
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal ion size profoundly affects H3glyox chelate chemistry†
Neha Choudhary ab,
Kendall E. Barrettc,
Manja Kubeild,
Valery Radchenkobe,
Jonathan W. Englec,
Holger Stephand,
María de Guadalupe Jaraquemada-Peláez*a and
Chris Orvig*a
ab,
Kendall E. Barrettc,
Manja Kubeild,
Valery Radchenkobe,
Jonathan W. Englec,
Holger Stephand,
María de Guadalupe Jaraquemada-Peláez*a and
Chris Orvig*a
aMedicinal Inorganic Chemistry Group, Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: orvig@chem.ubc.ca; mdgjara@chem.ubc.ca
bLife Sciences Division, TRIUMF, 4004 Wesbrook Mall, Vancouver, British Columbia V6T 2A3, Canada
cDepartment of Medical Physics, University of Wisconsin, 1111 Highland Avenue, Madison, WI 53711, USA
dInstitute of Radiopharmaceutical Cancer Research, Helmholtz-Zentrum Dresden-Rossendorf, Bautzner Landstraße 400, D-01328 Dresden, Germany
eDepartment of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada
First published on 27th April 2021
Abstract
The bisoxine hexadentate chelating ligand, H3glyox was investigated for its affinity for Mn2+, Cu2+ and Lu3+ ions; all three metal ions are relevant with applications in nuclear medicine and medicinal inorganic chemistry. The aqueous coordination chemistry and thermodynamic stability of all three metal complexes were thoroughly investigated by detailed DFT structure calculations and stability constant determination, by employing UV in-batch spectrophotometric titrations, giving pM values (pM = −log[Mn+]free when [Mn+] = 1 μM, [L] = 10 μM at pH 7.4 and 25 °C) – pCu (25.2) > pLu (18.1) > pMn (12.0). DFT calculated structures revealed different geometries and coordination preferences of the three metal ions; notable was an inner sphere water molecule in the Mn2+ complex. H3glyox labels [52gMn]Mn2+, [64Cu]Cu2+ and [177Lu]Lu3+ at ambient conditions with apparent molar activities of 40 MBq μmol−1, 500 MBq μmol−1 and 25 GBq μmol−1, respectively. Collectively, these initial investigations provide insight into the effects of metal ion size and charge on the chelation with the hexadentate H3glyox and indicate that further investigations of the Mn2+–H3glyox complex in 52g/55Mn-based bimodal imaging might be worthwhile.
Introduction
The radiotracer principle was first described by George de Hevesy, who stated that radiopharmaceuticals are administered at such low molar masses that they can participate in biological processes and facilitate imaging, but they do not perturb native biochemistry.1 Positron emission tomography (PET) relies on β+ emission of radionuclides; the emitted β+ collides with an electron leading to simultaneous emission of two γ-rays of 511 keV energy in opposite directions. This co-emission of two γ-rays leads to higher sensitivity and resolution of PET compared to single photon emission computed tomography (SPECT) which relies on the detection of unique emitted γ-rays in all directions. With the recent advent of advanced technologies in production and preparation of new radioisotopes, there is access to a large number of radiometals with varying physical (emission type and energies, half-lives) and chemical properties (oxidation state, metal hardness, size, coordination preferences) which can be harnessed for their applications in nuclear medicine.2–4ImmunoPET has emerged as an attractive imaging modality in the last decade because of the high affinity and specificity of the antibodies and their longer plasma half-life.5 Therefore, there has been burgeoning interest in longer-lived PET radiometals such as 89Zr (t1/2 = 78.4 h), 64Cu (t1/2 = 12.7 h) and 52gMn (t1/2 = 5.6 d) which match better with the biological half-lives of antibodies.6–10 Another advantage offered by manganese-based agents is their application for bimodal imaging using 55Mn (high spin quantum number, S = 5/2) which exhibits great potential in Mn-based magnetic resonance imaging (MRI) contrast agents.8,11,12 64Cu (t1/2 = 12.7 h, Eβ+ = 278 keV (19%), Eβ− = 190 keV (39%), EC (61%)) has been a radionuclide of great interest for five decades because of its unique decay scheme, which combines three types of decay (β+, β− and EC), and Auger electron emission with therapeutic potential.13 While 52gMn and 64Cu are two PET radiometals, 177Lu (t1/2 = 6.7 d) is a long lived radionuclide with its low energy β− emission (Eβ−avg = 134 keV, 100%) which makes it ideal for the treatment of small tumors using radiotherapy and the two useful γ-emissions (Eγ1 = 208 keV (10.41%) and Eγ2 = 113 keV (6.23%)) for SPECT imaging.14 Lutathera ([177Lu][Lu-DOTA-TATE]) is a recently FDA-approved 177Lu-based-radiopharmaceutical for the treatment of somatostatin receptor positive gastroentero-pancreatic neuroendocrine tumors.15
Previously, we reported a versatile bisoxine hexadentate chelator, H3glyox (Chart 1); its metal complexes (Sc3+, Ga3+ and In3+) displayed excellent thermodynamic stability in solution and against in vitro human serum challenge.16 Rapid radiolabeling of [44Sc]Sc3+, [68Ga]Ga3+ and [111In]In3+ at ambient conditions (RT, 15 min, pH 7) was observed with high specific activity and quantitative yields. Herein, we investigate the complexation ability of H3glyox with the divalent metal ions (Mn2+ and Cu2+) and trivalent metal ion (Lu3+) to gauge the effect of metal ion size, oxidation state and varying coordination preferences on the sequestering capabilities of H3glyox. All three metal ions (Mn2+, Cu2+ and Lu3+) are medicinally relevant but have very distinct chemical properties. Manganese and copper are first row transition metal ions with ionic radii of 0.83 Å (Mn2+, CN 6, high spin) and 0.73 Å (Cu2+, CN 6) respectively; while Lu3+ is the largest metal ion studied with H3glyox with an ionic radius 0.86 Å (CN 6).17 These results are compared with the previous study16 to draw conclusions across the periodic table for the applicability of this unique ligand.
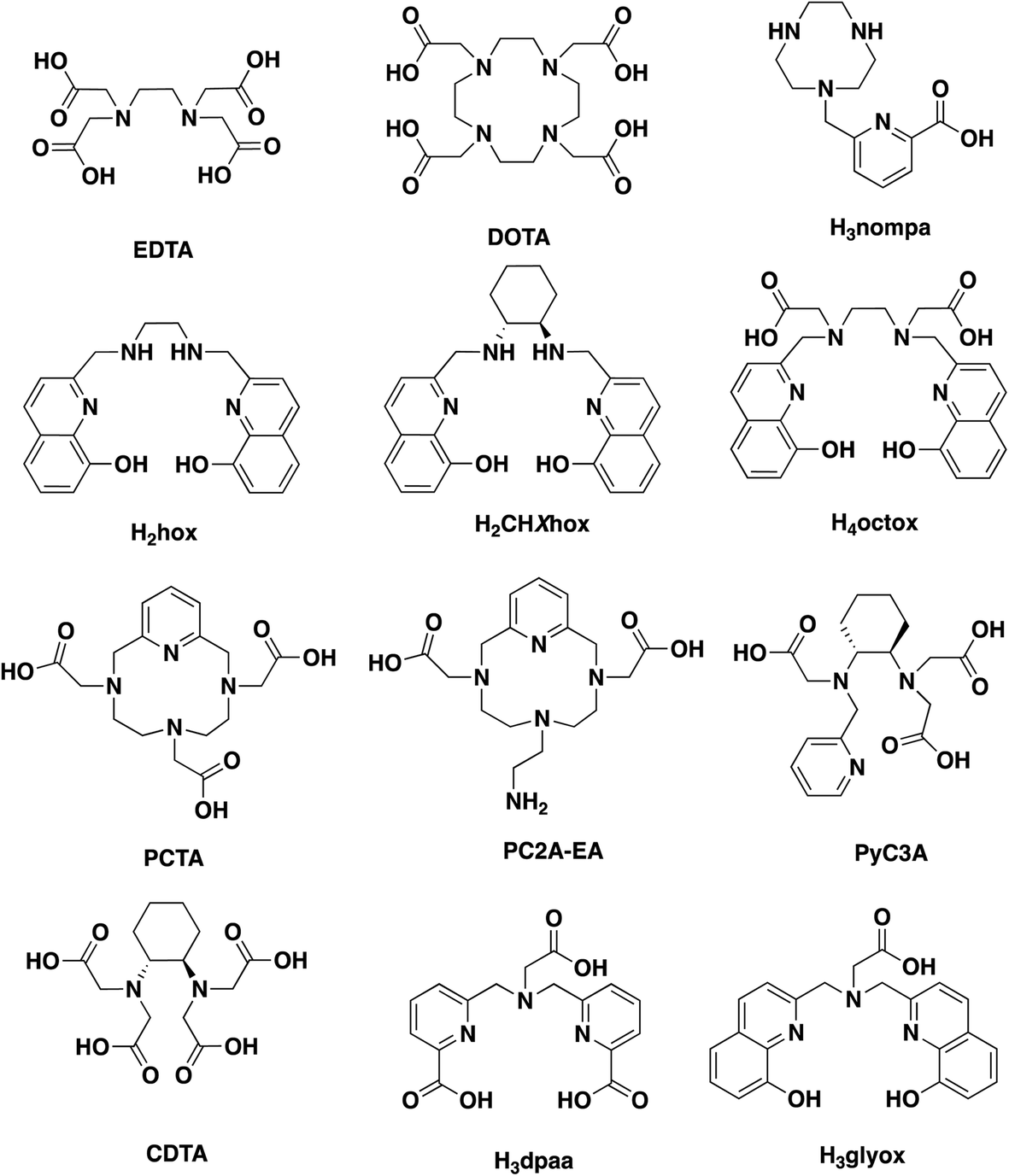 | ||
| Chart 1 Structures of discussed chelators. | ||
Results and discussion
Synthesis and characterisation
An easier modular higher yielding synthetic route is reported here for H3glyox from commercially available starting materials (Scheme 1). The compound 2-(bromomethyl)quinolin-8-ol (2) was prepared in two steps which involved a reduction of the 8-hydroxyquinoline-2-aldehyde using NaBH4 to give compound 1, which was then brominated using PBr3. This synthetic scheme provides a convenient method for the preparation of the alkylating compound 2 and overcomes the limitations of free radical bromination.18,19 Another advantage offered by this synthetic route is that it makes possible functionalised derivatives of the ligand by using other amino acids such as p-NO2–phenylalanine and N-alkylation using 2 in presence of base (K2CO3) and catalytic amount of KI. Compound 2 is not very stable at room temperature and is prone to decomposition; therefore, it is recommended to use it immediately. No additional column purification was required for the synthesis of compound 2 and the reactions provided high yields. H3glyox was prepared in overall 90% yield by SN2 reaction of glycine ethyl ester and compound 2 in basic conditions, and subsequent ethyl group deprotection under acidic conditions.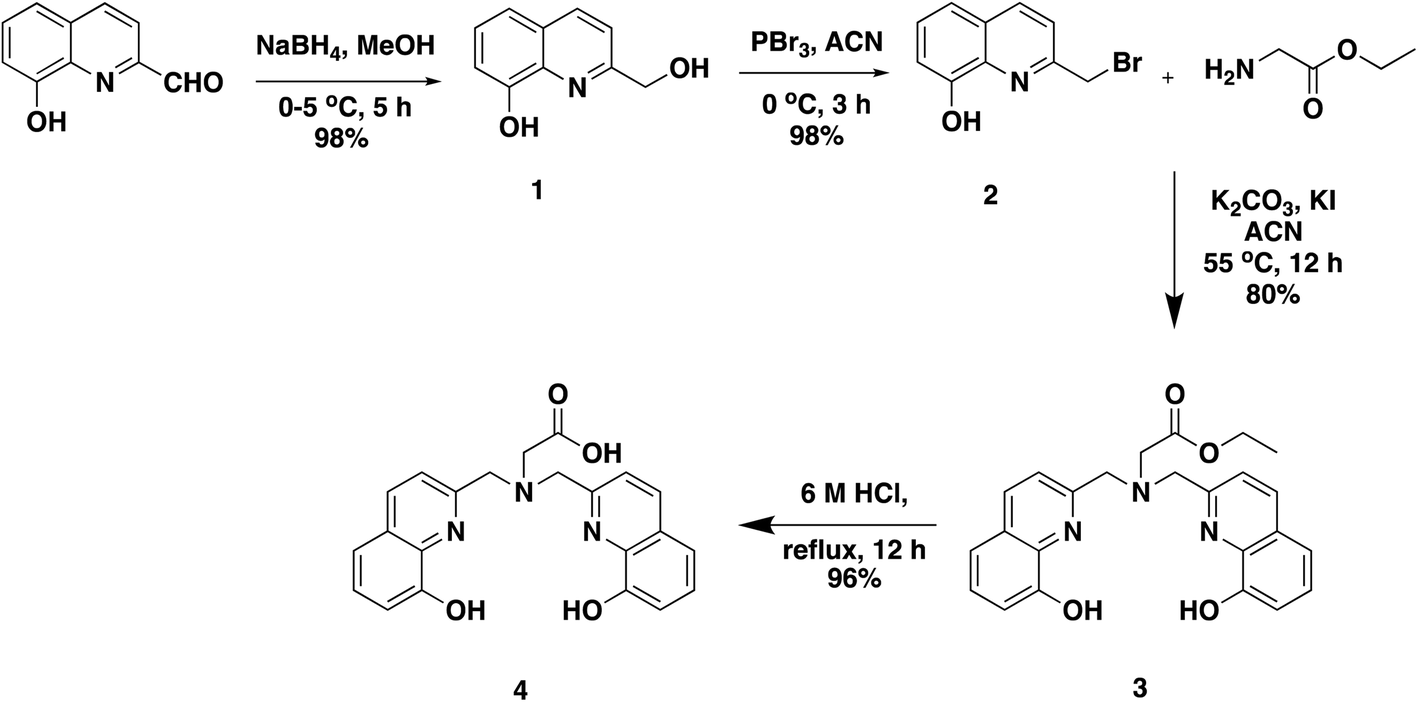 | ||
| Scheme 1 Improved high yield modular synthetic scheme for H3glyox, 4. | ||
Metal complexes
Equimolar 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 concentration solutions of the ligand H3glyox and the corresponding metal salts (MnCl2, CuSO4 and Lu(NO3)3) dissolved in water/D2O were mixed together and the pH was adjusted to 7 using 0.1 M NaOH to give the metal complexes. LR-MS and HR-MS were used to confirm the formation of the complexes, along with the change in fluorescence of the ligand observed under UV light. The addition of CuSO4 to the ligand solution led to the quenching of the fluorescence which was expected on Cu2+ complexation,20 meanwhile no changes in fluorescence intensity were observed on addition of MnCl2,21 but there was a drastic increase in fluorescence intensity on complexation of Lu3+ ion. In Fig. 1, the changes in chemical shift of the protons of the ligand on complexation with Lu3+ ion can be noted in the 1H NMR spectrum of the Lu3+–H3glyox complex but the complete coordination of Lu3+ ion remains inconclusive; there was no success in obtaining 13C NMR or 2D NMR spectra due to the poor solubility of the complex, leading to precipitation over time. Comparing the 1H NMR spectra of the Ga3+–H3glyox and Lu3+–H3glyox systems, it can be hypothesized that due to the larger ionic size of Lu3+ (0.86 Å) as compared to smaller Ga3+ (0.62 Å) ion, the Lu3+ ion does not fit well in the cavity of the ligand and forms an asymmetric complex.16
:1 concentration solutions of the ligand H3glyox and the corresponding metal salts (MnCl2, CuSO4 and Lu(NO3)3) dissolved in water/D2O were mixed together and the pH was adjusted to 7 using 0.1 M NaOH to give the metal complexes. LR-MS and HR-MS were used to confirm the formation of the complexes, along with the change in fluorescence of the ligand observed under UV light. The addition of CuSO4 to the ligand solution led to the quenching of the fluorescence which was expected on Cu2+ complexation,20 meanwhile no changes in fluorescence intensity were observed on addition of MnCl2,21 but there was a drastic increase in fluorescence intensity on complexation of Lu3+ ion. In Fig. 1, the changes in chemical shift of the protons of the ligand on complexation with Lu3+ ion can be noted in the 1H NMR spectrum of the Lu3+–H3glyox complex but the complete coordination of Lu3+ ion remains inconclusive; there was no success in obtaining 13C NMR or 2D NMR spectra due to the poor solubility of the complex, leading to precipitation over time. Comparing the 1H NMR spectra of the Ga3+–H3glyox and Lu3+–H3glyox systems, it can be hypothesized that due to the larger ionic size of Lu3+ (0.86 Å) as compared to smaller Ga3+ (0.62 Å) ion, the Lu3+ ion does not fit well in the cavity of the ligand and forms an asymmetric complex.16
 | ||
| Fig. 1 1H NMR spectra (300 MHz, room temperature, D2O, pD = 7) of the ligand H3glyox (top in red) and the Lu3+–H3glyox complex (bottom in blue). | ||
Density functional theory calculations
In the absence of crystal structures for the Ga3+, Mn2+, Cu2+ and Lu3+ complexes, and inconclusive 1H NMR of the Lu3+–H3glyox complex, DFT calculations were undertaken in order to study the coordination environment differences of the metal complexes in solution. The calculated structures are presented in Fig. 2 along with their coordination spheres. Fig. 2A demonstrates the distorted octahedral geometry (N2O4) around Ga3+ in the calculated structure of [Ga(glyox)(H2O)], the two oxine arms bind Ga3+ in a slightly distorted plane with axial coordination from the carboxylic group and trans water molecule, analogous to the crystal structure of [Ga(dpaa)(H2O)] without coordination of the tertiary N atom (Ga–N3 = 2.644 Å).22 The average Ga–O(ox) bond distances are similar for the reported crystal structure of [Ga(hox)]+ and the calculated structure of [Ga(glyox)(H2O)] with average bond distances of 1.959 Å and 2.000 Å, respectively.23 Meanwhile, the calculated average Ga–N(ox) bond distances in [Ga(glyox)(H2O)], are slightly longer than in [Ga(hox)]+ with a difference of 0.257 Å.23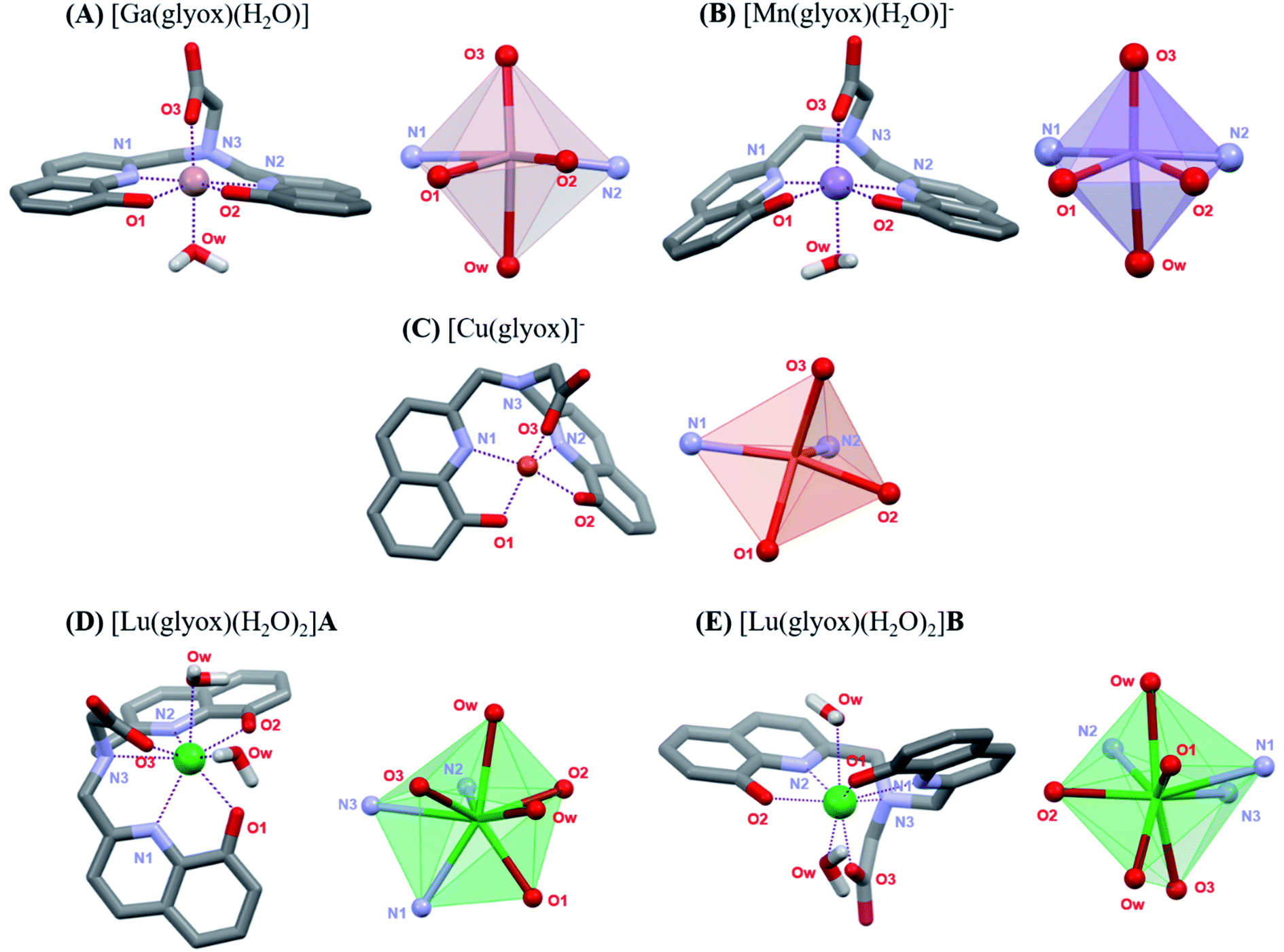 | ||
| Fig. 2 DFT calculated structures of (A) [Ga(glyox)(H2O)], (B) [Mn(glyox)(H2O)]−, (C) [Cu(glyox)]−, (D) [Lu(glyox)(H2O)2]A and (E) [Lu(glyox)(H2O)2]B; bond distances are found in Tables S5 and S6.† | ||
The calculated structure in Fig. 2B reveals that the geometry around Mn2+ is distorted octahedral with five donor atoms of the ligand and an additional O-donor atom of a water molecule. On comparing bond distances in the calculation of [Mn(glyox)(H2O)]− with those in the reported crystal structure of [Sc(glyox)(H2O)],16 it is noted that the average Mn–O(ox) and Mn–N(ox) bond distances are slightly shorter in [Mn(glyox)(H2O)]− with an average bond distance of 2.067 Å and 2.180 Å, respectively, despite the bigger size of Mn2+ (0.83 Å, CN 6)17 vs. Sc3+ (0.74 Å, CN 6).17 The bond distance of the amine Mn–N3 is somewhat longer (2.764 Å) than the analogous amine Sc–N (2.515 Å); the former is not considered as a coordinated donor atom.16 Additionally, comparison of the calculated structure of [Mn(glyox)(H2O)]− with the reported crystal structure of analogous [Mn(dpaa)(H2O)]− complex24 reveals that the average Mn–O(ox) and Mn–N(ox) bond distances are shorter than the Mn–O(pa) and Mn–N(pa) bond distances with a difference of 0.199 Å and 0.079 Å, respectively.24 It is noteworthy that the DFT calculated structures for both [Mn(glyox)(H2O)]− and [Mn(dpaa)(H2O)]− consist of an additional water molecule coordinated to the metal centre.32
Cu2+ prefers five-fold coordination with H3glyox, leading to a distorted square pyramidal geometry (Fig. 2C), in which the donor atoms N1, O1, O2 and O3 define the basal plane and the apical position is occupied by the N2 atom of the oxine arm. The inclusion of either one or three explicit water molecules in the calculations did not converge to a six-coordinate environment in either case (Fig. S17 and S18†). It has been previously reported that solvated Cu2+ ion prefers penta-coordination vs. the general assumption of octahedral solvation.25 Similarly, Cu2+ complexes of monopicolinate derivatives of cyclen and cyclam, and [Cu(nompa)]+ (Chart 1) all exhibit five-coordinate Cu2+ ion.26,27 Table S5† presents the calculated bond distances from the DFT structures. Compared to the Cu–O(ox) bond distance of 2.142 Å in the reported [Cu(hox)] structure,28 the Cu–O(ox) bond is slightly shorter in [Cu(glyox)]− complex with an average Cu–O(ox) bond distance of 2.006 Å. Due to the strong affinity of Cu2+ for the 8-hydroxyquinoline scaffold, the coordination uses the N2O2 donor set of the two oxine arms of the ligand and the O atom of the carboxylic group. The preorganization and rigidity of H3glyox leads to the longer distances for the coordination of the amine N-atom and this is reflected in the long Cu–N3 bond distance of 2.781 Å which is not considered as coordinated.
Fig. 2D and E illustrates the two geometrical isomers for [Lu(glyox)(H2O)2] (cis- and trans-diaqua) obtained through DFT calculations and the two structures exhibit a small energy difference (ΔG = 3.85 kJ mol−1); bond distances are tabulated in Table S6.† As expected, Lu3+ forms an eight-coordinate complex with the hexadentate glyox3− and two water molecules, in agreement with the reported crystal structure of the analogous [Lu(dpaa)(H2O)2] complex.29 There are no significant differences in the bond lengths for the calculated cis [Lu(glyox)(H2O)2] complex (Fig. 2D) and crystal structure of [Lu(dpaa)(H2O)2]. The calculated mean bond distances for Lu–Oox and Lu–Nox in cis [Lu(glyox)(H2O)2] are 2.256 Å and 2.431 Å, respectively; compared to the mean bond distances of 2.576 Å and 2.398 Å reported for Lu–Opa and Lu–Npa in [Lu(dpaa)(H2O)2] crystal structure.29 Due to the rigid and distorted planar structure of the ligand,16 the largest of the three metal ions studied here, Lu3+, is exposed to coordination from solvent molecules. Hence, a lesser kinetic inertness of the Lu3+–H3glyox complex can be predicted from the incorporation of the two cis or trans water molecules completing the Lu3+–H3glyox coordination sphere.
Solution thermodynamics
The evaluation of a chelator for its use in radiometal-based radiopharmaceuticals heavily relies on the determination of the thermodynamic formation constant (logKML) for the metal complex and the protonation constants of H3glyox (Table 1).16 Herein, the aim was to determine the thermodynamic stability of the Mn+–H3glyox complexes (Mn+ = Mn2+, Cu2+ and Lu3+). UV in-batch spectrophotometric titrations were applied to determine the stability constants and the results from the equilibrium studies are summarised in Table 2. Aqueous solutions of equimolar concentrations of H3glyox and each of three different metal ions were prepared at varying pH using standardised HCl and NaOH solutions, and were allowed to reach equilibrium before measuring the UV-spectra.
| Equilibrium reaction | log βa | log Ka |
|---|---|---|
| a From ref. 16.b In-batch UV spectrophotometric titrations at I = 0.16 M NaCl. | ||
| L3− + H+ ⇆ HL2− | 10.66(1)b | 10.66(1) |
| HL2− + H+ ⇆ H2L− | 20.36(1)b | 9.70(1) |
| H2L− + H+ ⇆ H3L | 27.87(2)b | 7.51(2) |
| H3L + H+ ⇆ H4L+ | 33.28(1)b | 5.41(1) |
| H4L+ + H+ ⇆ H5L2+ | 36.61(1)b | 3.33(1) |
| H5L2+ + H+ ⇆ H6L3+ | 39.27(1)b | 2.66(1) |
On comparing the extensive spectral changes in the electronic spectra of the metal complexes with the spectra of the ligand,16 the complexation is observed directly (Fig. S9–S11†). The stability constants of the metal complexes were determined using HypSpec2014 (ref. 30) by analysing the experimental data along with the molar absorptivity of the different protonated species of Hxglyox (Table 1).16 The speciation plots for the Mn2+–H3glyox, Cu2+–H3glyox, and Lu3+–H3glyox systems are presented in Fig. 3. In the aqueous environment, there is always competition between protons (H+) and the metal ions (Mn+) for the basic donor atoms of the ligands. Therefore, the pM value31 (pM = −log[Mn+]free where [Mn+] = 1 μM, [L] = 10 μM at pH = 7.4) is a reliable thermodynamic parameter to evaluate the sequestrating ability of the ligand for a specific metal ion at physiological pH and allows for a better comparison of the affinity of different ligands with different denticities, basicity and metal complex stoichiometries for a specific metal ion.
 | ||
| Fig. 3 Speciation diagrams of the Mn+–H3glyox complexes where Mn+ = Mn2+, Cu2+ and Lu3+; dashed line indicates pH 7.4. | ||
For the Mn2+–H3glyox system, the complex formation equilibria studies showed a trio of species Mn(Hglyox), [Mn(glyox)]− and [Mn(glyox)(OH)]2−. The complexation begins around pH 3.5 from the free [Mn(H2O)6]2+ to form the monoprotonated Mn(Hglyox) complex. Around pH 5, the formation of the [Mn(glyox)]− species can be observed as there is an increase in the absorbance bands at λmax = 255 and 370 nm characteristic of quinolinate metal complexes.16,23,32 The formation of the metal-hydroxo species [Mn(glyox)(OH)]2− starts after pH 8 and predominates at basic pH without the presence of free metal in solution (Fig. S9†). The absorbance band with λmax = 255 nm (Fig. S9C†) demonstrates the complexation of Mn2+ by the ligand in a manner similar to the UV-Vis spectrum for Mn(oxinate)2 complex formation.8
Consistent with the lack of ligand field stabilisation in the 3d5 configuration of Mn2+, [Mn(glyox)]− is the least thermodynamically stable metal complex of H3glyox among all six metal ions studied.16 But on the comparison (Table 3) with other macrocyclic and non-macrocyclic aminopolycarboxylate chelators (Chart 1) studied for Mn2+ complexation, and in particular, with the highest pMn reported for the macrocycle PCTA, H3glyox has a pMn value33 (pMn = −log[Mnn+]free when [Mnn+] = [L] = 10 μM at pH 7.4, unique pMn conditions different than for pM above with all other metal ions) just 1.71 units lower, notwithstanding having both nearly the same thermodynamic stability (logKML = 16.75(1) for H3glyox and logKML = 16.83 for PCTA).34 On the other hand, the logKMnL value for [Mn(glyox)]− is 3.56 units higher than for the structurally analogous [Mn(dpaa)]− which indeed reflects the effect of the oxine vs. picolinic acid groups on complexation of Mn2+ ion.24 In Fig. 4B, pMn31 (pMn = −log[Mnn+]free when [Mnn+] = 1 μM; [L] = 10 μM at pH 7.4) vs. pH are plotted for the most relevant Mn chelators to demonstrate the effect of the overall basicity of the ligand on the metal scavenging ability at different pH. The most significant effect can be observed at physiological pH where, even though the stability constant of [Mn(glyox)]− (logKML = 16.75) stands within the highest reported (Tables 3 and S7†), the high overall basicity of H3glyox with respect to the other ligands (Table S7†), mainly imparted by the two basic oxine–OH in the arms, makes the competition higher at physiological pH and consequently, lowers the conditional stability. Nevertheless, we see that still at higher pH the pMn continues to increase as the metal is secured by the [Mn(glyox)(OH)]2− species which reaches its maxima at pH ∼12. Additionally, since the pH-dependent Mn2+ scavenging profile is very close (and even superior at higher pH) to that of the also non-macrocyclic chelator PyC3A,11 it is worth to further explore the potential of this ligand for Mn2+ medicinal applications.
KML), corresponding stepwise protonation constants (logKMHL) and pMn values33 of relevant Mn2+ chelators
| Chelator | H3glyoxa | H3dpaab | PyC3Ac | PC2A-EAd | PCTAd |
|---|---|---|---|---|---|
| a This work.b From ref. 24.c From ref. 11.d From ref. 34.e pMn33 defined as −log[Mn2+]free when [Mn2+] = [L] = 10 μM at pH 7.4. | |||||
| logKML |
16.75(1) | 13.19(5) | 14.14 | 19.01 | 16.83 |
| logKMHL |
7.17(1) | 2.90(6) | 2.43 | 6.88 | 1.96 |
| logKM(OH)L |
10.45(2) | 11.97(6) | — | — | — |
| pMne | 8.03 | 8.98 | 8.17 | 9.27 | 9.74 |
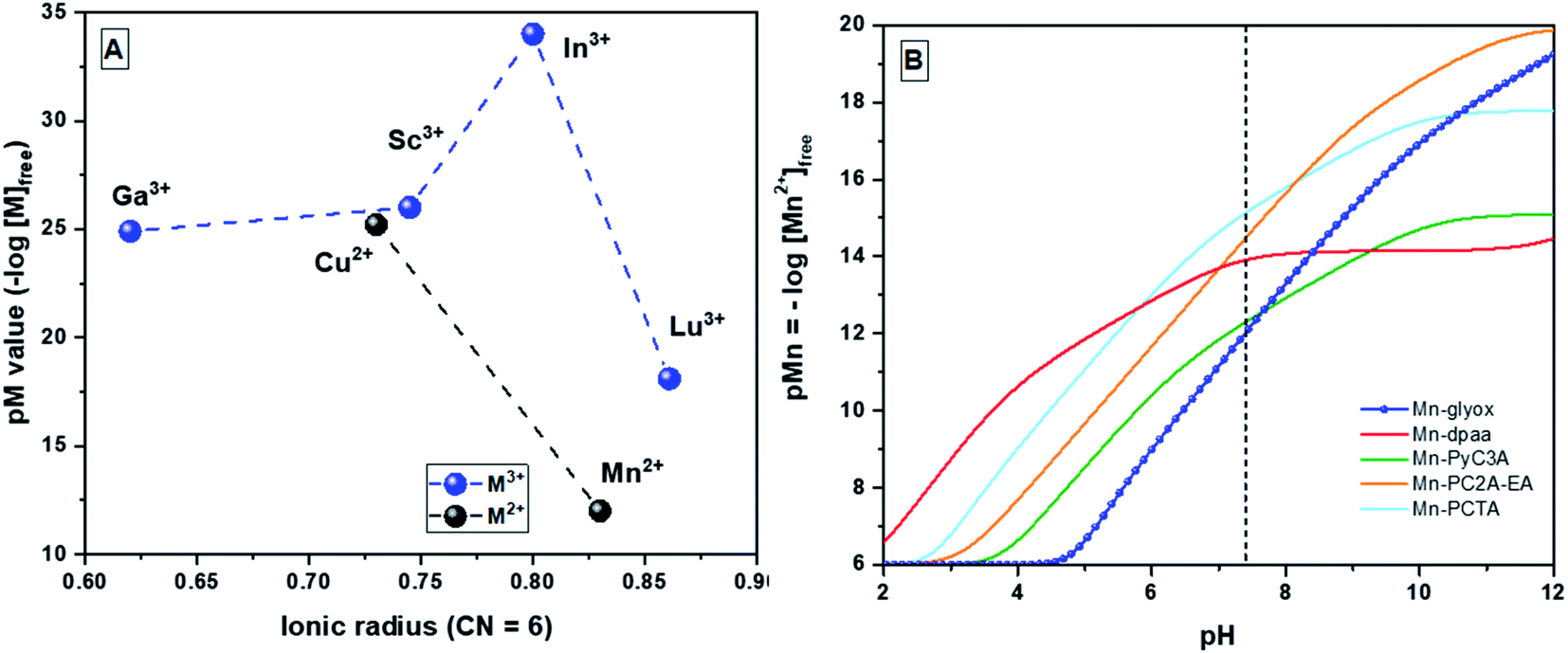 | ||
| Fig. 4 (A) pM values31 vs. ionic radii17 for Mn+–H3glyox complexes (n = 2 and 3) (CN = 6), pM = −log[Mn+]free when [Mn+] = 1 μM and [L] = 10 μM at pH 7.4; (B) Mn2+ scavenging ability of different chelators as the pH is raised from 2 to 12, when [Mn2+] = 1 μM and [L] = 10 μM;31 dashed line represents physiological pH 7.4. | ||
For each of the Cu2+–H3glyox and Lu3+–H3glyox systems, three metal–ligand species were observed (Fig. 3) – the protonated metal complex, Cu(Hglyox) and [Lu(Hglyox)]+, the metal complex, [Cu(glyox)]− and Lu(glyox), and the hydroxo–metal complex, [Cu(glyox)(OH)]2− and [Lu(glyox)(OH)]−. At neutral pH 7, the binary metal complexes [Cu(glyox)]− and Lu(glyox) are completely formed.
For the Cu2+–H3glyox system, different isosbestic points (Fig. S10†) can be seen in both the regions (225–300 nm and 300–450 nm) of the spectra with the increase in pH from 0.37 to 11.07 which indicates presence of different metal complex species in equilibrium. On comparing pM values of the Cu2+ complexes of H3glyox, H2hox and H2CHXhox, all belonging to the same “ox” family with varying backbone, they follow the trend [Cu(glyox)]− (pCu = 25.2, logKML = 30.30) > Cu(CHXhox) (pCu = 23.9, logKML = 29.78) > Cu(hox) (pCu = 22.8, logKML = 22.80).28 This comparison demonstrates that the pre-organisation of the ligand leads to stronger complexation of Cu2+.
For the Lu3+–H3glyox complexation equilibria, as pH increases from 3.74 to 6.54, there is an increase in absorption spectra at λmax = 260 nm with new isosbestic points at λ = 252, 338 nm (Fig. S11†). Although both H3glyox (hexadentate) and H4octox (octadentate) differ in their denticity, they have quite similar affinities for Lu3+; pM = 18.1 for Lu(glyox) and pM = 18.2 for [Lu(octox)]− and the stability constants (logKML) are 23.04 and 24.66, respectively.32
Fig. 4A summarizes the effects of the metal ion size and charge on chelation with H3glyox. As reported, the medium sized trivalent metal ions (Sc3+, Ga3+ and In3+) form thermodynamically stable and transchelation-resistant charge neutral complexes.16 Meanwhile, the larger trivalent lanthanide, Lu3+ is a poor match for the H3glox cavity size compared to the other smaller metals. Both the divalent metal ions Mn2+ and Cu2+, however, form thermodynamically stable complexes (logKMnL = 16.75 and logKCuL = 30.21) which suggested that radiolabeling studies might be rewarding.
Radiolabeling with [64Cu]CuCl2
Preliminary radiolabeling experiments were done in order to determine the complexation kinetics and radiolabeling efficiency of H3glyox with [64Cu]CuCl2. The first set of experiments involved radiolabeling in various buffer solutions (0.1 M MES, 0.1 M NH4OAc and 0.1 M NaOAc) at pH 5.5 and 6; quantitative radiolabeling (RCY > 95%) was achieved in both 0.1 M MES and 0.1 M NaOAc within 15 minutes at pH 6 and [L] = 10−5 M with the apparent molar activity of 500 MBq μmol−1. The second set of experiments varied temperature and reaction time vs. radiolabeling efficiency. No significant changes in radiolabeling efficiency were observed when radiolabeling with [64Cu]CuCl2 was done at room temperature, 37 °C and 80 °C, while the radiochemical yields (RCY) observed for the time dependent reactions are summarised in Table 4. Further, the ligand solution concentration was varied to determine the ligand concentration dependant radiolabeling efficiency, [H3glyox] = 10−4 to 10−7 M and the results are summarised in Table 5. The radiochemical yields were obtained by integrating the area under the peaks observed for TLC (Fig. S13†) when eluted with 0.1 M EDTA (pH 5) ([64Cu][Cu(glyox)]−, Rf < 0.1; [64Cu]Cu-EDTA, Rf > 0.7), consistent with the well-resolved peaks in the HPLC trace (Fig. S12†). Although H3glyox labels copper-64 at ambient conditions, it was found that the [64Cu(glyox)]− complex is unstable under acidic conditions (0.1% TFA in MQ water) as seen in the radio-HPLC trace. This is consistent with the observations and inferences made from both DFT calculations and solution studies due the protonation of the carboxylate arm at pH below 5.Radiolabeling with [52Mn]MnCl2
A radiochemically isolated [52gMn]Mn2+ sample35 was titrated against varying concentrations of H3glyox (0–100000 ng mL−1) yielding apparent molar activities of 30 MBq μmol−1 at pH 6.5 and 40 MBq μmol−1 at pH 7. The labelling of [52gMn]Mn2+ by H3glyox was compared with the “gold standard” chelator – DOTA at similar conditions (30 min, 85 °C, pH 7) and it was found that DOTA yields apparent molar activity of 60 MBq μmol−1, slightly higher than that for [52gMn]Mn2+–H3glyox (40 MBq μmol−1). It was observed that the molar activity of [52gMn]Mn2+–H3glyox increased with the use of higher activity of [52gMn]Mn2+ per reaction. Radiolabeling was not attempted at pH > 7 to avoid hydrolysis of the Mn2+ ion. The radiochemical yields (RCY%) of chelated [52gMn]Mn2+ were determined via TLC (silica gel plates) eluted with 0.1 M sodium citrate (Fig. S14†). The “free” [52gMn]Mn2+ travels up the plate as [52gMn]Mn2+–citrate complex (Rf > 0.8) while the [52gMn]Mn2+–DOTA and [52gMn]Mn2+–H3glyox complex stayed at baseline (Rf < 0.1). It has been noted previously that a rigid scaffold such as bispidine around the Mn2+ ion endows the manganese complex with increased kinetic inertness and similar to H3glyox, the radiolabeling of [52gMn]Mn2+ with a bispidine-based ligand was done at pH 7, 70 °C and 1 h.36 Meanwhile, radiolabeling of [52gMn]Mn2+ with the open chain chelator, CDTA (trans-1,2-diamino-cyclohexane-N,N,N′,N′-tetraacetic acid) is reported to be performed at pH 6, room temperature and 30 minutes ([CDTA] = 64 mM, ∼8 MBq [52gMn]MnCl2).10 No data on apparent molar activities for 52gMn-labeled complexes were found in the literature for other chelators discussed here for comparison.10,36
Radiolabeling with [177Lu]LuCl3
Encouraged by the relatively high pM value (pLu = 18.1), preliminary radiolabeling studies were done to investigate the complexation kinetics of the hexadentate chelator H3glyox with [177Lu]LuCl3. Although Lu3+ ion is a larger trivalent metal ion with an ionic radius of 86–103 pm and coordination number 6–9,17 the mismatch with the cavity size of H3glyox is evident from the observed RCYs. Table 6 summarizes the RCYs (n = 3) and as expected, quantitative radiolabeling (RCY > 95%) was not achieved even by varying conditions – pH 7.0 and 8.5 at 15 and 30 minutes. The decrease in RCY was observed when the labelling was performed at pH 8.5 after 30 minutes compared to the RCY at 15 minutes, this can be attributed to the labile nature of the [177Lu]Lu3+–H3glyox complex and subsequent transchelation of Lu3+ ion by EDTA (pH 7, mobile phase) leading to lower RCYs. The radiochemical yields were determined by integrating the area under the peaks observed in the iTLC plates (Fig. S15,† [177Lu]Lu(glyox), Rf < 0.1; [177Lu]Lu(EDTA), Rf > 0.8). A high apparent molar activity of 25 GBq μmol−1 was achieved by using 10−6 M ligand concentration at ambient conditions (15 min, RT, and pH 7).Conclusions
It has been an ongoing challenge for the medicinal inorganic chemistry community to stabilize the labile coordination complexes of endogenous metal ions such Mn2+ and Cu2+ in relevant biological environments. These results indicate that further investigation of the preorganized H3glyox as a potential chelator for 52g/55Mn-based dual MRI-PET tracer application is worthwhile, supported by the DFT calculated structure of [Mn(glyox)(H2O)]− and efficient radiolabeling of [52gMn]Mn2+. In-batch UV spectrophotometric titrations were conducted to access the thermodynamic stability of the metal complexes by determining the formation constants, speciation diagrams and the pM values, which are in the order pCu (25.2) > pLu (18.1) > pMn (12.0). Although the thermodynamic stability of the Cu2+–H3glyox complex is higher than that of the Mn2+ complex and in agreement with the Irving–William series, the [64Cu][Cu(glyox)]− was found to decompose under mild acidic conditions due to the formation of a protonated Cu(Hglyox) species at pH < 5. However, radiolabeling of H3glyox with [52gMn]Mn2+, [64Cu]Cu2+ and [177Lu]Lu3+ yielded apparent molar activities of 40 MBq μmol−1, 500 MBq μmol−1 and 25 GBq μmol−1, respectively. DFT calculations of the structures of the three complexes revealed that the Mn2+ is hexacoordinated and is bound to a water molecule similar to the reported [Mn(dpaa)(H2O)]− structure,24 which makes the Mn2+–H3glyox complex potentially relevant to Mn-based contrast agents. Based on these interesting and encouraging preliminary studies, the next step would be detailed studies to determine the kinetic inertness, water exchange rate and relaxivity measurements for application of H3glyox in Mn-based MRI contrast agents. Meanwhile, it can be concluded that the highly preorganized and rigid binding cavity of the ligand H3glyox is suitable for neither the smaller Cu2+ nor the larger Lu3+ ion.Experimental section
Materials and methods
All solvents and reagents were purchased from commercial suppliers (Sigma-Aldrich, TCI America, Fischer Scientific, Alfa Aesar) and were used as received. Reactions were monitored by TLC (MERCK Kieselgel 60 F254, aluminum sheet). Flash chromatography was performed using SilicaFlash F60 silica gel (40–63 μM particle size), RediSep Rf HP silica columns, and a CombiFlash Rf column machine. Water used was ultrapure (18.2 MΩ cm−1 at 25 °C, Milli-Q, Millipore, Billerica, MA).1H and 13C NMR spectroscopy was performed using Bruker Avance 300 and Bruker AV III HD 400 MHz spectrometers. Chemical shifts (δ) are quoted in ppm relative to residual solvent peaks as appropriate; coupling constants (J) are provided in Hertz (Hz). 1H NMR signals were designated as follows: s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), sxt (sextet), spt (septet), m (multiplet), or a combination of these, with br representing a broad signal. Low resolution ESI-MS was performed on a Waters 2965 HPLC-MS with the sample prepared in methanol or acetonitrile (ACN). Results are labeled with m/z (abundance percentage) values − [M + X]±. High resolution ESI-MS was performed on a Waters/Micromass LCT TOF-MS with the sample prepared in methanol. Results are labeled with m/z (abundance percentage) values. Semipreparative reverse phase high-performance liquid chromatography (HPLC) for H3glyox was performed on a Phenomenex Synergi hydro-RP 80 A, 250 × 21.2 mm column connected to a Waters 600 controller, a Waters 2487 dual wavelength absorbance detector, and a Waters delta 600 pump. The HPLC solvents were (A) H2O containing 0.1% trifluoroacetic acid (TFA) and (B) CH3CN containing 0.1% TFA. Silica gel impregnated TLC plates (MERCK Kieselgel 60 F254, aluminum sheet) were used to analyse [64Cu]Cu2+ and [177Lu]Lu3+ radiolabeling reaction progress and the complex stability tests were counted on a BioScan System 200 imaging scanner equipped with a BioScan Autochanger 1000. For the evaluation of [52gMn]Mn2+, TLC plates were scanned on a Packard Cyclone™ phosphor-storage plate equipped with Opti-quant™ software. Linear extrapolation was used to determine the amount of chelator needed to complex 50% and 100% of [52gMn]Mn2+.
[177Lu]LuCl3 was purchased from Isotope Technologies Garching (ITG). All the isotopes used were no-carrier added (n.c.a.). The production of [64Cu]CuCl2 was performed at a TR-Flex cyclotron from Advanced Cyclotron Systems Inc (ACSI, Canada) according to a reported procedure via 64Ni(p,n)64Cu nuclear reaction and module-assisted separation as described in detail recently.37 No-carrier-added [52gMn]Mn2+ was produced by irradiation of a metallic chromium target of natural isotopic abundance (83.8% 52Cr; 9.5% 53Cr; 4.3% 50Cr; and 2.4% 54Cr) with 16 MeV protons as described previously in a report.35 Deionized water was filtered through the PURELAB Ultra Mk2 system.
Synthesis and characterisation
:1 EtOAc/hexane). On completion of reaction after 5 hours, the solvent was removed under vacuum and the crude solid was dissolved in 10 mL distilled water and the product was extracted using DCM (20 mL × 3). The combined organic layers were dried over anhydrous MgSO4 and the solvent was removed under vacuum to give white solid product 1 (0.35 g, 2.00 mmol, 98%). 1H NMR (300 MHz, CD3OD, RT): δ 8.21 (d, J = 8.5 Hz, 1H), 7.54 (d, J = 8.5 Hz, 1H), 7.38 (m, 2H), 7.11 (d, J = 7.1 Hz, 1H), 4.90 (s, 2H). 13C NMR (75 MHz, CD3OD, RT): δ 158.8, 152.8, 137.6, 136.7, 128.3, 126.9, 118.9, 117.7, 110.9, 64.6. LR-ESI-MS calcd for C10H9NO2: 175.2. Found: 176.3 [M + H]+.:1 EtOAc/hexane) indicated the completion of reaction. The reaction was quenched by addition of saturated Na2CO3 (30 mL) and the mixture was stirred for another 10 minutes. The reaction mixture was transferred to a separating flask and DCM (3 × 20 mL) was used to extract out the product. The organic layers were combined and dried over anhydrous MgSO4 and the solvent was removed under vacuum to slightly yellow colored product 2 (0.40 g, 1.78 mmol, 98%). 1H NMR (300 MHz, CD3OD, RT): δ 8.17 (d, J = 8.5 Hz, 1H), 7.61 (d, J = 8.5 Hz, 1H), 7.49 (t, J = 7.9 Hz, 1H), 7.36 (d, J = 9.4 Hz, 1H), 7.22 (d, J = 7.6 Hz, 1H) 4.73 (s, 2H). 13C NMR (75 MHz, CD3OD, RT): δ 157.8, 151.7, 137.4, 127.9, 127.6, 124.1, 119.4, 118.2, 111.1, 64.9. LR-ESI-MS calcd for C10H879BrNO: 238.0. Found: 239.1 [M + H]+.:1 (L:M) solutions of ligand and Lu(NO3)3 prepared in D2O (10 mM) and 0.1 M NaOD was added to increase the pD of the solution to 7. 1H NMR (300 MHz, RT, D2O): δ 8.38 (d, J = 8.6 Hz, 0.5H), 8.28 (d, J = 8.6 Hz, 1H), 7.61 (t, J = 8.5 Hz, 0.5H), 7.42 (t, J = 8.4 Hz, 6H), 7.13 (m, 2H), 4.68 (s, 2H), 4.12 (s, 2H), 3.26 (s, 2H). HR-ESI-MS calcd for C22H16175LuN3O4: 561.0548. Found: 562.0546 [M + H]+.Radiolabeling
For concentration-dependent labeling of copper-64, different aliquots were added from the stock solution of H3glyox (2 mg dissolved in 1 mL 0.1 M HCl and 2 mL MQ H2O, ∼10−3 M), mixed with [64Cu]CuCl2 (25 MBq) and diluted to a final volume of 500 μL using 0.1 M NaOAc buffer solution (pH 6). After incubation for 15 min at RT, the radiolabeling yields were determined using iTLC (silica gel plates developed with aqueous 0.1 M EDTA solution, pH 5; [64Cu][Cu(glyox)]−, Rf < 0.1; [64Cu]Cu–EDTA, Rf > 0.7). For labeling of lutetium-177, a total volume of 500 μL of the reaction mixture was obtained by addition of an aliquot from the ligand stock solution (10−5 M), [177Lu]LuCl3 (2 MBq) and 0.1 M NaOAc/HOAc buffer (pH 7, 8.5). The RCYs were obtained by spotting an aliquot from reaction mixture after 15 min and 30 min on a TLC plate (silica gel) and developing with aqueous EDTA solution (0.1 M, pH 7). The [177Lu]Lu(glyox) complex stayed on the baseline (Rf < 0.1) while “free” [177Lu]Lu3+ travelled up the plate as [177Lu]Lu3+–EDTA complex (Rf > 0.7). For labeling with 52gMn, a reported procedure was followed.35 In summary, 100 μL of chelator solution was added to 1 mL vials in varying concentrations (0–100000 ng mL−1) and 100 μL of [52gMn]Mn2+ solution (∼2.7 MBq) was added to each vial. Finally, the pH of the solution was adjusted to pH 7 with addition of 100–200 μL of 1 M HEPES/NaOH buffer (pH 7). It was observed that the molar activity of [52gMn]Mn2+–H3glyox complex increased with the use of higher activity of [52gMn]Mn2+ per reaction. The apparent molar activity for [52gMn]Mn2+–H3glyox complex determined from titration of 52gMn solutions with H3glyox can be expressed as twice the ratio of the 52gMn over moles of chelator required to chelate 50% of the [52gMn]Mn2+. Radiolabeling was not attempted at pH > 7 in order to avoid hydrolysis of the Mn2+ ion. Vials were agitated, assayed and heated for 30 minutes at 85 °C. The percentage of chelated [52gMn]Mn2+ was determined via TLC (silica gel plates; Fig. S14†) eluted with 0.1 M aqueous sodium citrate (pH 5.5). The “free” [52gMn]Mn2+ travels up the plate as [52gMn]Mn2+–citrate complex (Rf > 0.8) while the [52gMn]Mn2+–DOTA and [52gMn]Mn2+–H3glyox complexes stayed at baseline (Rf < 0.1).
Solution thermodynamics
Metal stability constants were calculated from UV spectrophotometric titration data obtained using a Cary 60 UV-Vis spectrophotometer in the spectral range of 200–450 nm. The path length was 1 cm for all samples. Individual samples (5 mL) containing the ligand (H3glyox, [L] = 2.5 × 10−5 M) and the corresponding metal complexes (M = Mn2+, Cu2+ and Lu3+; [ML] = 2.5 × 10−5 M) were prepared in MQ water by adjusting the pH using standardized HCl or NaOH solutions, and NaCl was added to maintain a constant 0.16 M ionic strength over the pH range ∼0.37–11.07. A Ross combination pH electrode was calibrated for hydrogen ion concentration using HCl as described before38 and the results were analysed by the Gran39 procedure. For the most acidic samples, the equilibrium H+ concentration was calculated from solution stoichiometry and was not measured with the glass electrode. For the samples containing the metal complexes, the measurements were performed only when equilibrium was achieved. The stability constants for the metal complexes were calculated from the experimental data using the HypSpec2014 program.30 Proton dissociation constants corresponding to hydrolysis of Mn2+, Cu2+, and Lu3+ aqueous ions included in the calculations were taken from Baes and Mesmer.40 The species formed in the studied systems are characterized by the general equilibrium in eqn (1).| pM + qH + rL ↔ MpHqLr (charges omitted) | (1) |
By convention, a complex containing a metal ion M, proton H, and ligand L has the general formula MpHqLr. The stoichiometric index p might also be 0 in the case of ligand protonation equilibria, and negative values of q refer to proton removal or hydroxide ion addition during formation of the complex. The overall equilibrium constant for the formation of the complexes MpHqLr from its components is designated as log β. Stepwise equilibrium constants log K correspond to the difference in log units between the overall constants of sequentially protonated (or hydroxide) species. pM value is defined as −log[Mn+]free calculated at specific conditions ([Mn+] = 1 μM, [Lx−] = 10 μM, pH 7.4)31 except where pMn values amongst ligands are compared ([Mn2+] = 10 μM, [Lx−] = 10 μM, pH 7.4).33
DFT calculations
All DFT simulations were performed as implemented in the Gaussian 16 revision c01.41 Each structure was optimised and free energies were calculated using DFT with the hybrid meta generalized gradient approximation with the TPSSh exchange-correlation functional42 and the def2-TZVP basis set43 for all non-metal atoms. Effective core potentials (ECPs) were used to account for scalar relativistic effects in metal core electrons (Mn and Cu,44,45 Ga46,47 and Lu48,49). Metal basis sets were downloaded from the Basis Set Exchange website.50–52 The integral equation formalism of the polarisable continuum model (IEFPCM) was used as an implicit water model in all calculations to simulate the average dielectric effects of the solvent. Default IEFPCM parameters were used, as implemented in Gaussian (ε = 78.36, van der Waals surface without “added spheres”). For Cu2+–H3glyox, calculations included 0, 1 and 3 explicit water molecules while for Mn just one explicit water molecule was included. Lu3+–H3glyox calculations included 2 explicit water molecules. Before optimisation, the randomly placed water molecules were placed closer to the metal with the aim to try to coordinate it to Cu2+, Mn2+ or Lu3+ or to hydrogen bond centres, such as oxinates and carboxyl groups. Normal self-consistent field (SCF) and geometry convergence criteria were conducted for all the calculations. The calculated structures were visualized using GaussView version 6.0 (ref. 53) and Avogadro version 1.2.0.54Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
We gratefully acknowledge both the Canadian Institutes for Health Research (CIHR) and the Natural Sciences and Engineering Research Council (NSERC) of Canada for grant support, NSERC for an IsoSiM CREATE at TRIUMF studentship (NC), and MITACS for a Globalink Graduate fellowship (NC). We thank WestGrid and Compute Canada for access to their computational resources. Karin Landrock is acknowledged for excellent technical assistance, as is the HZDR for supporting the IsoSiM International Research Experience programme; TRIUMF receives federal funding via a contribution agreement with the National Research Council of Canada.References
- A. B. Garrett, J. Chem. Educ., 1963, 40, 36–37 CrossRef.
- C. Hoehr, F. Bénard, K. Buckley, J. Crawford, A. Gottberg, V. Hanemaayer, P. Kunz, K. Ladouceur, V. Radchenko, C. Ramogida, A. Robertson, T. Ruth, N. Zacchia, S. Zeisler and P. Schaffer, Phys. Procedia, 2017, 90, 200–208 CrossRef CAS.
- T. I. Kostelnik and C. Orvig, Chem. Rev., 2019, 119, 902–956 CrossRef CAS PubMed.
- E. Boros and A. B. Packard, Chem. Rev., 2019, 119, 870–901 CrossRef CAS.
- E. Boros and J. P. Holland, J. Labelled Compd. Radiopharm., 2018, 61, 652–671 CrossRef CAS.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed.
- S. Sarkar, N. Bhatt, Y. S. Ha, P. T. Huynh, N. Soni, W. Lee, Y. J. Lee, J. Y. Kim, D. N. Pandya, G. Il An, K. C. Lee, Y. Chang and J. Yoo, J. Med. Chem., 2018, 61, 385–395 CrossRef CAS PubMed.
- P. Gawne, F. Man, J. Fonslet, R. Radia, J. Bordoloi, M. Cleveland, P. Jimenez-Royo, A. Gabizon, P. J. Blower, N. Long and R. T. M. De Rosales, Dalton Trans., 2018, 47, 9283–9293 RSC.
- I. Verel, G. W. M. Visser, R. Boellaard, M. Stigter-van Walsum, G. B. Snow and G. A. M. S. van Dongen, J. Nucl. Med., 2003, 44, 1271–1281 CAS.
- C. Vanasschen, M. Brandt, J. Ermert and H. H. Coenen, Dalton Trans., 2016, 45, 1315–1321 RSC.
- E. M. Gale, I. P. Atanasova, F. Blasi, I. Ay and P. Caravan, J. Am. Chem. Soc., 2015, 137, 15548–15557 CrossRef CAS.
- F. K. Kálmán, F. K. Kálmán, V. Nagy, B. Váradi, Z. Garda, E. Molnár, G. Trencsényi, J. Kiss, S. Même, W. Même, É. Tóth and G. Tircsó, J. Med. Chem., 2020, 63, 6057–6065 CrossRef PubMed.
- P. J. Blower, J. S. Lewis and J. Zweit, Nucl. Med. Biol., 1996, 23, 957–980 CrossRef CAS PubMed.
- S. Banerjee, M. R. A. Pillai and F. F. Knapp, Chem. Rev., 2015, 115, 2934–2974 CrossRef CAS PubMed.
- FDA approves lutetium Lu-177 dotatate for treatment of GEP-NETS, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-lutetium-lu-177-dotatate-treatment-gep-nets, accessed 13 July 2020.
- N. Choudhary, M. d. G. Jaraquemada-Peláez, K. Zarschler, X. Wang, V. Radchenko, M. Kubeil, H. Stephan and C. Orvig, Inorg. Chem., 2020, 59, 5728–5741 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- G. K. Walkup and B. Imperiali, J. Org. Chem., 1998, 63, 6727–6731 CrossRef CAS.
- N. Choudhary, A. Dimmling, X. Wang, L. Southcott, V. Radchenko, B. O. Patrick, P. Comba and C. Orvig, Inorg. Chem., 2019, 58, 8685–8693 CrossRef CAS PubMed.
- D. Brox, A. Kiel, S. J. Wörner, M. Pernpointner, P. Comba, B. Martin and D. P. Herten, PLoS One, 2013, 8, e58049 CrossRef CAS PubMed.
- L. Prodi, F. Bolletta, M. Montalti and N. Zaccheroni, Coord. Chem. Rev., 2000, 205, 59–83 CrossRef CAS.
- D. M. Weekes, C. F. Ramogida, M. d. G. Jaraquemada-Pelaez, B. O. Patrick, C. Apte, T. I. Kostelnik, J. F. Cawthray, L. Murphy and C. Orvig, Inorg. Chem., 2016, 55, 12544–12558 CrossRef CAS PubMed.
- X. Wang, M. d. G. Jaraquemada-Peláez, Y. Cao, J. Pan, K. S. Lin, B. O. Patrick and C. Orvig, Inorg. Chem., 2019, 58, 2275–2285 CrossRef CAS PubMed.
- A. Forgács, R. Pujales-Paradela, M. Regueiro-Figueroa, L. Valencia, D. Esteban-Gómez, M. Botta and C. Platas-Iglesias, Dalton Trans., 2017, 46, 1546–1558 RSC.
- A. Pasquarello, I. Petri, P. S. Salmon, O. Parisel, R. Car, É. Tóth, D. H. Powell, H. E. Fischer, L. Helm and A. E. Merbach, Science, 2001, 291, 856–859 CrossRef CAS PubMed.
- L. M. P. Lima, D. Esteban-Gómezgómez, R. Delgado, C. Platas-Iglesias and R. L. Tripier, Inorg. Chem., 2012, 51, 6916–6927 CrossRef CAS PubMed.
- B. A. Vaughn, A. M. Brown, S. H. Ahn, J. R. Robinson and E. Boros, Inorg. Chem., 2020, 59, 16095–16108 CrossRef CAS PubMed.
- X. Wang, New Chelators for Radiopharmaceutical Chemistry, doctoral dissertation, University of British Columbia, Vancouver, Canada, 2019 Search PubMed.
- A. Nonat, P. H. Fries, J. Pécaut and M. Mazzanti, Chem.–Eur. J., 2007, 13, 8489–8506 CrossRef CAS PubMed.
- P. Gans, A. Sabatini and A. Vacca, Anal. Chim. Acta, 1999, 89, 45–49 CAS.
- W. R. Harris, C. J. Carrano and K. N. Raymond, J. Am. Chem. Soc., 1979, 101, 2213–2214 CrossRef CAS.
- X. Wang, M. d. G. Jaraquemada-Peláez, C. Rodríguez-Rodríguez, Y. Cao, C. Buchwalder, N. Choudhary, U. Jermilova, C. F. Ramogida, K. Saatchi, U. Hafeli, B. O. Patrick and C. Orvig, J. Am. Chem. Soc., 2018, 140, 15487–15500 CrossRef CAS PubMed.
- B. Drahoš, J. Kotek, P. Hermann, I. Lukeš and É. Tóth, Inorg. Chem., 2010, 49, 3224–3238 CrossRef PubMed.
- R. Botár, E. Molnár, G. Trencsényi, J. Kiss, F. K. Kálmán and G. Tircsó, J. Am. Chem. Soc., 2020, 142, 1662–1666 CrossRef PubMed.
- K. E. Barrett, E. Aluicio-Sarduy, A. P. Olson, C. J. Kutyreff, P. A. Ellison, T. E. Barnhart, R. J. Nickles and J. W. Engle, Appl. Radiat. Isot., 2019, 146, 99–103 CrossRef CAS PubMed.
- D. Ndiaye, M. Sy, A. Pallier, S. Même, I. de Silva, S. Lacerda, A. M. Nonat, L. J. Charbonnière and É. Tóth, Angew. Chem., Int. Ed., 2020, 59, 11958–11963 CrossRef CAS PubMed.
- M. Kreller, H. Pietzsch, M. Walther, H. Tietze, P. Kaever, T. Knieß, F. Füchtner, J. Steinbach and S. Preusche, Instruments, 2019, 3, 1–9 CrossRef.
- M. d. G. Jaraquemada-Peláez, X. Wang, T. J. Clough, Y. Cao, N. Choudhary, K. Emler, B. O. Patrick and C. Orvig, Dalton Trans., 2017, 46, 14647–14658 RSC.
- G. Gran, Analyst, 1952, 77, 661–670 RSC.
- C. F. Baes Jr and R. E. Mesmer, in The Hydrolysis of Cations, John Wiley & Sons, New York, 1976 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1986, 86, 866–872 CrossRef.
- J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408–3420 CrossRef CAS.
- T. Leininger, A. Berning, A. Nicklass, H. Stoll, H.-J. Werner and H.-J. Flad, Chem. Phys., 1997, 217, 19–27 CrossRef CAS.
- A. Bergner, M. Dolg, W. Küchle, H. Stoll and H. Preuß, Mol. Phys., 1993, 80, 1431–1441 CrossRef CAS.
- X. Cao and M. Dolg, J. Chem. Phys., 2001, 115, 7348–7355 CrossRef CAS.
- R. Gulde, P. Pollak and F. Weigend, J. Chem. Theory Comput., 2012, 8, 4062–4068 CrossRef CAS PubMed.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS.
- B. P. Pritchard, D. Altarawy, B. Didier, T. D. Gibson and T. L. Windus, J. Chem. Inf. Model., 2019, 59, 4814–4820 CrossRef CAS PubMed.
- D. Feller, J. Comput. Chem., 1996, 17, 1571–1586 CrossRef CAS.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView, Version 6.0.16, Semichem Inc., Shawnee Mission KS, 2016 Search PubMed.
- M. D. Hanwell, D. E. Curtis, D. C. Lonie, T. Vandermeerschd, E. Zurek and G. R. Hutchison, J. Cheminf., 2012, 4, 1–17 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra of compounds 1–4, high resolution mass spectra, solution thermodynamic results, radiolabeling studies, density functional theory (DFT) calculations. See DOI: 10.1039/d1ra01793d |
| This journal is © The Royal Society of Chemistry 2021 |