
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The graphite-catalyzed ipso-functionalization of arylboronic acids in an aqueous medium: metal-free access to phenols, anilines, nitroarenes, and haloarenes†
Anshu Dandia*a,
Ruchi Sharmaa,
Pratibha Sainia,
Ranveer Singh Badgotia,
Kuldeep S. Rathoreb and
Vijay Parewa *a
*a
aCentre of Advanced Studies, Department of Chemistry, University of Rajasthan, Jaipur, India. E-mail: dranshudandia@yahoo.co.in; parewavijay.parewa@gmail.com; parewavijay@uniraj.ac.in
bDepartment of Physics, Arya College of Engineering and IT, Jaipur, India. E-mail: kuldeep_ssr@yahoo.com
First published on 19th May 2021
Abstract
An efficient, metal-free, and sustainable strategy has been described for the ipso-functionalization of phenylboronic acids using air as an oxidant in an aqueous medium. A range of carbon materials has been tested as carbocatalysts. To our surprise, graphite was found to be the best catalyst in terms of the turnover frequency. A broad range of valuable substituted aromatic compounds, i.e., phenols, anilines, nitroarenes, and haloarenes, has been prepared via the functionalization of the C–B bond into C–N, C–O, and many other C–X bonds. The vital role of the aromatic π-conjugation system of graphite in this protocol has been established and was observed via numerous analytic techniques. The heterogeneous nature of graphite facilitates the high recyclability of the carbocatalyst. This effective and easy system provides a multipurpose approach for the production of valuable substituted aromatic compounds without using any metals, ligands, bases, or harsh oxidants.
1. Introduction
Arylboronic acids and their derivatives have attracted substantial research interest from chemists since they have immense value in chemical synthesis and materials science.1 They have also been used as synthons for the further construction of therapeutic molecules and value-added chemicals.2 Furthermore, arylboronic acids have been extensively used as ubiquitous structural motifs for various functional group transformations via ipso-functionalization.3Phenols are a well-known class of compounds for their biological applications, including antibacterial, antitumor, antiviral, pro-oxidant, cardioprotective, and antimutagenicity activities.4 They have also been employed as synthetic intermediates in various chemical transformations.5 Due to the wide-ranging applications of phenols, dedicated efforts have been made to advance the preparation of phenols.6 Among them, the ipso-hydroxylation of arylboronic acid has been the most-studied strategy in the recent past.7–9
This transformation is usually carried out with several equivalents of an oxidant, like hypervalent iodine, tert-butyl hydroperoxide (TBHP), ozone, PhI(OAc)2, benzoquinone, or hydrogen peroxide (H2O2), which can be explosive or have a hazardous environmental impact. These reactions can also entail high temperatures, strongly basic conditions, and the use of metal catalysts (Pd, Au, Cu, Ru, Ag, etc.) and microwave irradiation, and they often require the use of expensive specialized ligands. As phenols are susceptible to oxidants, the quantity of oxidant needs to be cautiously determined. Moreover, the removal of trace quantities of metal-containing catalysts from the final compounds can be complicated. Therefore, the development of sustainable protocols for the above transformation presents a difficult task for chemists.
To meet the requirements of green chemistry, carbon-based materials, such as ordered mesoporous carbon, activated carbon, graphene-based materials, graphite, and carbon nanotubes, open up opportunities in the field of catalysis due to their inimitable physical and chemical properties.10 In recent years, many scientists have established that carbon-based materials can show elevated catalytic efficiencies in organic synthesis compared to traditional metal catalysts.11 The sustainable nature of these carbon-based materials has been well documented in the literature.12 Among them, the use of graphite as an environmentally benign catalyst in organic synthesis has taken on vast significance.13
Continuing our interest in developing sustainable protocols for organic synthesis,14 herein, we evaluate the catalytic activities of various carbon-based substances for the preparation of a library of phenol derivatives via the ipso-hydroxylation of arylboronic acid, utilizing water as an environmentally friendly solvent under sustainable conditions. Commercially available graphite was found to be the best metal-free carbocatalyst for the above transformation. Furthermore, we have also extended our strategy to the synthesis of anilines, nitroarenes, and haloarenes via transforming the C–B bond of arylboronic acid to C–N and many other C–X bonds (Scheme 1).
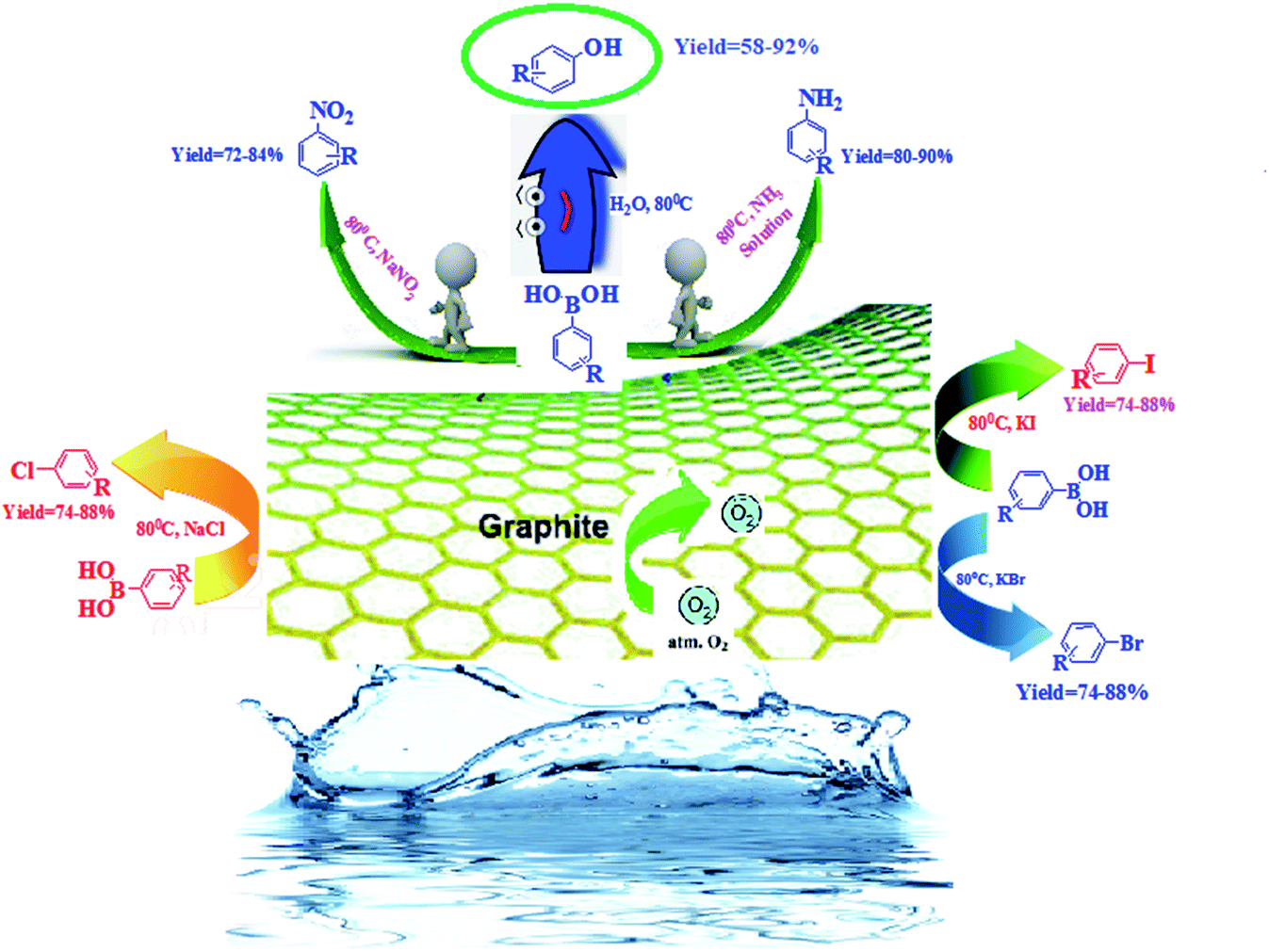 | ||
| Scheme 1 The synthesis of substituted phenols, anilines, nitroarenes, and haloarenes from arylboronic acids. | ||
2. Results and discussion
Firstly, phenylboronic acid has been selected as a model reactant for ipso-hydroxylation in an aqueous medium under air at 80 °C (Table 1). No transformation of phenylboronic acid into phenol was noticed in the absence of catalyst. This outcome forced us to further optimize the reaction conditions. Recently, we have developed a series of carbon-based catalysts for various organic transformations.15 Therefore, from a sustainable point of view, the catalytic competences of various carbocatalysts have been studied. To our surprise, graphite was found to be the best catalyst for this transformation in terms of the turnover frequency (TOF15b,16). Moreover, inferior yields were observed upon using other carbon-based materials as catalysts. GO and rGO gave poor product yields. Approximately similar catalytic activities were observed when the reaction was tried with various graphite samples purchased from different vendors.| S. no. | Catalyst | Solvent | Time | Yieldb (%) | TOF (×10−3 mol g−1 min−1) |
|---|---|---|---|---|---|
| a Reactions were carried out with phenylboronic acid (2 mmol) in H2O at 80 °C under air.b Isolated yield.c Multi-walled carbon nanotubes.d Conversion conducted under a N2 atmosphere. | |||||
| 1 | — | H2O | 24 h | — | n.c. |
| 2 | Graphite (20 wt%) | H2O | 24 h | 92 | 0.021 |
| 3 | Activated carbon (20 wt%) | H2O | 24 h | 74 | 0.017 |
| 4 | MWCNTs (20 wt%)c | H2O | 24 h | 72 | 0.016 |
| 5 | GO (20 wt%) | H2O | 24 h | 21 | 0.005 |
| 6 | rGO (20 wt%) | H2O | 24 h | 48 | 0.011 |
| 7 | Graphite (5 wt%) | H2O | 24 h | 64 | 0.069 |
| 8 | Graphite (10 wt%) | H2O | 24 h | 79 | 0.041 |
| 9 | Graphite (20 wt%)d | H2O | 24 h | — | — |
| 10 | Graphite (20 wt%) | CH3CN | 24 h | 82 | 0.019 |
| 11 | Graphite (20 wt%) | MeOH | 24 h | 69 | 0.016 |
| 12 | Graphite (20 wt%) | THF | 24 h | 44 | 0.010 |
| 13 | Graphite (20 wt%) | DMSO | 24 h | 38 | 0.009 |
An improvement in the yield of the desired product to an immense extent (36–92%) was observed when the catalyst loading was increased from 5 to 20 wt% (Table 1). We have tried different catalyst loadings (20, 30, 40, and 50 wt%), however further increases in catalyst loading did not affect the yield, and the optimum loading of catalyst was 20 wt%. The desired product was not formed when the model reaction was conducted under a nitrogen atmosphere. These results show that, from a sustainable point of view, air is an effective and environmentally benign oxidant. The desirable transformation was not observed in the absence of graphite, which shows that graphite plays a vital role in the further activation of oxygen molecules from air for the catalytic reaction.
A series of trials was carried out using a variety of solvents. Methanol and acetonitrile were found to give moderate yields among the various organic solvents. Compared to conventional organic solvents, H2O was found to be a best solvent for the above transformation from an economical perspective. The outcome can be attributed to the excellent dispersion of the catalyst in H2O. Additionally, the substrates show good dissolving capacities in water. The model reaction was also performed at different temperatures (60 °C, 80 °C, and 100 °C). It was concluded that, to achieve maximum conversion to the product, 80 °C was required.
To recognize the responsible catalytic sites in carbon-based catalysts, various organic compounds were employed as catalysts for the above transformation. Compounds with various oxygen functionalities showed poor catalytic activities. These outcomes confirmed that oxygen-containing functional groups may not be the catalytic sites in the carbon materials for hydroxylation. It was recognized from various catalytic tests that compounds containing aromatic π-conjugation systems demonstrated reasonable catalytic activities (Table 2). We suppose that the aromatic π-conjugation system has intrinsic activity for the above transformation. Thus, carbon materials that have significant oxygen functionality showed poor results compared to graphite.
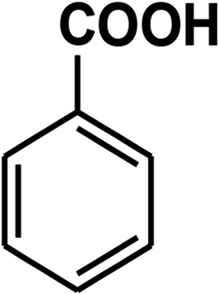
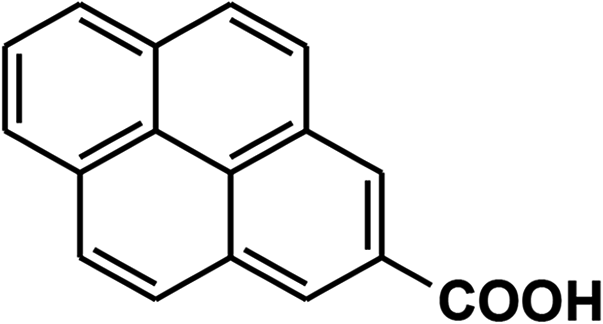
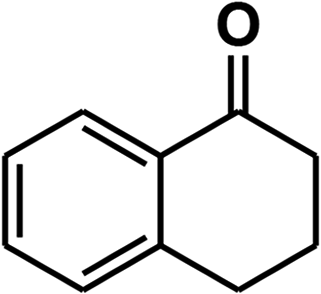




Carbon materials illustrate metallic features and activate O2 due to the presence of delocalized π-electrons.17 The introduction of any surface defects into a carbon material decreases its catalytic activity because this diminishes the mobility of delocalized π-electrons.18 Activated charcoal showed lower catalytic activity due to its decreased mass transfer efficiency because of its high degree of defects and its microporous structure.19 It has been revealed in the literature that sp2-hybridized carbon atoms are vital centers for oxygen adsorption and activation.17 These activated oxygen atoms can move on graphitic planes. As we already showed the necessity of oxygen for the above transformation, we proposed that the activation of oxygen was a decisive step in this transformation. The graphitic planes of the catalyst transform atmospheric O2 to short-lived O2˙− species that are stored in the carbon pores.17
There is a possibility that the observed catalytic properties of graphite could be attributed to the presence of trace metal-ion impurities. To establish the presence of trace amounts of metal ions, we have studied graphite catalyst samples (fresh and reused) via various analytical techniques. EDAX analysis confirmed the absence of any transition metals in the graphite catalyst, which ruled out catalytic assistance from any metal impurities. Moreover, the carbon![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oxygen atomic ratios of fresh and reused graphite were comparable, showing the stable nature of the graphite catalyst during the reaction (see ESI, Fig. S1 and S2†).
:oxygen atomic ratios of fresh and reused graphite were comparable, showing the stable nature of the graphite catalyst during the reaction (see ESI, Fig. S1 and S2†).
The deconvoluted C 1s XPS spectrum of fresh graphite shows a peak at 284.4 eV. Additionally, the O 1s spectrum illustrates a broad peak from 530 to 535 eV. The intensities and energies of the peaks of reused graphite in both XPS spectra were approximately the same (Fig. 1).
 | ||
| Fig. 1 The XPS spectra of fresh and reused graphite: (a) C 1s spectra; (b) O 1s spectra; and (c) the complete XPS spectra. | ||
In the Fourier-transform infrared (FT-IR) spectrum of reused graphite, absorption peak fluctuations from 1443 to 1573 cm−1 are noted because of aromatic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching (Fig. 2a). We have also carried out solid-state 13C-NMR spectroscopy analysis of reused graphite (see ESI, Fig. S5†). The spectrum showed broad resonance associated with sp2-hybridized carbon atoms.20 There were no additional peaks from any oxygen functionalities in the FT-IR and solid-state 13C-NMR spectra of reused graphite.
C stretching (Fig. 2a). We have also carried out solid-state 13C-NMR spectroscopy analysis of reused graphite (see ESI, Fig. S5†). The spectrum showed broad resonance associated with sp2-hybridized carbon atoms.20 There were no additional peaks from any oxygen functionalities in the FT-IR and solid-state 13C-NMR spectra of reused graphite.
 | ||
| Fig. 2 (a) The FT-IR spectrum of reused graphite and (b) Raman spectra of fresh and reused graphite. | ||
The Raman spectra of fresh and reused catalyst graphite samples comprise many overlapping bands (Fig. 2b). The D band, G band, and 2D band arise at around 1341 cm−1, 1570 cm−1, and 2690 cm−1, respectively.
In SEM images, the graphite sheets displayed crumpled and wrinkled surfaces of thin lamellae (Fig. 3a, see ESI, Fig. S3†). The average particle size of the catalyst is about 9.8 μm. The morphology and average particle size of the reused catalyst remain unchanged, as shown by SEM images of the reused graphite catalyst after four runs. The XRD pattern of recycled graphite was similarly documented, indicating that the structural integrity remains unchanged after the transformation reaction (Fig. 3b).
 | ||
| Fig. 3 (a) SEM images and (b) XRD patterns of fresh and reused graphite. | ||
These analytical results proved that no oxidation of the catalyst was observed during the reaction, and the aromatic nature of the catalyst remains constant throughout the process. These results demonstrate that the utilized graphite catalyst is a stable and reusable catalyst for the above transformation.
To further confirm the role of the catalyst in the activation of the oxygen, EPR measurements were carried out with DMPO (see ESI, Fig. S5†). When the EPR experiment was performed with catalyst, peaks from the spin adduct of the DMPO superoxide radical were observed,21 while in the absence of catalyst, these peaks were absent. This result indicated that the generation of superoxide radicals was indispensable for the reaction to proceed.
These results suggest that a radical process might be involved in the above transformation. We also conducted the reaction with H2O18 as a solvent under optimized conditions. The incorporation of 18O into the product was not viewed, which ruled out the possibility of water acting as an oxygen source (Scheme 2).
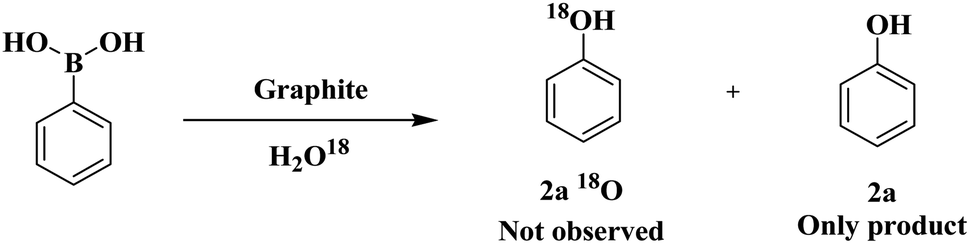 | ||
| Scheme 2 Isotopic experiment results. | ||
In most cases, literature reports reveal the requirement for a base/metal in the reaction when phenylboronic acid reacts with reactive oxygen species.22 The mechanism of the reaction is ambiguous, particularly in light of the fact that the reaction can occur in the absence of any base/metal under air. Via control experiments, we have already confirmed that a radical process might be involved in the transformation. Due to the presence of delocalized π-electrons, graphitic planes convert O2 to short-lived O2˙− species that assemble in the carbon pores.17 This short-lived O2˙− was the active sustainable oxidant for the above transformation. Based on the above information and literature reports,23 we considered that the reaction occurred via a radical nucleophilic substitution mechanism24 through an unexplored process (to be the subject of future investigations). Owing to the presence of a delocalized π-electron system, graphite also stabilizes the radical intermediates for better reaction performance.
To elaborate on the possibilities of this approach, numerous electronically diversified phenylboronic acids were used for hydroxylation under the established optimum conditions (Table 3). This approach shows broad functional-group tolerance. Arylboronic acids containing electron-withdrawing and -donating substituents at the para position gave excellent phenol yields. The use of sterically demanding ortho-substituted arylboronic acids did not influence the yields drastically, and the final products were achieved in good yields. meta-Substituted arylboronic acids gave analogous outcomes. Heteroaryl boronic acids also gave good yields in the optimized reaction environment.
|
|---|
| a Reactions are performed with various phenylboronic acids in water at 80 °C under air. |
 |

Encouraged by the above results, we would like to examine whether the formation of anilines would happen when aqueous NH3 solution was inserted into the system due to nucleophilic competition between the nitrogen and oxygen centers. To our delight, the formation of aniline was observed instead of phenol. A probable reason for this is that the superior nucleophilic power of the nitrogen center compared to the oxygen center led to the construction of aniline instead of phenol (Scheme 3). Therefore, the reaction proceeds through the more nucleophilic center (in this case the nitrogen center) instead of the oxygen center. Similarly, when we added NaNO2 to the system instead of aqueous NH3, nitroarenes were formed.
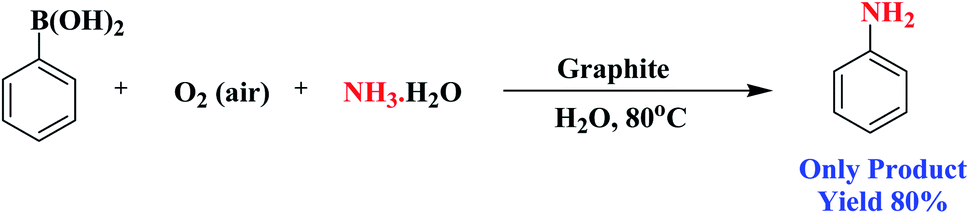 | ||
| Scheme 3 Intermolecular competition between the oxygen and nitrogen centres during the functionalization of phenylboronic acid. | ||
Inspired by these outcomes, we used the same sustainable protocol for the iodination of arylboronic acids, using KI as an iodide source. As iodide ions have more nucleophilic power, iodobenzene was formed as the sole product. In the same way, when we used KBr in the system, bromobenzene was formed as the desired product. Unfortunately, KCl failed to give the halogenated product under these conditions. However, NaCl withstands the reaction conditions and gives the chlorinated product in good yield. Since the functionalization of phenylboronic acids at the nitrogen center was successful in the presence of an oxygen center, a competitive trial was executed to determine which phenylboronic acid functionalization would happen when three halogen reactants were added at the same time (Scheme 4). Under these conditions, iodobenzene was formed as a major product, with the order I > Br > Cl.
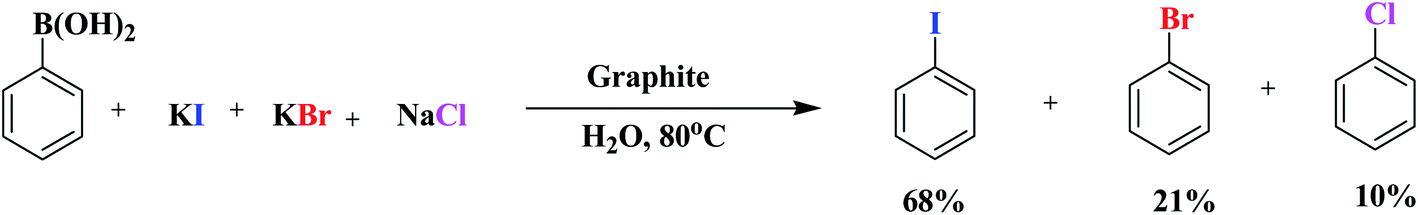 | ||
| Scheme 4 Intermolecular competition between various halide centres during the functionalization of phenylboronic acid. | ||
This protocol gave excellent outcomes when using NH3·H2O, NaNO2, KI, KBr, and NaCl as the functional-group sources. 1.5 eq. of these reagents was adequate for the reaction and the addition of additional amounts did not affect the yields remarkably. It was found that a temperature of 80 °C with a loading of 10 wt% catalyst was appropriate to give quantitative amounts of anilines, nitroarenes, and haloarenes. We employed our established conditions with the model substrates described in Scheme 5. The formation of anilines, nitroarenes, and haloarenes was not identified in the absence of atmospheric air (under a N2 environment), and the outcome designated that an oxidative route was involved in the synthesis of these useful compounds. To demonstrate the opportunity and boundaries of the above approach, we used a broad range of arylboronic acids. The reactions proceeded efficiently with electron-withdrawing and electron-donating arylboronic acids, but lower and more variable yields were obtained in the case of chlorobenzenes.
 | ||
| Scheme 5 The synthesis of substituted anilines, nitroarenes, and haloarenes from arylboronic acids. | ||
The heterogeneous character and excellent recyclability of graphite was confirmed (see ESI†). For recycling experiments, arylboronic acid was selected as the model substrate for all transformations. The used graphite was simply isolated via filtration, cleaned with ethanol, and reused for an additional four cycles. As shown in Table 4, graphite was found to be a very stable carbocatalyst for all these transformations.
|
||||
|---|---|---|---|---|
| Functional entity | First cycle [%] | Second cycle [%] | Third cycle [%] | Fourth cycle [%] |
| a Isolated product yield. | ||||
| Ph-OH | 92 | 88 | 86 | 86 |
| Ph-NH2 | 80 | 78 | 77 | 77 |
| Ph-NO2 | 72 | 70 | 68 | 68 |
| Ph-I | 76 | 72 | 72 | 71 |
| Ph-Br | 73 | 71 | 71 | 68 |
| Ph-Cl | 72 | 67 | 66 | 66 |

3. Experimental
3.1. General
See ESI.†3.2. Preparation of substituted phenols (2)
A round-bottomed flask was loaded with arylboronic acid (2 mmol) and 20 wt% graphite in 20 mL of H2O, and the mixture was stirred at 80 °C until complete transformation was achieved (checked via TLC). The used graphite was removed simply via filtration after the transformation was complete. The filtrate was permitted to cool to atmospheric temperature and was then extracted with ethyl acetate (3 × 2 mL). The solution was then concentrated under reduced pressure and purified via column chromatography on silica gel (eluent, hexane:EtOAc) to furnish the pure final product.
3.3. Preparation of substituted anilines (3)
A round-bottomed flask was loaded with arylboronic acid (2 mmol), NH3 solution (1.5 eq.) and 20 wt% graphite in 20 mL of H2O, and the mixture was stirred at 80 °C until the complete transformation was achieved (checked via TLC). The used graphite catalyst was separated simply via filtration after the completion of the transformation. The filtrate was permitted to cool to atmospheric temperature and was then extracted with ethyl acetate (3 × 2 mL). The solution was then concentrated under reduced pressure and purified via column chromatography on silica gel (eluent, hexane:EtOAc) to deliver the required pure product.
3.4. Preparation of substituted nitroarenes (4)
A round-bottomed flask was loaded with arylboronic acid (2 mmol), NaNO2 (1.5 eq.) and 20 wt% graphite in 20 mL of H2O, and the mixture was stirred at 80 °C until the conversion was complete (monitored via TLC). The used graphite was separated simply via filtration after the completion of the transformation. The filtrate was permitted to cool to atmospheric temperature and was then extracted with ethyl acetate (3 × 2 mL). The solution was then concentrated under reduced pressure and purified via column chromatography on silica gel (eluent, hexane:EtOAc) to supply the final pure product.
3.5. Preparation of substituted haloarenes (5–7)
A round-bottomed flask was loaded with arylboronic acid (2 mmol), KCl (1.5 eq.)/KI (1.5 eq.)/KBr (1.5 eq.), and 20 wt% graphite in 20 mL of H2O, and the mixture was stirred at 80 °C until complete transformation was achieved (checked via TLC). Graphite was isolated simply via filtration after the transformation was complete. The filtrate was permitted to cool to atmospheric temperature and was then extracted with ethyl acetate (3 × 2 mL). The solution was then concentrated under reduced pressure and purified via column chromatography on silica gel (eluent, hexane:EtOAc) to offer the desired pure product.
4. Conclusions
A versatile and simple graphite-catalyzed process has been illustrated for the conversion of numerous functional groups on aromatic rings in an aqueous medium under air via the ipso-functionalization of phenylboronic acids. Various important aromatic compounds, e.g., phenols, anilines, nitroarenes, and haloarenes, have been synthesized without using any metals, ligands, bases, or oxidants. The vital role of the aromatic π-conjugation system of graphite in assisting the reaction has been confirmed via various analytic techniques, viz., FT-IR, Raman, XPS, SEM, and XRD analysis. Via control experiments, we have offered a plausible mechanistic pathway for the above transformation. The main advantage of this protocol is the use of economical and abundant graphite as an efficient carbocatalyst without any prior treatment. Overall, this strategy offers numerous advantages, such as the recyclability of the catalyst, operational simplicity, inexpensiveness, environmental friendliness, a broad substrate scope, and high yields.Conflicts of interest
There are no conflicts to declare.Acknowledgements
DST-SERB, C.S.I.R., and U.G.C., New Delhi, are acknowledged for financial assistance. The authors are thankful to MNIT, Jaipur, for assistance with the spectral studies.References
- D. Hall, Boronic Acids: Preparation and Applications in Organic Synthesis, Medicine and Materials, ed. D. G. Hall, Wiley-VCH, Weinheim, 2nd edn, 2011 Search PubMed.
- (a) C. Kleeberg, L. Dang, Z. Lin and T. B. Marder, Angew. Chem., 2009, 121, 5454–5458 CrossRef; (b) M. Murata, T. Oyama, S. Watanabe and Y. Masuda, J. Org. Chem., 2000, 65, 164–168 CrossRef CAS PubMed; (c) W. Zhu and D. Ma, Org. Lett., 2006, 8, 261–263 CrossRef CAS PubMed; (d) D. A. Wilson, C. J. Wilson, B. M. Rosen and V. Percec, Org. Lett., 2008, 10, 4879–4882 CrossRef CAS PubMed; (e) F. Mo, Y. Jiang, D. Qiu, Y. Zhang and J. Wang, Angew. Chem., 2010, 49, 1846–1849 CrossRef CAS PubMed; (f) J. Y. Cho, M. K. Tse, D. Holmes, R. E. Maleczka and M. R. Smith, Science, 2002, 295, 305–308 CrossRef CAS PubMed; (g) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura and N. R. Anastasi, Science, 2002, 295, 305–308 CrossRef PubMed; (h) T. Ishiyama, J. Takagi, K. Ishida, N. Miyaura, N. R. Anastasi and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 390–391 CrossRef CAS PubMed; (i) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- C. Zhua and R. F. John, Adv. Synth. Catal., 2014, 356, 2395–2410 CrossRef PubMed.
- (a) J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier, New York, 1996 Search PubMed; (b) Z. Rappoport, The Chemistry of Phenols, Wiley-VCH, Weinheim, Germany, 2003 CrossRef.
- (a) R. W. Owen, A. Giacosa, R. Haubner, B. Spiegel-Hader and H. Bartsch, Eur. J. Cancer., 2000, 36, 1235–1247 CrossRef CAS PubMed; (b) L. Pilato, React. Funct. Polym., 2013, 73, 270–277 CrossRef CAS.
- (a) C. Hoarau and T. R. R. Pettus, Synlett, 2003, 1, 127–137 Search PubMed; (b) P. Hanson, J. R. Jones, A. B. Taylor, P. H. Walton and A. W. Timms, J. Am. Chem. Soc., 2002, 2, 1135–1150 Search PubMed; (c) T. George, R. Mabon, G. Sweeney, J. B. Sweeney and A. Tavassoli, J. Am. Chem. Soc., 2000, 1, 2529–2574 Search PubMed; (d) T. Schulz, C. Torborg and B. Schaffner, Angew. Chem., Int. Ed., 2009, 48, 918–921 CrossRef CAS PubMed; (e) K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128, 10694–10695 CrossRef CAS PubMed.
- (a) B. A. Dar, P. Bhatti, A. P. Singh, A. Lazar, P. R. Sharma, M. Sharma and B. Singh, Appl. Catal., A, 2013, 466, 60–67 CrossRef CAS; (b) D. Yang, B. An, W. Wei, M. Jiang, J. You and H. Wang, Tetrahedron, 2014, 70, 3630–3634 CrossRef CAS; (c) H. Yang, Y. Li, M. Jiang, J. Wang and H. Fu, Chem.–Eur. J., 2011, 17, 5652–5660 CrossRef CAS PubMed; (d) A. Affrose, I. A. Azath, A. Dhakshinamoorthy and K. Pitchumani, J. Mol. Catal. A: Chem., 2014, 395, 500–505 CrossRef CAS; (e) J. Xu, X. Wang, C. Shao, D. Su, G. Cheng and Y. Hu, Org. Lett., 2010, 12, 1964–1967 CrossRef CAS PubMed; (f) K. Inamoto, K. Nozawa, M. Yonemoto and Y. Kondo, Chem. Commun., 2011, 47, 11775–11777 RSC; (g) A. D. Chowdhury, S. M. Mobin, S. Mukherjee, S. Bhaduri and G. K. Lahiri, Eur. J. Inorg. Chem., 2011, 21, 3232–3239 CrossRef; (h) J. Zheng, S. Lin, X. Zhu, B. Jiang, Z. Yang and Z. Pan, Chem. Commun., 2012, 48, 6235–6237 RSC; (i) L. Wang, W. Zhang, D. Sheng Su, X. Meng and F.-S. Xiao, Chem. Commun., 2012, 48, 5476–5478 RSC; (j) N. Gogoi, P. K. Gogoi, G. Borah and U. Bora, Tetrahedron Lett., 2016, 57, 4050–4052 CrossRef CAS; (k) T. Begum, A. Gogoi, P. K. Gogoi and U. Bora, Tetrahedron Lett., 2015, 56, 95–97 CrossRef CAS; (l) T. Toyao, N. Ueno, K. Miyahara, Y. Matsui, T.-H. Kim, Y. Horiuchi, H. Ikeda and M. Matsuoka, Chem. Commun., 2015, 51, 16103–16106 RSC; (m) M.-J. Zhang, H.-X. Li, H.-Y. Li and J.-P. Lang, Dalton Trans., 2016, 45, 17759–17769 RSC.
- (a) G. K. S. Prakash, S. Chacko, C. Panja, T. E. Thomas, L. Gurung, G. Rasul, T. Mathew and G. A. Olah, Adv. Synth. Catal., 2009, 351, 1567–1574 CrossRef CAS; (b) L. Wang, D.-Y. Dai, Q. Chen and M.-Y. He, J. Fluorine Chem., 2014, 158, 44–47 CrossRef CAS; (c) S. Gupta, P. Chaudhary, V. Srivastava and J. Kandasamy, Tetrahedron Lett., 2016, 57, 2506–2510 CrossRef CAS; (d) S. Guo, L. Lu and H. Cai, Synlett, 2013, 24, 1712–1714 CrossRef CAS; (e) D.-S. Chen and J.-M. Huang, Synlett, 2013, 24, 499–501 CrossRef CAS; (f) K. S. Webb and D. Levy, Tetrahedron Lett., 1995, 36, 5117–5118 CrossRef CAS; (g) B. R. Travis, B. P. Ciaramitaro and B. Borhan, Eur. J. Org. Chem., 2002, 2002, 3429–3434 CrossRef; (h) R. E. Maleczka Jr, F. Shi, D. Holmes and M. R. Smith III, J. Am. Chem. Soc., 2003, 125, 7792–7793 CrossRef PubMed; (i) G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 623–630 CrossRef CAS PubMed; (j) E. Kianmehr, M. Yahyaee and K. Tabatabai, Tetrahedron Lett., 2007, 48, 2713–2715 CrossRef CAS; (k) C. Zhu, R. Wang and J. R. Falck, Org. Lett., 2012, 14, 3494–3497 CrossRef CAS PubMed; (l) Y. K. Bommegowda, N. Mallesha, A. C. Vinayaka and M. P. Sadashiva, Chem. Lett., 2016, 45, 268–270 CrossRef CAS; (m) K. Murugan and S. Palaniswamy, ACS Sustainable Chem. Eng., 2019, 7, 9028–9034 CrossRef.
- (a) A. Paul, D. Chatterjee, H. T. Rajkamal, S. Banerjee and S. Yadav, Tetrahedron Lett., 2015, 56, 2496–2499 CrossRef CAS; (b) N. Chatterjee, H. Chowdhury, K. Sneh and A. Goswami, Tetrahedron Lett., 2015, 56, 172–174 CrossRef CAS; (c) J. S. Ruso, N. Rajendiran and R. S. Kumaran, Tetrahedron Lett., 2014, 55, 2345–2347 CrossRef CAS; (d) G. Cheng, X. Zeng and X. Cui, Synthesis, 2014, 46, 295–300 Search PubMed; (e) P. Gogoi, P. Bezboruah, J. Gogoi and R. C. Boruah, Eur. J. Org. Chem., 2013, 32, 7291–7294 CrossRef; (f) K. Hosoi, Y. Kuriyama, S. Inagi and T. Fuchigami, Chem. Commun., 2010, 46, 1284–1286 RSC; (g) H.-L. Qi, D.-S. Chen, J.-S. Ye and J.-M. Huang, J. Org. Chem., 2013, 78, 7482–7487 CrossRef CAS PubMed; (h) H. Jiang, L. Lykke, S. U. Pedersen, W.-J. Xiao and K. A. Jørgensen, Chem. Commun., 2012, 48, 7203–7205 RSC; (i) A. N. Cammidge, V. H. M. Goddard, C. P. J. Schubert, H. Gopee, D. L. Hughes and D. Gonzalez-Lucas, Org. Lett., 2011, 13, 6034–6037 CrossRef CAS PubMed; (j) H. Kotoučová, I. Strnadová, M. Kovandová, J. Chudoba, H. Dvořáková and R. Cibulka, Org. Biomol. Chem., 2014, 12, 2137–2142 RSC; (k) X. Yu and S. M. Cohen, Chem. Commun., 2015, 51, 9880–9883 RSC; (l) J. Luo, X. Zhang and J. Zhang, ACS Catal., 2015, 5, 2250–2254 CrossRef CAS; (m) S. P. Pitre, C. D. McTiernan, H. Ismaili and J. C. Scaiano, J. Am. Chem. Soc., 2013, 135, 13286–13289 CrossRef CAS PubMed; (n) Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 124, 802–812 Search PubMed; (o) J. A. Johnson, J. Luo, X. Zhang, Y.-S. Chen, M. D. Morton, E. Echeverría, F. E. Torres and J. Zhang, ACS Catal., 2015, 5, 5283–5291 CrossRef CAS; (p) W. Yina, X. Pana, W. Lenga, J. Chenb and H. Hec, Green Chem., 2019, 21, 4614–4618 RSC.
- (a) L. Xien and L. Dai, Nat. Rev. Mater., 2016, 1, 1–12 Search PubMed; (b) E. Pérez-Mayoral, V. Calvino-Casildaa and E. Soriano, Catal. Sci. Technol., 2016, 6, 1265–1291 RSC; (c) E. Lam and H. T. John Luong, ACS Catal., 2014, 4, 3393–3410 CrossRef CAS.
- (a) M. B. Gawande, P. Fornasiero and R. Zbořil, ACS Catal., 2020, 10, 2231–2259 CrossRef CAS; (b) Z. Liu, B. Zhang, H. Yu, K. H. Wu and X. Huang, Front. Chem., 2020, 8, 308 CrossRef PubMed.
- (a) K. P. Gopinath, D. V. N. Vo, D. G. Prakash, A. A. Joseph, S. Viswanathan and J. Arun, Environ. Chem. Lett., 2020, 1–26 CAS; (b) D. R. Dreyer and C. W. Bielawski, Chem. Sci., 2011, 2, 1233–1240 RSC; (c) S. S. Shang and G. Shuang, ChemCatChem, 2019, 11, 3730–3744 CrossRef CAS; (d) S. Chenliang and K. P. Loh, Acc. Chem. Res., 2013, 46, 2275–2285 CrossRef PubMed.
- (a) M. Nagai, R. Isoe, K. Ishiguro, H. Tominaga and M. Shimizu, Chem. Eng. J., 2012, 207, 938–942 CrossRef; (b) B. H. Han, D. H. Shin and S. Y. Cho, Tetrahedron Lett., 1985, 26, 6233–6234 CrossRef CAS; (c) H. K. Kadam, S. Khan, R. A. Kunkalkar and S. G. Tilve, Tetrahedron Lett., 2013, 54, 1003–1007 CrossRef CAS; (d) M. Kodomari, Y. Suzuki and K. Yoshida, Chem. Commun., 1997, 1567–1568 RSC; (e) M. R. Acocella, L. D'Urso, M. Maggio and G. Guerra, ChemCatChem, 2016, 8, 1915–1920 CrossRef CAS; (f) K. N. Nandeesh, G. M. Raghavendra, C. N. Revanna, T. A. J. Vijay, K. S. Rangappa and K. Mantelingu, Synth. Commun., 2014, 44, 1103–1110 CrossRef CAS.
- (a) A. Dandia, V. Parewa, A. K. Jain and K. S. Rathore, Green Chem., 2011, 13, 2135–2145 RSC; (b) A. Dandia, V. Parewa, S. Kumari, S. Bansal and A. Sharma, Green Chem., 2016, 18, 2488–2499 RSC; (c) A. Dandia, S. L. Gupta, A. Indora, P. Saini, V. Parewa and K. S. Rathore, Tetrahedron Lett., 2017, 58, 1170–1175 CrossRef CAS; (d) A. Dandia, S. Bansal, R. Sharma, D. K. Mahawar, K. S. Rathore, M. L. Meena and V. Parewa, J. Photochem. Photobiol., A, 2020, 389, 112242 CrossRef CAS; (e) A. Dandia, S. Khan, R. Sharma, S. Parihar and V. Parewa, ChemistrySelect, 2017, 2, 9684–9690 CrossRef CAS; (f) A. Dandia, S. Parihar, R. Sharma, K. S. Rathore and V. Parewa, Nanocatalysis in green organic synthesis, in Green Sustainable Process for Chemical and Environmental Engineering and Science, Elsevier, 2020, pp. 71–103 Search PubMed; (g) A. Dandia, P. Saini, R. Sharma and V. Parewa, Green organic synthesis by photochemical protocol, in Green Sustainable Process for Chemical and Environmental Engineering and Science, Elsevier, 2020, pp. 155–198 Search PubMed; (h) A. Dandia, R. Sharma, A. Indora and V. Parewa, ChemistrySelect, 2018, 3, 8285–8290 CrossRef CAS.
- (a) A. Dandia, S. Parihar, P. Saini, K. S. Rathore and V. Parewa, Catal. Lett., 2019, 149, 3169–3175 CrossRef CAS; (b) A. Dandia, D. K. Mahawar, R. Sharma, R. S. Badgoti, K. S. Rathore and V. Parewa, Appl. Organomet. Chem., 2019, 33, e5232 CrossRef CAS; (c) A. Dandia, S. Bansal, R. Sharma, K. S. Rathore and V. Parewa, RSC Adv., 2018, 8, 30280–30288 RSC; (d) A. Dandia, A. Sharma, A. Indora, K. S. Rathore, A. Jain and V. Parewa, Mol. Catal., 2018, 459, 97–105 CrossRef CAS.
- A. Dandia, A. Sharma, V. Parewa, B. Kumawat, K. S. Rathore and A. Sharma, RSC Adv., 2015, 5, 91888–91902 RSC.
- F. Atamny, J. Blocker, A. Dubotzky, H. Kurt, O. Timpe and G. Loose, Mol. Phys., 1992, 4, 851–886 CrossRef.
- H. Yu, F. Peng, J. Tan, X. W. Hu, H. J. Wang and J. Yang, Angew. Chem., 2011, 123, 4064–4068 CrossRef.
- (a) A. Corma, M. T. Navarro, L. Nemeth and M. Renz, Chem. Commun., 2001, 2190–2191 RSC; (b) J. Rivera-Utrilla and M. Sáchez-Polo, Appl. Catal., B, 2002, 39, 319–329 CrossRef CAS.
- (a) A. S. Mazur, M. A. Vovk and P. M. Tolstoy, Fullerenes, Nanotubes, Carbon Nanostruct., 2020, 28(3), 202–213 CrossRef CAS; (b) M. A. Vieira, G. R. Gonçalves, D. F. Cipriano, M. A. Schettino Jr, E. A. Silva Filho, A. G. Cunha and J. C. Freitas, Carbon, 2016, 98, 496–503 CrossRef CAS.
- H. Li, F. Qin, Z. P. Yang, X. M. Cui, J. F. Wang and L. Z. Zhang, J. Am. Chem. Soc., 2017, 139, 3513–3521 CrossRef CAS PubMed.
- (a) A. Ding, Y. Zhang, Y. Chen, R. Rios, J. Hu and H. Guo, Tetrahedron Lett., 2019, 60, 660–663 CrossRef CAS; (b) Y. L. Ren, X. Z. Tian, C. Dong, S. Zhao, J. Wang, M. Yan, X. Qi and G. Liu, Catal. Commun., 2013, 32, 15–17 CrossRef CAS; (c) R. Gujjarappa, N. Vodnala, A. Garg, C. K. Hazra, S. Gupta and C. C. Malakar, ChemistrySelect, 2020, 5, 2419–2423 CrossRef CAS.
- (a) I. B. Seiple, S. Shun, R. A. Rodriguez, R. Gianatassio, Y. Fujiwara, A. L. Sobel and P. S. Baran, J. Am. Chem. Soc., 2010, 132, 13194–13196 CrossRef CAS PubMed; (b) N. R. Patel and R. A. Flowers, J. Am. Chem. Soc., 2013, 135, 4672–4675 CrossRef CAS PubMed; (c) A. S. Demir, O. Reis and M. Emrullahoglu, J. Org. Chem., 2003, 68, 578–580 CrossRef CAS PubMed; (d) G. Yan, M. Yang and X. Wu, Org. Biomol. Chem., 2013, 11, 7999–8008 RSC.
- (a) G. A. Molander and L. N. Cavalcanti, J. Org. Chem., 2011, 76, 7195–7203 CrossRef CAS PubMed; (b) R. W. Alder, J. Chem. Soc., Chem. Commun., 1980, 24, 1184–1186 RSC; (c) S. Hashimoto, I. Kurimoto, Y. Fujii and R. Noyori, J. Am. Chem. Soc., 1985, 107, 1427–1429 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01940f |
| This journal is © The Royal Society of Chemistry 2021 |