DOI:
10.1039/D0RA09737C
(Paper)
RSC Adv., 2021,
11, 18026-18039
Eco-friendly synthesis of chromeno[4,3-b]chromenes with a new photosensitized WO3/ZnO@NH2-EY nanocatalyst†
Received
16th November 2020
, Accepted 29th April 2021
First published on 18th May 2021
Abstract
A new heterogeneous photoredox nanocatalyst WO3/ZnO@NH2-EY (EY: eosin Y) was fabricated and characterized employing some instrumental techniques such as XRD, FT-IR, ICP, TGA, and SEM. The photocatalytic efficiency of the prepared material was investigated in the preparation of various chromeno[4,3-b]chromenes via a simple and practical method. The chromene derivatives were prepared through the condensation of aromatic aldehydes, dimedone, and coumarin under an open-air atmosphere in the presence of a green LED under solventless conditions. The significant advantages of this new method include low reaction time, easy work-up, cost-effective, wide substrate scope, excellent yield, and complete atom economy of the final products. Moreover, the prepared photocatalyst could be frequently recovered up to four times with only a little decrease in the catalytic activity. Furthermore, the progress of the condensation reaction is demonstrated to occur via a radical mechanism, which shows that reactive species such as ˙O2− and OH˙ together with h+ would be involved in the photocatalytic process. Stability and reusability studies also warranty good reproducibility of the nanocatalyst for at least 4 runs. Eventually, a hot filtration test ensured that the nanohybrid catalyst is stable in the reaction medium and its catalytic activity originates from the whole undecomposed conjugated composite.
1 Introduction
Multi-component reactions have been known to be powerful tools over conventional multi-step reactions and have emerged as a novel promotion in organic synthesis.1,2 These reactions offer a direct fast route and enable the assembly of highly complicated and diversified molecules in a one-pot single-step process with enhanced atom economy.3–7 Chromenes are crucial oxygenated compounds with different biological activities such as antihypertensive,8 antioxidative,9 antitumor,10 antiviral,11 antibacterial,12 antileishmanial,13 anti-tubulin,14 and anticoagulant,15 and enable to turn on potassium channels to inhibit dihydrofolate reductase and phosphodiesterase.16 In the area of anti-cancer chemotherapy, 4H-chromenes have been known as a new class of molecules that bind to Bcl-2 anti-apoptotic proteins and promote the apoptotic processes in the cancerous cells.17–20 Therefore, novel chromenes should be further progressed as potential therapeutic species against cancer cell lines such as glioma, liver, melanoma, and prostate.19 In addition, the utilization of chromenes is well known in industrial areas such as pigments, biodegradable agrochemicals, cosmetics, optical brightener, fluorescence markers, and laser dyes.21–23
Considering the emerging properties of chromene derivatives, the progress of advanced, clean, and uncomplicated methodologies for the efficient catalytic synthesis of these compounds with accessible reagents is of great importance. Some routes have been reported for the synthesis of chromeno[4,3-b]chromenes by the reaction of cyclic-1,3-dicarbonyl compounds, arylaldehydes, and 4-hydroxycoumarine in the presence of SnCl2·2H2O,24 ZnO nanoparticles,25 Mg(ClO4)2,26 molybdic acid-magnetic nanoparticles,27 p-toluene sulfonic acid,28 nano-CuFe2O4@SO3H,29 I2/HOAc,30 heteropolyacids,31 and Zn (L-proline)2.32
Although most of the reported routes have some benefits, however, some disadvantages are also combined with many of them using environmentally toxic organic solvents, expensive mediators, long reaction time, corrosive nature, tedious work-up, non-recyclable catalysts, limited substrate scope, high temperature, and low yields. Therefore, the development of effective and environmental benign methods is desirable for the synthesis of chromene compounds.
Sunlight is an accessible, plentiful, and clean energy source for chemical reactions. Photochemical reactions using visible light are attractive areas in organic synthesis, which provide ubiquitous, non-toxic, eco-friendly, sustainable, inexpensive, and a universally available energy source.33 However, due to the increasing attention on the usage of green platforms in organic synthesis, scholars are trying to redesign the synthetic methods by means of visible light.34–37 Visible light involves a major part of the arriving solar radiation, which can be used to drive various photochemical transformations. Since most organic compounds do not absorb visible light, the application of photochemical reactions has been restricted. Therefore, the development of new and efficient visible light photocatalysts towards chemical transformations is a necessity. Photoredox catalysts absorb visible light to provide the required energy to initiate a photochemical reaction. Among metal-free organic dyes, EY has been used as an economic and environment-friendly alternative to many transition metal photoredox catalysts.
The photochemistry of EY has been well examined via excitation by visible light and this molecule goes through a fast intersystem crossing to the lowest energy states.38 EY can easily absorb green light; therefore, various applications have emerged for this organic photosensitizer in visible light-promoted organic synthesis.39–41 However, because EY has harmful effects on the environment, a solution is required. One of the most efficient ways to solve this problem is to prepare a heterogeneous catalyst from this material using suitable catalytic substrates. There is an example of the heterogenization of EY on TiO2 and its application in the degradation of diclofenac via the solar activation of titanium dioxide.42 In addition, a new metal–organic framework photocatalyst modified by EY “EY@UiO-66-NH2” has been synthesized and used in the C–H activation of tertiary amines.43 Also, the reduction of 4-nitrophenol has been achieved by handling solar irradiation under green conditions by utilizing an effective polymer-supported EY photocatalyst.44 Further transformations involving oxidation of thioethers and phenylboronic acids45 as well as C–C and C–P coupling reactions46 were also performed by EY-supported photocatalysts.
As far as we know, semiconducting metal oxides have a very crucial impact in a wide domain of chemical industries such as lithium ion batteries,47 sensors,48 optoelectronics,49 and photocatalysts.50 Among various types of these semiconductors, zinc oxide and tungsten(VI) oxide are very appropriate to be applied as photocatalysts because of their proper bandgap (3.2–3.35 eV).51 However, semiconductor composites can improve the electron transitions between the (e−)/(h+) pairs in the large energy gaps and can be regarded as effectual photocatalysts.52 To date, ZnO-WO3,53 CuO/WO3/TiO2,54 ZnO–TiO2,55 and ZnO/CeO2 (ref. 56) are prepared and used as effective photocatalysts in a number of routine transformations under visible light irradiation.
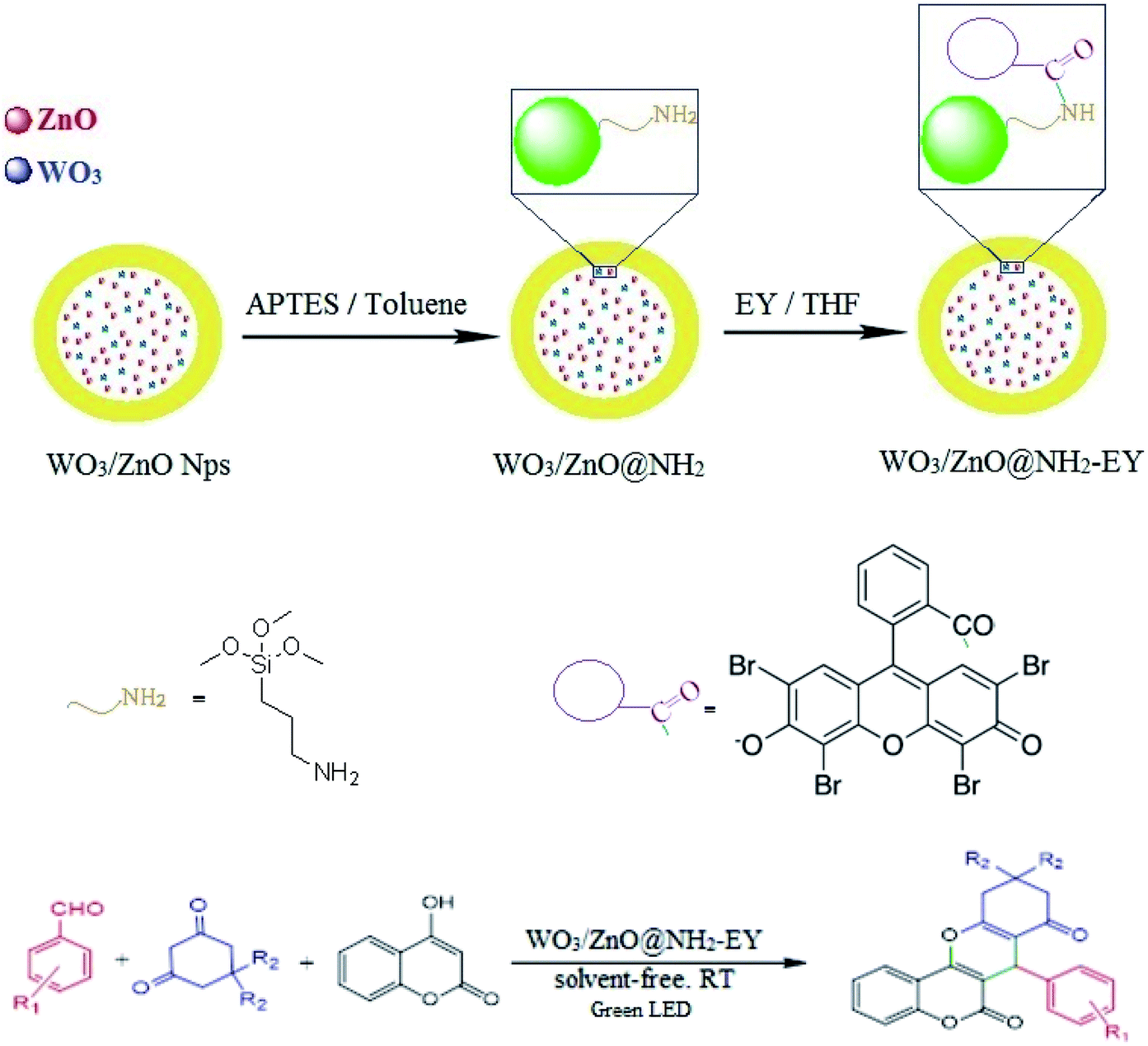
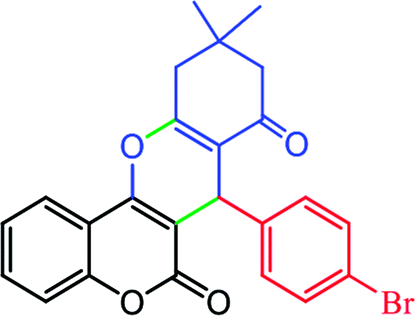
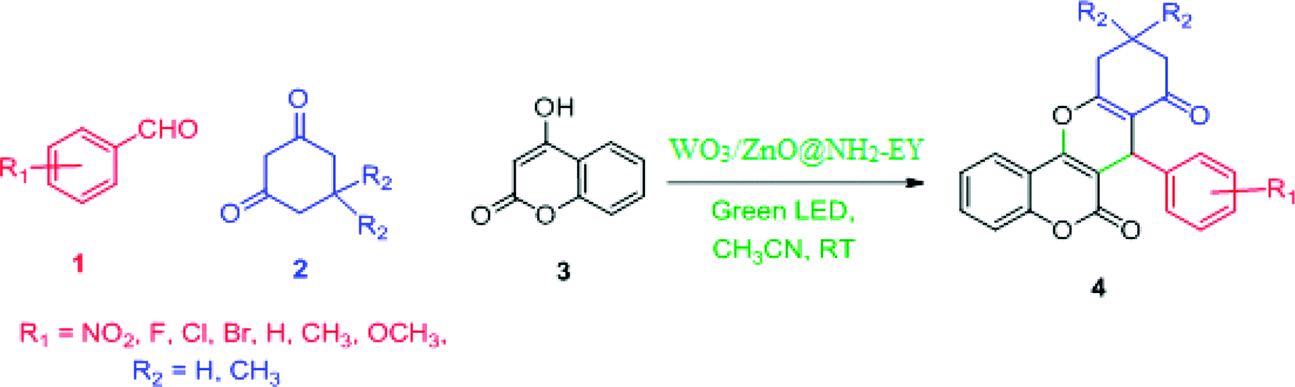
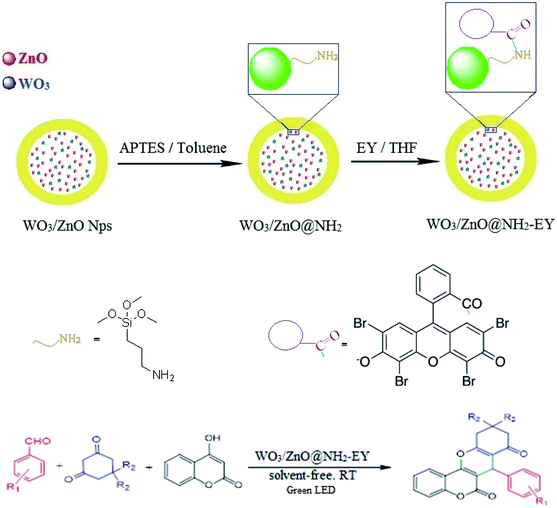
In the extension of our previous studies on the development of green synthetic protocols to achieve important heterocyclic compounds57–59 and to recognize the extensive applications of chromene derivatives, herein, we report a simple and convenient new method to synthesize different substituted chromeno[4,3-b]chromene derivatives by means of a green LED through the multicomponent condensation of aryl aldehydes, 1,3-cyclohexanedione, and 4-hydroxylcoumarin, using an EY-supported nanocomposite (WO3/ZnO) as a green heterogeneous nanocatalyst under solventless conditions (Scheme 1).
 |
| | Scheme 1 A general route for the preparation of different substituted chromeno[4,3-b]chromenes. | |
The interaction of EY with the solid support material can occur via three routes including hydrophobic, hydrogen bond, and Lewis acid–base interactions. The hydrophobic interaction refers to the adsorption affinity of EY via its non-hydrophobic functional groups. This interaction is not too strong, however, it should be considered. Hydrogen bonding also widely happens in the adsorption of this polar compound to the support material through the lone-pair hydrogen bonding donors on EY with the acceptor sites on the support. Finally, Lewis acid–base interaction occurs via the carboxyl group of EY with the basic sites such as the pendant amino group of the alkyl chain. Therefore, all of the above interactions could be regarded as electronic interactions between EY and WO3/ZnO@NH2.60–63 However, precise adsorption isotherm experiments are necessary to elucidate the exact mechanism of adsorption.
2 Experimental section
2.1 Materials and methods
All chemicals including zinc chloride, sodium hydroxide, zinc acetate, sodium tungstate, EY, benzaldehyde, dimedone, 4-hydroxycoumarin, (3-aminopropyl)triethoxysilane (APTES), and solvents were purchased from Sigma-Aldrich, Merck, and Fluka, and utilized as received without further purification. The morphology of the synthesized nanophotocatalyst was studied by a Mira 3-XMU field emission scanning electron microscope (FE-SEM). 1H- and 13C-NMR spectra were recorded on a 300 MHz Bruker AVANCEC III spectrometer using TMS as the internal reference. Melting points were attained on a Stuart BI Branstead Electrothermal IA9200 apparatus. Fourier transform infrared (FT-IR) spectra were recorded on a Shimadzu 8700 Fourier transform spectrometer in the range of 400–4000 cm−1 with KBr pellets. UV-visible spectra were obtained using a Photonix UV-visible array spectrophotometer. X-ray diffraction patterns (XRD) were attained on an Xpert MPD diffractometer with Cu Kα radiation at 30 mA and 40 keV and a scanning rate of 3° min−1 in the 2θ domain from 5° to 80°. Thermogravimetric analysis was carried out with a TGA 92 Setaram at 10 °C min−1. The chemical composition of the catalyst was determined by an inductively coupled plasma spectrometer (ICP-MS; model VARIAN VISTA-PRO).
2.2 Preparation of WO3/ZnO nanoparticles
About 30 mL NaOH (4 M) was added drop-wise into a 20 mL mixture of zinc acetate (1 M) and 2 mL of Na2WO4·2H2O (0.5 M). Then, the produced suspension remained at room temperature under mild stirring for 3 h to generate the desired precursor. Thereafter, the obtained suspension was subsequently transferred to a 250 mL ground-glass stopped conical flask and aged at 95 °C for 10 h. Finally, the deposited precipitate at the bottom of the conical flask was gathered, completely washed with water, and air-dried under ambient conditions.
2.3 Surface modification of WO3/ZnO via the anchoring of APTES
800 mg WO3/ZnO was dispersed in an anhydrous solution of toluene (60 mL). To this suspension, 1.6 mL APTES was added and the obtained mixture was refluxed for 24 h. Finally, the attained precipitate was filtered and dried in an oven at 60 °C for 12 h.
2.4 Surface modification of WO3/ZnO@NH2 with EY
WO3/ZnO@NH2-EY was supplied through a well-known wet-impregnation method. According to this procedure, 20 × 10−3 mL EY was dissolved in THF. Then, 0.5 g of WO3/ZnO@NH2 was gradually added to this solution for 12 h in a dark atmosphere. Eventually, the resulting precipitate was dried at 80 °C under air for 24 h.
2.5 A general procedure for the synthesis of substituted chromeno[4,3-b]chromene
Benzaldehyde (100 mg, 1 mmol), dimedone (140 mg, 1 mmol), 4-hydroxycoumarin (162 mg, 1 mmol), and the desired amount of WO3/ZnO@NH2-EY (30 mg) were successively added to a test tube equipped with a magnet. Then, the reaction mixture was irradiated with a green light-emitting diode (λmax = 535 nm) at room temperature in an open-air atmosphere and completion of the reaction was monitored by TLC (n-hexane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate, 3:1). After completion of the reaction, the mixture was quenched with H2O (3 mL) and the desired chromene product was dissolved in EtOAc. Then, the EtOAc layer was removed and the aqueous fraction was again extracted with EtOAc (3 × 3 mL). Finally, the attained organic layer was dried over sodium sulfate and concentrated in vacuo. The crude accomplished product was purified through column chromatography (100–200 mesh silica gel; EtOAc/hexane) to afford the pure chromene.
:ethyl acetate, 3:1). After completion of the reaction, the mixture was quenched with H2O (3 mL) and the desired chromene product was dissolved in EtOAc. Then, the EtOAc layer was removed and the aqueous fraction was again extracted with EtOAc (3 × 3 mL). Finally, the attained organic layer was dried over sodium sulfate and concentrated in vacuo. The crude accomplished product was purified through column chromatography (100–200 mesh silica gel; EtOAc/hexane) to afford the pure chromene.
2.6 Spectral data
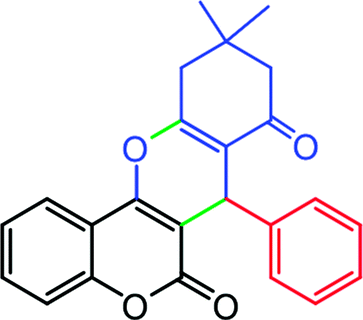
2.6.1 10,10-Dimethyl-7-phenyl-10,11-dihydrochromeno[4,3-b]chromene-6,8(7H,9H)-dione (Table 4, entry 1). 1H NMR (300 MHz, DMSO-d6); δ: 7.83 (1H, d, J = 7.8 Hz), 7.53 (1H, d, J = 7.2 Hz), 7.05–7.35 (7H, m), 4.69 (1H, s), 2.66 (2H, s), 2.24 (1H, d, J = 15.9 Hz), 2.12 (1H, d, J = 16.2 Hz), 1.07 (3H, s), 0.97 (3H, s) ppm; 13C NMR (75 MHz, DMSO-d6); δ: 195.8, 162.6, 160.2, 154.0, 152.4, 143.0, 132.7, 128.7, 128.3, 127.0, 124.7, 122.8, 116.5, 114.5, 113.5, 106.3, 50.6, 33.4, 32.3, 29.0, 27.3 ppm.
2.6.2 10,10-Dimethyl-7-(4-nitrophenyl)-10,11-dihydrochromeno[4,3-b]chromene-6,8(7H,9H)-dione (Table 4, entry 2). 1H NMR (300 MHz, DMSO-d6); δ: 8.00 (2H, d, J = 8.7 Hz), 7.89 (1H, s), 7.84 (1H, d, J = 7.8 Hz), 7.55 (1H, d, J = 7.2 Hz), 7.47 (1H, d, J = 8.7 Hz), 7.34 (1H, t, J = 7.5 Hz), 7.26 (1H, d, J = 8.1 Hz), 4.82 (1H, s), 2.68 (2H, s), 2.24 (1H, d, J = 16.2 Hz), 2.11 (1H, d, J = 16.2 Hz), 1.07 (3H, s), 0.96 (3H, s) ppm; 13C NMR (75 MHz, MSO-d6); δ: 195.8, 163.0, 160.2, 154.5, 152.6, 150.2, 146.7, 133.0, 129.9, 124.8, 123.4, 122.9, 116.8, 13.6, 113.3, 105.0, 50.5, 33.8, 32.3, 28.9, 27.4 ppm.
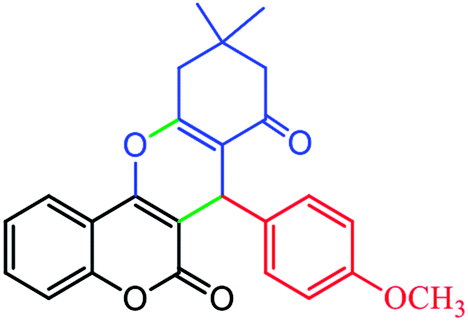
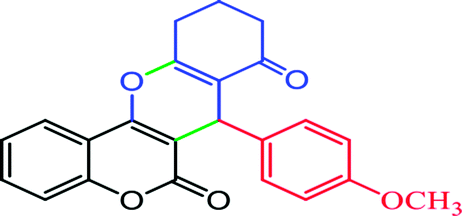
2.6.3 10,10-Dimethyl-7-(4-methoxyphenyl)-10,11-dihydrochromeno[4,3-b]chromene-6,8(7H,9H)-dione (Table 4, entry 4). 1H NMR (300 MHz, DMSO-d6); δ: 7.82–7.85 (1H, m), 7.52–7.57 (1H, m), 7.24–7.35 (2H, m), 7.14 (2H, d, J = 9.0 Hz), 7.04 (1H, d, J = 9.0 Hz), 6.62–6.70 (1H, m), 4.66 (1H, s), 3.63 (3H, s), 2.66 (2H, s), 2.16 (1H, d, J = 16.2 Hz), 2.04 (1H, d, J = 16.2 Hz), 1.08 (3H, s), 0.95 (3H, s) ppm; 13C NMR (75 MHz, DMSO-d6); δ: 195.9, 161.5, 160.5, 158.5, 153.6, 152.3, 135.1, 132.5, 129.6, 129.3, 124.6, 122.7, 116.6, 115.6, 113.6, 113.3, 106.3, 55.1, 50.7, 32.3, 32.1, 29.1, 27.3 ppm.
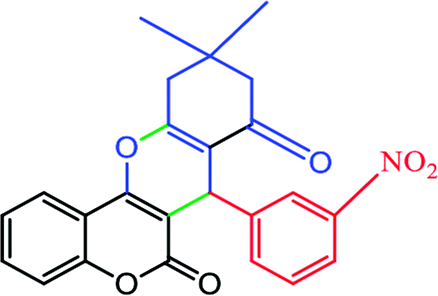
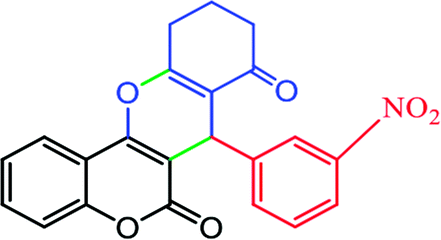
2.6.4 10,10-Dimethyl-7-(3-nitrophenyl)-10,11-dihydrochromeno[4,3-b]chromene-6,8(7H,9H)-dione (Table 4, entry 5). 1H NMR (300 MHz, DMSO-d6); δ: 8.01 (1H, s), 7.87 (1H, d, J = 8.1 Hz), 7.78 (1H, d, J = 7.8 Hz), 7.59 (1H, d, J = 6.6 Hz), 7.49 (1H, t, J = 7.5 Hz), 7.24–7.35 (2H, m), 7.18 (1H, d, J = 8.1 Hz), 4.81 (1H, s), 2.66 (1H, d, J = 18 Hz), 2.58 (1H, d, J = 18 Hz), 2.19 (1H, d, J = 16.5 Hz), 2.08 (1H, d, J = 16.5 Hz), 1.03 (3H, s), 0.93 (3H, s) ppm; 13C NMR (75 MHz, DMSO-d6); δ: 195.6, 163.0, 159.8, 154.1, 152.1, 147.5, 144.7, 135.0, 132.8, 129.4, 124.6, 123.1, 122.7, 121.7, 116.5, 113.0, 104.6, 49.9, 38.8, 33.2, 31.9, 28.4, 26.8 ppm.
2.6.5 7-(3-Nitrophenyl)-10,11-dihydrochromeno[4,3-b]chromene-6,8(7H,9H)-dione (Table 4, entry 6). 1H NMR (300 MHz, DMSO-d6); δ: 7.99–8.02 (2H, m), 7.87–7.90 (2H, m), 7.54–7.60 (1H, m), 7.23–7.45 (3H, m), 5.04 (1H, s), 2.77–2.95 (2H, m), 2.39–2.44 (2H, m), 2.04–2.17 (2H, m), ppm; 13C NMR (75 MHz, DMSO-d6); δ: 195.9, 164.3, 152.7, 144.7, 136.0, 132.7, 129.0, 124.5, 122.8, 122.6, 122.2, 117.0, 115.3, 113.3, 105.4, 36.7, 33.5, 27.1, 20.1 ppm.
2.6.6 7-(4-Methoxyphenyl)-10,11-dihydrochromeno[4,3-b]chromene-6,8(7H,9H)-dione (Table 4, entry 7). 1H NMR (300 MHz, DMSO-d6); δ: 8.04 (1H, d, J = 7.8 Hz), 7.73 (1H, t, J = 7.5 Hz), 7.43–7.54 (2H, m), 7.33 (2H, d, J = 8.4 Hz), 6.87 (2H, d, J = 7.8 Hz), 3.82 (3H, s), 4.87 (1H, s), 2.88–3.06 (2H, m), 2.50 (2H, t, J = 6.3 Hz), 2.15–2.27 (2H, m) ppm; 13C NMR (75 MHz, DMSO-d6); δ: 195.5, 163.3, 160.2, 159.3, 153.4, 152.5, 135.3, 132.6, 129.6, 124.7, 122.8, 120.8, 116.7, 116.1, 113.7, 113.3, 111.7, 106.5, 55.2, 36.9, 32.4, 26.9, 20.2 ppm.
3 Results and discussion
3.1 Characterization and physicochemical properties of WO3/ZnO@NH2-EY nanoparticles
The morphology and physicochemical properties of the prepared WO3/ZnO@NH2-EY nanocatalyst were investigated by means of FT-IR, SEM, ICP, UV-Vis, and XRD. FT-IR and UV-Vis techniques are the most informative techniques to examine the surface interaction of EY with the surface functional groups of WO3/ZnO@NH2.
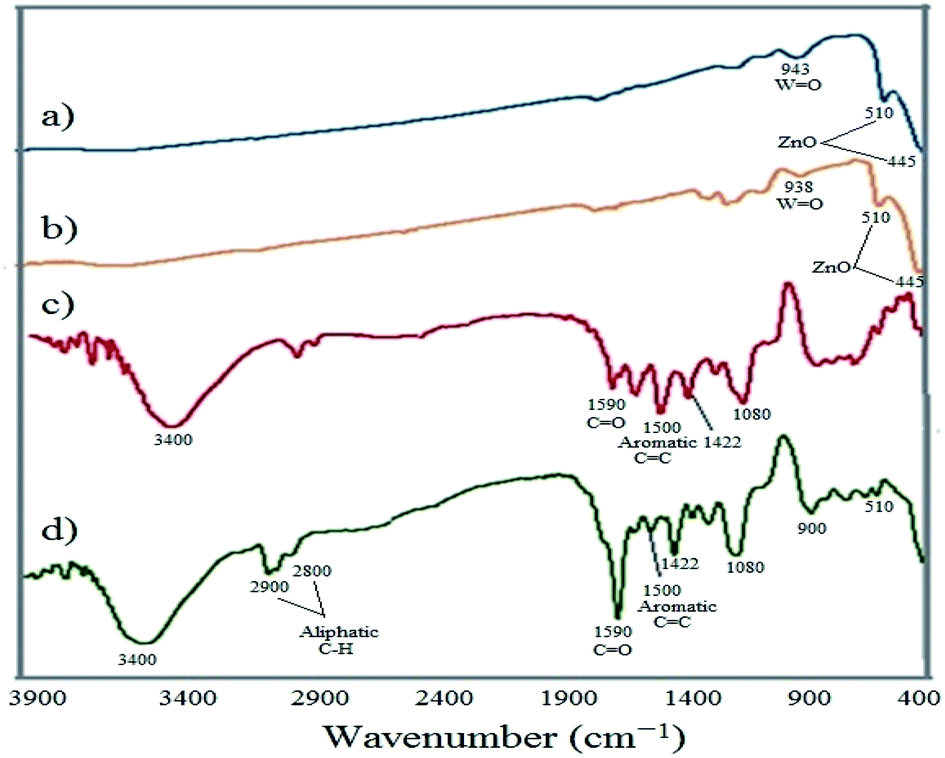
3.1.1 FT-IR spectroscopy. The FT-IR spectra of the fabricated samples in the range of 400–4000 cm−1 are shown in Fig. 1. To ensure the binding of EY on the surface of WO3/ZnO@NH2-EY, the FT-IR of all the fragments including WO3/ZnO (a), WO3/ZnO@NH2 (b), EY (c), and WO3/ZnO@NH2-EY (d) were studied. This study proved the anchoring of EY onto the surface of WO3/ZnO@NH2-EY. The typical absorption bands in the region of 400–600 cm−1 are due to ZnO species. The FT-IR of WO3/ZnO and WO3/ZnO@NH2 revealed a strong absorption band at ∼445 cm−1 with a shoulder at ∼510 cm−1 corresponding to the Zn–O stretching vibrations. Moreover, the weak bands at 943 and 948 cm−1 are related to W![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in the octahedral WO3.64,65 The grafting of the amine group on the surface of WO3/ZnO can be approved by the observation of the corresponding aliphatic bands in 2900 and 3400 cm−1.66 The appearance of the EY characteristic bands in the FT-IR of WO3/ZnO@NH2-EY proved that the structure of EY is unchanged after immobilization. The FT-IR spectrum of WO3/ZnO@NH2-EY was also compared to that of the parent EY. The observation of the absorption bands at 1590 and 1500 cm−1 due to the carboxyl functional group and the CC of aromatic phenyl rings, respectively, unambiguously confirmed the loading of EY onto the surface of the WO3/ZnO@NH2 framework.67,68 However, there were no detectable peaks due to the WO3 species and it seems that a little surface segregated WO3 had no apparent effects on the infrared spectrum of ZnO nanoparticles.
O bonds in the octahedral WO3.64,65 The grafting of the amine group on the surface of WO3/ZnO can be approved by the observation of the corresponding aliphatic bands in 2900 and 3400 cm−1.66 The appearance of the EY characteristic bands in the FT-IR of WO3/ZnO@NH2-EY proved that the structure of EY is unchanged after immobilization. The FT-IR spectrum of WO3/ZnO@NH2-EY was also compared to that of the parent EY. The observation of the absorption bands at 1590 and 1500 cm−1 due to the carboxyl functional group and the CC of aromatic phenyl rings, respectively, unambiguously confirmed the loading of EY onto the surface of the WO3/ZnO@NH2 framework.67,68 However, there were no detectable peaks due to the WO3 species and it seems that a little surface segregated WO3 had no apparent effects on the infrared spectrum of ZnO nanoparticles.
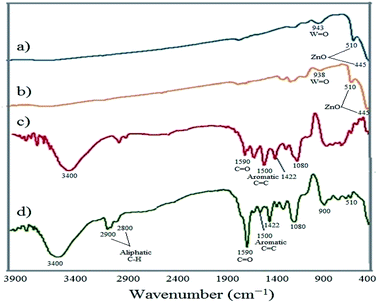 |
| | Fig. 1 FT-IR spectra of (a) WO3/ZnO, (b) WO3/ZnO@NH2, (c) EY, and (d) WO3/ZnO@NH2-EY. | |
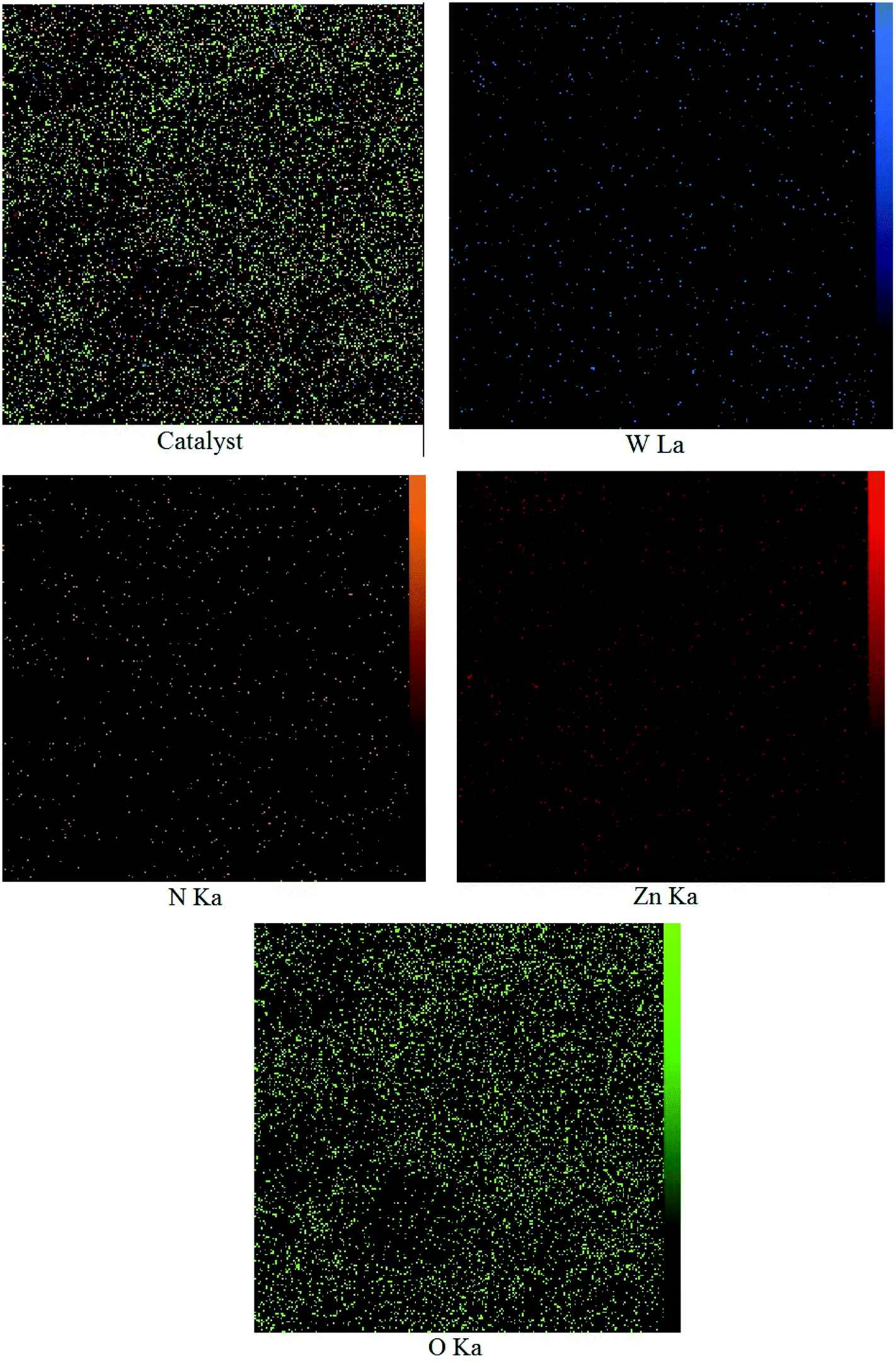
3.1.2 FESEM of WO3/ZnO@NH2-EY. The surface morphology of WO3/ZnO@NH2-EY was further studied by FE-SEM (Fig. 2). From this figure, it can be concluded that the prepared nanocatalyst has an approximately nanosheet framework. Moreover, the diameter of these nanosheets was about 25–35 nm. In addition, the FE-SEM images of WO3/ZnO@NH2-EY with 5 mm particle size were achieved to investigate the distribution of tungsten and zinc (Fig. 3).69 As can be seen from these pictures, the presence and steady distribution of these elements in the WO3/ZnO@NH2-EY nanocatalyst can be easily proven.
 |
| | Fig. 2 The FE-SEM images of WO3/ZnO@NH2-EY. | |
 |
| | Fig. 3 The FE-SEM map pictures of WO3/ZnO@NH2-EY. | |
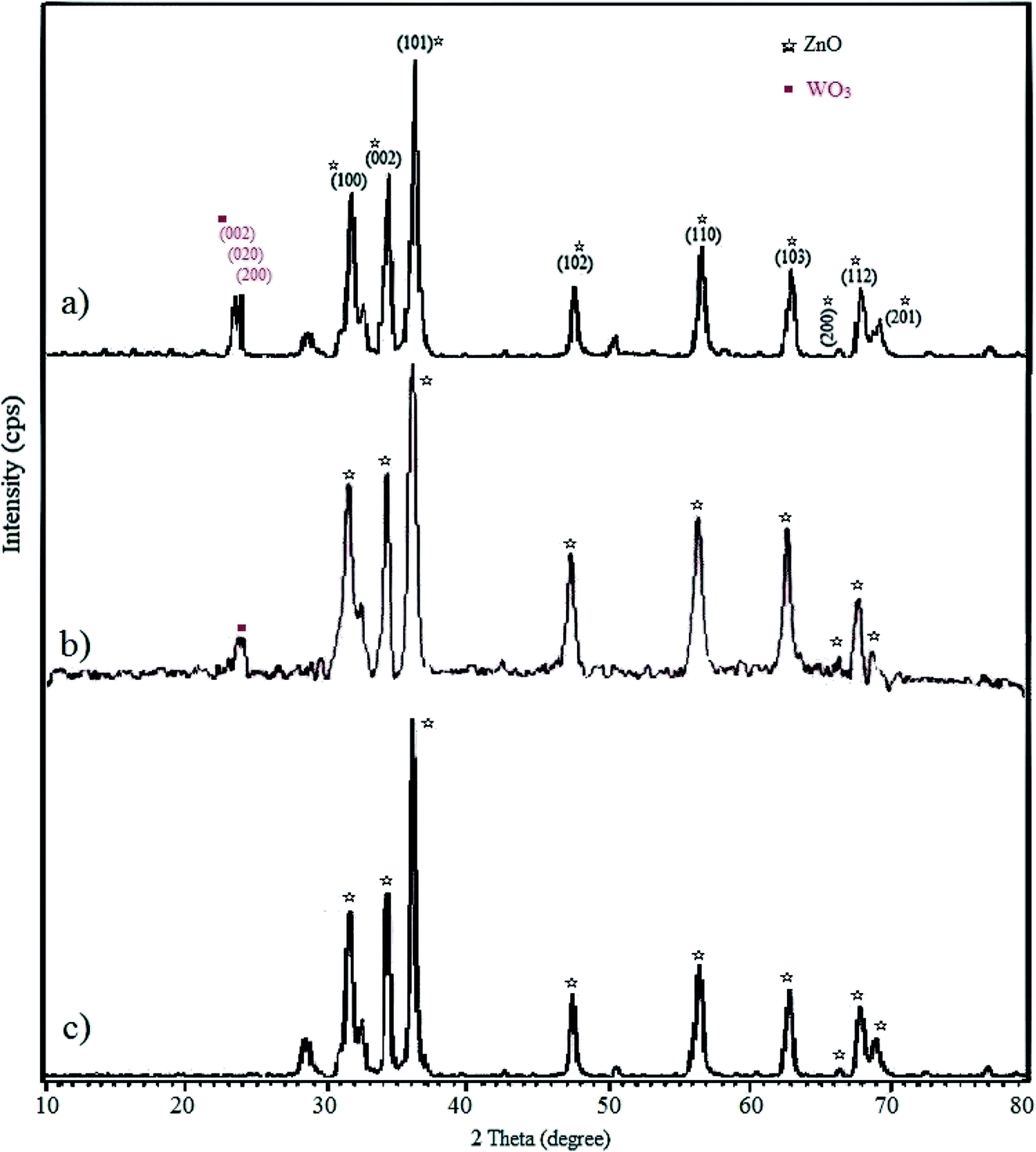
3.1.3 XRD. The crystallinity of the fabricated WO3/ZnO, WO3/ZnO@NH2, and WO3/ZnO@NH2-EY was explored through wide-angle XRD (Fig. 4). In the XRD pattern of WO3/ZnO, obvious diffraction peaks at the 2θ of 23.1°, 23.7°, and 24.2° corresponded to the (002), (020), and (200) crystal planes of WO3, respectively, which appeared as broad peaks.70 However, there were no detectable peaks related to WO3 in the XRD pattern of WO3/ZnO@NH2-EY due to its low amount or amorphous nature on the surface of ZnO. The diffraction patterns of the synthesized WO3/ZnO@NH2-EY showed that the material is a crystalline material and major parts of the XRD pattern were fitted well with the standard data of the ZnO structure. Low-intensity peaks at the 2θ of 31.6, 34.4, 36.3, 47.6, 56.7, 62.9, 66.3, 67.8, 69.1, 72.6, and 76.9° corresponded to the (100), (002), (101), (102), (110), (103), (200), (112), (201), (004), and (202) planes, respectively. These results confirmed that WO3/ZnO@NH2-EY has a very close topology to ZnO.68 Moreover, the half-value crystallite scale can be calculated using the XRD data in the Debye–Scherrer formula; thus, the half-value crystallite scale of WO3/ZnO@NH2-EY was attained as ∼31 nm, which is in accordance with the FESEM results.
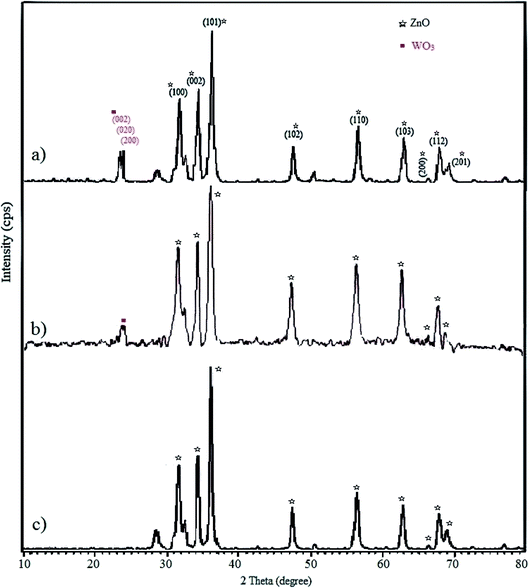 |
| | Fig. 4 Wide-angle XRD pattern of (a) WO3/ZnO, (b) WO3/ZnO@NH2, and (c) WO3/ZnO@NH2-EY. | |
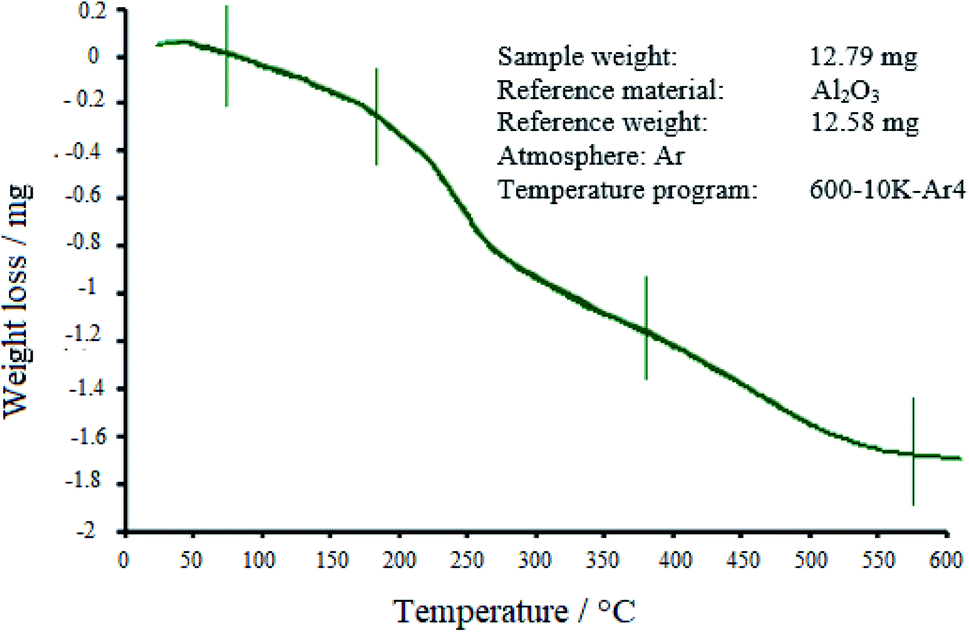
3.1.4 TGA. The thermal stability of the WO3/ZnO@NH2-EY nanophotocatalyst was explored by TGA (Fig. 5). It seems that a little weight-gain at the temperature of about 40 °C would be due to the differences in the thermal conductivity and the heat capacity of the purging gas.71 Following the graph, three stages of decomposition can be seen. The first gradual weight-loss of about 1.79% was observed below 180 °C, which is most likely due to water desorption from the surface of nanoparticles and loss of moisture content.72 Then, the EY moiety started to be destroyed at about 200–380 °C with an average weight loss of about 6.64%. Further weight loss (4.37%) at higher temperatures would probably be due to the burning of alkyl-chain remnants, which was accompanied by the crystallization of ZnO and WO3 up to 500 °C.
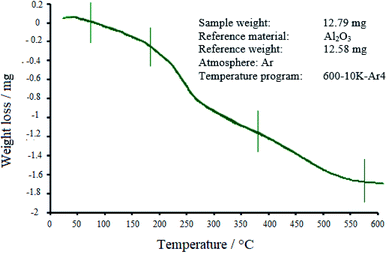 |
| | Fig. 5 TGA profile for WO3/ZnO@NH2-EY. | |
3.1.5 UV-vis and ICP. The amount of EY loaded onto WO3/ZnO@NH2 was monitored by UV-vis spectroscopy of the solution involving EY during the immobilization process. To achieve the degree of loaded EY, the UV-vis spectra of the solutions of WO3/ZnO@NH2 were attained before and after the addition of EY. Thus, 0.5 g of WO3/ZnO@NH2 was suspended in 5 mL THF solution having 100 mg of EY. Then, the heterogeneous suspension was stirred for 7 h (Fig. 6). Obviously, most of the EY content was adsorbed on the surface of WO3/ZnO@NH2 after 5 h. However, the amount of EY loaded onto to the surface of the solid material was precisely detected by ICP and the weight% of 15 was attained. It means that ∼0.21 mmol of EY was grafted per gram of the WO3/ZnO@NH2 photocatalyst. Thus, most of the vanished EY (as documented by the UV-vis study) was physisorbed onto the surface of the photocatalyst.
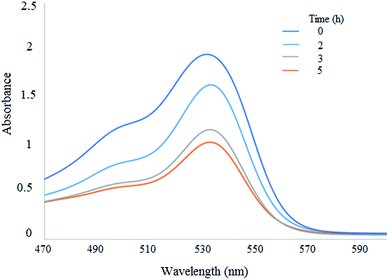 |
| | Fig. 6 UV-vis spectral changes under the loading of EY on WO3/ZnO@NH2. | |
3.2 Catalytic tests
To achieve the best reaction conditions, we performed a model condensation reaction using benzaldehyde (1 mmol), dimedone (1 mmol), and 4-hydroxycoumarin (1 mmol) in the presence of WO3/ZnO@NH2-EY (0.03 g) under solventless conditions. Then, the reaction mixture was irradiated with a green light-emitting diode (λmax, 535 nm) at room temperature in an open-air container. Thereafter, the desired chromeno[4,3-b]chromene was formed in good yield after 45 min (Table 1). This study demonstrated that if we remove any of the aforementioned requirements such as EY, WO3/ZnO@NH2-EY, and LED, the reaction yield will be significantly diminished.
Table 1 Optimization of the reaction conditions for the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarina
| Entry |
Light |
Light source |
WO3/ZnO@NH2-EY (g) |
Time (min) |
Yield (%) |
| Reaction conditions: dimedone (1.0 mmol), benzaldehyde (1.0 mmol), and 4-hydroxycoumarin (1.0 mmol) under solvent-free conditions in an open-air atmosphere at room temperature. |
| 1 |
— |
— |
— |
45 |
4 |
| 2 |
Green LED |
25 W, λ = 535 nm |
— |
45 |
9 |
| 3 |
Dark |
— |
0.03 |
45 |
43 |
| 4 |
White LED |
20 × 1 W |
0.03 |
45 |
55 |
| 5 |
Green LED |
25 W, λ = 535 nm |
0.03 |
45 |
97 |
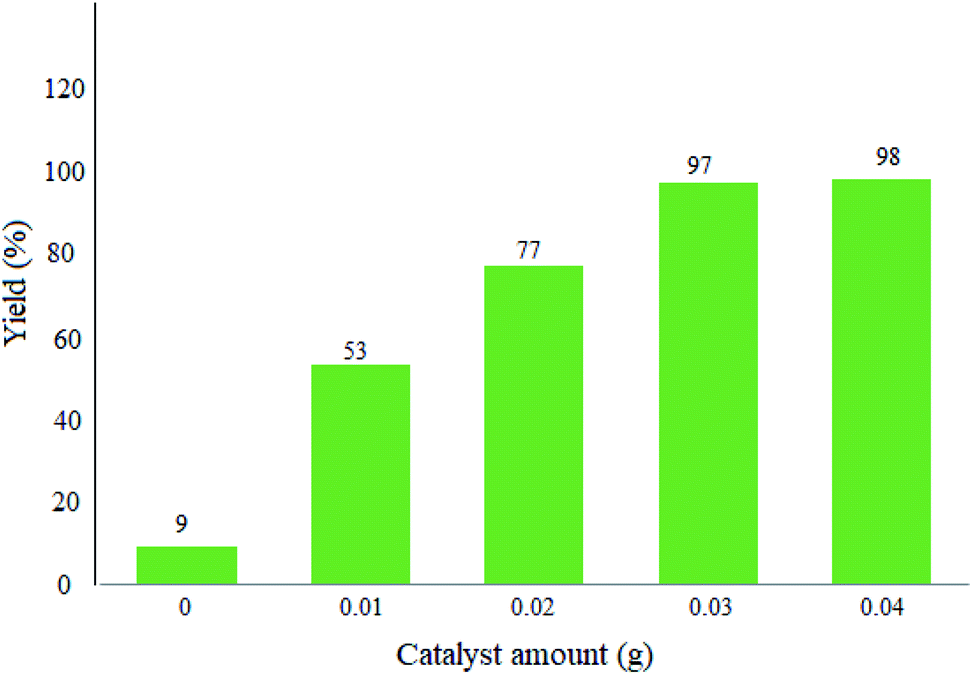
3.2.1 Studying the effect of the catalyst amount. Herein, the reactions were performed with different amounts of the catalyst in order to optimize the exact quantity of WO3/ZnO@NH2-EY. As seen in Fig. 7, 0.03 g of WO3/ZnO@NH2-EY was appropriate and gave 97% yield under the selected reaction conditions. However, a further increment of the catalyst amount affected neither the yield% nor the reaction time, while a lower yield of the desired product was attained on decreasing the catalyst amount (Fig. 7).
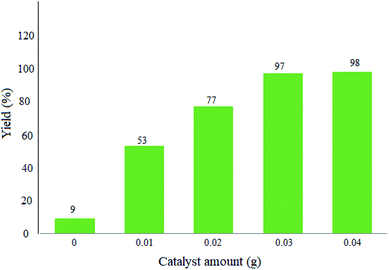 |
| | Fig. 7 Effect of the catalyst amount on the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarin under the selected reaction conditions below Table 1 in the presence of a green LED (25 W, λ = 535 nm) after 45 min. | |
3.2.2 Effect of different solvents. In this step, we shifted our focus on the impact of the solvent in the reaction progress (Table 2). Thus, the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarin under optimum conditions was performed with some selected solvents such as CH3CN, THF, toluene, and ethanol. According to the obtained data in Table 2, the solvent-free condition was the best choice between the tried solvents and provided the maximum yield of 97% after 45 min.
Table 2 Optimization of the solvent on the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarin using a green LED (25 W)a
| Entry |
WO3/ZnO@NH2-EY (g) |
Solvent |
Time (min) |
Yield (%) |
| Reaction conditions are described below Table 1. 3 mL solvent was used. |
| 1 |
0.03 |
— |
45 |
97 |
| 2 |
0.03 |
H2O |
180 |
45 |
| 3 |
0.03 |
Toluene |
180 |
62 |
| 4 |
0.03 |
Ethanol |
180 |
77 |
| 5 |
0.03 |
CH3CN |
180 |
95 |
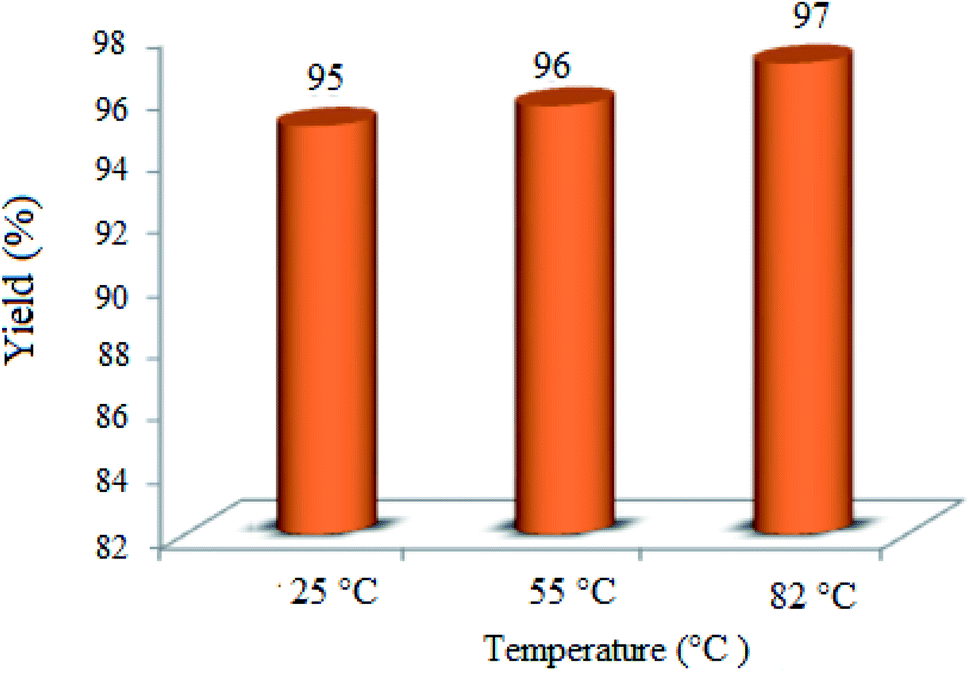
3.2.3 Effect of the reaction temperature. The impact of the reaction temperature was speculated in the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarin to achieve the best reaction conditions (Fig. 8). Interestingly, the yield% was only a little affected by the reaction temperature and the best result was attained at RT (∼25 °C) after 3 h. Therefore, the temperature of 25 °C was the optimum temperature for all the runs.
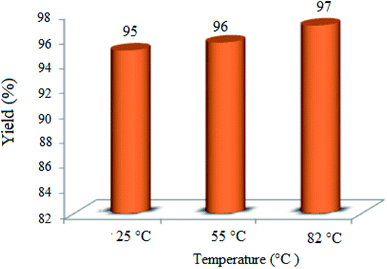 |
| | Fig. 8 Effect of the temperature on the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarin in the presence of 0.03 g of WO3/ZnO@NH2-EY, green LED, and CH3CN (3 mL). The reaction time was selected as 3 h. | |
3.2.4 Effect of the reaction time. The effect of the reaction time was investigated to explore the minimum time needed to acquire the best yield. As Fig. 9 shows, 45 min was adequate to reach the maximum yield of 97%. Noteworthily, a little decrease in the yield% was attained by extending the reaction time, which would be due to the poisoning of the catalyst surface. The above experiments declared that the best condition can be solvent-free under an open air atmosphere at room temperature in the presence of 0.03 g of the WO3/ZnO@NH2-EY nanocatalyst.
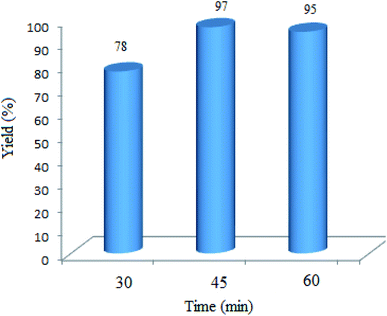 |
| | Fig. 9 Effect of the reaction time on the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarin. Green LED (25 W) was used under solvent-free conditions in an open atmosphere at room temperature. | |
3.2.5 Effect of the light source, presence of oxygen, and EY on the reaction progress. The catalytic activity of EY, WO3/ZnO@NH2, and WO3/ZnO@NH2-EY nanoparticles was separately investigated for the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarin under the standard reaction conditions (Table 3). This investigation demonstrated that the condensation reaction is greatly dependent on the presence of light and oxygen. In addition, the best yield was attained with the nanocomposite WO3/ZnO@NH2-EY and WO3/ZnO@NH2 showed a lower catalytic activity.
Table 3 Studying the catalytic activity and some contributing experimental parameters on the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarina
| Entry |
Green LED (25 W, λ = 535 nm) |
Catalyst |
Air |
Time (min) |
Yield (%) |
| Reaction conditions are described below Table 1. 0.03 g of the catalyst was used under solvent-free conditions. |
| 1 |
+ |
— |
+ |
45 |
9 |
| 2 |
+ |
EY |
+ |
45 |
72 |
| 3 |
+ |
WO3/ZnO@NH2 |
+ |
45 |
42 |
| 4 |
+ |
WO3/ZnO@NH2-EY |
+ |
45 |
97 |
| 5 |
− |
WO3/ZnO@NH2-EY |
+ |
45 |
43 |
| 6 |
+ |
WO3/ZnO@NH2-EY |
N2 |
45 |
18 |
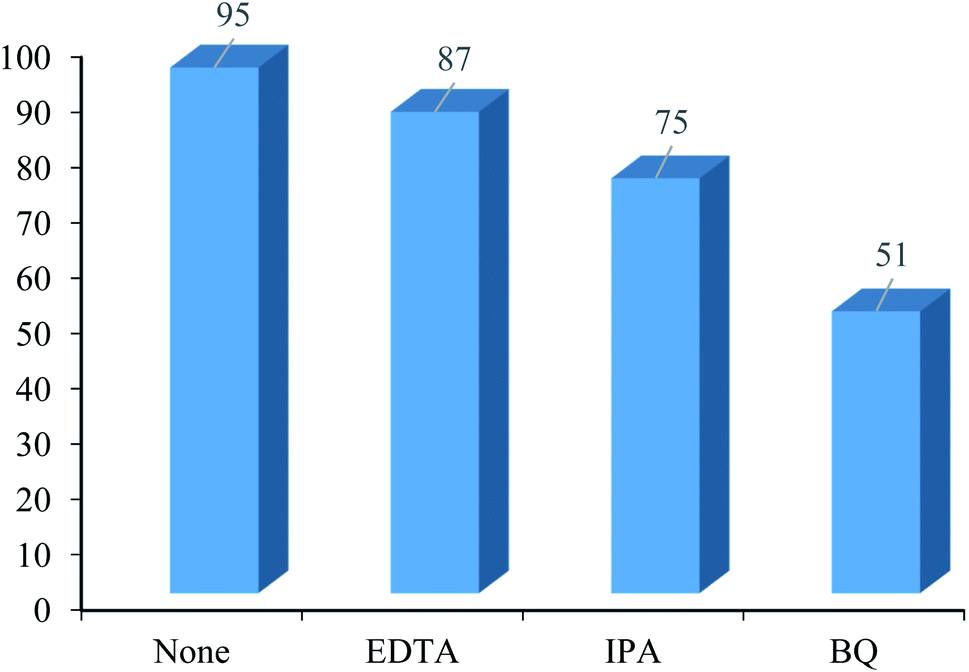
3.2.6 Role of hole and electron scavengers. The effect of various hole and electron scavengers was monitored in the photocatalytic preparation of chromene derivatives in the presence of the heterogeneous nanocatalyst WO3/ZnO@NH2-EY (Fig. 10). Generally, effective radical species such as hydroxyl radical, holes, and superoxide anion-radical are responsible for the photocatalytic reaction. Therefore, some familiar scavengers were separately included in the present reaction mixture. In these experiments, isopropyl alcohol (IPA) was employed to scavenge the hydroxyl free radical.73 In this case, the yield% was decreased from 95 to 75%. In addition, a great decrease in the yield% (44%) in the presence of p-benzoquinone (BQ) (as an ˙O2− scavenger)74 proved that ˙O2− would be involved in the reaction mechanism; therefore, O2 strongly affects the photocatalytic process. Meanwhile, an 8% decrease in the yield% in the presence of ethylenediaminetetraacetic acid (EDTA) as an h+ scavenger75 also confirmed the possible contribution of h+ in the photocatalytic reaction. As a result, reactive species such as OH˙ and ˙O2− along with h+ were expected to be concerned in the present photocatalytic reaction.
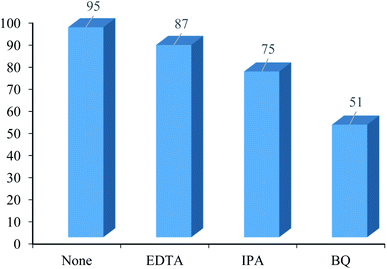 |
| | Fig. 10 Effect of some familiar scavengers on the reaction progress under the standard reaction conditions described below Table 1. | |
3.2.7 Synthesis of different chromeno[4,3-b]chromene catalyzed by WO3/ZnO@NH2-EY. The versatility, limitations, and general scope of the present methodology were identified for the synthesis of a series of chromeno[4,3-b]chromene derivatives using a variety of aromatic aldehydes. Most of the performed aldehydes provided good to excellent yields in a short period of time (Table 4). Aromatic aldehydes with different electron withdrawing/releasing substituents generated the desired products in good yields with superior selectivity. Although most of the electron-rich and electron-poor aldehydes afforded the desired products in high yields; however, in agreement with previous reports,76,77 our experiments revealed that aldehyde-bearing electron-withdrawing groups react with better efficacy than those bearing electron-donating substituents.
Table 4 Substrate scope for the condensation of dimedone, benzaldehyde, and 4-hydroxycoumarina
3.2.8 Superiority of the present method over some reported catalysts. To date, a number of similar reports have accounted for the preparation of chromeno[4,3-b]chromene under different reaction conditions. The condensation yield of dimedone, benzaldehyde, and 4-hydroxycoumarin was contrasted with that reported in the literature, as shown in Table 5. In this experiment, the comparison was focused in terms of the most important experimental variables such as the reaction time, catalyst mol%, and yield%. Interestingly, the present methodology showed a few advantages using a very low amount of an environmental benign catalyst under mild reaction conditions and with an acceptable generality. Thus, this methodology is a special case, offering a new approach for the one-pot single-step preparation of the desired chromenes under milder conditions and, especially, under photochemical conditions with environment-friendly LED irradiation, which consumes a very low amount of electrical energy.
Table 5 The comparison of the catalytic activity of WO3/ZnO@NH2-EY with some reported catalysts in the synthesis of chromeno[4,3-b]chromenea
| Catalyst |
Catalyst amount |
Time (h) |
Temp (°C) |
Solvent |
Yield (%) |
| Reaction conditions are described below Table 1. |
| Fe(DS)3 |
10 mol% |
1.5 |
70 |
H2O (2.5 mL) |
90 (ref. 76) |
| Fe3O4@SiO2@(CH2)3OMoO3H |
0.002 g |
0.75 |
80 |
— |
90 (ref. 27) |
| Nano-CuFe2O4@SO3H |
(0.05 g) |
2.5 |
70 |
EtOH (5 mL) |
90 (ref. 29) |
| t-ZrO2 np |
10 mol%, 12.3 mg |
0.5 |
80 |
H2O (5 mL) |
91 (ref. 82) |
| Eozin Y |
2 mol% |
3.5 |
r.t green LED |
CH3CN (3 mL) |
77 (ref. 83) |
| WO3/ZnO@NH2-EY |
30 mg (∼0.21 mmol EY per g of photocatalyst) |
0.75 |
r.t. green LED |
— |
97 (this work) |
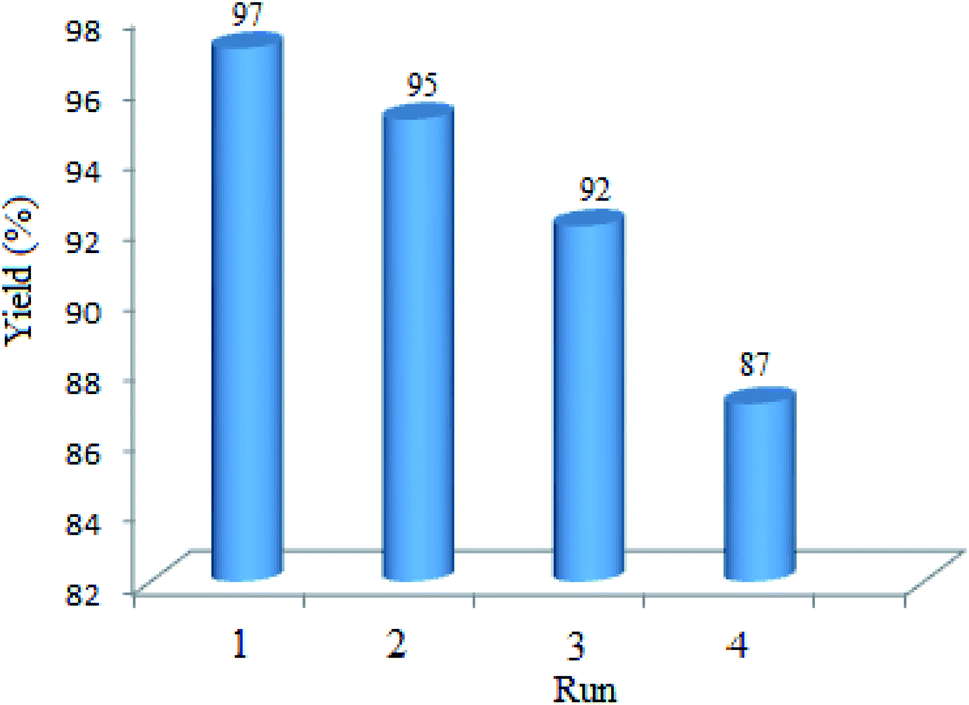
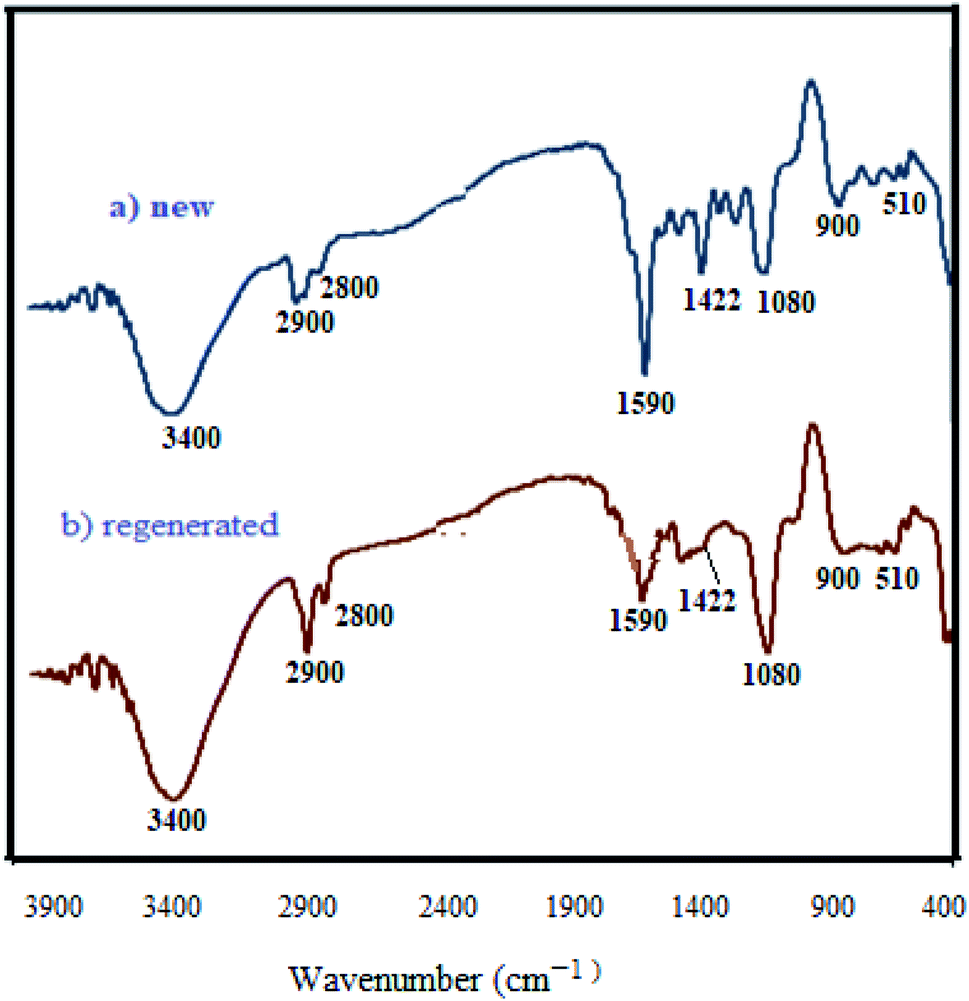
3.2.9 Studying the stability and reusability of WO3/ZnO@NH2-EY. To study the stability and reusability of WO3/ZnO@NH2-EY, the photocatalytic nanoparticles were removed after the completion of the first run by simple filtration. Then, the filtered catalyst was washed with a copious amount of toluene, well-dried at room temperature, and prepared for the next run. Therefore, the nanocatalyst was subsequently examined for four runs to prove its good stability and reusability (Fig. 11). This experiment revealed that the reactivity of the photocatalyst declined such that the yield% was decreased ∼10% after the 4th run, which may be due to the poisoning of the catalyst surface or a little destruction of the photocatalyst framework. However, the comparison of the infrared spectra of WO3/ZnO@NH2-EY before and after the 4th run proved the existence of the main characteristic bands at about 1200–1800 cm−1, assuring that the structure of WO3/ZnO@NH2 remained almost intact during the reaction (Fig. 12). In addition, typical experiments were repeated to warrant the reproducibility of the condensation reaction in the synthesis of chromeno[4,3-b]chromene and the acquired yields were repeatable with ±4% variation. Therefore, according to this experiment together with the consideration of the solvent-free conditions, it seems that the present protocol has some significant advantages compared to the reported protocols, especially with that of Singh et al.83 The major improvements include the heterogeneity and recyclability of the catalyst.
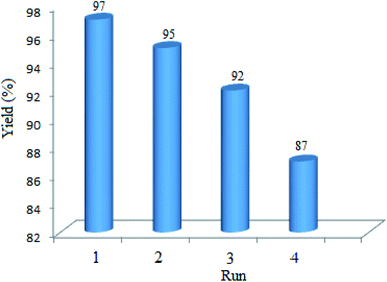 |
| | Fig. 11 Yield% as a function of the reusability of nano WO3/ZnO@NH2-EY. | |
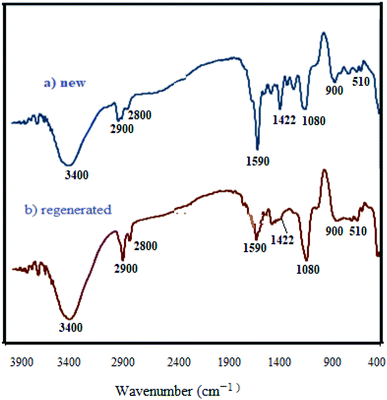 |
| | Fig. 12 FT-IR spectra of new (a) and regenerated (b) WO3/ZnO@NH2-EY. | |
3.2.10 Hot filtration test. The hot filtration test is recommended to ensure that the catalytic activity of the photocatalyst originates from the conjugated composite WO3/ZnO@NH2-EY and not from the drained segments, especially EY, in the reaction medium. Considering the high activity of EY in the preparation of chromeno[4,3-b]chromene, we specifically studied the leaching of this species from WO3/ZnO@NH2-EY. For this purpose, WO3/ZnO@NH2-EY (2 mg) was added to a mixture of dimedone, benzaldehyde, and 4-hydroxycoumarin, and the reaction was started under the standard conditions for 1.5 h. Then, the catalyst was separated off and the product yield of 58% was attained after removing the produced chromene. After that, the reaction was continued for another 1.5 h and the improved yield of 65% was attained based on the separated chromene. Interestingly, no further increase in the yield% was observed by prolonging the time to 5 h. Clearly, this experiment showed that an ∼8% increase in the yield% should be due to the leached EY or other fragments from the mother active nanocomposite WO3/ZnO@NH2-EY.
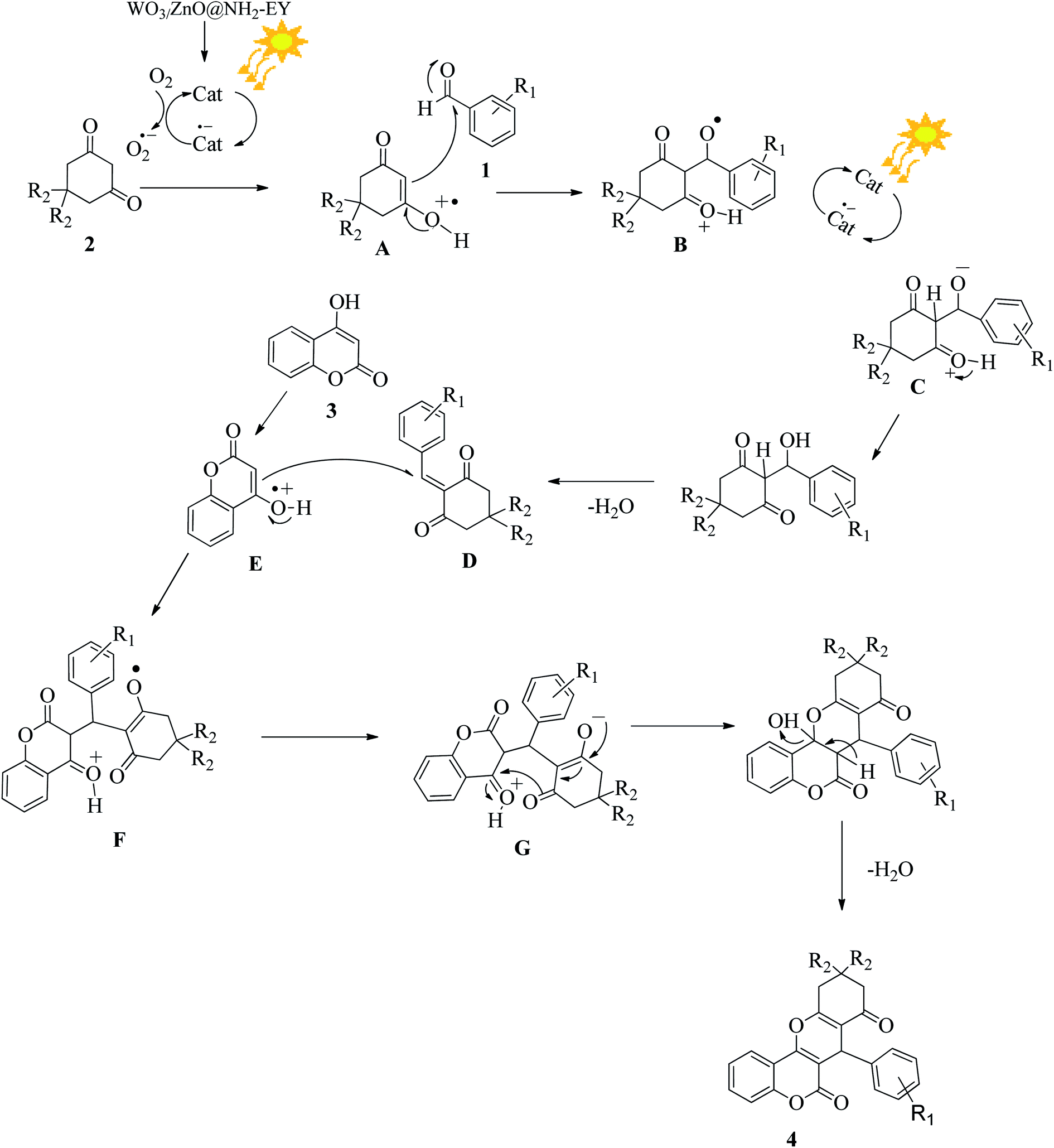
3.2.11 Suggesting a reasonable reaction pathway. On the basis of the literature survey,58 a probable mechanism is illustrated in Fig. 13. Upon absorption of visible light, the photoredox catalyst can be converted into its excited state and provides a superoxide species to promote the conversion of the 1,3-dicarbonyl compound (2) into a radical cation (A). The generated radical cation (A) can interact with the aromatic aldehyde (1) to form an intermediate (B). Again, the photocatalyst participates in the reaction and facilitates the transformation of compound (B) to (C) via an electron transfer process. Eventually, the dehydration of compound (C) produces the β-dicarbonyl-enone (D), which can act as a Michael acceptor. Simultaneously, 4-hydroxycoumarin (3) can be converted into the radical-cation (E) via photochemical electron transfer and initiates a Michael-type addition reaction with β-dicarbonyl-enone (D) to furnish the desired compound (F). The continuation of the photochemical electron transfer reactions by the mediation of the nanophotocatalyst achieves the conversion of (F) into (G). Finally, (G) undergoes a simple dehydration reaction and generates the target chromene product (4).
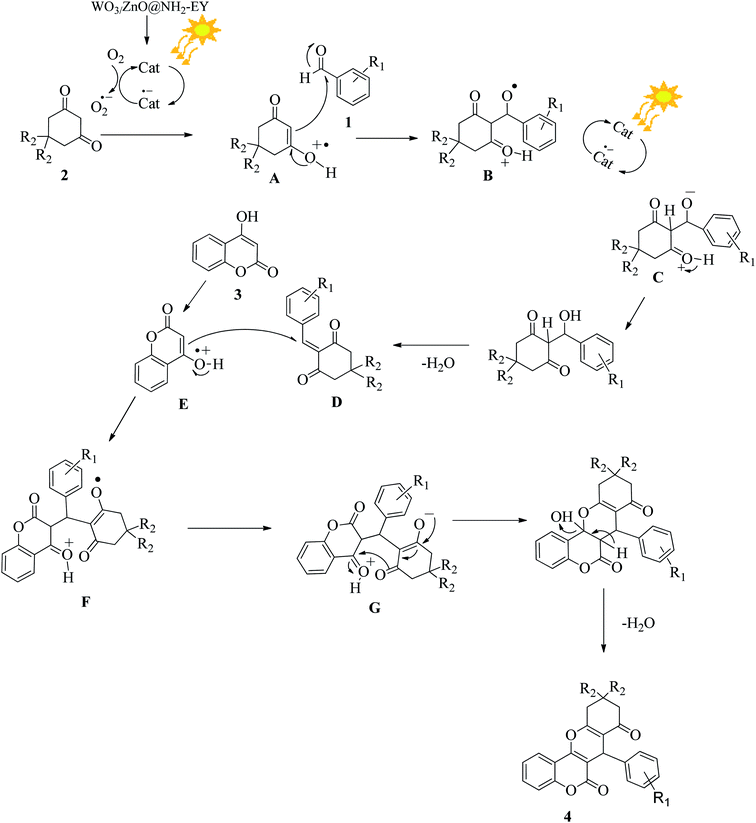 |
| | Fig. 13 A plausible reaction pathway for the fabrication of the desired chromene via consecutive electron transfer reactions. | |
4 Conclusion
In conclusion, we have reported an ecofriendly and efficient method to achieve visible light-promoted synthesis of chromeno[4,3-b]chromenes under mild conditions. The photoredox catalyst WO3/ZnO@NH2-EY is used as a superior green alternative catalyst for the desired heterocyclic condensation reaction. The heterogeneous photoredox nanocatalyst was characterized by means of XRD, FT-IR, ICP, TGA, and SEM. The mild reaction conditions, wide substrate tolerance, solventless reactor, easy workup, high atom economy, cost-effectiveness, and good to excellent yields are the main features of this new photocatalytic system. The WO3/ZnO@NH2-EY nanocomposite exhibits its photocatalytic functions via a series of electron transfer reactions in the presence of visible light through a radical mechanism, as proven using various scavengers. This investigation showed that the reactive species such as ˙O2− and OH˙ along with h+ are the main reactive species involved in the photochemical synthesis of chromenes. The WO3/ZnO@NH2-EY photocatalyst could be frequently recovered over four times without the loss of the catalytic activity. Eventually, a hot filtration test ensured that the nanohybrid catalyst is stable in the reaction mixture and its catalytic activity originates from the whole undecomposed nanocomposite.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors/creators would like to acknowledge the financial support of Ministry of Science, Research and Technology for this project under grant number 12-99-02-000029. Also, the work has been supported by Hakim Sabzevari University.
References
- R. C. Cioc, E. Ruijter and R. V. Orru, Green Chem., 2014, 16, 2958–2975 RSC.
- M. B. Gawande, V. D. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, Chem. Soc. Rev., 2013, 42, 5522–5551 RSC.
- X. Guo and W. Hu, Acc. Chem. Res., 2013, 46, 2427–2440 CrossRef CAS PubMed.
- A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef PubMed.
- F. Wang, Y. Shen, H. Hu, X. Wang, H. Wu and Y. Liu, J. Org. Chem., 2014, 79, 9556–9566 CrossRef CAS PubMed.
- C. Simon, T. Constantieux and J. Rodriguez, Eur. J. Org. Chem., 2004, 2004, 4957–4980 CrossRef.
- E. Ruijter, R. Scheffelaar and R. V. Orru, Angew. Chem., Int. Ed., 2011, 50, 6234–6246 CrossRef CAS PubMed.
- A. Amr, M. Abdalla, S. Essaouy, M. Areef, M. Elgamal, T. Nassear and A. Haschich, Russ. J. Gen. Chem., 2017, 87, 1826–1833 CrossRef CAS.
- S. W. Ng, L. H. Chung, C. F. Yeung, H. S. Lo, H. L. Shek, T. S. Kang, C. H. Leung, D. L. Ma and C. Y. Wong, Chem.–Eur. J., 2018, 24, 1779–1783 CrossRef CAS PubMed.
- W. H. Zhang, S. Chen, X. L. Liu, X. W. Liu and Y. Zhou, Bioorg. Med. Chem. Lett., 2020, 30, 127410 CrossRef CAS PubMed.
- I. V. Ilyina, O. S. Patrusheva, V. V. Zarubaev, M. A. Misiurina, A. V. Slita, I. L. Esaulkova, D. V. Korchagina, Y. V. Gatilov, S. S. Borisevich and K. P. Volcho, Bioorg. Med. Chem. Lett., 2020, 31, 127677 CrossRef PubMed.
- N. Baral, D. R. Mishra, N. P. Mishra, S. Mohapatra, B. P. Raiguru, P. Panda, S. Nayak, M. Nayak and P. S. Kumar, J. Heterocycl. Chem., 2020, 57, 575–589 CrossRef CAS.
- A. Neghra, M. Lecsö, M. J. Butel, L. S. Espindola, B. Deguin and E. Seguin, Nat. Prod. Commun., 2017, 12, 1459–1463 CrossRef.
- K. Haider, S. Rahaman, M. S. Yar and A. Kamal, Expert Opin. Ther. Pat., 2019, 29, 623–641 CrossRef CAS PubMed.
- I. Zghab, B. Trimeche, M. B. Mansour, M. Hassine, D. Touboul and H. B. Jannet, Arabian J. Chem., 2017, 10, S2651–S2658 CrossRef CAS.
- F. Borges, F. Roleira, N. Milhazes, L. Santana and E. Uriarte, Curr. Med. Chem., 2005, 12, 887–916 CrossRef CAS PubMed.
- M. Mahdavi, J. Davoodi, M. R. Zali and A. Foroumadi, Biomed. Pharmacother., 2011, 65, 175–182 CrossRef CAS PubMed.
- J.-L. Wang, D. Liu, Z.-J. Zhang, S. Shan, X. Han, S. M. Srinivasula, C. M. Croce, E. S. Alnemri and Z. Huang, Proc. Natl. Acad. Sci., India, 2000, 97, 7124–7129 CAS.
- S. A. Patil, J. Wang, X. S. Li, J. Chen, T. S. Jones, A. Hosni-Ahmed, R. Patil, W. L. Seibel, W. Li and D. D. Miller, Bioorg. Med.
Chem. Lett., 2012, 22, 4458–4461 CrossRef CAS PubMed.
- A. M. Shestopalov, Y. M. Litvinov, L. A. Rodinovskaya, O. R. Malyshev, M. N. Semenova and V. V. Semenov, ACS Comb. Sci., 2012, 14, 484–490 CrossRef CAS PubMed.
- G. A. Kraus and I. Kim, J. Org. Chem., 2003, 68, 4517–4518 CrossRef CAS PubMed.
- J. G. Tangmouo, A. L. Meli, J. Komguem, V. Kuete, F. N. Ngounou, D. Lontsi, V. P. Beng, M. I. Choudhary and B. L. Sondengam, Tetrahedron Lett., 2006, 47, 3067–3070 CrossRef CAS.
- R. O. S. Kitamura, P. Romoff, M. C. M. Young, M. J. Kato and J. H. G. Lago, Phytochemistry, 2006, 67, 2398–2402 CrossRef PubMed.
- M. Kamali, Synth. Commun., 2020, 51, 1–9 CrossRef.
- H. Anaraki-Ardakani, Russ. J. Gen. Chem., 2017, 87, 1820–1825 CrossRef CAS.
- H. Emtiazi and M. A. Amrollahi, S. Afr. J. Chem., 2014, 67, 175–179 Search PubMed.
- F. Khosravian, B. Karami and M. Farahi, New J. Chem., 2017, 41, 11584–11590 RSC.
- H. Anaraki-Ardakani, R. Ghanavatian and M. Akbari, World Appl. Sci. J., 2013, 22, 802–808 CAS.
- S. Vajar and M. Mokhtary, Polycyclic Aromat. Compd., 2019, 39, 111–123 CrossRef CAS.
- X.-J. Sun, J.-F. Zhou and S.-J. Zhi, Synth. Commun., 2012, 42, 1987–1994 CrossRef CAS.
- R. Tayebee, A. Pejhan, H. Ramshini, B. Maleki, N. Erfaninia, Z. Tabatabaie and E. Esmaeili, Appl. Organomet. Chem., 2018, 32, e3924 CrossRef.
- B. Maleki, S. Babaee and R. Tayebee, Appl. Organomet. Chem., 2015, 29, 408–411 CrossRef CAS.
- S. K. Sahoo, Renewable Sustainable Energy Rev., 2016, 59, 927–939 CrossRef.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. L. Belser and A. V. von Zelewsky, Coord. Chem. Rev., 1998, 84, 85–277 CrossRef.
- P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 1360–1363 CrossRef CAS PubMed.
- D. A. Nagib, M. E. Scott and D. W. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed.
- M. A. Ischay, Z. Lu and T. P. Yoon, J. Am. Chem. Soc., 2010, 132, 8572–8574 CrossRef CAS PubMed.
- Y. Zhang, C. Ye, S. Li, A. Ding, G. Gu and H. Guo, RSC Adv., 2017, 7, 13240–13243 RSC.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 RSC.
- D. P. Hari and B. König, Chem. Commun., 2014, 50, 6688–6699 RSC.
- S. Yadav, M. Srivastava, P. Rai, B. P. Tripathi, A. Mishra, J. Singh and J. Singh, New J. Chem., 2016, 40, 9694–9701 RSC.
- N. Hashim, S. Thakur, M. Patang, F. Crapulli and A. K. Ray, Environ. Technol., 2017, 38, 933–944 CrossRef CAS PubMed.
- G. Kumar, P. Solanki, M. Nazish, S. Neogi, R. I. Kureshy and H. K. Noor-ul, J. Catal., 2019, 371, 298–304 CrossRef CAS.
- S. Gazi and R. Ananthakrishnan, Appl. Catal., B, 2011, 105, 317–325 CrossRef CAS.
- A. Sridhar, R. Rangasamy and M. Selvaraj, New J. Chem., 2019, 43, 17974–17979 RSC.
- P. Li, G.-W. Wang, X. Zhu and L. Wang, Tetrahedron, 2019, 75, 3448–3455 CrossRef CAS.
- L. Wang, S. Yue, Q. Zhang, Y. Zhang, Y. R. Li, C. S. Lewis, K. J. Takeuchi, A. C. Marschilok, E. S. Takeuchi and S. S. Wong, ACS Energy Lett., 2017, 2, 1465–1478 CrossRef CAS.
- R. Celiesiute, A. Ramanaviciene, M. Gicevicius and A. Ramanavicius, Crit. Rev. Anal. Chem., 2019, 49, 195–208 CrossRef CAS PubMed.
- X. Yu, T. J. Marks and A. Facchetti, Nat. Mater., 2016, 15, 383–396 CrossRef CAS PubMed.
- A. K. Arora, V. S. Jaswal, K. Singh and R. Singh, Orient. J. Chem., 2016, 32, 2035 CrossRef CAS.
- M. J. Limo, A. Sola-Rabada, E. Boix, V. Thota, Z. C. Westcott, V. Puddu and C. C. Perry, Chem. Rev., 2018, 118, 11118–11193 CrossRef CAS PubMed.
- M. B. Tahir, G. Nabi and N. Khalid, Mater. Sci. Semicond. Process., 2018, 84, 36–41 CrossRef CAS.
- E. Mugunthan, M. Saidutta and P. Jagadeeshbabu, J. Photochem. Photobiol., A, 2019, 383, 111993 CrossRef CAS.
- F. Akhlaghian and A. Najafi, Sci. Iran., 2018, 25, 3345–3353 Search PubMed.
- N. Geetha, S. Sivaranjani, A. Ayeshamariam, M. K. Micheal, D. Saravankkumar, S. Fowziya, A. Mohideen and M. Jayachandran, J. Adv. Microsc. Res., 2018, 13, 3–11 CrossRef.
- Z. Xiong, Z. Lei, Z. Xu, X. Chen, B. Gong, Y. Zhao, H. Zhao, J. Zhang and C. Zheng, J. CO2 Util., 2017, 18, 53–61 CrossRef CAS.
- B. Maleki, H. Natheghi, R. Tayebee, H. Alinezhad, A. Amiri, S. A. Hossieni and S. M. M. Nouri, Polycyclic Aromat. Compd., 2020, 40, 633–643 CrossRef CAS.
- B. Maleki, M. Chahkandi, R. Tayebee, S. Kahrobaei, H. Alinezhad and S. Hemmati, Appl. Organomet. Chem., 2019, 33, e5118 CrossRef.
- R. Tayebee, M. Fattahi Abdizadeh, N. Erfaninia, A. Amiri, M. Baghayeri, R. M. Kakhki, B. Maleki and E. Esmaili, Appl. Organomet. Chem., 2019, 33, e4959 CrossRef.
- H. Zhao, X. Liu, Z. Cao, Y. Zhan, X. Shi, Y. Yang, J. Zhou and J. Xu, J. Hazard. Mater., 2016, 310, 235–245 CrossRef CAS PubMed.
- Y. Liu, J. Xu, Z. Cao, R. Fu, C. Zhou, Z. Wang and X. Xu, J. Colloid Interface Sci., 2020, 559, 215–225 CrossRef CAS PubMed.
- X. Liu, J. Xu, Y. Zhao, H. Shi and C.-H. Huang, Chemosphere, 2019, 226, 726–735 CrossRef CAS PubMed.
- J. Xu, Z. Cao, Y. Zhang, Z. Yuan, Z. Lou, X. Xu and X. Wang, Chemosphere, 2018, 195, 351–364 CrossRef CAS PubMed.
- K. S. Babu, A. R. Reddy, C. Sujatha, K. V. Reddy and A. Mallika, J. Adv. Ceram., 2013, 2, 260–265 CrossRef.
- B. Li, R. Tayebee, E. Esmaeili, M. S. Namaghi and B. Maleki, RSC Adv., 2020, 10, 40725–40738 RSC.
- X. Z. Yajun Wang, L. Duan, F. Wang, H. Niu, W. Guo and A. Ali, Mater. Sci. Semicond. Process., 2015, 29, 372–379 CrossRef.
- D. Jaiswal, A. Mishra, P. Rai, M. Srivastava, B. P. Tripathi, S. Yadav, J. Singh and J. Singh, Res. Chem. Intermed., 2018, 44, 231–246 CrossRef CAS.
- D. A. Young, T. B. Freedman, E. D. Lipp and L. A. Nafie, J. Am. Chem. Soc., 1986, 108, 7255–7263 CrossRef CAS.
- A. Chiolerio, A. Chiodoni and P. Allia, Thin Solid Films, 2008, 516, 8453–8461 CrossRef CAS.
- F. Liu, X. Chen, Q. Xia, L. Tian and X. Chen, RSC Adv., 2015, 5, 77423–77428 RSC.
- H. Alinezhad, M. Tarahomi, B. Maleki and A. Amiri, Appl. Organomet. Chem., 2019, 33, e4661 CrossRef.
- V. Kandathil, B. D. Fahlman, B. Sasidhar, S. A. Patil and S. A. Patil, New J. Chem., 2017, 41, 9531–9545 RSC.
- R. K. Sharma, B. Arora, S. Sharma, S. Dutta, A. Sharma, S. Yadav and K. Solanki, Mater. Chem. Front., 2020, 4, 605–620 RSC.
- Z. Yin, M. Han, Z. Hu, L. Feng, Y. Liu, Z. Du and L. Zhang, Chem. Eng. J., 2020, 390, 124532 CrossRef CAS.
- X. Zheng, J. Yuan, J. Shen, J. Liang, J. Che, B. Tang, G. He and H. Chen, J. Mater. Sci.: Mater. Electron., 2019, 30, 5986–5994 CrossRef CAS.
- K. Pradhan, S. Paul and A. R. Das, Tetrahedron Lett., 2013, 54, 3105–3110 CrossRef CAS.
- F. Adibian, A. R. Pourali, B. Maleki, M. Baghayeri and A. Amiri, Polyhedron, 2020, 175, 114179 CrossRef.
- Z. Chen, Q. Zhu and W. Su, Tetrahedron Lett., 2011, 52, 2601–2604 CrossRef CAS.
- K. T. Patil, L. Walekar, S. Undare, G. Kolekar, M. B. Deshmukh, P. Choudhari and P. V. Anbhule, Indian J. Chem., 2016, 55, 1151–1159 Search PubMed.
- A. Shafiee, R. Motamedi, O. Firuzi, S. Meili, A. R. Mehdipour and R. Miri, Med. Chem. Res., 2011, 20, 466–474 CrossRef CAS.
- R. Motamedi, S. Baghbani and F. F. Bamoharram, Synth. Commun., 2012, 42, 1604–1612 CrossRef CAS.
- A. Saha, S. Payra and S. Banerjee, RSC Adv., 2015, 5, 101664–101671 RSC.
- A. K. Sharma, J. Tiwari, D. Jaiswal, S. Singh, J. Singh and J. Singh, Curr. Organocatal., 2019, 6, 222–230 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09737c |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Reza Tayebee
,
Reza Tayebee