
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of indazole derivatives as anti-cancer agents†
Wei Wei‡
a,
Zhihao Liu‡a,
Xiuli Wua,
Cailing Gana,
Xingping Sua,
Hongyao Liua,
Hanyun Quea,
Qianyu Zhangb,
Qiang Xuea,
Lin Yuea,
Luoting Yu *a and
Tinghong Ye*a
*a and
Tinghong Ye*a
aSichuan University-University of Oxford Huaxi Joint Centre for Gastrointestinal Cancer, State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, China. E-mail: yeth1309@scu.edu.cn; yuluot@scu.edu.cn
bResearch Center for Public Health & Preventive Medicine, West China School of Public Health & Healthy Food Evaluation Research Center, West China Fourth Hospital, Sichuan University, Chengdu, Sichuan 610041, China
First published on 27th April 2021
Abstract
Several FDA approved small molecule anti-cancer drugs contain indazole scaffolds. Here, we report the design, synthesis and biological evaluation of a series of indazole derivatives. In vitro antiproliferative activity screening showed that compound 2f had potent growth inhibitory activity against several cancer cell lines (IC50 = 0.23–1.15 μM). Treatment of the breast cancer cell line 4T1 with 2f inhibited cell proliferation and colony formation. 2f dose-dependently promoted the apoptosis of 4T1 cells, which was connected with the upregulation of cleaved caspase-3 and Bax, and downregulation of Bcl-2. 2f also decreased the mitochondrial membrane potential and increased the levels of reactive oxygen species (ROS) in 4T1 cells. Additionally, treatment with 2f disrupted 4T1 cells migration and invasion, and the reduction of matrix metalloproteinase metalloproteinase-9 (MMP9) and increase of tissue inhibitor matrix metalloproteinase 2 (TIMP2) were also observed. Moreover, 2f could suppress the growth of the 4T1 tumor model without obvious side effects in vivo. Taken together, these results identified 2f as a potential small molecule anti-cancer agent.
1. Introduction
Cancer is one of the most common causes of death and thus is a significant public health burden. The International Agency for Research on Cancer estimated that there were 19.3 million new cases of cancer globally in 2020, mainly due to female breast cancer, lung cancer, and colorectal cancer, which each accounted for ≥10% of the total cases.1 In addition, the same report estimated that 10.0 million cancer associated-deaths occurred in 2020.1 Despite major advances in targeted therapies such as immunotherapy,2 gene therapy,3 and small molecule drugs,4 for relapsed or refractory cancers, many of them still lack effective treatment strategies. Metastasis, an important contributor to poor prognosis, also challenges the existing therapies and hinders blocking of cancer progression although the knowledge of underlying mechanisms is growing.5 Thus, it remains problematic to improve the efficacy of therapeutic regimens, and there is an urgent need to develop more treatment options to control the progression of cancer.An indazole motif can be applied as not only a functional substituent but also the scaffold to small molecule drugs. Indazole-containing bioactive molecules have pharmacological effects on a wide range of diseases,6–8 including infectious, inflammatory, neurodegenerative disease and cancer.9–11 In particular, some indazole derivatives that are developed as kinase inhibitors, have significant anti-cancer activity in vitro and in vivo. For instance, the multi-kinase inhibitor pazopanib (1-1, Fig. 1),12,13 the c-Met inhibitor merestinib (1-2),14–16 the tropomyosin receptor kinases/proto-oncogene tyrosine-protein kinase ROS1/anaplastic lymphoma kinase inhibitor entrectinib (1-3),17 and the vascular endothelial growth factor receptor/platelet-derived growth factor receptor inhibitor axitinib (1-4)18,19 are approved for the treatment of different cancer types. Furthermore, the fibroblast growth factor receptor inhibitor LY2874455 (1-5)20,21 and the polo-like kinase 4 inhibitor CFI-400945 (1-6)22–24 are investigated in clinical trials of acute myeloid leukemia (in combination with merestinib, NCT03125239)25 and breast cancer (NCT03624543),26 respectively (Fig. 1). Additionally, indazole derivatives have various anti-cancer mechanisms besides inhibiting kinase signaling pathways.27–29 The effective anti-cancer activity of these small molecules prompted us to further design and characterize indazole derivatives as potential therapeutic agents.
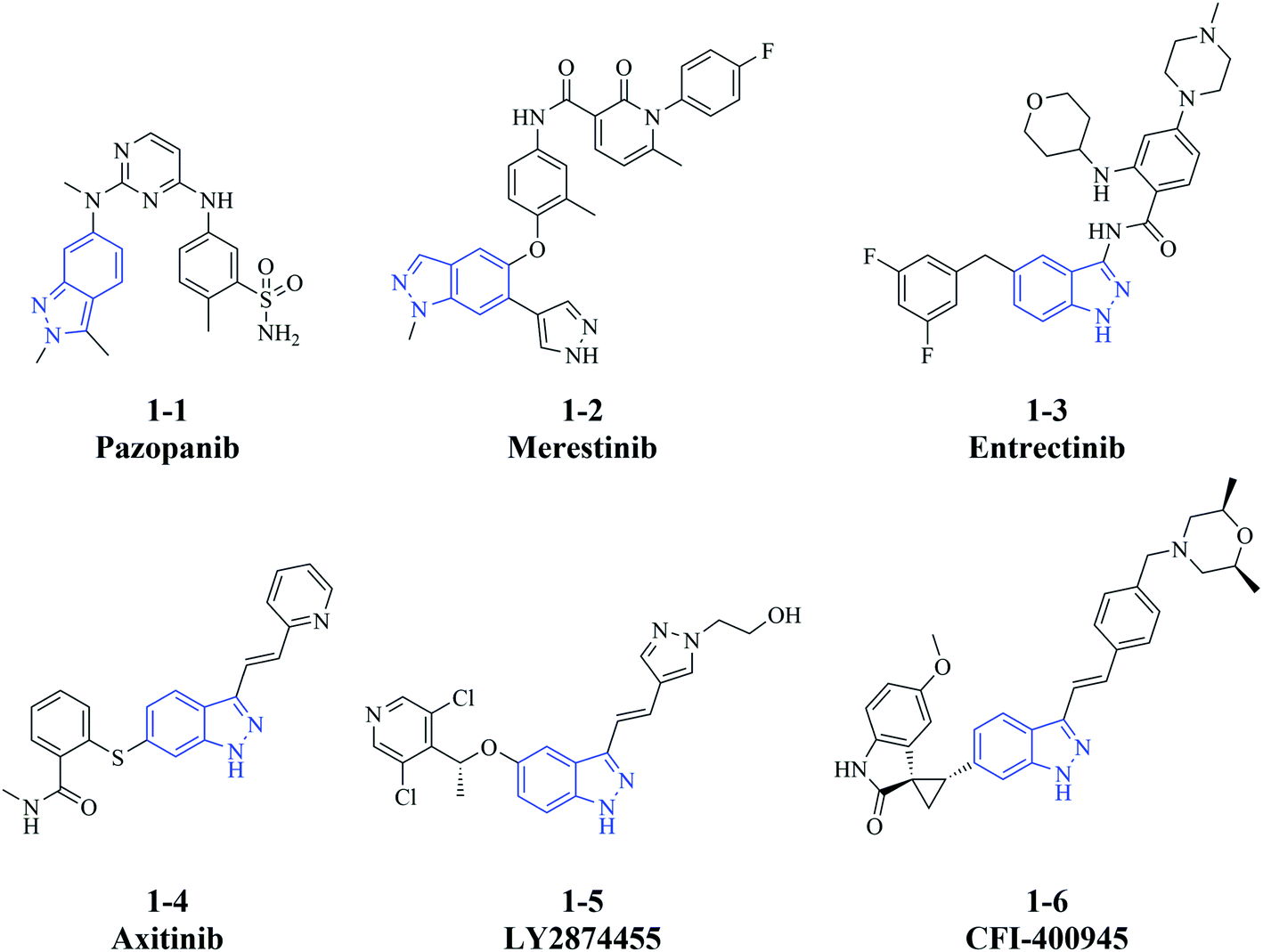 | ||
| Fig. 1 Structures of kinase inhibitors containing an indazole moiety. | ||
To enrich our compound library for screening of anti-cancer agents, we focused on building novel molecules bearing an indazole motif as the scaffold. The effect of a hydrophobic group at the indazole C6 position has already been elucidated by compounds found in some reports and our previous work, and some of them also have a hydrophilic group at the C3 position like 1-6.23,30–35 In this study, we investigated whether exchanging the groups at C3 and C6 would retain their anti-cancer activities. Thus, we synthesized a series of compounds in which a hydrophobic group based on substituted phenyl was linked to the indazole C3 position (R2) and a hydrophilic group was introduced at indazole C6 (R1; Fig. 2).
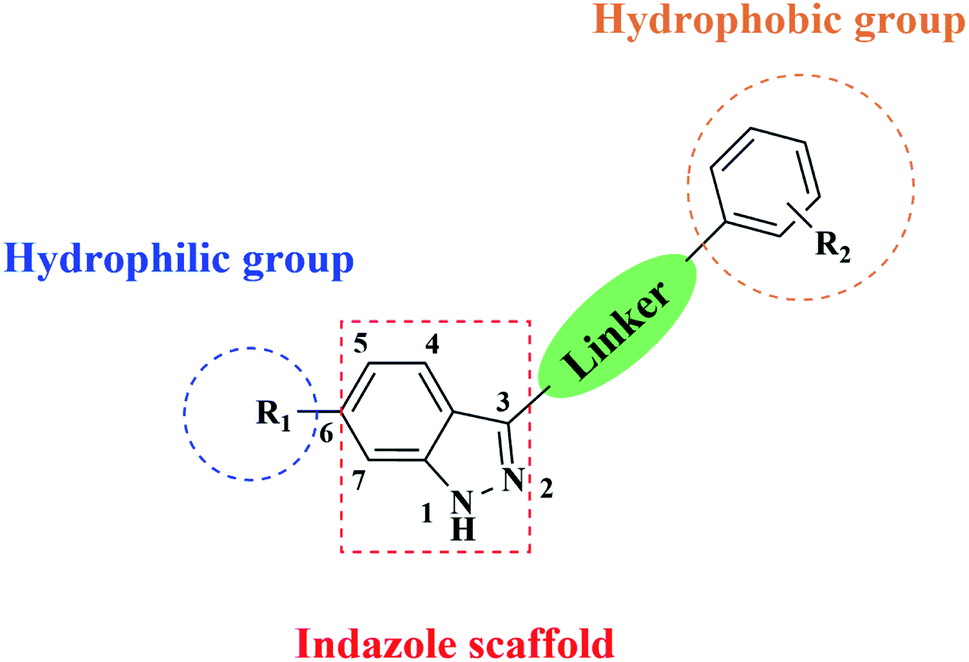 | ||
| Fig. 2 Structure of our indazole derivatives. | ||
2. Results and discussion
2.1 Chemistry
The synthetic routes of our compounds, 2a–2p, are shown in Scheme 1. The Heck coupling between 1-bromo-3,5-dimethoxybenzene and 4,4,5,5-tetramethyl-2-vinyl-1,3,2-dioxaborolane produced intermediate 2-1. Then, 2-1 was dissolved in DMF and reacted with N-chlorosuccinimide at 85 °C to afford 2-2. Intermediate 2-3 was prepared from the iodination of 6-bromo-1H-indazole, and reacted with 2-1 or 2-2 through Suzuki coupling to give key intermediates 2-4 and 2-5, respectively. The target products 2a–2i and 2k were obtained from reactions between 2-4 or 2-5 and corresponding boronic acid pinacol esters. The hydrogenation of compound 2f generated compound 2p. Compounds 2j and 2l–2n were synthesized by another synthesis route (Scheme 2). The preparation of intermediates 2-6–2-9 were conducted through Suzuki coupling between (1H-indazol-6-yl)boronic acid and corresponding halides. 2-6–2-9 reacted with iodine in the presence of K2CO3 to afford key intermediates 2-10–2-13 respectively. Reactions between intermediates 2-1 and 2-10–2-13 produced target compounds 2j and 2l–2n, respectively. | ||
| Scheme 1 General procedures for the synthesis of compounds 2a–2i, 2k and 2o–2p. (a) Pd2(dba)3, DIPEA, P(t-Bu)3·HBF4, toluene, 95 °C, 6 h, 78%; (b) N-chlorosuccinimide, DMF, 85 °C, 5 h, 47%; (c) iodine, KOH, DMF, r.t., 3 h, 71%; (d) Pd(dppf)Cl2, Cs2CO3, dioxane/H2O, 100 °C, 8 h, 46–67%; (e) Pd(dppf)Cl2, K2CO3, dioxane/H2O, 95 °C, 8 h, 17–31%; (f) H2, Pd/C, methanol, r.t., overnight, 57%. | ||
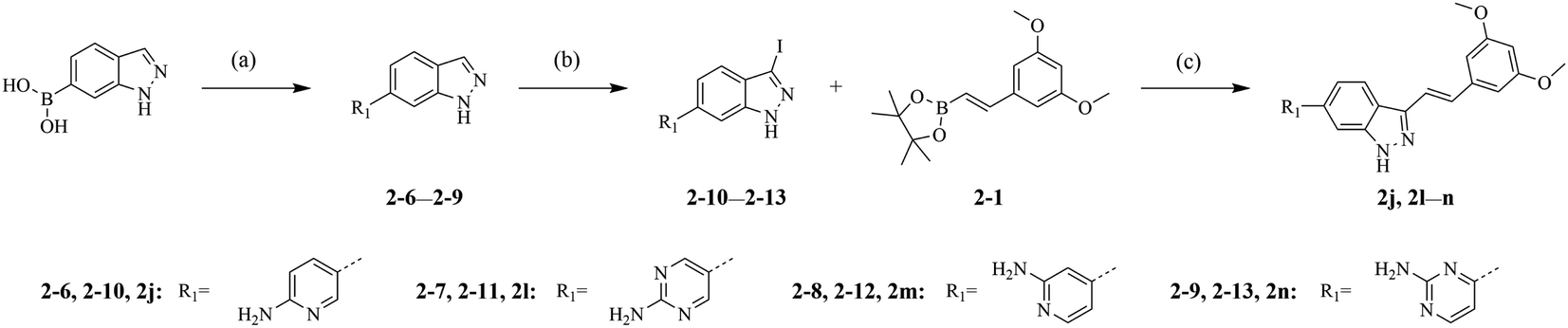 | ||
| Scheme 2 General procedures for the synthesis of compounds 2j and 2l–2n. (a) Pd(dppf)Cl2, K2CO3, dioxane/H2O, 100 °C, overnight, 56–62%; (b) iodine, K2CO3, DMF, 65 °C, 10 h, 80–88%; (c) Pd(dppf)Cl2, Cs2CO3, dioxane/H2O, 100 °C, 8 h, 12–30%. | ||
2.2 Antiproliferative activities screening in vitro
To evaluate the anti-cancer activities of synthesized compounds, we first examined their ability to inhibit the proliferation of several cell lines representative of cancers with high morbidity; namely, A549 lung cancer cells, HepG2 hepatocellular carcinoma cells, MCF-7 breast cancer cells, and HCT116 colorectal cancer cells, all of which are of human origin. We also examined the mouse breast cancer cell line 4T1, which has a propensity to metastasize. We found that most compounds displayed moderate inhibitory activities against all five cancer cell lines (Table 1). Compounds 2a–2n all contained (E)-3,5-dimethoxystyryl at indazole C3, allowing us to investigate the structure–activity relationship of R1 substituents. When R1 was substituted by 4-(4-methylpiperazin-1-yl)phenyl, compound 2a exhibited unobvious inhibitory activities against A549, 4T1 and HepG2 cell lines even at a relatively high concentration (IC50 > 10 μM), but the proliferation of MCF-7 and HCT116 cells was inhibited by 2a with IC50 values of 1.15 μM and 4.89 μM, respectively. Replacing 4-methylpiperazin-1-yl of 2a with N,N-dimethylsulfonamide (2b) or 4-sulfonylmorpholine (2c) improved the potencies against A549 and 4T1 cells, while placing (4-methylpiperazin-1-yl)methyl or 4-methylmorpholine at the para-position of the phenyl (2d and 2e, respectively) maintained or enhanced the activities against all five cell lines, indicating that introducing a sulfonyl or methylene group into phenyl at R1 was tolerated.| Compd | Antiproliferative activitya (IC50, μM) | ||||
|---|---|---|---|---|---|
| A549 | 4T1 | HepG2 | MCF-7 | HCT116 | |
| a Data were the mean ± SD from three independent tests. | |||||
| 2a | >10 | >10 | >10 | 1.15 ± 0.13 | 4.89 ± 0.29 |
| 2b | 6.59 ± 1.11 | 0.91 ± 0.09 | >10 | 1.10 ± 0.04 | 7.97 ± 1.42 |
| 2c | 8.06 ± 1.53 | 1.49 ± 0.19 | >10 | 1.01 ± 0.28 | 8.63 ± 1.85 |
| 2d | 1.40 ± 0.22 | 0.46 ± 0.02 | 1.16 ± 0.21 | 0.25 ± 0.01 | 2.46 ± 0.27 |
| 2e | 3.90 ± 1.01 | 1.70 ± 0.43 | 5.99 ± 1.91 | 1.01 ± 0.09 | 4.96 ± 1.52 |
| 2f | 0.95 ± 0.12 | 0.23 ± 0.03 | 0.80 ± 0.05 | 0.34 ± 0.02 | 1.15 ± 0.10 |
| 2g | 1.57 ± 0.31 | 0.64 ± 0.11 | 1.90 ± 0.35 | 0.79 ± 0.15 | 3.00 ± 0.72 |
| 2h | >10 | 2.62 ± 0.65 | 9.59 ± 1.14 | 2.10 ± 0.32 | 8.28 ± 1.40 |
| 2i | 3.95 ± 0.78 | 0.97 ± 0.09 | 9.52 ± 1.07 | 1.06 ± 0.18 | >10 |
| 2j | 0.88 ± 0.09 | >10 | 8.44 ± 1.29 | >10 | 3.51 ± 0.86 |
| 2k | 1.50 ± 0.17 | 0.20 ± 0.06 | 2.20 ± 0.53 | 0.78 ± 0.23 | 2.88 ± 0.24 |
| 2l | 1.23 ± 0.38 | 3.42 ± 0.49 | 9.41 ± 1.12 | 8.22 ± 1.06 | 8.50 ± 1.25 |
| 2m | 4.16 ± 0.58 | 3.45 ± 0.39 | 9.62 ± 1.58 | 3.28 ± 0.57 | 7.85 ± 1.03 |
| 2n | 8.39 ± 1.23 | 9.15 ± 1.60 | 9.35 ± 1.74 | 7.56 ± 1.31 | >10 |
| 2o | 2.10 ± 0.36 | 0.59 ± 0.08 | 5.16 ± 0.67 | 0.79 ± 0.14 | 3.31 ± 0.66 |
| 2p | 3.22 ± 0.61 | 1.15 ± 0.46 | 8.77 ± 1.41 | 1.69 ± 0.48 | 4.88 ± 0.63 |
| Doxorubicin | 6.50 ± 1.33 | 0.98 ± 0.16 | 0.62 ± 0.03 | 0.75 ± 0.26 | 0.19 ± 0.07 |
Compared with 2a, the pyridyl analogue 2f displayed improved antiproliferative activity, especially against 4T1, HepG2, and MCF-7 cells (IC50 values, 0.23 μM, 0.80 μM, and 0.34 μM, respectively). In contrast, either no substitution (2g) or a bulky substituent (2h) at the piperazinyl N4 position reduced the suppression of cell growth compared with 2f, implying that suitable size of alkyl substituent at that position was favorable. 2i, containing 4-morpholine at the pyridyl C2 position, was also inferior to 2f, but an amino at that position (2j) dramatically reduced the antiproliferative activity against all cell lines except A549 (IC50 = 0.88 μM). Changing the R1 substituent to 2-(4-ethylpiperazin-1-yl)pyrimidine (2k) only maintained the activity compared with 2f against 4T1 cells. Similar to 2j, an amino substituent at pyrimidine C2 position (2l) also significantly decreased the activity against all cell lines except A549, suggesting that 2j and 2l might have distinct mechanisms of action compared with the other synthesized indazole derivatives. Moving the amino group to the meta-position of indazole (2m and 2n) did not improve their potencies compared with 2f.
To investigate R2 substitutions, we retained 6-(4-methylpiperazin-1-yl)pyridin-3-yl at the indazole C6 position. Adding chlorine to 3,5-dimethoxystyryl (2o) led to a reduction in potency compared with 2f, and a similar effect was observed when the linker was transformed from vinyl to ethyl (2p). Based on the analysis of structure–activity relationship, 2f, with optimum antiproliferative efficacy in vitro, was selected for further evaluation.
2.3 The effects of 2f on 4T1 cell proliferation
To validate the inhibitory activity of 2f on cancer cell growth and proliferation, we performed the MTT assay and colony formation assay. As shown in Fig. 3A, 2f decreased 4T1 cell proliferation in a dose- and time-dependent manner. In addition, exposure to 2f significantly inhibited colony formation of 4T1 cells, and treatment with 2f at 1.25 μM led to a complete inhibition (Fig. 3B and C), confirming that 2f had a strong cytostatic effect on 4T1 cells. | ||
| Fig. 3 Effects of 2f on 4T1 breast cancer cells. (A) MTT proliferation assay performed after incubation of cells for 1–3 days with the indicated concentrations of 2f. (B, C) Colony formation measured after incubation of cells for 10 days with the indicated concentrations of 2f. Data were the mean ± SD. ***p < 0.001. | ||
2.4 The effects of 2f on 4T1 cell apoptosis
To determine whether the antiproliferative activity of 2f was associated with activation of the programmed cell death pathways, we examined 4T1 cell apoptosis using the annexin V-FITC/propidium iodide (PI) dual-labeling flow cytometry assay. After 2f treatment for 24 h, we observed a dose-dependent increase in the percentage of apoptotic cells from 3.7% in the presence of 2f at 0 μM to 53.2% in the presence of 2f at 5 μM (Fig. 4A).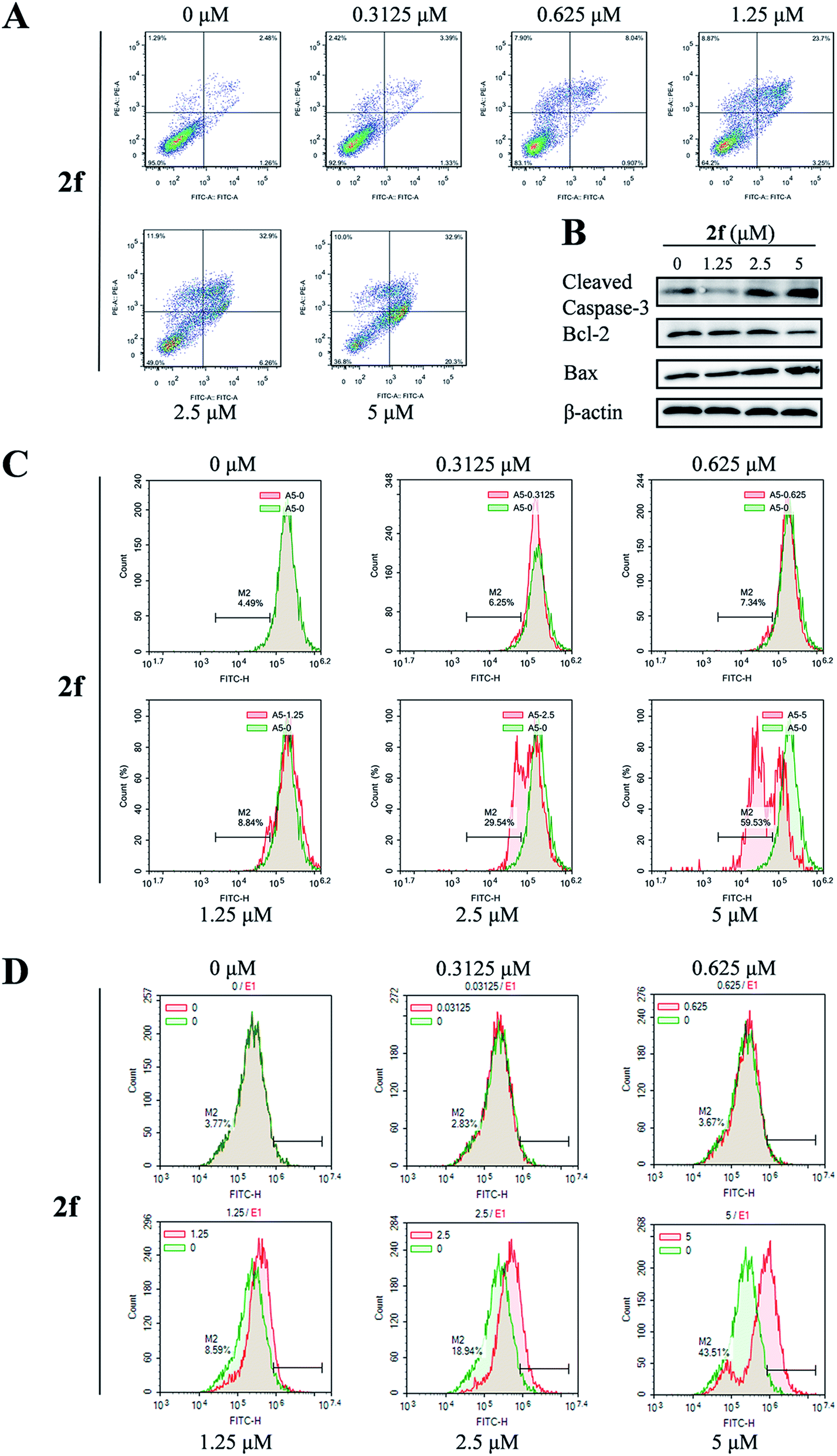 | ||
| Fig. 4 Effects of 2f on cell apoptosis. (A) Flow cytometric assay of apoptosis performed by annexin V-FITC/PI dual staining of cells after incubation with the indicated concentrations of 2f for 24 h. (B) Western blot analysis of cleaved caspase-3, Bcl-2, Bax, and β-actin (loading control) after incubation of cells with 2f for 24 h. (C) 2f decreased the mitochondrial membrane potential (ΔΨm) in 4T1 cells. 4T1 cells were treated with various concentrations of 2f for 24 h and then stained with 5 μg ml−1 Rh123 to detect the change of ΔΨm by FCM. (D) 2f increased the levels of ROS in 4T1 cells. After treatment with various concentrations of 2f for 24 h, 4T1 cells were incubated with 10 μM DCFH-DA, and then the ROS levels were measured by DCF fluorescence using FCM. | ||
To further investigate the pro-apoptotic effect of 2f, expression of the pro-apoptotic proteins Bax and cleaved caspase-3 and the anti-apoptotic protein Bcl-2 was examined by western blot analysis. As shown in Fig. 4B, incubation of 4T1 cells with 2f decreased the expression of Bcl-2 and increased that of cleaved caspase-3 and Bax, suggesting that 2f might induce apoptosis via the mitochondrial apoptotic pathway. Then we measured the effect of 2f on the mitochondrial membrane potential (ΔΨm) of 4T1 cells by a flow cytometric assay using a green fluorochrome Rh123. As shown in Fig. 4C, 2f caused a dose-dependent loss of ΔΨm potential. We also detected ROS levels using the redox-sensitive fluorescent dye 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), and exposure to 2f resulted in a dose-dependent increase of ROS levels (Fig. 4D). These results implied that the inhibition of 4T1 cells viability after treatment with 2f was mediated by the mitochondria-mediated apoptotic pathway.
2.5 The effects of 2f on 4T1 cell migration and invasion
To evaluate the potential of 2f for retarding cell migration and invasion, we performed Transwell assays on 4T1 cells. As shown in Fig. 5A and B, 2f-treated 4T1 cells exhibited significant and dose-dependent reductions in migration and invasion, and similar results were obtained when migration was evaluated by wound-healing assays. We investigated whether the levels of matrix metalloproteinase metalloproteinase-9 (MMP9) and tissue inhibitor of matrix metalloproteinase 2 (TIMP2), which were considered to be correlated with cell migration and invasion, were affected by 2f treatment. Western blot analysis demonstrated that 2f diminished MMP9 levels and increased TIMP2 levels in 4T1 cells (Fig. 5C). Taken together, these findings demonstrated that 2f restrained the migration and invasion of 4T1 cells.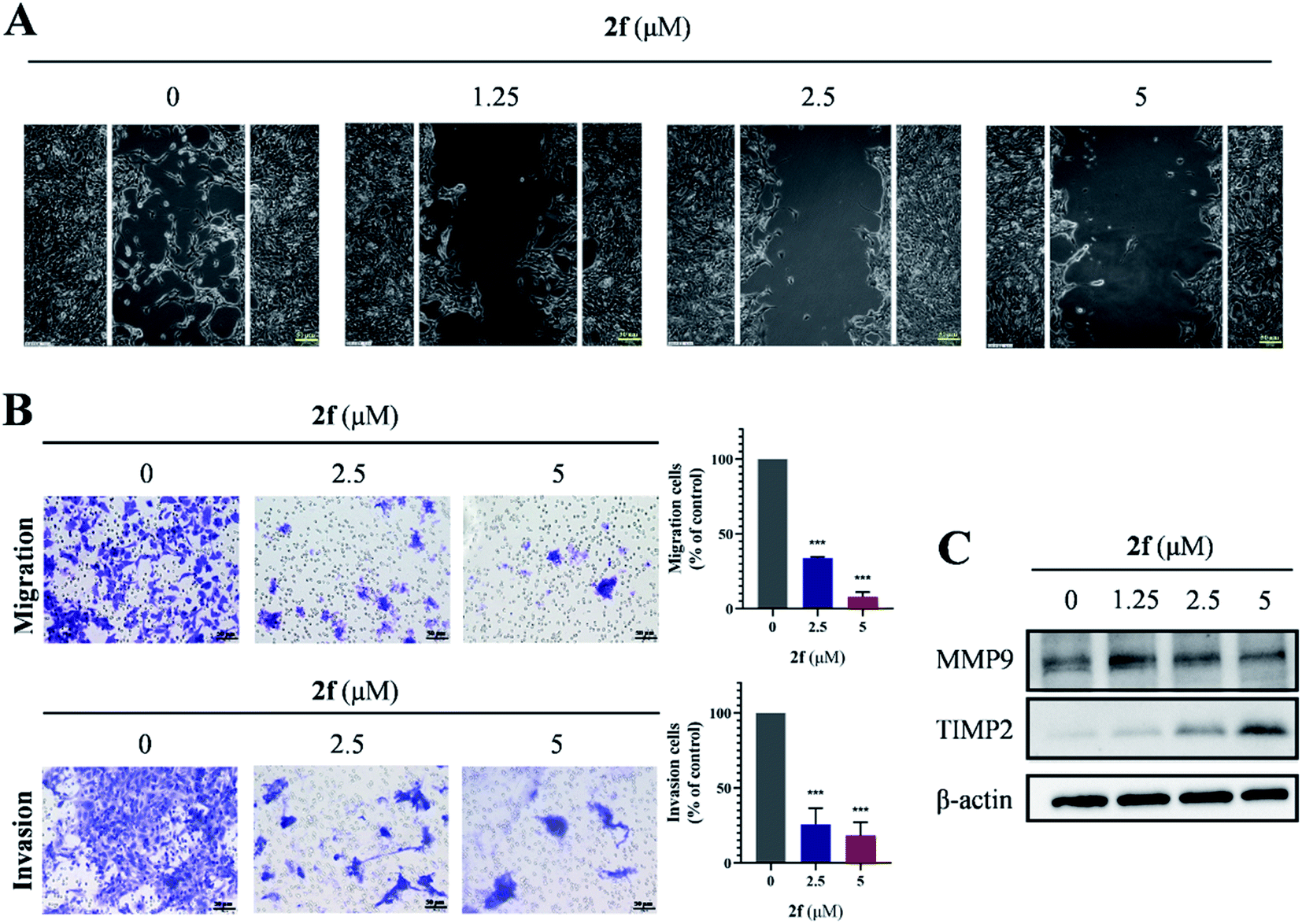 | ||
| Fig. 5 Effects of 2f on cell migration and invasion. (A) Cells were seeded in 6 well culture plates, and the cell monolayer was wounded manually. Then, the images were taken after treated with 2f for 24 h. The scale bar is 50 μm. (B) Images of cells (left) and quantification of data (right) from Transwell migration assays (upper panels) and invasion assays (lower panels) after 24 h treatment of cells with 2f. The scale bar is 50 μm. (C) Western blot analysis of MMP9, TIMP2, and β-actin (loading control) after incubation of cells with 2f for 24 h. Data were the mean ± SD. ***p < 0.001. | ||
2.6 Anti-tumor activity of 2f in vivo
Next, a preliminary assessment of the anti-tumor activity of 2f in vivo was performed in a mice model. 4T1 tumor-bearing mice were treated with 2f at doses of 12.5 mg kg−1 and 25 mg kg−1 once daily. As shown in Fig. 6A and B, 2f dose-dependently suppressed tumor growth compared with the control treatment without any significant effect on body weights. Considering that tumor volumes are sometimes not very accurate, photographs of tumor size at the end of in vivo study were also presented in Fig. S1.† We performed immunohistochemical staining to investigate the underlying mechanism of 2f anti-tumor activity in this 4T1 model. Notably, tumors from 2f-treated mice exhibited reduced expression of the cell proliferation marker Ki67 and of MMP9, while expression of cleaved caspase-3 was increased (Fig. 6C). Routine analyses of blood samples and microscopic examination of the major organs of the treated mice revealed no obvious abnormalities in blood markers or morphology of the heart, liver, spleen, lung, and kidney (Fig. S2 and S3†). These results indicated that 2f potentially reduces the growth of 4T1 breast tumors by promoting cell apoptosis, and that 2f has an acceptable safety profile in mice. We noted that the effect of 2f on tumor growth in vivo was modest, which was probably due to suboptimal administration route or dosage, and another possibility was that the tumor was too large at the beginning of administration. Further study will be necessary to enhance the anti-tumor and anti-metastatic activities of 2f in vivo. | ||
| Fig. 6 Anti-tumor effects of 2f in vivo. (A–C) Groups of female BALB/c mice (n = 5/group) were subcutaneously injected with 4T1 cells and received intraperitoneally injection (i.p.) of 2f 12.5 mg kg−1, 25 mg kg−1 or vehicle, respectively once daily for 16 days. (A) Tumor volumes. (B) Body weights. (C) Immunohistochemical staining of Ki67, cleaved caspase-3, and MMP9 in sections of tumors. The scale bar is 50 μm. Data were the mean ± SD, n.s. not significant, *p < 0.05, **p < 0.01. | ||
3. Conclusion
In summary, we designed and synthesized a series of indazole derivatives. 2f, which was identified as the most potent compound, inhibited cell proliferation and colony formation, as well as migration and invasion in vitro. Treatment with 2f also promoted apoptosis via the ROS-mitochondrial apoptotic pathway. Additionally, 2f suppressed tumor growth in the 4T1 mouse model without obvious side effects. Moreover, Swiss Target Prediction tool was applied to predict the potential target for 2f, and the results pointed to some tyrosine kinases. However, 2f only had moderate inhibitory activities against those kinases with inhibition rate <30% at 1 μM (data not shown), which implied that 2f is probably more like a multitarget inhibitor than a specific kinase inhibitor. Thus, further studies are required to elucidate the precise targets and mechanisms of anti-cancer activity of 2f, which enable extensive optimization of this compound. Besides, the evaluation of anti-cancer activity in mice xenograft models is also warranted. Overall, 2f will serve as a potential lead for developing anti-cancer agents.4. Experimental section
4.1 Chemistry
All commercially available reagents were used without further purification or dried, unless otherwise specified. Thin layer chromatography was carried out on silica gel plates GF254. Column chromatography was performed with silica gel (200–300 mesh). NMR spectra were recorded on a Bruker AV-400 spectrometer with tetramethylsilane (TMS) as an internal standard. The chemical shifts δ and coupling constants J were in ppm and Hz units, respectively. The intermediates were confirmed by Bruker amazon SL LRMS, and the target compounds were identified by Waters Q-TOF Premier HRMS, with ESI ion source. Target compounds were of >95% purity, which was analyzed on a Dionex Ultimate 3000 HPLC, with a Kromasil C18 reversed-phase column (4.6 mm × 250 mm, 5 μm).4.1.1.1 (E)-2-(3,5-Dimethoxystyryl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2-1). To a solution of 1-bromo-3,5-dimethoxybenzene (20 mmol, 1.0 equiv.) in dried toluene (60 mL) was added Pd2(dba)3 (1 mmol, 5 mol%), P(t-Bu)3·HBF4 (2 mmol, 10 mol%), DIPEA (40 mmol, 2.0 equiv.) and 4,4,5,5-tetramethyl-2-vinyl-1,3,2-dioxaborolane (22 mmol, 1.1 equiv.). Then the mixture was stirred at 95 °C under N2 atmosphere. After 6 h the reaction was monitored by TLC. The solvent was removed under vacuum. To the residue was added water, and extracted with ethyl acetate. The organic layer was dried over anhydrous sodium sulfate and concentrated. The intermediate 2-1 was obtained by column chromatography as a pale-yellow solid (yield 78.1%). MS m/z (ESI): 291.2 [M + H]+, 1H NMR (400 MHz, DMSO-d6) δ 7.94 (d, J = 8.0 Hz, 2H), 7.72 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 18.4 Hz, 1H), 6.31 (d, J = 18.5 Hz, 1H), 3.86 (s, 3H), 1.26 (s, 12H).
4.1.1.2 (E)-2-(2,6-Dichloro-3,5-dimethoxystyryl)-4,4,5,5-tetramethyl-1,3,2-dioxaborolane (2-2). N-Chlorosuccinimide (10 mmol, 2.0 equiv.) was added to the solution of compound 2-1 (5 mmol, 1.0 equiv.) in DMF (15 mL). Then the mixture was stirred at 85 °C under N2 atmosphere. After 5 h the reaction was monitored by TLC. The solvent was removed under vacuum. To the residue was added water, and extracted with ethyl acetate. The organic layer was dried and concentrated. The intermediate 2-2 was obtained by column chromatography as an offwhite solid (yield 47.2%). MS m/z (ESI): 359.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 7.20 (d, J = 18.8 Hz, 1H), 6.89 (s, 1H), 5.98 (d, J = 18.8 Hz, 1H), 3.92 (s, 6H), 1.26 (s, 12H).
4.1.1.3 6-Bromo-3-iodo-1H-indazole (2-3). To a solution of 6-bromo-1H-indazole (10 mmol, 1.0 equiv.) in DMF was added KOH (20 mmol, 2.0 equiv.). Then the solution of I2 (15 mmol, 1.5 equiv.) in DMF (8 mL) was added dropwise into mixture, and stirred at room temperature for 3 h. After that, the reaction mixture was poured into the aqueous solution of Na2S2O4 and K2CO3, with a white solid precipitated. The solid was filtered and dried to give intermediate 2-3 as a white solid (yield 71.2%). MS m/z (ESI): 322.9 [M + H]+, 1H NMR (400 MHz, DMSO-d6) δ 13.62 (s, 1H), 7.82 (d, J = 1.5 Hz, 1H), 7.40 (d, J = 8.6 Hz, 1H), 7.33 (dd, J = 8.6, 1.6 Hz, 1H).
4.1.1.4 (E)-6-Bromo-3-(3,5-dimethoxystyryl)-1H-indazole (2-4). The intermediates 2-1 (2.4 mmol, 1.2 equiv.), 2-3 (2 mmol, 1.0 equiv.), Pd(dppf)Cl2 (0.1 mmol, 5 mol%) and cesium carbonate (3 mmol, 1.5 equiv.) were added to a mixed solvent of water and dioxane (1
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4, 30 mL). Then the mixture was stirred at 100 °C under N2 atmosphere. After 8 h the reaction was monitored by TLC. The solvent was evaporated under reduced pressure. The residue was dissolved in dichloromethane and methanol, and filtered with silica. The filtrate was concentrated and purified by column chromatography to afford 2-4. Pale-yellow solid (yield 67.5%). MS m/z (ESI): 359.1 [M + H]+.1H NMR (400 MHz, DMSO-d6) δ 13.28 (s, 1H), 8.18 (d, J = 8.6 Hz, 1H), 7.77 (d, J = 1.6 Hz, 1H), 7.63–7.41 (m, 2H), 7.33 (dd, J = 8.7, 1.7 Hz, 1H), 6.91 (d, J = 2.3 Hz, 2H), 6.44 (t, J = 2.2 Hz, 1H), 3.80 (s, 6H).
:4, 30 mL). Then the mixture was stirred at 100 °C under N2 atmosphere. After 8 h the reaction was monitored by TLC. The solvent was evaporated under reduced pressure. The residue was dissolved in dichloromethane and methanol, and filtered with silica. The filtrate was concentrated and purified by column chromatography to afford 2-4. Pale-yellow solid (yield 67.5%). MS m/z (ESI): 359.1 [M + H]+.1H NMR (400 MHz, DMSO-d6) δ 13.28 (s, 1H), 8.18 (d, J = 8.6 Hz, 1H), 7.77 (d, J = 1.6 Hz, 1H), 7.63–7.41 (m, 2H), 7.33 (dd, J = 8.7, 1.7 Hz, 1H), 6.91 (d, J = 2.3 Hz, 2H), 6.44 (t, J = 2.2 Hz, 1H), 3.80 (s, 6H).
4.1.1.5 (E)-6-Bromo-3-(2,6-dichloro-3,5-dimethoxystyryl)-1H-indazole (2-5). The preparation of intermediate 2-5 is the same as 2-4. Yellow solid (yield 46.2%). MS m/z (ESI): 427.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 13.25 (s, 1H), 8.03 (d, J = 8.6 Hz, 1H), 7.82 (d, J = 1.6 Hz, 1H), 7.46–7.30 (m, 3H), 6.93 (s, 1H), 3.96 (s, 6H).
4.1.2.1 The intermediates 2-4 or 2-5 (0.5 mmol, 1.0 equiv.). Pd(dppf)Cl2 (0.1 mmol, 5 mol%), potassium carbonate (1 mmol, 2.0 equiv.) and corresponding boronic acid pinacol esters (0.6 mmol, 1.2 equiv.) were added to a mixed solvent of water and dioxane (1
:4, 15 mL). Then the mixture was stirred at 95 °C under N2 atmosphere. After 8 h the reaction was monitored by TLC. The solvent was evaporated under reduced pressure. The residue was dissolved in ethyl acetate, and washed with water. The organic layer was dried over anhydrous sodium sulfate and concentrated. The crude products were purified by preparative thin layer chromatography to give target compounds.
4.1.2.2 (E)-3-(3,5-Dimethoxystyryl)-6-(4-(4-methylpiperazin-1-yl)phenyl)-1H-indazole (2a). Pale-yellow solid (yield 21.1%). HRMS (ESI-TOF) m/z calcd for C28H30N4O2 [M + H]+: 455.2443, found: 455.2449; 1H NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 8.21 (d, J = 8.5 Hz, 1H), 7.68–7.60 (m, 3H), 7.57 (d, J = 16.7 Hz, 1H), 7.51–7.42 (m, 2H), 7.04 (d, J = 8.7 Hz, 2H), 6.92 (d, J = 2.2 Hz, 2H), 6.44 (t, J = 2.2 Hz, 1H), 3.81 (s, 6H), 3.21 (d, J = 4.9 Hz, 4H), 2.47 (t, J = 5.0 Hz, 4H), 2.23 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 161.16, 150.92, 142.69, 142.56, 139.73, 138.96, 130.80, 129.88, 128.07, 121.78, 121.64, 120.57, 119.87, 115.97, 107.06, 104.82, 100.57, 55.73, 55.02, 48.27, 46.26.
4.1.2.3 (E)-4-(3-(3,5-Dimethoxystyryl)-1H-indazol-6-yl)-N,N-dimethylbenzenesulfonamide (2b). Pale-yellow solid (yield 31.0%). HRMS (ESI-TOF) m/z calcd for C25H25N3O4S [M + Na]+: 486.1459, found: 486.1458; 1H NMR (400 MHz, DMSO-d6) δ 13.36 (s, 1H), 8.34 (d, J = 8.5 Hz, 1H), 8.05 (d, J = 8.3 Hz, 2H), 7.92–7.81 (m, 3H), 7.66–7.56 (m, 2H), 7.50 (d, J = 16.6 Hz, 1H), 6.93 (d, J = 2.2 Hz, 2H), 6.45 (d, J = 2.2 Hz, 1H), 3.82 (s, 6H), 2.68 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 161.17, 145.20, 142.73, 142.31, 139.63, 137.15, 134.07, 130.23, 128.71, 128.49, 122.15, 121.39, 121.06, 120.96, 109.30, 104.91, 100.66, 55.74, 38.08.
4.1.2.4 (E)-4-((4-(3-(3,5-Dimethoxystyryl)-1H-indazol-6-yl)phenyl)sulfonyl)morpholine (2c). Pale-yellow solid (yield 27.3%). HRMS (ESI-TOF) m/z calcd for C27H27N3O5S [M + Na]+: 528.1564, found: 528.1569; 1H NMR (400 MHz, DMSO-d6) δ 13.38 (s, 1H), 8.35 (d, J = 8.5 Hz, 1H), 8.09–8.04 (m, 2H), 7.90–7.81 (m, 3H), 7.64–7.57 (m, 2H), 7.50 (d, J = 16.7 Hz, 1H), 6.94 (d, J = 2.2 Hz, 2H), 6.45 (t, J = 2.2 Hz, 1H), 3.82 (s, 6H), 3.66 (t, J = 4.7 Hz, 4H), 2.99–2.90 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 161.17, 145.56, 142.73, 142.29, 139.63, 137.08, 133.69, 130.24, 128.84, 128.60, 122.16, 121.39, 121.10, 120.99, 109.37, 104.91, 100.66, 65.79, 55.74, 46.39.
4.1.2.5 (E)-3-(3,5-Dimethoxystyryl)-6-(4-((4-methylpiperazin-1-yl)methyl)phenyl)-1H-indazole (2d). Light grey solid (yield 24.8%). HRMS (ESI-TOF) m/z calcd for C29H32N4O2 [M + H]+:469.2599, found: 469.2603; 1H NMR (400 MHz, DMSO-d6) δ 13.23 (s, 1H), 8.27 (d, J = 8.5 Hz, 1H), 7.77–7.67 (m, 3H), 7.58 (d, J = 16.7 Hz, 1H), 7.54–7.38 (m, 4H), 6.92 (d, J = 2.3 Hz, 2H), 6.44 (t, J = 2.3 Hz, 1H), 3.81 (s, 6H), 3.51 (s, 2H), 2.38 (d, J = 21.4 Hz, 8H), 2.17 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 161.17, 139.68, 139.49, 138.89, 138.13, 130.03, 129.97, 128.35, 127.43, 121.80, 121.61, 120.99, 120.38, 108.24, 104.85, 100.60, 62.12, 55.74, 55.11, 52.89, 46.05.
4.1.2.6 (E)-4-(4-(3-(3,5-Dimethoxystyryl)-1H-indazol-6-yl)benzyl)morpholine (2e). Pale-yellow solid (yield 22.3%). HRMS (ESI-TOF) m/z calcd for C28H29N3O3 [M + H]+: 456.2283, found: 456.2281; 1H NMR (400 MHz, DMSO-d6) δ 13.23 (s, 1H), 8.27 (d, J = 8.5 Hz, 1H), 7.72 (d, J = 7.8 Hz, 3H), 7.59 (d, J = 16.7 Hz, 1H), 7.53–7.39 (m, 4H), 6.92 (d, J = 2.3 Hz, 2H), 6.44 (t, J = 2.2 Hz, 1H), 3.81 (s, 6H), 3.60 (t, J = 4.6 Hz, 4H), 3.52 (s, 2H), 2.39 (t, J = 4.6 Hz, 4H); 13C NMR (101 MHz, DMSO-d6) δ 161.17, 142.62, 142.49, 139.69, 139.58, 138.85, 137.73, 130.04, 127.47, 121.81, 121.64, 120.96, 120.42, 108.26, 104.86, 100.61, 66.70, 62.54, 55.73, 53.68.
4.1.2.7 (E)-3-(3,5-Dimethoxystyryl)-6-(6-(4-methylpiperazin-1-yl)pyridin-3-yl)-1H-indazole (2f). Pale-yellow solid (yield 17.9%). HRMS (ESI-TOF) m/z calcd for C27H29N5O2 [M + H]+: 456.2395, found: 456.2393; 1H NMR (400 MHz, DMSO-d6) δ 13.18 (s, 1H), 8.54 (d, J = 2.6 Hz, 1H), 8.24 (d, J = 8.5 Hz, 1H), 7.95 (dd, J = 8.9, 2.6 Hz, 1H), 7.68 (s, 1H), 7.57 (d, J = 16.6 Hz, 1H), 7.51–7.42 (m, 2H), 7.02–6.86 (m, 3H), 6.44 (t, J = 2.2 Hz, 1H), 3.81 (s, 6H), 3.56 (dd, J = 7.1, 3.1 Hz, 4H), 2.43 (t, J = 5.0 Hz, 4H), 2.24 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 161.16, 158.80, 146.37, 142.59, 139.70, 136.66, 136.45, 129.96, 125.50, 121.85, 121.68, 120.26, 120.04, 107.49, 107.03, 104.86, 100.58, 55.73, 54.81, 46.23, 45.05.
4.1.2.8 (E)-3-(3,5-Dimethoxystyryl)-6-(6-(piperazin-1-yl)pyridin-3-yl)-1H-indazole (2g). Light grey solid (yield 29.1%). HRMS (ESI-TOF) m/z calcd for C26H27N5O2 [M + H]+:442.2239, found: 442.2239; 1H NMR (400 MHz, DMSO-d6) δ 13.25 (s, 1H), 8.59 (d, J = 2.6 Hz, 1H), 8.29 (d, J = 8.5 Hz, 1H), 8.05–7.95 (m, 1H), 7.73 (s, 1H), 7.62 (d, J = 16.8 Hz, 1H), 7.57–7.46 (m, 2H), 6.97 (d, J = 2.4 Hz, 3H), 6.49 (t, J = 2.3 Hz, 1H), 3.87 (s, 6H), 3.54 (t, J = 4.7 Hz, 4H), 2.96–2.77 (m, 4H), 1.28 (s, 1H); 13C NMR (101 MHz, DMSO-d6) δ 161.16, 146.35, 142.67, 142.51, 139.70, 136.58, 136.50, 129.96, 125.22, 121.84, 121.62, 120.25, 120.00, 107.31, 107.03, 104.85, 100.58, 55.73, 46.24, 45.82.
4.1.2.9 tert-Butyl (E)-4-(5-(3-(3,5-dimethoxystyryl)-1H-indazol-6-yl)pyridin-2-yl)piperazine-1-carboxylate (2h). Light brown solid (yield 18.3%). HRMS (ESI-TOF) m/z calcd for C31H35N5O4 [M + Na]+: 564.2582, found: 564.2594; 1H NMR (400 MHz, DMSO-d6) δ 13.19 (s, 1H), 8.56 (d, J = 2.6 Hz, 1H), 8.24 (d, J = 8.5 Hz, 1H), 7.98 (dd, J = 8.9, 2.6 Hz, 1H), 7.71–7.66 (m, 1H), 7.57 (d, J = 16.7 Hz, 1H), 7.51–7.43 (m, 2H), 6.96 (d, J = 8.9 Hz, 1H), 6.92 (d, J = 2.2 Hz, 2H), 6.44 (t, J = 2.2 Hz, 1H), 3.81 (s, 6H), 3.57 (dd, J = 6.9, 3.7 Hz, 4H), 3.46 (dd, J = 6.6, 3.8 Hz, 4H), 1.44 (s, 9H); 13C NMR (101 MHz, DMSO-d6) δ 161.16, 150.93, 142.68, 142.56, 139.73, 138.97, 138.23, 130.81, 129.88, 128.08, 121.77, 121.64, 120.57, 120.37, 119.88, 115.98, 107.04, 104.83, 100.57, 55.73, 55.02, 48.28, 46.26.
4.1.2.10 (E)-4-(5-(3-(3,5-Dimethoxystyryl)-1H-indazol-6-yl)pyridin-2-yl)morpholine (2i). Offwhite solid (yield 31.1%). HRMS (ESI-TOF) m/z calcd for C26H26N4O3 [M + H]+: 443.2079, found: 443.2081; 1H NMR (400 MHz, DMSO-d6) δ 13.19 (s, 1H), 8.57 (d, J = 2.6 Hz, 1H), 8.24 (d, J = 8.5 Hz, 1H), 7.98 (dd, J = 8.8, 2.6 Hz, 1H), 7.69 (t, J = 1.0 Hz, 1H), 7.57 (d, J = 16.7 Hz, 1H), 7.50–7.43 (m, 2H), 7.01–6.85 (m, 3H), 6.44 (t, J = 2.2 Hz, 1H), 3.81 (s, 6H), 3.77–3.70 (m, 4H), 3.57–3.46 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 161.16, 158.94, 146.29, 142.58, 142.56, 139.69, 136.75, 136.35, 129.99, 126.00, 121.87, 121.63, 120.30, 120.07, 107.51, 107.15, 104.86, 100.58, 66.43, 55.73, 45.64.
4.1.2.11 (E)-3-(3,5-Dimethoxystyryl)-6-(2-(4-ethylpiperazin-1-yl)pyrimidin-5-yl)-1H-indazole (2k). Light grey solid (yield 18.5%). HRMS (ESI-TOF) m/z calcd for C27H30N6O2 [M + H]+: 471.2504, found: 471.2503; 1H NMR (400 MHz, DMSO-d6) δ 13.24 (s, 1H), 8.80 (s, 2H), 8.26 (d, J = 8.5 Hz, 1H), 7.73 (s, 1H), 7.57 (d, J = 16.7 Hz, 1H), 7.51–7.43 (m, 2H), 6.92 (d, J = 2.2 Hz, 2H), 6.44 (d, J = 2.3 Hz, 1H), 3.81 (s, 10H), 2.46 (s, 4H), 2.39 (d, J = 7.4 Hz, 2H), 1.05 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 161.16, 161.01, 156.63, 142.66, 142.46, 139.66, 133.72, 130.06, 122.72, 122.04, 121.54, 120.27, 119.89, 107.08, 104.88, 100.58, 55.73, 52.55, 52.10, 43.90, 12.28.
4.1.2.12 (E)-3-(2,6-Dichloro-3,5-dimethoxystyryl)-6-(6-(4-methylpiperazin-1-yl)pyridin-3-yl)-1H-indazole (2o). Pale-yellow solid (yield 24.4%). HRMS (ESI-TOF) m/z calcd for C27H27Cl2N5O2 [M + H]+: 524.1616, found: 524.1616; 1H NMR (400 MHz, DMSO-d6) δ 13.32 (s, 1H), 8.54 (d, J = 2.6 Hz, 1H), 8.10 (d, J = 8.5 Hz, 1H), 7.95 (dd, J = 8.9, 2.6 Hz, 1H), 7.75–7.70 (m, 1H), 7.51 (dd, J = 8.5, 1.5 Hz, 1H), 7.45 (d, J = 16.9 Hz, 1H), 7.38 (d, J = 17.0 Hz, 1H), 6.99–6.90 (m, 2H), 3.96 (s, 6H), 3.56 (t, J = 5.0 Hz, 4H), 2.42 (t, J = 5.0 Hz, 4H), 2.23 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 158.81, 154.94, 148.73, 146.37, 142.68, 136.63, 135.54, 129.66, 125.35, 124.22, 121.31, 120.81, 119.83, 114.25, 112.98, 107.50, 107.19, 97.86, 57.18, 54.79, 46.21, 45.03.
4.1.4.1 5-(1H-Indazol-6-yl)pyridin-2-amine (2-6). 5-Bromopyridin-2-amine (20 mmol, 1.0 equiv.), (1H-indazol-6-yl)boronic acid (24 mmol, 1.2 equiv.), Pd(dppf)Cl2 (1 mmol, 5 mol%) and potassium carbonate (30 mmol, 1.5 equiv.) were added to a mixed solvent of dioxane and H2O (4
:1, 50 mL). Then the reaction was heated to 100 °C and stirred overnight. The solvent was evaporated under reduced pressure, and the residue was dissolved in ethyl acetate, then filtered with silica. The filtrate was concentrated and purified by column chromatography to afford 2-6. Light brown solid (yield 62.3%). MS m/z (ESI): 211.1 [M + H]+.
4.1.4.2 5-(1H-Indazol-6-yl)pyrimidin-2-amine (2-7). The preparation of 2-7 was similar to 2-6. Light brown solid (yield 59.7%). MS m/z (ESI): 212.0 [M + H]+.
4.1.4.3 4-(1H-Indazol-6-yl)pyridin-2-amine (2-8). The preparation of 2-8 was similar to 2-6. Light brown solid (yield 60.5%). MS m/z (ESI): 211.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 13.23 (s, 1H), 8.13 (s, 1H), 8.00 (d, J = 5.4 Hz, 1H), 7.86 (d, J = 8.4 Hz, 1H), 7.75 (s, 1H), 7.39 (d, J = 8.4 Hz, 1H), 6.94–6.74 (m, 2H), 6.05 (s, 2H).
4.1.4.4 4-(1H-Indazol-6-yl)pyrimidin-2-amine (2-9). The preparation of 2-9 was similar to 2-6. Light brown solid (yield 56.0%). MS m/z (ESI): 212.1 [M + H]+; 1H NMR (400 MHz, DMSO-d6) δ 13.17 (s, 1H), 8.41 (d, J = 5.2 Hz, 1H), 8.32 (d, J = 5.1 Hz, 1H), 8.10 (s, 1H) 7.86 (d, J = 8.6 Hz, 1H), 7.71 (d, J = 8.6 Hz, 1H), 7.64 (d, J = 5.3 Hz, 1H), 6.73 (s, 2H).
4.1.4.5 5-(3-Iodo-1H-indazol-6-yl)pyridin-2-amine (2-10). To a solution of intermediate 2-6 (10 mmol, 1.0 equiv) in DMF (15 mL) was added K2CO3 (20 mmol, 2 equiv) and iodine (15 mmol, 1.5 equiv) successively. After heated at 65 °C for 10 h, the mixture was poured into the aqueous solution of Na2S2O4 and K2CO3, with a white solid precipitated. The solid was filtered and dried to give intermediate 2-10 as a white solid (yield 83.7%). MS m/z (ESI): 359.0 [M + Na]+;
4.1.4.6 5-(3-Iodo-1H-indazol-6-yl)pyrimidin-2-amine (2-11). The preparation of 2-11 was similar to 2-10. Offwhite solid (yield 85.0%). MS m/z (ESI): 359.0 [M + Na]+;
4.1.4.7 4-(3-Iodo-1H-indazol-6-yl)pyridin-2-amine (2-12). The preparation of 2-12 was similar to 2-10. Pale-yellow solid (yield 80.1%). MS m/z (ESI): 337.0 [M + H]+.
4.1.4.8 4-(3-Iodo-1H-indazol-6-yl)pyrimidin-2-amine (2-13). The preparation of 2-13 was similar to 2-10. Pale-yellow solid (yield 88.0%). MS m/z (ESI): 360.1 [M + Na]+; 1H NMR (400 MHz, DMSO-d6) δ 13.20 (s, 1H), 8.30 (d, J = 5.0 Hz, 1H), 7.85 (d, J = 8.6 Hz, 1H); 7.66 (s, 1H), 7.41 (s, 1H), 6.79 (d, J = 5.1 Hz, 1H), 6.74 (s, 2H).
4.1.5.1 (E)-5-(3-(3,5-Dimethoxystyryl)-1H-indazol-6-yl)pyridin-2-amine (2j). The intermediates 2-10 (1 mmol, 1 equiv.) and 2-1 (1.2 mmol, 1.2 equiv.) was added to a mixed solvent of dioxane and H2O (4
:1, 15 mL), Pd(dppf)Cl2 (0.05 mmol, 5 mol%) and cesium carbonate (2 mmol, 2.0 equiv.) was added as well. Then the reaction mixture was heated to 100 °C under N2 atmosphere. After 8 h the solvent was evaporated under reduced pressure. The residue was dissolved in dichloromethane and methanol, and filtered with silica. The filtrate was concentrated and purified by preparative thin layer chromatography to give compound 2j as a pale-yellow solid (yield 12.8%). HRMS (ESI-TOF) m/z calcd for C22H20N4O2 [M + H]+: 373.1660, found: 373.1659; 1H NMR (400 MHz, DMSO-d6) δ 13.14 (s, 1H), 8.34 (d, J = 2.4 Hz, 1H), 8.22 (d, J = 8.5 Hz, 1H), 7.80 (dd, J = 8.6, 2.6 Hz, 1H), 7.64–7.53 (m, 2H), 7.50–7.40 (m, 2H), 6.92 (d, J = 2.3 Hz, 2H), 6.58 (d, J = 8.6 Hz, 1H), 6.44 (d, J = 2.3 Hz, 1H), 6.11 (s, 2H), 3.81 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 161.16, 159.60, 146.36, 142.64, 142.58, 139.70, 136.95, 136.45, 129.91, 124.60, 121.76, 121.69, 120.15, 108.63, 106.65, 104.83, 100.58, 99.98, 55.73.
4.1.5.2 (E)-5-(3-(3,5-Dimethoxystyryl)-1H-indazol-6-yl)pyrimidin-2-amine (2l). The preparation of 2l was similar to 2j. Light brown solid (yield 30.1%). HRMS (ESI-TOF) m/z calcd for C21H19N5O2 [M + H]+: 374.1613, found: 374.1611; 1H NMR (400 MHz, DMSO-d6) δ 13.21 (s, 1H), 8.67 (s, 2H), 8.25 (d, J = 8.5 Hz, 1H), 7.69 (s, 1H), 7.57 (d, J = 16.6 Hz, 1H), 7.50–7.43 (m, 2H), 6.92 (d, J = 2.2 Hz, 2H), 6.80 (s, 2H), 6.43 (t, J = 2.2 Hz, 1H), 3.81 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 163.41, 161.16, 156.81, 153.71, 142.49, 139.68, 134.10, 130.00, 122.82, 121.98, 121.57, 120.17, 119.79, 106.82, 104.87, 100.60, 55.73.
4.1.5.3 (E)-4-(3-(3,5-Dimethoxystyryl)-1H-indazol-6-yl)pyridin-2-amine (2m). The preparation of 2m was similar to 2j. Light brown solid (yield 27.5%). HRMS (ESI-TOF) m/z calcd for C22H20N4O2 [M + H]+: 373.1660, found: 373.1660; 1H NMR (400 MHz, DMSO-d6) δ 13.33 (s, 1H), 8.31 (d, J = 8.5 Hz, 1H), 8.01 (d, J = 5.3 Hz, 1H), 7.76 (s, 1H), 7.61 (d, J = 16.7 Hz, 1H), 7.54–7.43 (m, 2H), 6.94 (d, J = 2.2 Hz, 2H), 6.89 (dd, J = 5.4, 1.6 Hz, 1H), 6.82 (d, J = 1.6 Hz, 1H), 6.45 (t, J = 2.2 Hz, 1H), 6.03 (s, 2H), 3.82 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 161.16, 160.91, 148.95, 148.88, 142.70, 142.25, 139.64, 137.19, 130.18, 121.98, 121.46, 121.19, 120.31, 110.93, 108.46, 105.96, 104.87, 100.65, 55.73.
4.1.5.4 (E)-4-(3-(3,5-Dimethoxystyryl)-1H-indazol-6-yl)pyrimidin-2-amine (2n). The preparation of 2n was similar to 2j. Light brown solid (yield 29.7%). HRMS (ESI-TOF) m/z calcd for C21H19N5O2 [M + H]+: 374.1613, found: 374.1613; 1H NMR (400 MHz, DMSO-d6) δ 13.42 (s, 1H), 8.35 (d, J = 5.2 Hz, 1H), 8.33–8.22 (m, 2H), 7.90 (dd, J = 8.6, 1.4 Hz, 1H), 7.61 (d, J = 16.7 Hz, 1H), 7.49 (d, J = 16.7 Hz, 1H), 7.25 (d, J = 5.2 Hz, 1H), 6.93 (d, J = 2.3 Hz, 2H), 6.73 (s, 2H), 6.44 (t, J = 2.3 Hz, 1H), 3.82 (s, 6H); 13C NMR (101 MHz, DMSO-d6) δ 164.28, 164.08, 161.16, 159.59, 142.76, 142.03, 139.61, 135.65, 130.21, 122.31, 121.49, 121.36, 120.02, 109.27, 106.72, 104.90, 100.67, 55.74.
4.2 Biology experiments
:1, 60 μL per well, BD Biosciences, USA). After Matrigel polymerization, the bottom chambers were filled with 600 μL medium containing 10% FBS. 5 × 104 4T1 cells in 100 μL serum-free medium were added in the top part of each transwell and treated with different concentrations of 2f. After incubation for 24 h, non-migrated cells on the upper side of the filter were removed with a cotton swab, and migrated cells were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet and counted under a light microscope. The percentage of migrated cells inhibited by 2f was expressed on the basis of control wells.Author contributions
Wei Wei, Zhihao Liu and Tinghong Ye developed the idea for the study, performed the research, analyzed data and wrote the paper; Xiuli Wu, Cailing Gan, Xingping Su, Hongyao Liu Hanyun Que and Qianyu Zhang provided data; Qiang Xue and Lin Yue revised the manuscript; Luoting Yu and Tinghong Ye supervised the research, acquired funding and revised the manuscript. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Shanghai pharmaceutical group co. LTD (16H0017), the Natural Science Foundation of China (No. 81903441), and Post-Doctor Research Project, West China Hospital, Sichuan University, China (No. 2018HXBH009). We thank Anne M. O'Rourke, PhD, from Liwen Bianji, Edanz Group China (www.liwenbianji.cn/ac), for editing the English text of a draft of this manuscript.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021 DOI:10.3322/caac.21660.
- L. A. Emens, P. A. Ascierto, P. K. Darcy, S. Demaria, A. M. M. Eggermont, W. L. Redmond, B. Seliger and F. M. Marincola, Eur. J. Cancer, 2017, 81, 116–129 CrossRef CAS.
- K. A. High and M. G. Roncarolo, N. Engl. J. Med., 2019, 381, 455–464 CrossRef CAS PubMed.
- K. D. Miller, L. Nogueira, A. B. Mariotto, J. H. Rowland, K. R. Yabroff, C. M. Alfano, A. Jemal, J. L. Kramer and R. L. Siegel, Ca-Cancer J. Clin., 2019, 69, 363–385 CrossRef PubMed.
- L. Wan, K. Pantel and Y. Kang, Nat. Med., 2013, 19, 1450 CrossRef CAS.
- D. D. Gaikwad, A. D. Chapolikar, C. G. Devkate, K. D. Warad, A. P. Tayade, R. P. Pawar and A. J. Domb, Eur. J. Med. Chem., 2015, 90, 707–731 CrossRef CAS.
- I. Denya, S. F. Malan and J. Joubert, Expert Opin. Ther. Pat., 2018, 28, 441–453 CrossRef CAS.
- S. G. Zhang, C. G. Liang and W. H. Zhang, Molecules, 2018, 23 Search PubMed.
- J. Dong, Q. Zhang, Z. Wang, G. Huang and S. Li, ChemMedChem, 2018, 13, 1490–1507 CrossRef CAS PubMed.
- S. Yan, Y. Dong, Q. Peng, Y. Fan, J. Zhang and J. Lin, RSC Adv., 2013, 3, 5563–5569 CAS.
- G. Sandeep Reddy, S. Mohanty, J. Kumar and B. Venteswar Rao, Russ. J. Gen. Chem., 2018, 88, 2394–2399 CrossRef CAS.
- P. A. Harris, A. Boloor, M. Cheung, R. Kumar, R. M. Crosby, R. G. Davis-Ward, A. H. Epperly, K. W. Hinkle, R. N. Hunter 3rd, J. H. Johnson, V. B. Knick, C. P. Laudeman, D. K. Luttrell, R. A. Mook, R. T. Nolte, S. K. Rudolph, J. R. Szewczyk, A. T. Truesdale, J. M. Veal, L. Wang and J. A. Stafford, J. Med. Chem., 2008, 51, 4632–4640 CrossRef CAS PubMed.
- S. Miyamoto, S. Kakutani, Y. Sato, A. Hanashi, Y. Kinoshita and A. Ishikawa, Jpn. J. Clin. Oncol., 2018, 48, 503–513 CrossRef PubMed.
- B. W. Konicek, A. R. Capen, K. M. Credille, P. J. Ebert, B. L. Falcon, G. L. Heady, B. K. R. Patel, V. L. Peek, J. R. Stephens, J. A. Stewart, S. L. Stout, D. E. Timm, S. L. Um, M. D. Willard, I. H. Wulur, Y. Zeng, Y. Wang, R. A. Walgren and S. C. Betty Yan, Oncotarget, 2018, 9, 13796–13806 CrossRef PubMed.
- S. B. Yan, S. L. Um, V. L. Peek, J. R. Stephens, W. Zeng, B. W. Konicek, L. Liu, J. R. Manro, V. Wacheck and R. A. Walgren, Invest. New Drugs, 2018, 36, 536–544 CrossRef CAS PubMed.
- A. R. He, R. B. Cohen, C. S. Denlinger, A. Sama, A. Birnbaum, J. Hwang, T. Sato, N. Lewis, M. Mynderse, M. Niland, J. Giles, J. Wallin, B. Moser, W. Zhang, R. Walgren and E. R. Plimack, Oncologist, 2019, 24, e930–e942 CrossRef CAS PubMed.
- Z. T. Al-Salama and S. J. Keam, Drugs, 2019, 79, 1477–1483 CrossRef PubMed.
- Y. Umeyama, Y. Shibasaki and H. Akaza, Future Oncol., 2017, 13, 1839–1852 CrossRef CAS PubMed.
- A. Bellesoeur, E. Carton, J. Alexandre, F. Goldwasser and O. Huillard, Drug Des., Dev. Ther., 2017, 11, 2801–2811 CrossRef CAS PubMed.
- G. Zhao, W. Y. Li, D. Chen, J. R. Henry, H. Y. Li, Z. Chen, M. Zia-Ebrahimi, L. Bloem, Y. Zhai, K. Huss, S. B. Peng and D. J. McCann, Mol. Cancer Ther., 2011, 10, 2200–2210 CrossRef CAS PubMed.
- M. Michael, Y. J. Bang, Y. S. Park, Y. K. Kang, T. M. Kim, O. Hamid, D. Thornton, S. C. Tate, E. Raddad and J. Tie, Target. Oncol., 2017, 12, 463–474 CrossRef PubMed.
- J. M. Mason, D. C. Lin, X. Wei, Y. Che, Y. Yao, R. Kiarash, D. W. Cescon, G. C. Fletcher, D. E. Awrey, M. R. Bray, G. Pan and T. W. Mak, Cancer Cell, 2014, 26, 163–176 CrossRef CAS PubMed.
- P. B. Sampson, Y. Liu, B. Forrest, G. Cumming, S. W. Li, N. K. Patel, L. Edwards, R. Laufer, M. Feher, F. Ban, D. E. Awrey, G. Mao, O. Plotnikova, R. Hodgson, I. Beletskaya, J. M. Mason, X. Luo, V. Nadeem, X. Wei, R. Kiarash, B. Madeira, P. Huang, T. W. Mak, G. Pan and H. W. Pauls, J. Med. Chem., 2015, 58, 147–169 CrossRef CAS PubMed.
- Y. Zhao and X. Wang, J. Cancer Res. Clin. Oncol., 2019, 145, 2413–2422 CrossRef PubMed.
- Combination Merestinib and LY2874455 for Patients With Relapsed or Refractory Acute Myeloid Leukemia, https://www.clinicaltrials.gov/ct2/show/NCT03125239?term=NCT03125239%26draw=2%26rank=1, accessed Feb. 9, 2020.
- CFI-400945 in Patients With Advanced/Metastatic Breast Cancer, https://www.clinicaltrials.gov/ct2/show/NCT03624543?term=NCT03624543%26draw=2%26rank=1, accessed Feb. 9, 2021.
- R. Sheng, S. Li, G. Lin, S. Shangguan, Y. Gu, N. Qiu, J. Cao, Q. He, B. Yang and Y. Hu, RSC Adv., 2015, 5, 81817–81830 RSC.
- L. Yang, Y. Chen, J. He, E. M. Njoya, J. Chen, S. Liu, C. Xie, W. Huang, F. Wang, Z. Wang, Y. Li and S. Qian, Bioorg. Med. Chem., 2019, 27, 1087–1098 CrossRef CAS PubMed.
- L. N. Solano, G. L. Nelson, C. T. Ronayne, S. Jonnalagadda, S. K. Jonnalagadda, K. Kottke, R. Chitren, J. L. Johnson, M. K. Pandey, S. C. Jonnalagadda and V. R. Mereddy, Sci. Rep., 2020, 10, 17969 CrossRef CAS PubMed.
- R. Laufer, B. Forrest, S.-W. Li, Y. Liu, P. Sampson, L. Edwards, Y. Lang, D. E. Awrey, G. Mao, O. Plotnikova, G. Leung, R. Hodgson, I. Beletskaya, J. M. Mason, X. Luo, X. Wei, Y. Yao, M. Feher, F. Ban, R. Kiarash, E. Green, T. W. Mak, G. Pan and H. W. Pauls, J. Med. Chem., 2013, 56, 6069–6087 CrossRef CAS PubMed.
- P. B. Sampson, Y. Liu, N. K. Patel, M. Feher, B. Forrest, S.-W. Li, L. Edwards, R. Laufer, Y. Lang, F. Ban, D. E. Awrey, G. Mao, O. Plotnikova, G. Leung, R. Hodgson, J. Mason, X. Wei, R. Kiarash, E. Green, W. Qiu, N. Y. Chirgadze, T. W. Mak, G. Pan and H. W. Pauls, J. Med. Chem., 2015, 58, 130–146 CrossRef CAS PubMed.
- J. Liu, X. Peng, Y. Dai, W. Zhang, S. Ren, J. Ai, M. Geng and Y. Li, Org. Biomol. Chem., 2015, 13, 7643–7654 RSC.
- B. Zhao, Y. Li, P. Xu, Y. Dai, C. Luo, Y. Sun, J. Ai, M. Geng and W. Duan, ACS Med. Chem. Lett., 2016, 7, 629–634 CrossRef CAS PubMed.
- J. Cui, X. Peng, D. Gao, Y. Dai, J. Ai and Y. Li, Bioorg. Med. Chem. Lett., 2017, 27, 3782–3786 CrossRef CAS PubMed.
- Z. Liu, Q. Lei, W. Wei, L. Xiong, Y. Shi, G. Yan, C. Gao, T. Ye, N. Wang and L. Yu, RSC Adv., 2017, 7, 27737–27746 RSC.
- T. Ye, X. Wei, T. Yin, Y. Xia, D. Li, B. Shao, X. Song, S. He, M. Luo, X. Gao, Z. He, C. Luo, Y. Xiong, N. Wang, J. Zeng, L. Zhao, G. Shen, Y. Xie, L. Yu and Y. Wei, Breast Cancer Res. Treat., 2014, 143, 435–446 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01147b |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |