Wang, X. Z. Yuan, H. Li and M. H. Shao, Electrochemical Nitrogen Reduction on Ruthenium, ACS Energy Lett., 2019, 4, 1336–1341 CrossRef CAS.
- K. Imamura and J. Kubota, Electrochemical Membrane Cell for NH3 Synthesis from N2 and H2O by Electrolysis at 200 to 250 °C Using a Ru Catalyst, Hydrogen-Permeable Pd Membrane and Phosphate-Based Electrolyte, Sustain, Energy Fuels, 2018, 2, 1278–1286 CAS.
- K. Kugler, M. Luhn, J. A. Schramm, K. Rahimi and M. Wessling, Galvanic Deposition of Rh and Ru on Randomly Structured Ti Felts for the Electrochemical NH3 Synthesis, Phys. Chem. Chem. Phys., 2015, 17, 3768–3782 RSC.
- J. Wang, L. Yu, L. Hu, G. Chen, H. L. Xin and X. F. Feng, Ambient Ammonia Synthesis via Palladium-Catalyzed Electrohydrogenation of Dinitrogen at Low Overpotential, Nat. Commun., 2018, 9, 1795 CrossRef PubMed.
- S. J. Li, D. Bao, M. M. Shi, B. R. Wulan, J. M. Yan and Q. Jiang, Amorphizing of Au Nanoparticles by CeOx-RGO Hybrid Support towards Highly Efficient Electrocatalyst for N2 Reduction under Ambient Conditions, Adv. Mater., 2017, 29, 1700001 CrossRef PubMed.
- M. M. Shi, D. Bao, B. R. Wulan, Y. H. Li, Y. F. Zhang, J. M. Yan and Q. Jiang, Au Sub-Nanoclusters on TiO2 toward Highly Efficient and Selective Electrocatalyst for N2 Conversion to NH3 at Ambient Conditions, Adv. Mater., 2017, 29, 1606550 CrossRef PubMed.
- H. M. Liu, S. H. Han, Y. Zhao, Y. Y. Zhu, X. L. Tian, J. H. Zeng, J. X. Jiang, B. Y. Xia and Y. Chen, Surfactant-Free Atomically Ultrathin Rhodium Nanosheet Nanoassemblies for Efficient Nitrogen Electroreduction, J. Mater. Chem. A, 2018, 6, 3211–3217 RSC.
- X. R. Zhao, F. X. Yin, N. Liu, G. R. Li, T. X. Fan and B. H. Chen, Highly Efficient Metal-Organic-Framework Catalysts for Electrochemical Synthesis of Ammonia from N2 (Air) and Water at Low Temperature and Ambient Pressure, J. Mater. Sci., 2017, 52, 10175–10185 CrossRef CAS.
- K. Kim, C. Y. Yoo, J. N. Kim, H. C. Yoon and J. I. Han, Electrochemical Synthesis of Ammonia from Water and Nitrogen in Ethylenediamine under Ambient Temperature and Pressure, J. Electrochem. Soc., 2016, 163, F1523–F1526 CrossRef CAS.
- D. S. Yang, T. Chen and Z. J. Wang, Electrochemical Reduction of Aqueous Nitrogen (N2) at a Low Overpotential on (110)-Oriented Mo Nanofilm, J. Mater. Chem. A, 2017, 5, 18967–18971 RSC.
- Y. C. Hao, Y. Guo, L. W. Chen, M. Shu, X. Y. Wang, T. A. Bu, W. Y. Gao, N. Zhang, X. Su, X. Feng, J. W. Zhou, B. Wang, C. W. Hu, A. X. Yin, R. Si, Y. W. Zhang and C. H. Yan, Promoting Nitrogen Electroreduction to Ammonia with Bismuth Nanocrystals and Potassium Cations in Water, Nat. Catal., 2019, 2, 448–456 CrossRef CAS.
- R. Manjunatha and A. Schechter, Electrochemical Synthesis of Ammonia Using Ruthenium-Platinum Alloy at Ambient Pressure and Low Temperature, Electrochem. Commun., 2018, 90, 96–100 CrossRef CAS.
- L. Zhang, X. Q. Ji, X. Ren, Y. J. Ma, X. F. Shi, Z. Q. Tian, A. M. Asiri, L. Chen, B. Tang and X. P. Sun, Electrochemical Ammonia Synthesis via Nitrogen Reduction Reaction on a MoS2 Catalyst: Theoretical and Experimental Studies, Adv. Mater., 2018, 30, 1800191 CrossRef PubMed.
- X. P. Zhang, R. M. Kong, H. T. Du, L. Xia and F. L. Qu, Highly Efficient Electrochemical Ammonia Synthesis via Nitrogen Reduction Reactions on a VN Nanowire Array under Ambient Conditions, Chem. Commun., 2018, 54, 5323–5325 RSC.
- S. M. Chen, S. Perathoner, C. Ampelli, C. Mebrahtu, D. S. Su and G. Centi, Electrocatalytic Synthesis of Ammonia at Room Temperature and Atmospheric Pressure from Water and Nitrogen on a Carbon nanotube-Based electrocatalyst, Angew. Chem., Int. Ed., 2017, 56, 2699–2703 CrossRef CAS PubMed.
- Y. M. Liu, Y. Su, X. Quan, X. F. Fan, S. Chen, H. T. Yu, H. M. Zhao, Y. B. Zhang and J. J. Zhao, Facile Ammonia Synthesis from Electrocatalytic N2 Reduction under Ambient Conditions on N-Doped Porous Carbon, ACS Catal., 2018, 8, 1186–1191 CrossRef CAS.
- C. Lv, Y. Qian, C. Yan, Y. Ding, Y. Liu, G. Chen and G. Yu, Defect Engineering Metal-Free Polymeric Carbon Nitride Electrocatalyst for Effective Nitrogen Fixation under Ambient Conditions, Angew. Chem., Int. Ed., 2018, 57, 10246–10250 CrossRef CAS PubMed.
- Y. Abghoui and E. Skulason, Computational Predictions of Catalytic Activity of Zincblende (110) Surfaces of Metal Nitrides for Electrochemical Ammonia Synthesis, J. Phys. Chem. C, 2017, 121, 6141–6151 CrossRef CAS.
- S. Mukherjee, D. A. Cullen, S. Karakalos, K. X. Liu, H. Zhang, S. Zhao, H. Xu, K. L. More, G. F. Wang and G. Wu, Metal-Organic Framework-Derived Nitrogen-Doped Highly Disordered Carbon for Electrochemical Ammonia Synthesis Using N2 and H2O in Alkaline Electrolytes, Nano Energy, 2018, 48, 217–226 CrossRef CAS.
- Y. Yao, J. Wang, U. B. Shahid, M. Gu, H. J. Wang, H. Li and M. H. Shao, Electrochemical Synthesis of Ammonia from Nitrogen Under Mild Conditions: Current Status and Challenges, Electrochem. Energy Rev., 2020, 3, 239–270 CrossRef CAS.
- R. Manjunatha, A. Karajic, M. Liu, Z. B. Zhai, L. Dong, W. Yan, D. P. Wilkinson and J. J. Zhang, A Review of Composite/Hybrid Electrocatalysts and Photocatalysts for Nitrogen Reduction Reactions: Advanced Materials, Mechanisms, Challenges and Perspectives, Electrochem. Energy Rev., 2020, 3, 506–540 CrossRef CAS.
- G. Ertl, Surface Science and Catalysis-Studies on the Mechanism of Ammonia Synthesis: The P. H. Emmett Award Address, Catal. Rev., 1980, 21, 201–223 CrossRef CAS.
- Y. Yao, S. Q. Zhu, H. J. Wang, H. Li and M. H. Shao, A Spectroscopic Study on the Nitrogen Electrochemical Reduction Reaction on Gold and Platinum Surfaces, J. Am. Chem. Soc., 2018, 140, 1496–1501 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the Computational Design of Solid Catalysis, Nat. Chem., 2009, 1, 37–46 CrossRef.
- J. K. Nørskov, F. Abild-Pedersen, F. Studt and T. Bligaard, Density Functional Theory in Surface Chemistry and Catalysis, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 937–943 CrossRef PubMed.
- E. Skúlason, T. Bligaard, S. Gudmundsdóttir, F. Studt, J. Rossmeisl, F. Abild-Pederson, T. Vegge, H. Jósson and J. K. Nørskov, A Theoretical Evaluation of Possible Transition Metal Electro-Catalysts for N2 Reduction, Phys. Chem. Chem. Phys., 2012, 14, 1235–1245 RSC.
- H. Y. Li, Z. F. Zhao, Q. H. Cai, L. C. Yin and J. X. Zhao, Nitrogen Electroreduction Performance of Transition Metal Dimers Embedded into N-Doped Graphene: A Theoretical Prediction, J. Mater. Chem. A, 2020, 8, 4533–4543 RSC.
- G. Rostamikia, A. J. Mendoza, M. A. Hickner and M. J. Janik, First-Principles Based Microkinetic Modeling of Borohydride
Oxidation on a Au(111) Electrode, J. Power Sources, 2011, 196, 9228–9237 CrossRef CAS.
- X. W. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Selectivity of CO2 Reduction on Copper Electrodes: The Role of the Kinetics of Elementary Steps, Angew. Chem., Int. Ed., 2013, 52, 2459–2462 CrossRef CAS PubMed.
- S. A. Akhade, N. J. Bernstein, M. R. Esopi, M. J. Regula and M. J. Janik, Electrochemical Reduction of Carbon Dioxide by Heterogenous and Homogeneous Catalysts: Experiment and Theory, Catal. Today, 2017, 288, 63–73 CrossRef CAS.
- S. Maheshwari, G. Rostamikia and M. J. Janik, Elementary Kinetics of Nitrogen Electroreduction on Fe Surfaces, J. Chem. Phys., 2019, 150, 041708 CrossRef.
- G. Rostamikia, S. Maheshwari and M. J. Janik, Elementary Kinetics of Nitrogen Electroreduction to Ammonia on Late Transition Metals, Catal. Sci. Technol., 2019, 9, 174–181 RSC.
- D. Bao, Q. Zhang, F. L. Meng, H. X. Zhong, M. M. Shi, Y. Zhang, J. M. Yang, Q. Jiang and X. B. Zhang, Electrochemical Reduction of N2 under Ambient Conditions for Artificial N2 Fixation and Renewable Energy Storage Using N2/NH3 Cycle, Adv. Mater., 2017, 29, 1604799 CrossRef PubMed.
- L. H. Ou, J. X. Chen, Y. D. Chen and J. L. Jin, Mechanistic Study of Pt-Catalyzed Electrooxidation of HCOOH in Acid Medium: Kinetic Considerations on the Effect of Solvation, J. Phys. Chem. C, 2018, 122, 24871–24884 CrossRef CAS.
- L. H. Ou, Theoretical Insights into the Effect of Solvation and Sublayer Ru on Pt-Catalytic CH3OH Oxidation Mechanisms in the Aqueous Phase, J. Phys. Chem. C, 2018, 122, 14554–14565 CrossRef CAS.
- L. H. Ou, J. X. Chen, Y. D. Chen and J. L. Jin, Mechanistic Study on Cu-catalyzed CO2 Electroreduction into CH4 at Simulated Low Overpotentials Based on an Improved Electrochemical Model, Phys. Chem. Chem. Phys., 2019, 21, 15531–15540 RSC.
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. Gudmundsdóttir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, Modeling the Electrochemical Hydrogen Oxidation and Evolution Reactions on the Basis of Density Functional Theory Calculations, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef.
- E. Skúlason, G. S. Karlberg, J. Rossmeisl, T. Bligaard, J. Greeley, H. Jónsson and J. K. Nørskov, Density Functional Theory Calculations for the Hydrogen Evolution Reaction in an Electrochemical Double Layer on the Pt(111) Electrode, Phys. Chem. Chem. Phys., 2007, 9, 3241–3250 RSC.
- J. Rossmeisl, E. Skúlason, M. E. Björketun, V. Tripkovic and J. K. Nørskov, Modeling the Electrified Solid-Liquid Interface, Chem. Phys. Lett., 2008, 466, 68–71 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadóttir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- G. S. Karlberg, T. F. Jaramillo, E. Skúlason, J. Rossmeisl, T. Bligaard and J. K. Nørskov, Cyclic Voltammograms for H on Pt(111) and Pt(100) from First Principles, Phys. Rev. Lett., 2007, 99, 126101 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, A. Logadottir, J. R. Kitchin, J. G. Chen, S. Pandelov and U. Stimming, Trends in the Exchange Current for Hydrogen Evolution, J. Electrochem. Soc., 2005, 152, J23–J26 CrossRef.
- F. Yang, Q. F. Zhang, Y. W. Liu and S. L. Chen, A Theoretical Consideration on the Surface Structure and Nanoparticle Size Effects of Pt in Hydrogen Electrocatalysis, J. Phys. Chem. C, 2011, 115, 19311–19319 CrossRef CAS.
- H. Mistry, R. Reske, Z. H. Zeng, Z. J. Zhao, J. Greeley, P. Strasser and B. R. Cuenya, Exceptional Size-Dependent Activity Enhancement in the Electroreduction of CO2 over Au Nanoparticles, J. Am. Chem. Soc., 2014, 136, 16473–16476 CrossRef CAS PubMed.
- A. A. Peterson, F. Abild-Pederson, F. Studt, J. Rossmeisl and J. K. Nørskov, How Copper Catalyzes the Electroduction of Carbon Dioxide into Hydrocarbon Fuels, Energy Environ. Sci., 2010, 3, 1311–1315 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01978c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Junling Jin* and
Yuandao Chen
*,
Junling Jin* and
Yuandao Chen
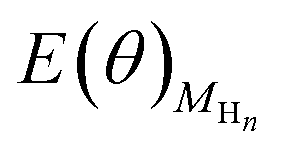 is the total energy of the Au surface with different coverage of adsorbed H atoms.
is the total energy of the Au surface with different coverage of adsorbed H atoms.
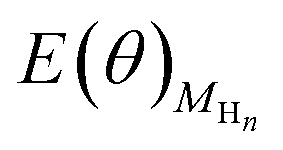 and
and  is directly available via DFT calculations. A series of values of
is directly available via DFT calculations. A series of values of 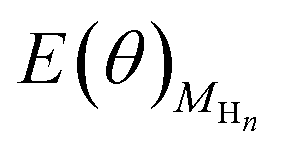 can be calculated by changing the coverage of surface adsorbed H atoms on Au(111). At the different coverages, the values of ΔE(θ) can be obtained by differentiating the plots of
can be calculated by changing the coverage of surface adsorbed H atoms on Au(111). At the different coverages, the values of ΔE(θ) can be obtained by differentiating the plots of  against θ on the basis of eqn (3).
against θ on the basis of eqn (3).