
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe polyphenolic phytoalexin polydatin inhibits amyloid aggregation of recombinant human prion protein
Preeti Rana Sirohi ab,
Anchala Kumaria,
Nikita Admanea,
Pallavi Somvanshi*cd and
Abhinav Grover*a
ab,
Anchala Kumaria,
Nikita Admanea,
Pallavi Somvanshi*cd and
Abhinav Grover*a
aSchool of Biotechnology, Jawaharlal Nehru University, New Delhi, 110067, India. E-mail: agrover@jnu.ac.in; abhinavgr@gmail.com; Fax: +91-11-26702040; Tel: +91-11-26738728
bDepartment of Biotechnology, TERI School of Advanced Studies, New Delhi, 110070, India
cSchool of Computational and Integrative Sciences, Jawaharlal Nehru University, New Delhi, 110067, India. E-mail: psomvanshi@gmail.com; Tel: +91-9899931682
dSpecial Centre of Systems Medicine, Jawaharlal Nehru University, New Delhi, 110067, India. E-mail: psomvanshi@gmail.com; Tel: +91-9899931682
First published on 28th July 2021
Abstract
Prion diseases involve misfolded and highly infectious aggregates of prion protein (PrPSc) which forms amyloid plaques leading to fatal neurodegeneration. The absence of clinically proven therapeutics makes the discovery of effective remedial interventions a prime concern. Herein, we report novel prion intervention by the polyphenolic phytoalexin, polydatin which binds with moderate affinity to the recombinant protease resistant core of human prion protein, encompassing the sequence 90–231 (rPrPres) and inhibits its conversion into the highly neurotoxic forms. An extensive evaluation using biophysical techniques revealed that polydatin incubated rPrPres samples generate off-pathway oligomers having reduced cross-β sheet signature, and relatively smaller in size than the native rPrPres oligomers. The detailed structural analysis using molecular dynamics simulations elucidated the induction of antagonistic mobilities in the β2–α2 loop, α3 helix and the N-terminal amyloidogenic region of prions. This study puts forward novel prion fibrillogenesis inhibitory potential of polydatin, specifically by stabilizing the N-terminal amyloidogenic region. Collectively our results affirm the importance of polydatin in crippling the prion pathogenesis and may serve as a structural scaffold for designing novel therapeutic agents targeting amyloidogenic transition in prions.
1. Introduction
Transmissible spongiform encephalopathies (TSE's), commonly known as prion diseases, belong to a class of neurodegenerative disorders represented by the aggregation of an infectious, misfolded prion protein (PrP) capable of forming amyloid plaques. Bovine spongiform encephalopathy (kuru), fatal familial insomnia (FFI), Gerstmann–Straussler–Scheinker disease, and Creutzfeldt–Jakob disease are some of the most commonly occurring prion diseases in humans.1–3 At present, no clinically proven therapeutics is available to treat these prion-borne encephalopathies, making the discovery of effective remedial interventions a prime concern.Glycosylphosphatidylinositol (GPI)-anchored cell-surface prion proteins (PrPC) are expressed in different organs and tissues, with maximum levels of expression in the peripheral and central nervous systems.4–6 The pathogenesis of rapidly progressing, dementia-causing, eventually fatal prion diseases usually involves converting the typical cellular form of a prion protein, PrPC (a soluble, helical protein), into an infectious and aggregation-prone scrapie form, PrPSc (an insoluble form-containing β-sheet).7,8
A number of cell models of prion diseases have been proposed, which have proven to be valuable in searching for novel drug leads. Most of the reported assays have focused on inhibiting the prion misfolding process, reducing the overall amount of cellular prion proteins, and stabilizing the natively-folded prion protein. The mechanism involved in catalyzing the translation of PrPC into PrPSc and the structure of PrPSc remain unknown, thus complicating the process of designing potential drugs against pathogenic isoforms. Therefore, the structural studies of prion protein and other and related peptides in non-native forms signify an imperative step in comprehending the molecular minutiae of prion proteins.
A mature human prion protein is composed of 208 amino acids (residues 23–230).9 The recombinant polypeptide form of this human prion protein encompassing the amino acid residues 90–231 (rPrPres), corresponds to the protease-resistant fragment originating after the truncation of amino-terminal. This rPrPres fragment has a flexible backbone that is similar to the mature cellular PrP, which can undergo structural rearrangement to form PrPSc-like conformation, thus representing an important model for studying the in vitro neurotoxicity of prions.10–12 Using this recombinant human prion protein fragment, we performed high throughput virtual screening of a library of natural compounds to discover molecules that bind to the highly conserved β2–α2 region of the 90–231 fragment of prion protein. Polydatin was found to be amongst the top scoring hits obtained.
Polydatin, or piceid (3,4′,5-trihydroxystilbene-3-β-D-glucoside, PD), is a monocrystalline stilbene derivative known to be a natural precursor of resveratrol and is derived from the roots of Polygonum cuspidatum.13,14 It has been found to exhibit anti-inflammatory, immunoregulatory, anti-oxidative, and anti-tumor activity.14–16 Polydatin is also known to have neuroprotective activity17–19 and is inhibitory to amyloid formation.20,21 But does not have any apparent cellular toxicity while being cytotoxic to various cancerous cells thus possessing anti-tumor activity.22
In our study, we utilized biophysical techniques, as well as in silico and in vitro data, and found polydatin to be a pharmacologically-significant scaffold that can bind to the rPrPres repertoire and inhibit its conversion to the highly infectious and neurotoxic PrPSc-like form. Hence, polydatin is a promising anti-prion drug lead.
An efficient anti-prion treatment will involve the amalgamation of drugs acting at various steps of the pathogenic process, leading to the effective suppression of neurotoxic signaling, along with reduced prion levels. This study provides, on the atomic level, the conformational transitions of the rPrPres monomer and the mechanism by which a natural, small molecule (polydatin) inhibits prion aggregation, thus providing new clues for the development of therapeutic candidates against prion-borne encephalopathies and related neurodegenerative disorders. The collective conclusion of our data shows polydatin to be a probable structural candidate for coherent drug discovery against prion diseases, which will be beneficial to both humans and animals.
2. Results and discussion
2.1 Polydatin specifically binds to rPrPres and alters its amyloidogenic transition
To assess the specific hetero-molecular interactions between rPrPres and polydatin, we examined the binding kinetics of rPrPres with polydatin using surface plasmon resonance (SPR) experiments. Analysis of dose–response sensorgrams generated during the interaction analysis suggests the presence of a favorable polydatin binding pocket in the rPrPres structure [Fig. 2C]. Corresponding dose–response curves were obtained by plotting the maximum response signal against different concentrations of polydatin [Fig. 2D]. The association (kon) and dissociation rate constants (koff) of the binding reaction were derived from the Langmuir global fitting model, which was later used to calculate the dissociation constants (KD) in each case [Table 2].The binding curves showed a plateau depicting the saturation of binding sites and the absence of non-specific binding. The fitting model yielded kon and koff values in the range of 9.091 × 103 and 4.14 × 10−3, respectively, suggesting that polydatin exhibits fast association with rPrPres, but its dissociation rate is significantly slower. The equilibrium dissociation constant (KD) of polydatin was in the range of 0.45 μM which signifies a good binding affinity. After affirming the binding affinity, flexible molecular docking was done for deducing the plausible binding mechanism. The top docking clusters showed engagement of polydatin in the rPrPres structure near the binding pocket encompassing the β2–α2 loop (residues Tyr162, Met166, Tyr169, Asp173, and Phe175) and C-terminal region of α3 helix (residues Gln217, Tyr218, Gln221, Ala224, Tyr225, and Arg228). The most favorable binding pose of the rPrPres and polydatin complex showed a docking score of −8.5 kcal mol−1 which shows a stringer binding when compared to epigallocatechin gallate (EGCG), a well-established inhibitor of prion amyloidogenesis as revealed by the glide score of −7.026 kcal mol−1.23–25
The chemical structure of polydatin and its molecular interactions with rPrPres have been demonstrated in Fig. 1A and B, respectively. Further analysis of the binding pose indicated a strong association stabilized by several hydrophobic interactions and hydrogen bonds. The hydrogen bonds with Arg164 and Gln172 of rPrPres and hydrophobic interactions with various critical aromatic and hydrophobic residues like Met 129, Gly 131, Met 134, Tyr 163, Met 166, Phe 175, Gln 217, Tyr 218 [Table 1 and Fig. 2A, B] indicated a strong enthalpy driven association of polydatin with rPrPres.
 | ||
| Fig. 1 Structural details of polydatin and rPrPres. (A) Stick representation of 2D and 3D chemical structure of polydatin. (B) rPrPres; cartoon representation showing secondary structural elements (PDB: 2KUN; human prion residues 90–231). | ||
| Compound | Polydatin |
| Library | FDA approved |
| Molecular weight | 390 g mol−1 |
| Glide docking score | −8.5 kcal mol−1 |
| H-Bonds | Arg 164, Gln 172 |
| Hydrophobic interactions | Met 129, Gly 131, Met 134, Tyr 163, Met 166, Phe 175, Gln 217, Tyr 218 |
| Association rate kon (M−1 s−1) | Dissociation rate koff (s−1) | Equilibrium affinity KD (μM) | |
|---|---|---|---|
| Polydatin | 9.091 × 103 | 4.14 × 10−3 | 0.45 |
 | ||
| Fig. 2 Interaction analysis of polydatin with rPrPres through molecular docking and SPR studies. (A) Non covalent bonding interactions of polydatin at the binding pocket shown through LigPlot image. (B) Molecular representation of the docking pose of polydatin at the interface of the rPrPres. SPR sensorgrams (C) and the saturation curve (D) of titration of polydatin on rPrPres monomer immobilized upon a gold surface chip with a density of 2700 RU. The rise in RUs from the baseline was measured and used for the calculation of Ka and Kd. Illustrated sensorgrams was obtained from different concentrations of ligand (different colors of the sensorgrams represent different concentrations ranging from 0.1–20 μM). | ||
To further examine the mechanism of conformation transitions consequent to the heteromolecular association of rPrPres with polydatin, molecular dynamics (MD) simulations were performed. The variations in both backbone RMSD and radius of gyration (Rg) were lowered in case of polydatin-complexed rPrPres (Fig. 3A and B) suggestive of a stable structure and induction of compaction in the prion protein. Moreover, the stability of RMSD trajectories of both the systems is implicative of their suitability for further analysis on mean residue fluctuations, intermolecular bonding, and conformational sampling. Interestingly, the root mean square fluctuation (RMSF) plot of prion–polydatin complex also denotes lower fluctuations in the N-terminal amyloidogenic region as compared to the unliganded prion structure. The RMSF in the β2–α2 loop regions and α3 helix also indicated stabilization upon binding to polydatin (Fig. 3D). The stabilization of these regions has been previously reported to abrogate the malicious structural transitions in the prion protein and further crippling the disease propagation.26–28
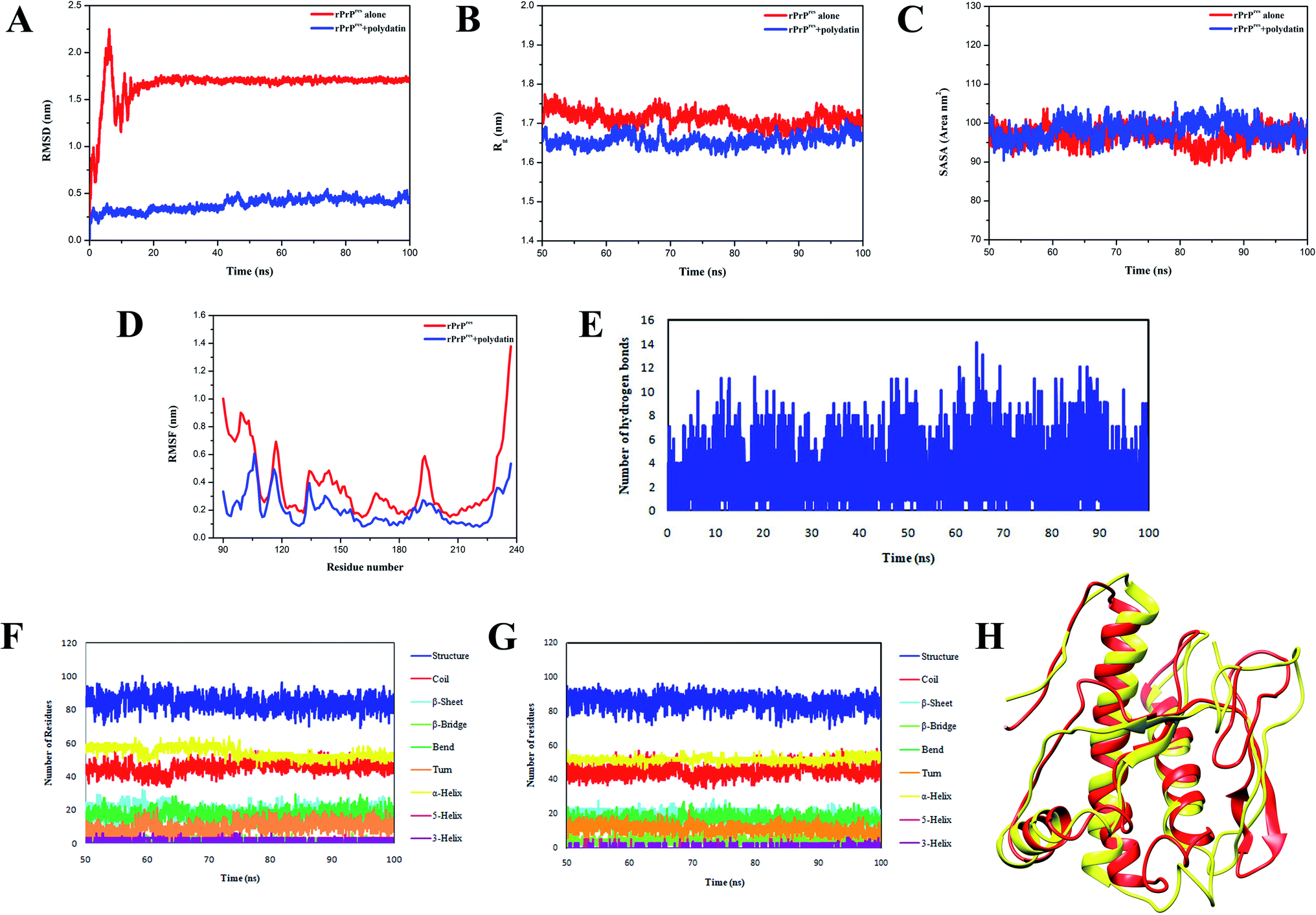 | ||
| Fig. 3 Analyzing molecular dynamics simulation trajectories for studying structural variations. (A) Depiction of backbone RMSD variations of rPrPres alone (red traces) and rPrPres polydatin complex (blue traces) for the simulation time period (100 ns). (B) and (C) Shows the variations in radius of gyration (Rg) and solvent accessible surface area (SASA), respectively, in case of rPrPres alone (red traces) and rPrPres polydatin complex (blue traces) for the stable time frame (last 50 ns). (D) Root mean square fluctuations (RMSF) plot for studying the residual fluctuations. (E) Variations in hydrogen bond numbers for the rPrPres-polydatin complex during the simulation trajectory. Secondary structural variations in rPrPres structure (F) and rPrPres-polydatin complex (G) observed for the stable trajectory of all-atomistic MD simulations. Colored traces represent various secondary structural elements. (H) Structural view of the prion protein through cartoon depiction of simulated and superposed rPrPres alone (red) and rPrPres-polydatin complex (yellow) depicting the changes observed in absence and presence of polydatin after 100 ns of MD simulation run. | ||
The variations in intramolecular H-bonds (depicted in Fig. 3E) show the formation of more than 10 intermolecular hydrogen bonds on an average which contribute in stabilizing the prion–polydatin complex. Further, the native prion structure and docked polydatin ensemble were examined for any changes in secondary structural elements. On assessing the variations in secondary structural content, we observed a decrease in β-sheet content in prion structure in the presence of polydatin (Fig. 3F and G). A comparison between the stabilized structure of prion protein alone and the polydatin ensemble obtained at the end of simulations was performed. The overlapping structures were analyzed and the conformational alterations have been illustrated in Fig. 3H showing conversion of β-sheet to random coil in the presence of polydatin. These results further motivated us to probe the effect of polydatin on the amyloid aggregation and fibrillization of rPrPres.
2.2 Polydatin inhibits rPrPres fibrillation stemming aggregates of lower order
The kinetics of rPrPres fibrillation incubated with and without polydatin was examined using thioflavin T (a benzothiazole dye) binding assay, which has a tendency to strongly fluoresce upon binding with amyloid fibrils' signature cross-β-sheet structures.29 rPrPres aggregating alone follows atypical sigmoidal kinetics initiated by a lag phase of 30 h followed by an exponential phase which saturates after almost 120 h, marking the formation of mature amyloid fibrils. rPrPres was incubated with increasing concentrations of polydatin (30–150 μM), resulting in a concentration-dependent decline in ThT fluorescence [Fig. 4A], this decline was evident even at lowest concentrations. There was almost a 12-fold decrease in the magnitude of ThT fluorescence upon binding with rPrPres aggregates which were formed in the absence of polydatin at the saturation phase of ThT kinetics [Fig. 4B]. The binding of polydatin plausibly affects the primary nucleation event in rPrPres aggregation, which is evident by a significant increase in the lag phase and a remarkable drop in the rate of aggregation kinetics.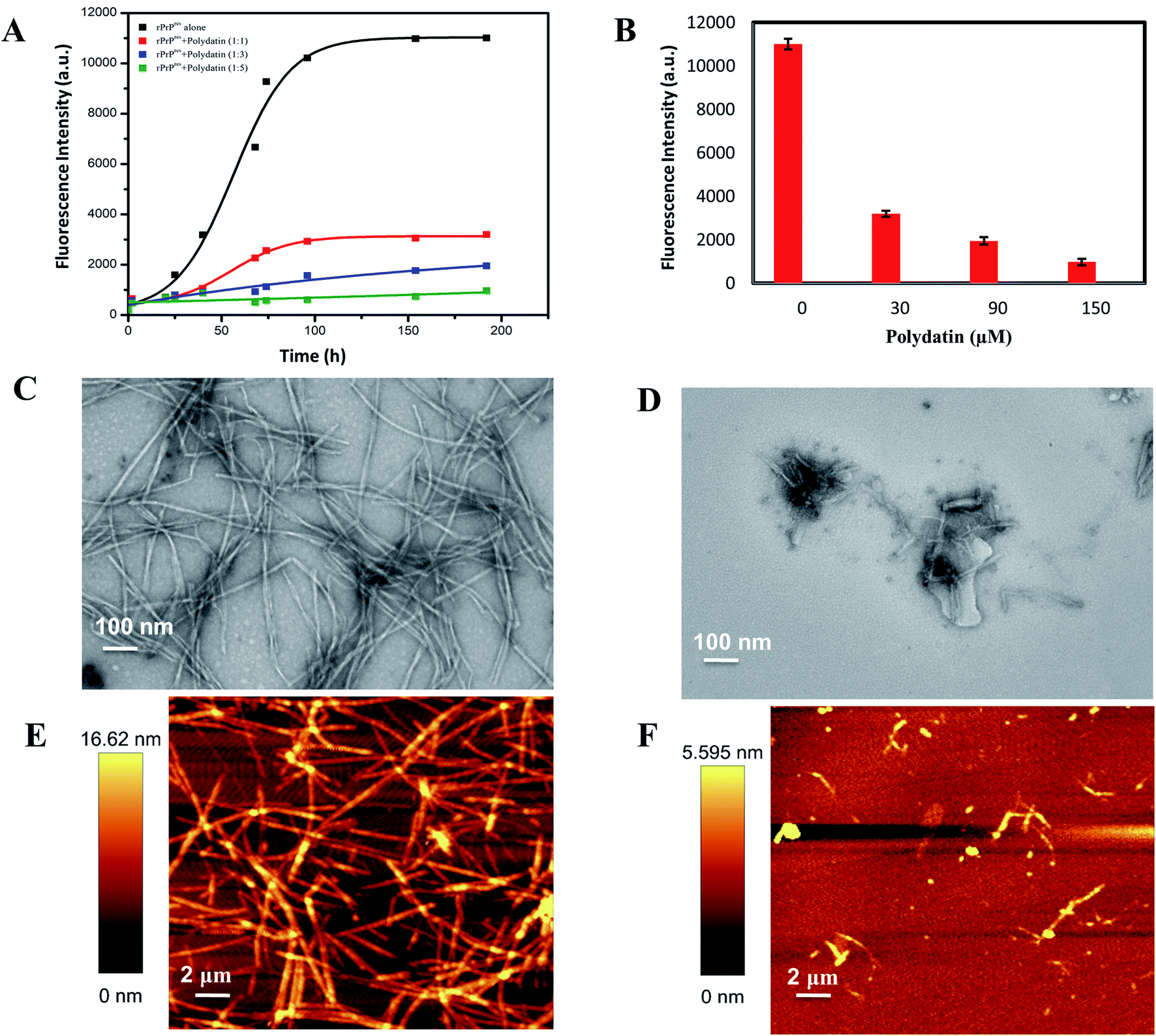 | ||
| Fig. 4 Effect on fibrillation kinetics and morphology. (A) Change in ThT fluorescence plotted as a function of time to represent aggregation kinetics of rPrPres alone (black traces) and in the presence of different concentrations of polydatin (color traces). The aggregation kinetics of rPrPres followed a sigmoidal transition and a dose-dependent abrogation in ThT fluorescence was observed in the presence of polydatin. (B) Corresponding suppression of ThT intensity upon increasing the concentration of polydatin. Electron micrographs of rPrPres aggregates formed (C) alone and (D) in the presence of polydatin. Corresponding AFM images of rPrPres aggregates formed (E) alone and (F) in the presence of polydatin after 192 h of incubation. Scale bars represented are 100 nm and 2 μm for TEM images and AFM images, respectively. | ||
To confirm that reduction in ThT fluorescence in presence of polydatin is because of inhibition of fibril formation rather than interference between polydatin and ThT, quenching of ThT was observed in presence of polydatin in reaction buffer, which showed insignificant fluorescence during fibrillation time-intervals. This negates the possibility of ThT fluorescence quenching in presence of polydatin.
The subdued ThT fluorescence in presence of polydatin motivated us to examine the structural and morphological alterations of rPrPres aggregates formed in presence of polydatin. This was studied by using transmission electron microscopy (TEM) and atomic force microscopy (AFM). A major decline in fibrillar load was evident upon incubating monomeric rPrPres with polydatin, which corroborates with the observed subdued ThT fluorescence. rPrPres aggregates formed in the presence and absence of polydatin were imaged using TEM and AFM, wherein a considerable reduction in the formation of mature amyloid fibrils was seen in the presence of polydatin. Interestingly, rPrPres aggregating alone, formed intertwined, laterally stacked, mature fibrils having an average height of ∼16.6 nm [Fig. 4C and E].
Conversely, no mature fibrils were observed in the presence of polydatin. Large, rod-shaped, feeble fibrillar structures having an average height of 5.6 nm were seen, along with sparsely populated off pathway oligomeric aggregates (Fig. 4D and F). A significant decrease in fibrillation of rPrPres by polydatin at sub-stoichiometric ratios and the recognized use of stilbene derivatives underline the relevance of these findings at physiological levels.
To further ascertain the inhibitory potential of polydatin and validate its effect on rPrPres fibrillation, we performed dynamic light-scattering (DLS) experiments. The DLS data collected from aggregating rPrPres alone indicated a gradual increment in the particle size with evidently distinguishable populations at 0 h and 192 h. The ones at 0 h have hydrodynamic radii (RH) of 0–5 nm and the end-stage aggregates obtained at saturation phase at 192 h have relatively higher RH, beyond 500 nm [Fig. 5A], depicting aggregates of higher-order. The DLS data substantiated our ThT kinetics and TEM data, where small oligomeric intermediates were observed at the end of the lag phase, which were later organized into large fibrils at the end of the exponential phase, confirming the on-pathway aggregation of rPrPres. Interestingly, the RH of rPrPres incubated with polydatin showed relatively slower diffusion into higher-order aggregates. At 192 h, the inhibitor-treated sample showed a heterogeneous particle size distribution of 50–500 nm [Fig. 5B]. Conclusively, the study of particle size distribution during rPrPres aggregation and gradual appearance of higher order aggregates corroborated their on-pathway nature that substantially gets averted in the presence of polydatin. These findings also authenticate the AFM and TEM data that negated the formation of higher order mature fibrillar species in polydatin incubated rPrPres samples.
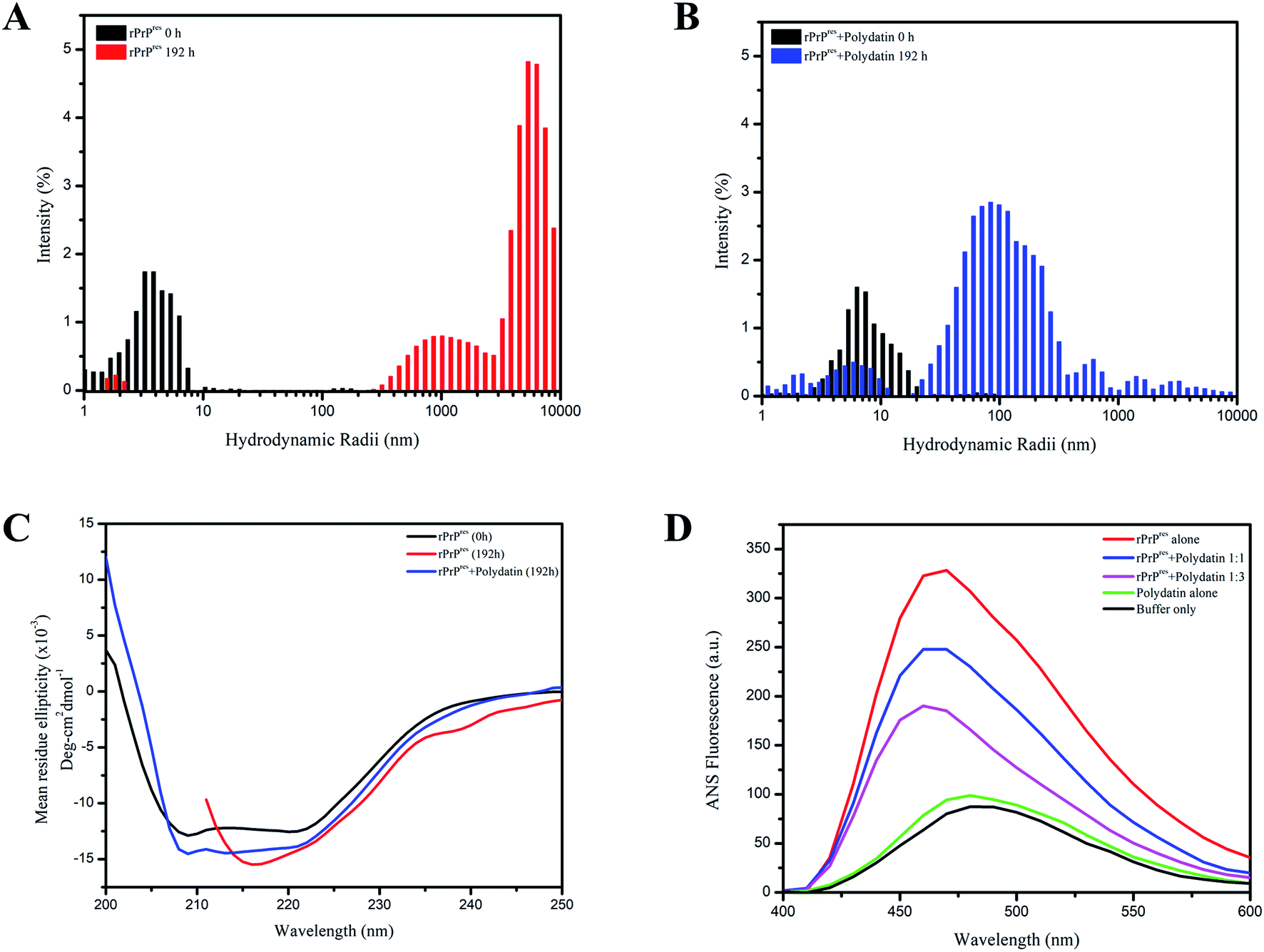 | ||
Fig. 5 Effect on size distribution, secondary structure and hydrophobicity. Particle size distributions of the aggregating species are portrayed as colored bars for rPrPres alone ((A); black 0 h, red 192 h) and in the presence of polydatin ((B); black 0 h, blue 192 h). (C) Far-UV CD spectra of native rPrPres and aggregates formed after 192 h incubation in the absence (black) and presence of polydatin (red). CD spectrum of native, non-aggregated rPrPres (blue) is also displayed. (D) Surface hydrophobicity assessment using ANS fluorescence of rPrPres alone (red trace) and in the presence of different concentrations of polydatin (blue and brown trace). ANS spectra of buffer only (black trace) and polydatin only (green trace) are also shown. The ratio of ANS![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :protein was 5:1 with an ANS excitement wavelength of 350 nm. The spectra were recorded with excitation and emission slit widths of 5 nm each between 400 and 600 nm and a scan rate of 50 nm min−1. :protein was 5:1 with an ANS excitement wavelength of 350 nm. The spectra were recorded with excitation and emission slit widths of 5 nm each between 400 and 600 nm and a scan rate of 50 nm min−1. | ||
2.3 Polydatin-induced off-pathway rPrPres aggregates exhibit low cross-β content and retain a lower surface hydrophobicity
The morphological and size distribution of polydatin incubated samples reveals heterogeneous aggregates indicating altered aggregation kinetics owing to stabilization of rPrPres native conformation polydatin. This ultimately impedes the conversion of native rPrPres structures into a misfolded and aggregation-prone state. It was now important to understand the secondary structural characteristics of this altered aggregation and resultant aggregates. Previous studies have authenticated that the scrapie-like conversion of prion protein is an outcome of conformational transition of its chief helical architecture (α1–α3) into a β-sheet rich structure.30 Thus, to establish any such secondary structural variations, we performed far-UV CD analysis of both types of aggregates. The far-UV CD spectra of monomeric and unaggregated rPrPres revealed a characteristic double minimum at 210 and 222 nm validating its majorly α-helical architecture.31 On the contrary, saturation phase rPrPres aggregation (192 h) displayed a single minimum at 218 nm representative of their β-sheet rich conformation, as observed in mature amyloid aggregates. The far-UV CD spectra of rPrPres monitored after 192 h of fibrillation showed a shift in the negative ellipticity, indicating a structural transition from the natively-unfolded conformation to a characteristic β-sheet structure. Interestingly, this change in secondary structure was not observed in presence of polydatin, which showed retention of the 222 nm peak indicating the presence of a residual helical-component and negated the formation of β-sheet rich structures [Fig. 5C].It is a well-known fact that amyloid fibrils have a characteristic feature of β-sheet formation. An important physicochemical property known to be involved in propelling the development of β-sheet structures is an increase in the hydrophobicity which can lead to the accumulation of oligomeric species that are highly toxic in nature.32 The amount of hydrophobic surface area exposed in case of rPrPres was estimated via ANS binding assay. 1-Anilino-8-naphthalenesulfonic acid (ANS) is a solvent-sensitive dye having a property of fluorescing and giving a blue-shift in emission when it binds with the hydrophobic surroundings. Elevated fluorescence intensity along with shifts towards lower wavelengths is due to the interaction of ANS with the hydrophobic patches of proteins that are solvent-exposed. ANS fluorescence spectra were measured in the absence and presence of polydatin, which showed a consequent rise in the intensity of fluorescence with a typical blue-shifted emission (λem at 470 nm) in non-treated rPrPres fibrils. Emission spectra of ANS achieved in case of polydatin treated rPrPres fibrils were shifted towards blue with a reduced intensity [Fig. 5D]. The intensity of ANS fluorescence was increased under fibrillating conditions which signifies the development of a globule-like molten form that is necessary for fibril formation by intrinsically disordered proteins. These were absent in polydatin treating rPrPres samples, thus demonstrating that an overall low surface hydrophobicity was maintained by the polydatin modulated species. Previous studies have reported the importance of hydrophobicity and aromaticity in amyloid assembly and fibrillation.33–36 The subdued hydrophobicity in presence of polydatin further validates its potential to be used as a prion intervening scaffold and therapeutic agent which may help in abrogating the amyloid assembly of aggregating monomers by binding to its aromatic and hydrophobic patches.
3. Materials and methods
3.1 Peptide preparation
The crystal structure of human prion protein was obtained in from RCSB Protein Data Bank, PDB code: 2KUN.37 Protein preparation was done using Protein Preparation Wizard available at Schrodinger, wherein overlapping coordinates have been corrected and missing atoms were added to obtain a monomeric form. Acetylation and amidation of the N-termini and C-termini of the peptide were carried out to reduce the risk of abrupt results due to uncapped and non-neutralized terminals.3.2 Molecular docking and scoring
Next, the receptor grid generation platform of Schrodinger's Glide module was used to generate a grid of size 20 × 20 × 20 Å around the literature reported binding pocket.38 Molecular docking of polydatin with the above-described grids was performed using the Glide module of Schrodinger.3.3 Molecular dynamics simulation
All-atomistic MD simulations were performed on the highest scoring ensemble obtained through docking using the GROMACS 5.0 package.39 The GROMACS compatible topology parameters for ligand polydatin were generated using PRODRG server.40 The system was immersed in a cubic box using simple point charge (SPC) water model and counter ions were added to neutralize the overall charge on the system. Energy minimization of the system was carried out using steepest descent algorithm for 50000 steps until it attained the maximum force <10.0 kJ mol−1 nm−1. The long-range electrostatic interactions were treated using particle mesh Ewald method.41 The energy minimized system was further subjected to NVT and NPT equilibration for 100 ps at a constant temperature set to 300 K and 1 bar constant pressure. During equilibration, v-rescale, a modified Berendsen thermostat42 was used for temperature coupling along with Parrinello–Rahman barostat for pressure coupling.43 The resulting system was subjected to 100 ns MD production run using leap-frog integrator44 with 2 fs integration time step. The linear constraint solver (LINCS) algorithm was used for restraining the bond lengths.45 The trajectories were saved every 10 ps and further structural analyses were done using various inbuilt GROMACS scripts, PyMol, chimera and LigPlot+.46–48
3.4 Protein purification
Human prion protein corresponding to the residues 90–231 (rPrPres), was expressed as a recombinant, his-tagged protein and purified by on-column refolding protocol. Briefly, the inclusion bodies were solubilized by sonication in lysis buffer (100 mM Na2PO4, 10 Tris–Cl, 8 M urea, and 10 mM reduced glutathione, pH 8.0) and loaded on pre-equilibrated Ni-NTA column. Following a slow decreasing gradient of urea (0–8 M) on-column, the folded protein was eluted in 0.5 M imidazole (pH 5.8). The eluted protein was dialyzed overnight against 100 mM Na2HPO4 (pH 5.8) followed by dialysis against Milli-Q. The dialyzed protein solution was filtered, lyophilized and stored in −20 °C until further use. The concentration of rPrPres was determined using the extinction coefficient at 280 nm (ε280 = 22015 M−1 cm−1).38,49
3.5 Amyloid aggregation experiments
The monomeric rPrPres was added at a 30 μM final concentration in aggregation buffer [50 mM phosphate buffer (pH 7.4); 2 M GdnHCl; 0.02% (v/v) NaN3] and incubated at 37 °C in 1.5 mL tubes with continuous agitation at 200 rpm. Aggregation of untreated control and in presence of increasing molar concentrations of polydatin was monitored by standard ThT fluorescence assay and electron microscopy.3.6 SPR binding kinetics analysis
The binding analysis of rPrPres and polydatin was carried out using a BIAcore X100 instrument operating at 25 °C temperature (GE Healthcare. USA). Polydatin with a purity of >95% was purchased from Sigma Life Sciences. A CM5 sensor chip (GE Healthcare) having a self-assembled monolayer of carboxymethylated dextran covalently attached to a gold surface was activated using an equal molar concentration mix of EDC [N-ethyl-N-(diethylaminopropyl)carbodiimide] and NHS (N-hydroxysuccinimide). Coupling of the ligand was carried out in presence of sodium acetate buffer at pH 5.8 and blocking was completed using ethanolamine as per manufacturers' protocol. The final coupling level achieved for rPrPres was around 2700 resonance units (RU). Two times priming of the system was done using a running buffer [20 mM phosphate buffer, pH 7.4] followed by regeneration which was performed using a 30 s pulse of the regeneration buffer [10 mM glycine, pH 2.5] with a flow rate maintained at 30 μL min−1. One of the two flow cells acted as a reference surface that was blocked using ethanolamine for monitoring the probable non-specific binding of the analyte (polydatin). The final dilution of polydatin was made in the running buffer and it was injected over both flow cells. Interaction analysis of polydatin with rPrPres was accomplished by injecting varying concentrations of the compound (0.1–20 μM). The monitoring of association kinetics was done for 150 s, which was followed by 180 s of dissociation kinetics. The response obtained from the control flow cell without immobilized ligand was subtracted from that obtained in the sample cell containing the ligand to get the final data. Dissociation constant values were assessed using BIA evaluation 4.1 software (BIAcore) wherein Langmuir 1:1 binding model was used for fitting the obtained data.3.7 Thioflavin T (ThT) fluorescence assay
The aggregation kinetics of rPrPres was examined via thioflavin T (ThT) fluorescence assay. A stock solution of 10 μM ThT was prepared in phosphate buffer (50 mM at pH 7.4). Aliquots of 10 μL samples were withdrawn at different time points and uniformly mixed with 90 μL of ThT solution. The mixture was incubated for 10 min at RT before taking measurements using a 1 cm pathlength quartz cuvette with excitation and emission wavelengths fixed at 450 nm and 485 nm (slit width for excitation and emission were set at 5 nm and 10 nm respectively) in a LS 55 fluorescence spectrometer (PerkinElmer, USA). For each reaction, average fluorescence from triplicate reactions was fitted using Boltzmann-sigmoidal equation in Origin software.3.8 Transmission electron microscopy (TEM)
TEM imaging to study morphology of the aggregates was done using a JEM-2100F transmission electron microscope (JEOL Inc., USA) operating at 200 kV. The samples were three-fold diluted prior to being adsorbed onto formvar-coated carbon grids (300 mesh, Ted Pella, Inc., USA). These were incubated for 2 min followed by negative staining for 30 s with a 2% (w/v) uranyl acetate solution. The sample-coated grids were then rinsed with Milli-Q water and imaged after air drying.3.9 Atomic force microscopy (AFM)
The topographical analysis of aggregates was carried out using a Bioscope Catalyst AFM (Bruker Corporation, Billerica, MA, USA). The samples were three-fold diluted and layered over a freshly cleaved mica surface. After an incubation period of 30 min, the samples were washed with Milli-Q and blown dried under a nitrogen stream. All images were acquired at ambient temperature using Scan Asyst Peak Force Tapping mode with 512 samples per line and a standard scan rate of 0.5 Hz. All images were processed using Nanoscope analysis, v.1.4.3.10 Dynamic light scattering (DLS)
All DLS measurements were performed on an Xtal-Spectrosize-300 (Marlowring, Germany) set at 25 °C. A correlation time defined as 20 seconds per run with 10 runs for every measurement was kept constant in all the samples. Samples collected at different time points were centrifuged (10000 g, 20 min) and transferred into DLS cuvette before each measurement. A total of 10 scans per sample were obtained, averaged and plotted as hydrodynamic radii (nm) versus intensity of distribution (%) of particles.
3.11 Far-UV circular dichroism (CD) measurements
Far-UV CD experiments were conducted to analyze variations in the secondary structure of protein at the end stage of aggregation. A spectrum of rPrPres (30 μg mL−1) was noted from 200–250 nm wavelengths at 25 °C (1 nm interval and a 50 nm min−1 scanning speed) using a peltier device equipped Jasco-815 CD spectropolarimeter (Jasco Inc., USA). A quartz cuvette with a path length of 1 mm was used for the experiments. An average of three scans was obtained and the data was plotted in mean residue ellipticity units (degrees square centimeters per decimole).3.12 Anilino-8-naphthalenesulfonate (ANS) binding assay
ANS binding emission spectra of fluorescence were scanned in a range of 400–600 nm at 25 °C and an excitation wavelength of 370 nm. The slit widths of excitation and emission were 5 nm. A final concentration of 5 μM ANS was added to the 90 μL aliquots of aggregated reaction mixture which were incubated at 25 °C for 5 min before taking the scan.4. Conclusion
Detailed analysis using biophysical assays and computational studies unraveled the important role of polydatin in prion intervention. The data demonstrated here implies that the selective inhibition of a specific, aggregated prion peptide is viable and helpful in deciphering the mechanisms underlying protein fibrillization, as well as in the therapeutic testing of neuro-degeneration models. This research may help in the development of new approaches that aid in rational drug designing to obstruct the prion pathogenesis specifically and amyloid inhibition in general.This study provides details of the varying effects of polydatin upon fibrillation pathway of human prion protein depicting that polydatin causes a considerable suppression of human prion fibrillation by abrogating the structural transitions in the N-terminal amyloidogenic region and inducing non-malicious transitions in the β2–α2 loop and α3-helix. PrP fibrillation inhibition property of polydatin can be justified through the formation of off-pathway species that are retarded kinetically and have an increased α-helical and random coil propensity. The polydatin-mediated modulation of PrP fibrillation here points to a significant role of polyphenols in disease pathogenesis suggesting that a thorough research on the effects of this modulator on fibrillation pathway of the above mentioned intrinsically disordered protein will be helpful in well understanding of the events that lead to the onset of prion-related disorders and contribute towards developing novel and productive intervention strategies.
Author contributions
PRS, AG and PS designed the methods and experimental setup. PRS performed and analyzed the biophysical experiments. AK conducted computational experiments; PRS and NA examined the data and wrote the manuscript. All authors reviewed the manuscript.Conflicts of interest
The authors declare that they have no conflicts of interest with the contents of this article.Acknowledgements
The authors thank the Jawaharlal Nehru University (JNU) and TERI School of Advanced Studies for infrastructural support. Authors are thankful to Advanced Instrumentation Research Facility (AIRF, JNU) for TEM, AFM and other facilities. PRS is thankful to Indian Council of Medical Research (ICMR), India for the senior research fellowship. AG is thankful to the University Grant Commission, India for the faculty recharge position.References
- B. Chesebro, Introduction to the transmissible spongiform encephalopathies or prion diseases, Br. Med. Bull., 2003, 66(1), 1–20 CrossRef CAS PubMed.
- I. Poggiolini, D. Saverioni and P. Parchi, Prion protein misfolding, strains, and neurotoxicity: an update from studies on Mammalian prions, Int. J. Cell Biol., 2013 DOI:10.1155/2013/910314.
- B. Caughey and B. Chesebro, Transmissible spongiform encephalopathies and prion protein interconversions, Adv. Virus Res., 2001, 56, 277–311 CrossRef CAS PubMed.
- H. A. Kretzschmar, S. B. Prusiner, L. E. Stowring and S. J. DeArmond, Scrapie prion proteins are synthesized in neurons, Am. J. Pathol., 1986, 122(1), 1–5 CAS.
- A. Aguzzi and A. M. Calella, Prions: protein aggregation and infectious diseases, Physiol. Rev., 2009, 89(4), 1105–1152 CrossRef CAS PubMed.
- R. Linden, V. R. Martins, M. A. M. Prado, M. Cammarota, I. Izquierdo and R. R. Brentani, Physiology of the prion protein, Physiol. Rev., 2008, 88(2), 673–728 CrossRef CAS PubMed.
- J. Tatzelt and K. F. Winklhofer, Folding and misfolding of the prion protein in the secretory pathway, Amyloid, 2004, 11(3), 162–172 CrossRef CAS PubMed.
- K. M. Pan, M. Baldwin, J. Nguyen, M. Gasset, A. Serban and D. Groth, et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins, Proc. Natl. Acad. Sci. U. S. A., 1993, 90(23), 10962–10966 CrossRef CAS PubMed.
- R. Zahn, A. Liu, T. Lührs, R. Riek, C. von Schroetter and F. López García, et al. NMR solution structure of the human prion protein, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(1), 145–150 CrossRef CAS PubMed.
- D. G. Donne, J. H. Viles, D. Groth, I. Mehlhorn, T. L. James and F. E. Cohen, et al. Structure of the recombinant full-length hamster prion protein PrP(29–231): The N terminus is highly flexible, Proc. Natl. Acad. Sci. U. S. A., 1997, 94(25), 13452–13457 CrossRef CAS PubMed.
- A. Corsaro, S. Thellung, V. Villa, M. Nizzari, A. Aceto and T. Florio, Recombinant human prion protein fragment 90-231, a useful model to study prion neurotoxicity, OMICS, 2012, 16(1–2), 50–59 CrossRef CAS PubMed.
- H. Liu, S. Farr-Jones, N. B. Ulyanov, M. Llinas, S. Marqusee and D. Groth, et al. Solution structure of Syrian hamster prion protein rPrP(90-231), Biochemistry, 1999, 38(17), 5362–5377 CrossRef CAS PubMed.
- X. Wu, Q. Li, Y. Feng and Q. Ji, Antitumor Research of the Active Ingredients from Traditional Chinese Medical Plant Polygonum Cuspidatum, Evid.-Based Complementary Altern. Med, 2018, 2018, 2313021 Search PubMed.
- S. De Maria, I. Scognamiglio, A. Lombardi, N. Amodio, M. Caraglia and M. Carteni, et al. Polydatin, a natural precursor of resveratrol, induces cell cycle arrest and differentiation of human colorectal Caco-2 cell, J. Transl. Med., 2013, 11, 264 CrossRef PubMed.
- G. Lanzilli, A. Cottarelli, G. Nicotera, S. Guida, G. Ravagnan and M. P. Fuggetta, Anti-inflammatory effect of resveratrol and polydatin by in vitro IL-17 modulation, Inflammation, 2012, 35(1), 240–248 CrossRef CAS PubMed.
- G. Ravagnan, A. De Filippis, M. Carteni, S. De Maria, V. Cozza and M. Petrazzuolo, et al. Polydatin, a natural precursor of resveratrol, induces beta-defensin production and reduces inflammatory response, Inflammation, 2013, 36(1), 26–34 CrossRef CAS PubMed.
- H. Ji, X. Zhang, Y. Du, H. Liu, S. Li and L. Li, Polydatin modulates inflammation by decreasing NF-κB activation and oxidative stress by increasing Gli1, Ptch1, SOD1 expression and ameliorates blood–brain barrier permeability for its neuroprotective effect in pMCAO rat brain, Brain Res. Bull., 2012, 87(1), 50–59 CrossRef CAS PubMed.
- R.-P. Li, Z.-Z. Wang, M.-X. Sun, X.-L. Hou, Y. Sun and Z.-F. Deng, et al. Polydatin protects learning and memory impairments in a rat model of vascular dementia, Phytomedicine, 2012, 19(8–9), 677–681 CrossRef CAS PubMed.
- Y. Gao, T. Chen, X. Lei, Y. Li, X. Dai and Y. Cao, et al. Neuroprotective effects of polydatin against mitochondrial-dependent apoptosis in the rat cerebral cortex following ischemia/reperfusion injury, Mol. Med. Rep., 2016, 14(6), 5481–5488 CrossRef CAS PubMed.
- C. Rivière, T. Richard, L. Quentin, S. Krisa, J.-M. Mérillon and J.-P. Monti, Inhibitory activity of stilbenes on Alzheimer's beta-amyloid fibrils in vitro, Bioorg. Med. Chem., 2007, 15(2), 1160–1167 CrossRef PubMed.
- C. Rivière, Y. Papastamoulis, P.-Y. Fortin, N. Delchier, S. Andriamanarivo and P. Waffo-Teguo, et al. New stilbene dimers against amyloid fibril formation, Bioorg. Med. Chem. Lett., 2010, 20(11), 3441–3443 CrossRef PubMed.
- D. Şöhretoğlu, M. Y. Baran, R. Arroo and A. Kuruüzüm-Uz, Recent advances in chemistry, therapeutic properties and sources of polydatin, Phytochem. Rev., 2018, 17(5), 973–1005 CrossRef.
- J.-H. Lee, J.-H. Moon, S.-W. Kim, J.-K. Jeong, U. M. Nazim and Y.-J. Lee, et al. EGCG-mediated autophagy flux has a neuroprotection effect via a class III histone deacetylase in primary neuron cells, Oncotarget, 2015, 6(12), 9701–9717 CrossRef PubMed.
- A. Freyssin, G. Page, B. Fauconneau and A. Rioux Bilan, Natural polyphenols effects on protein aggregates in Alzheimer's and Parkinson's prion-like diseases, Neural Regener. Res., 2018, 13(6), 955–961 CrossRef PubMed.
- B. E. Roberts, M. L. Duennwald, H. Wang, C. Chung, N. P. Lopreiato and E. A. Sweeny, et al. A synergistic small-molecule combination directly eradicates diverse prion strain structures, Nat. Chem. Biol., 2009, 5(12), 936–946 CrossRef CAS PubMed.
- E. Caldarulo, A. Barducci, K. Wüthrich and M. Parrinello, Prion protein β2–α2 loop conformational landscape, Proc. Natl. Acad. Sci. U. S. A., 2017, 114(36), 9617–9622 CrossRef CAS PubMed.
- T. D. Kurt, C. Bett, N. Fernández-Borges, S. Joshi-Barr, S. Hornemann and T. Rülicke, et al. Prion Transmission Prevented by Modifying the β2-α2 Loop Structure of Host PrPC, J. Neurosci., 2014, 34(3), 1022–1027 CrossRef CAS PubMed.
- C. Bett, N. Fernández-Borges, T. D. Kurt, M. Lucero, K. P. R. Nilsson and J. Castilla, et al. Structure of the β2-α2 loop and interspecies prion transmission, FASEB J., 2012, 26(7), 2868–2876 CrossRef CAS PubMed.
- H. LeVine III, Quantification of beta-sheet amyloid fibril structures with thioflavin T, Methods Enzymol., 1999, 309, 274–284 Search PubMed.
- P. K. Baral, J. Yin, A. Aguzzi and M. N. G. James, Transition of the prion protein from a structured cellular form (PrP(C)) to the infectious scrapie agent (PrP(Sc)), Protein Sci., 2019, 28(12), 2055–2063 CrossRef CAS PubMed.
- R. Banerjee and G. Basu, Direct evidence for alteration of unfolding profile of a helical peptide by far-ultraviolet circular dichroism aromatic side-chain contribution, FEBS Lett., 2002, 523(1), 152–156 CrossRef CAS PubMed.
- B. Mannini, E. Mulvihill, C. Sgromo, R. Cascella, R. Khodarahmi and M. Ramazzotti, et al. Toxicity of protein oligomers is rationalized by a function combining size and surface hydrophobicity, ACS Chem. Biol., 2014, 9(10), 2309–2317 CrossRef CAS PubMed.
- F. Chiti, in Relative Importance of Hydrophobicity, Net Charge, and Secondary Structure Propensities in Protein Aggregation BT - Protein Misfolding, Aggregation, and Conformational Diseases: Part A: Protein Aggregation and Conformational Diseases, ed. Uversky V. N. and Fink A. L., Springer, Boston, MA, USA, 2006, pp. 43–59 Search PubMed.
- J. H. M. van Gils, E. van Dijk, A. Peduzzo, A. Hofmann, N. Vettore and M. P. Schützmann, et al. The hydrophobic effect characterises the thermodynamic signature of amyloid fibril growth, PLoS Comput. Biol., 2020, 16(5), e1007767 CrossRef CAS PubMed.
- S. M. Tracz, A. Abedini, M. Driscoll and D. P. Raleigh, Role of Aromatic Interactions in Amyloid Formation by Peptides Derived from Human Amylin, Biochemistry, 2004, 43(50), 15901–15908 CrossRef CAS PubMed.
- K. E. Marshall, K. L. Morris, D. Charlton, N. O'Reilly, L. Lewis and H. Walden, et al. Hydrophobic, Aromatic, and Electrostatic Interactions Play a Central Role in Amyloid Fibril Formation and Stability, Biochemistry, 2011, 50(12), 2061–2071 CrossRef CAS PubMed.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- N. Admane, A. Srivastava, S. Jamal, B. Kundu and A. Grover, Protective Effects of a Neurohypophyseal Hormone Analogue on Prion Aggregation, Cellular Internalization, and Toxicity, ACS Chem. Neurosci., 2020, 2422–2430 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith and B. Hess, et al. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers, SoftwareX, 2015, 1–2, 19–25 CrossRef.
- D. M. F. van Aalten, R. Bywater, J. B. C. Findlay, M. Hendlich, R. W. W. Hooft and G. Vriend, PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules, J. Comput.-Aided Mol. Des., 1996, 10(3), 255–262 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, Particle mesh Ewald: An N.log(N) method for Ewald sums in large systems, J. Chem. Phys., 1993, 98(12), 10089–10092 CrossRef CAS.
- A. S. Lemak and N. K. Balabaev, On The Berendsen Thermostat, Mol. Simul., 1994, 13(3), 177–187 CrossRef CAS.
- M. Parrinello and A. Rahman, Polymorphic transitions in single crystals: A new molecular dynamics method, J. Appl. Phys., 1981, 52(12), 7182–7190 CrossRef CAS.
- W. F. Van Gunsteren and H. J. C. Berendsen, A Leap-frog Algorithm for Stochastic Dynamics, Mol. Simul., 1988, 1(3), 173–185 CrossRef.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, LINCS: A linear constraint solver for molecular simulations, J. Comput. Chem., 1997, 18(12), 1463–1472 CrossRef CAS.
- L. L. C. Schrodinger, The PyMOL Molecular Graphics System, Version 1.8, 2015 Search PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt and E. C. Meng, et al. UCSF Chimera--a visualization system for exploratory research and analysis, J. Comput. Chem., 2004, 25(13), 1605–1612 CrossRef CAS PubMed.
- R. A. Laskowski and M. B. Swindells, LigPlot+: multiple ligand-protein interaction diagrams for drug discovery, J. Chem. Inf. Model., 2011, 51(10), 2778–2786 CrossRef CAS PubMed.
- A. Srivastava, S. Sharma, S. Sadanandan, S. Gupta, J. Singh and S. Gupta, et al. Modulation of prion polymerization and toxicity by rationally designed peptidomimetics, Biochem. J., 2016, 474(1), 123–147 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |