
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImprovement in optical properties of Cs4PbBr6 nanocrystals using aprotic polar purification solvent
Zikuan Shi†
 a,
Yu Yang†a,
Xin-Yuan Sunb,
Feng Langa and
Liangwu Lin*a
a,
Yu Yang†a,
Xin-Yuan Sunb,
Feng Langa and
Liangwu Lin*a
aNational Key Laboratory of Science and Technology on High-strength Structural Materials, Central South University, Changsha, Hunan 410083, People's Republic of China. E-mail: linliangwu@csu.edu.cn
bDepartment of Physics, Jinggangshan University, Ji'an, Jiangxi 343009, People's Republic of China
First published on 4th May 2021
Abstract
We demonstrate the influence mechanism on the optical property of Cs4PbBr6 during purification of solution with different protonated levels and polarities. During the purification process, organic groups originating from oleic acid (OA) and PbBr impurity on the surface of Cs4PbBr6 nanocrystals can be removed using high polarity aprotic and protonic solvents, and the number of Br vacancies (VBr) can be reduced. The protonic polar solvent can not only etch the organic groups on the surface of nanocrystals, causing surface reconstruction and particle growth of nanocrystals, but also enter into the lattice of Cs4PbBr6 and react with the embedded CsPbBr3. However, aprotic polar solvent decreases the particle size of Cs4PbBr6 nanocrystals with the increase in the solvent polarity. The optical properties of Cs4PbBr6 can be effectively improved using aprotic polar solvents as a purification solvent, which is very significant to improve the luminescence efficiency of perovskites.
Introduction
Inorganic perovskites have attracted extensive attention and have become one of the research hotspots in the photovoltaic field since their genesis, owing to excellent photoelectric characteristics, such as high extinction coefficient, long charge carrier diffusion lengths, precisely tunable bandgaps, high absorption coefficients, and high defect tolerance.1–7 It is well known, CsPbX3 quantum dots (QDs) feature a fragile and unstable structure composed of [PbX6]4− octahedron with Cs+ cations filling. This structure has poor tolerance under moisture and heat because of the easy losing of halide anions. Diverse strategies have been used to overcome the above concerns, such as ligand passivation route8 and core-shelling method.9,10CsPbX3 is very sensitive to polar solutions. For example, CsPbX3 is easy to hydrolyze in water (high polarity) but can be preserved in n-hexane (low polarity). Anirban Dutta et al.11 found that CsPbBr3 tuned the phase upon changing the polarity of the reaction solvent from chloroform to hexane, while CsPbCl3 retained the favorable cubic phase, but their shape was tuned from a cube to the platelet. In the process, solvent-polarity-induced precursor activation led to growth controls and different shapes and phase-controlled perovskite nanocrystals. Wu et al.12 explored the effect of solvent polarity on the morphology and crystallization of perovskite films during the process of synthesis and found that the root mean square (RMS) roughness decreases, while crystallization increases with the enhancement of alcohol solvent polarity. Li et al.13 found that the size of perovskite QDs reduced with the increase of solvent polarity. Recently, some studies reported that Cs4PbX6, a derived member of Cs–Pb–X compounds with large band gaps,14 can be synthesized by adding excessive cesium ions15–17 and has better stability than CsPbX3. Zhang et al.18 found that the synthesized Cs4PbX6 phase 0-D nanocrystals could maintain good stability in some polar solvents (such as DMF) and air due to their high exciton binding energy (171 MeV). Chen et al.19 found that Cs4PbBr6 has excellent thermal stability, and the humidity resistance experiment shows that it still has good stability in the humidity environment. However, few scholars have studied the influence mechanism of different polar solutions on the optical performance of Cs4PbBr6 nanocrystals during the process of purification. Considering the polarity of the solution, we explore the influence on the optical properties of perovskite during the process of purification treatment from the perspective of polarity.
Here, we used common different polar reagents (n-hexane, ethyl acetate, acetone, and absolute ethanol) to purify Cs4PbBr6 nanocrystals. It was found that there were great changes in luminescent properties when the solvents with different protonated levels and polarities were used for purification. Then, we explored the influence mechanism of different protonated levels polar solutions on the optical properties of perovskites by analyzing the microstructure, morphology, defects, band gap, and so on. This work provides a reference for the purification of perovskites and lays a foundation for the application of this kind of material in the field of lighting.
Experimental section
Materials
Cesium bromide (CsBr, 99.5% metals basis), lead(II) bromide (PbBr2, AR, 99.0%), oleylamine (OM, 80–90%) and dimethyl sulfoxide (DMSO) were purchased from Aladdin®. Ethyl acetate (EA), n-hexane (n-H), oleic acid (OA, AR), absolute ethanol and acetone were purchased from Sinopharm Chemical Reagent. All reagents were used as received without any further purification.Synthesis of Cs4PbBr6 nanocrystals
Cs4PbBr6 nanocrystals were synthesized at room temperature without any protective gas. Briefly, 25 ml acetone was loaded into a 150 ml conical flask at 25 °C. Subsequently, 5 ml OA and 2.5 ml OM were added to the conical flask with stirring for 1 min to make it homogenized, and kept overnight at room temperature. After that, solid powders of 1 mmol PbBr2 (0.367 g dissolved in 4 ml DMSO) and 1 mmol CsBr (0.213 g dissolved in deionized 1 ml water) were added to the flask with vigorous stirring (1200 rpm) for 10 min. Through ultraviolet irradiation, we could observe strong green fluorescence. As such, Cs4PbBr6 was synthesized successfully.The purification process of Cs4PbBr6 nanocrystals
The crude solution was divided into four parts and centrifuged at 7000 rpm for 5 min. Nanocrystals were dispersed in 10 ml n-H, 10 ml EA, 10 ml acetone and 10 ml absolute ethanol individually, followed by ultrasonic cleaning for 5 min, and centrifuged at 7000 rpm for 5 min. Finally, they were dried in a vacuum oven for 12 h at room temperature, and the powder was prepared for characterization.Characterization
X-ray diffractometry (XRD) patterns were collected with an X-ray automatic diffractometer (Advance D8, BRUGG GROUP, Switzerland) using Cu Kα radiation (wavelength 1.55406 Å). XRD patterns were recorded in the range of 10° ≤ 2θ ≤ 60° with a scanning step of 0.02° and counting time 2 s per step. Transmission electron microscopy (TEM) was performed on a field emission scanning electron microscope (JEM-2100F, Japan). Photoluminescence (PL), photoluminescence excitation (PLE) spectra and PL decay spectra were acquired on a full-function fluorescence spectrometer (FLS-1000, Edinburgh, Britain) with a time range: 0–199.6 μs; step: 0.40 μs; frequency: 100.0 Hz. Ultraviolet and visible (UV-vis) absorption spectra were measured with a UV-3600 Ultraviolet-visible spectrophotometer (Shimadzu, Kyoto, Japan). X-ray photoelectron spectroscopy (XPS) was recorded with a K-ALPHA X-ray photoelectron spectrometer (Thermo Fisher Scientific, Britain) in the 1300–0 eV region. Fourier transform infrared reflection (FTIR) spectra were measured on a Nicolet instrument (iS5, Thermo, United States) in the region of 3800–400 cm−1. Raman spectra were measured on a Lab RAM HR800 spectrometer with a 633 nm laser light at room temperature and recorded in the region of 50–500 cm−1.Results and discussion
After synthesis, the as-synthesized Cs4PbBr6 nanocrystal solution was divided into four equal parts and separated from the mother liquor by centrifugation. Then, the obtained Cs4PbBr6 nanocrystals were dispersed in different polar solutions (n-hexane, ethyl acetate, acetone and ethanol) for further purification treatment. Sample A was dispersed in n-H after centrifuging, while Sample B, C and D were dispersed in ethyl acetate, acetone and ethanol, respectively, and all samples were centrifuged again (solvent polarity: A < B < C < D) (see Scheme 1). The order of polarity is in accordance with previous report.20 | ||
| Scheme 1 The synthesis and purification of Cs4PbBr6 nanocrystal. | ||
The precipitates of these samples were dried in a vacuum oven at room temperature. A significant color change of the nanocrystal powder occurred after purification, which indicated that the optical properties changed dramatically, as shown in Fig. 1. In our studies, we found that the optical photos of Cs4PbBr6 dispersed in toluene (aprotic inert solvent) were obviously different, as shown in Fig. 1a and b. Under daylight, the purified samples showed different degrees of yellow and the sample treated with acetone was even light green among these samples. PLE and PL (excitation: 360 nm) spectra obtained from powder are also shown in Fig. 1c and d. It can be seen that all PLE spectra consisted of an excitation peak at about 285 nm and a broad excitation band (320–500 nm), and the intensity first increased and then decreased with the increase of solvent polarity, as shown in Fig. 1c. We can also find, with the increase in the polarity, the PL peak position shifted from 512 nm (A) to 515 nm (B), 515 nm (C), 513 nm (D), and the PL intensity showed the same trend with PLE and PLQY as shown in Fig. 1e and f. These results suggested that the solvent polarity has a stronger influence on the PL peak position and PLQY optical properties of Cs4PbBr6 nanocrystals during the purification process. Considering the polarity of the solution, we explored the influence mechanism on the optical properties of perovskite Cs4PbBr6 nanocrystals during the process of solution treatment from the perspective of polarity.
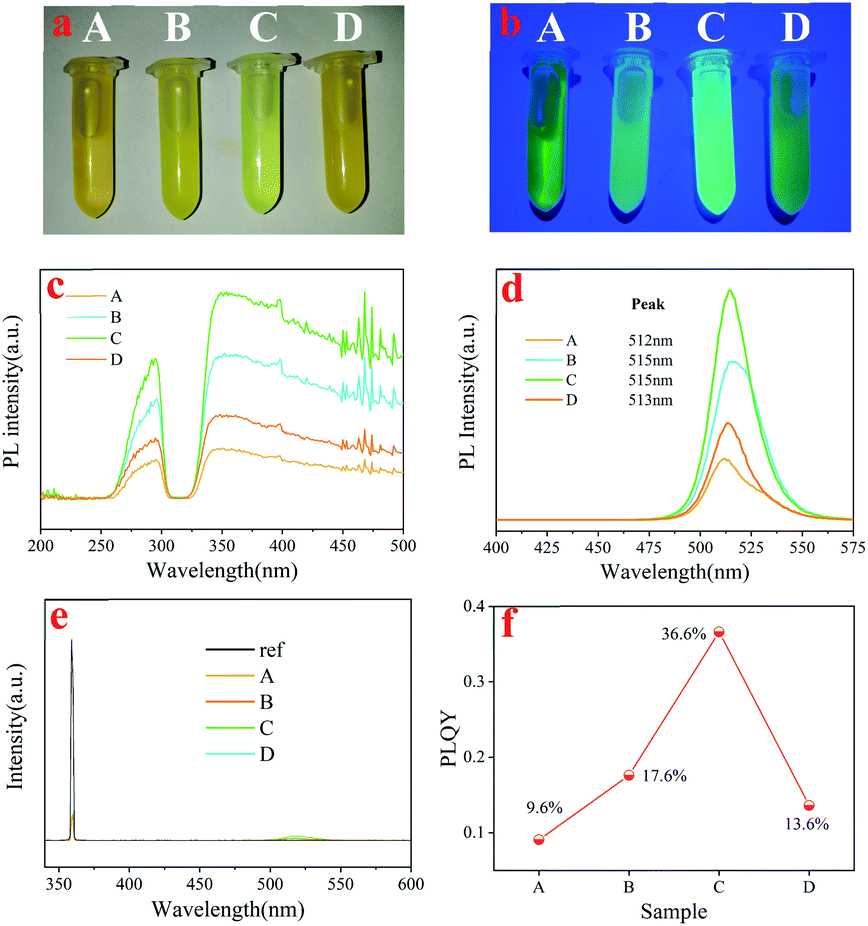 | ||
| Fig. 1 Images of Cs4PbBr6 in toluene (10 mg ml−1), washed by n-H (A), EA (B), acetone (C) and ethanol (D), respectively, under ambient light (a), and under UV lamp (b); the photoluminescence excitation (c) and the photoluminescence emission (d) spectra of sample A, B, C and D; (e and f) the PLQY of four samples. | ||
To confirm the influence mechanism, we explored its principle through the characterization of the microstructure. The TEM images shown in Fig. 2 demonstrate that Cs4PbBr6 nanocrystals show different sizes and morphologies after purifying with different polar solutions. It can be clearly seen that when the polarity is relatively low, the shape of nanocrystals is generally rectangular, and crystal dimension approximately equal to 10–20 nm, as shown in Fig. 2a. With the increase in polarity, the shape of nanocrystals changes accompanied by the appearance of a triangle, sphere, polygon and so on, and the size also decreased (5–10 nm), as shown in Fig. 2c. As the polarity continues to increase, nanocrystals turn into small spheres, and the size is also up to the quantum size (less than 5 nm), as shown in Fig. 2e. Then, as the polarity increases further, the morphology becomes is a regular rectangle, and the size increases sharply to more than 20 nm, as shown in Fig. 2g. However, the data of Hu et al. show that with the increase in the polarity of the solution, the particles gradually become smaller and uniform, and the phase of Cs4PbBr6 gradually transform into CsPbBr3. The size of nanocrystals in lower polarity solvents increases, compared to those with moderate polarity.11 When sample D was magnified, we could clearly see that the square phase was formed by spherical particles agglomerating together with the organic matter, as shown in the inset in Fig. 2g, which is not found by the previous research institute.
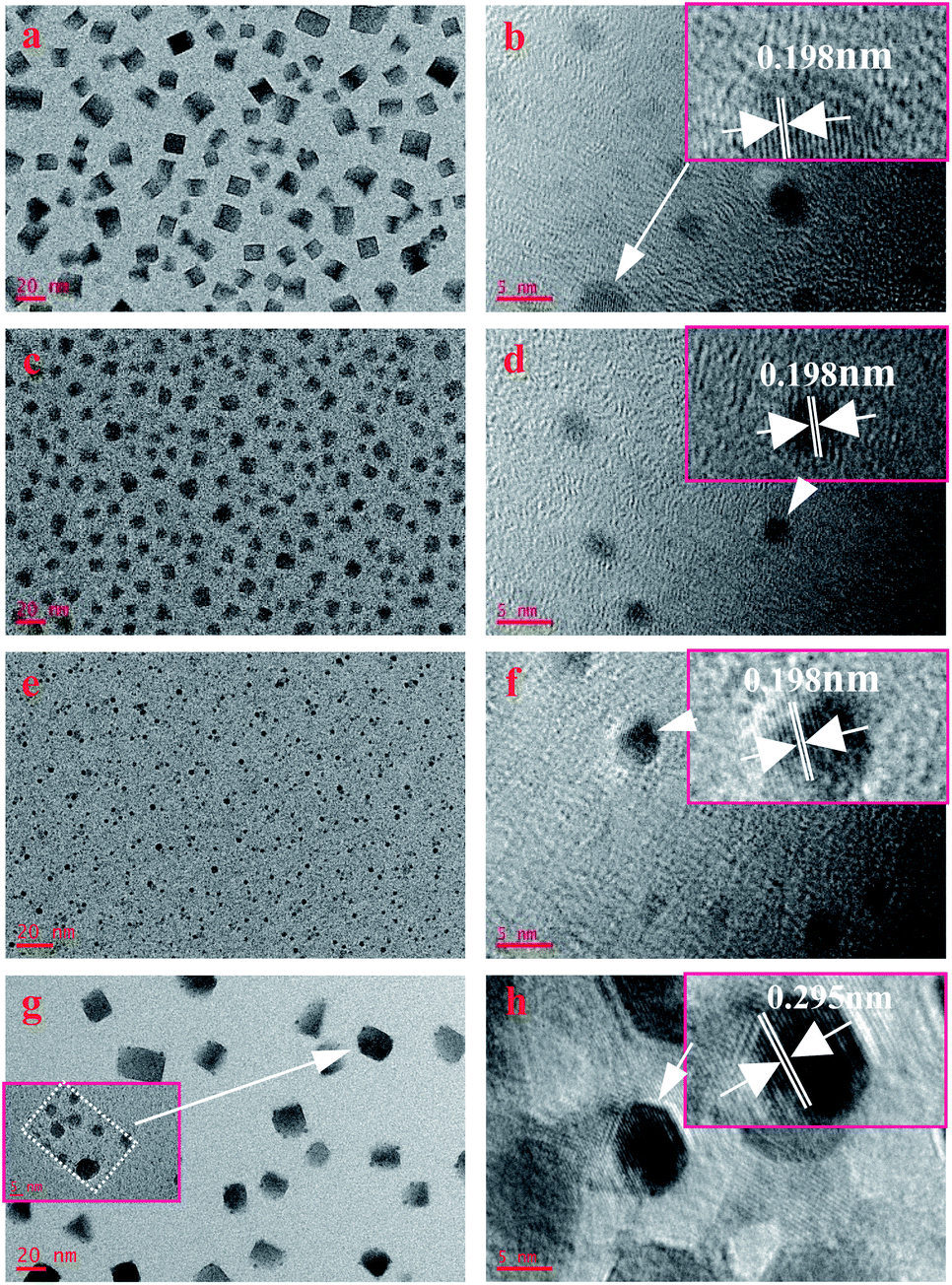 | ||
| Fig. 2 TEM images of samples prepared with different polarity solvent: (a) A; (c) B; (e) C; (g) D. HRTEM images of samples prepared with different polarity solvent: (b) A; (d) B; (f) C; (h) D. | ||
HRTEM was further carried out to measure the lattice spacing of the products, as shown in Fig. 2b, d, f and h. The lattice fringes (sample A, B, and C) are clearly observed with an interplanar spacing of 0.195 nm, corresponding to the (600) plane of hexagonal Cs4PbBr6 nanocrystals (d600 = 0.195 nm, JCPDS no.73-2478), while the interplanar spacing in sample D is 0.295 nm, corresponding the (223) plane of hexagonal Cs4PbBr6 nanocrystals (d223 = 0.295 nm, JCPDS no.73-2478).
From the results of TEM and HRTEM, we found an interesting phenomenon, compared to the (600) dominant growth plane of the former three samples, the plane of sample D changes to the (223) plane. At the same time, with the increase in polarity, the particle size of the former three samples decreased, but in the last sample, it increased significantly. This phenomenon suggests that the interaction between ethanol and Cs4PbBr6 nanocrystals was different from that in n-hexane, ethyl acetate and acetone during the purification process. It is worth noting that the solvents n-hexane (inert solvent), acetone and ethyl (aprotic polar solvent) acetate, ethanol (proton polar solvent) are different. During the purification process, protonated ethanol can etch organic groups on the surface of nanocrystals, improve surface energy, and cause surface reconstruction and particle growth of nanocrystals, which is well agreeing with the result shown in the inset in Fig. 2g. This is why the dominant growth plane of perovskite Cs4PbBr6 nanocrystals changes and the particle size increases when purified by ethanol. As ethyl acetate and acetone are aprotic polar solvents, the etching effect on the nanocrystalline surface is very weak so that they cannot cause surface reconstruction. However, the solvent effect is improved due to polarity, and finally decreases the size of Cs4PbBr6 nanocrystals. Additionally, the size of sample B and sample C are smaller than those previously reported in the literature21 and can produce a quantum size effect, which can strongly influence the tuneable PL properties.22
Fig. 3 shows X-ray diffraction (XRD) patterns of the above four samples. It can be seen that with the polarity increasing, the structure of Cs4PbBr6 nanocrystals does not change, which matched well with the standard diffraction peaks shown in JCPDS no. 73-2478. The illustration reports a detailed investigation of the peak at 31.9 degrees, which are matching well with the standard diffraction peaks shown in JCPDS no. 75-0412. These results confirmed the coexistence of Cs4PbBr6 (JCPDS no. 73-2478) and a tiny bit of CsPbBr3 (JCPDS no. 75-0412), which is consistent with previous studies.23–25 These results also indicated that CsPbBr3 are embedded in Cs4PbBr6 nanocrystals and are not easily removed by polar solvents during purification. We calculated the lattice parameter and lattice strain by Williamson–Hall and Scherrer's formulas, respectively, as shown in Table 1. It can be seen that the trends of the lattice parameter and the lattice strain are first increasing and then decreasing with the increase of the solution polarity. At the same time, the lattice strain can make PL red shift and the change of lattice strain is consistent with the change of PL peak shift, which indicates that the quantum size effect is not dominant in the process, and the change of PL peak shift is mainly controlled by the lattice strain.
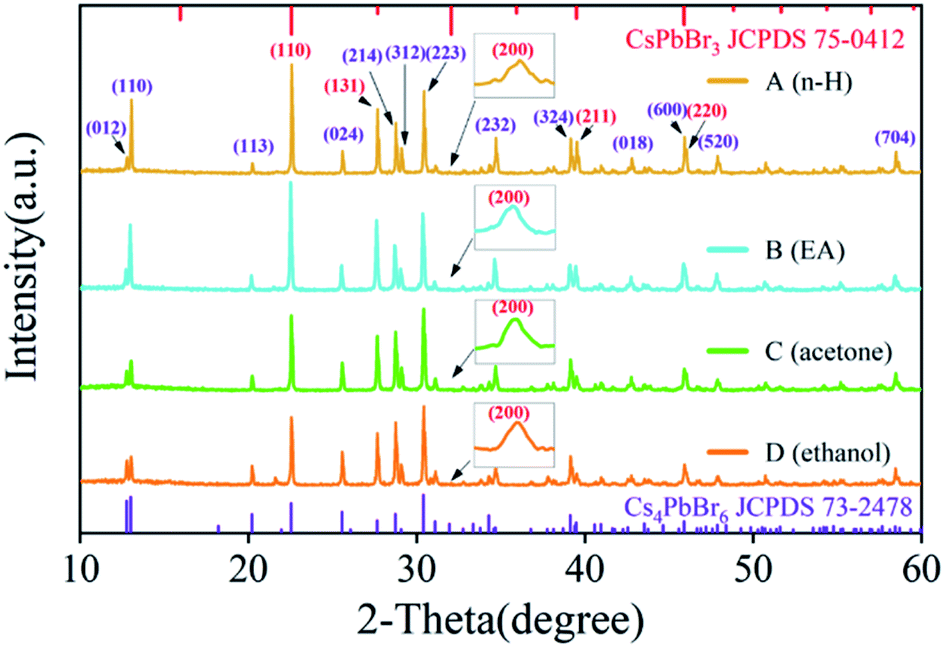 | ||
| Fig. 3 XRD patterns of sample A, sample B, sample C and sample D. the inset is the peak at 31.9 degrees. | ||
| Sample | A | B | C | D |
|---|---|---|---|---|
| Lattice strain (10−4) | 7.4153 | 9.4920 | 12.2000 | 7.9509 |
| Lattice parameter a and b (Å) | 13.7038 | 13.7286 | 13.7080 | 13.7050 |
| Lattice parameter c (Å) | 17.2870 | 17.3199 | 17.2851 | 17.2868 |
Note that, being similar to the phenomenon observed in previous studies,26 Cs4PbBr6 is easily damaged under irradiation of focused electron beam, which may due to its low lattice energy. However, it can be seen that the phase transition in nanocrystals does not occur after purification with different polar solutions, which indicates that Cs4PbBr6 nanocrystals prepared by us have better phase transition stability, suggesting that the obtained Cs4PbBr6 nanocrystals are a good candidate material for light-emitting applications.
Previous reports showed that purification could etch the surface of nanocrystals with the removal of the ligand.27 Two organic surfactants were used during the synthesis; oleylamine enhanced the solubility of lead salts, whereas oleic acid acted as a hydrophobic ligand. Using FTIR, the presence of surface ligands was analyzed (Fig. 4). The peaks located at 1470 cm−1 (–COO– stretch) mainly originated from the groups in OA molecules. The peaks at 2930 cm−1 (–CH2– stretch), 2850 cm−1 (–CH2– stretch) and 1410 cm−1 (–CH3 bending), which belong to OM and OA, are significantly weakened or even disappeared after purification using different polar solutions, suggesting that these groups can be eliminated easily by purification. The peaks appearing at 1090 cm−1 (–C–N stretch) and 1640 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O or N–H stretch) originated from the groups in OM molecules, and their intensity change range is relatively small.27 It is worth noting that the OH stretch caused by water is also apparent at about 3450 cm−1, and its strength has hardly changed after purification with different polar solutions. The reason is that for the special structure of Cs4PbBr6, some small molecules can easily enter the lattice, which can influence the structure and bandgap, leading to the change of optical properties. Wang et al. found that hydroxyl has a deep influence on the structure and bandgap of Cs4PbBr6 to form a sub-bandgap of about 2.6 eV.28 Pure Cs4PbBr6 nanocrystals have no photoluminescence efficiency in visible light emission. A very small amount of water, which is a proton polar solvent, can enter the lattice of Cs4PbBr6 nanocrystals and cause some Cs4PbBr6 nanocrystals to change into CsPbBr3.
O or N–H stretch) originated from the groups in OM molecules, and their intensity change range is relatively small.27 It is worth noting that the OH stretch caused by water is also apparent at about 3450 cm−1, and its strength has hardly changed after purification with different polar solutions. The reason is that for the special structure of Cs4PbBr6, some small molecules can easily enter the lattice, which can influence the structure and bandgap, leading to the change of optical properties. Wang et al. found that hydroxyl has a deep influence on the structure and bandgap of Cs4PbBr6 to form a sub-bandgap of about 2.6 eV.28 Pure Cs4PbBr6 nanocrystals have no photoluminescence efficiency in visible light emission. A very small amount of water, which is a proton polar solvent, can enter the lattice of Cs4PbBr6 nanocrystals and cause some Cs4PbBr6 nanocrystals to change into CsPbBr3.
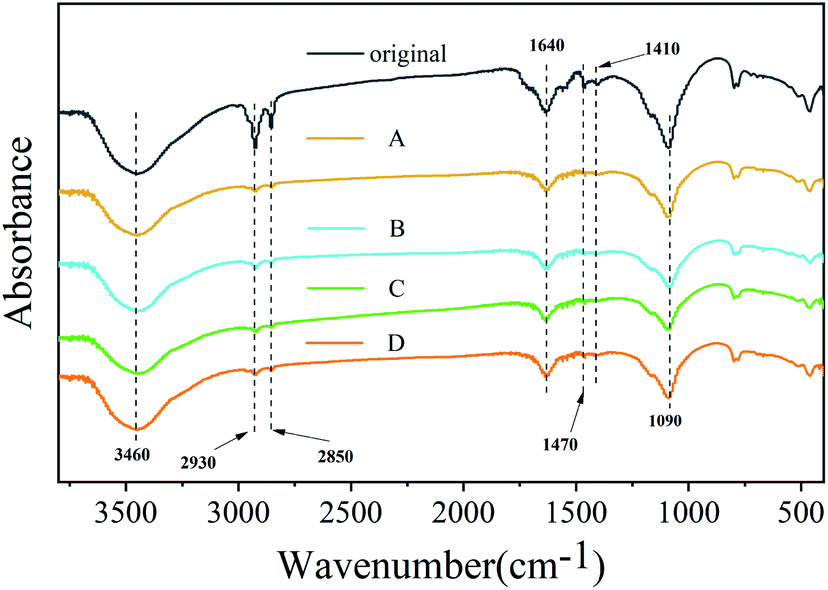 | ||
| Fig. 4 FTIR spectra of sample original, A, B, C and D. | ||
Fig. 5 shows the HRTEM of CsPbBr3 embedded in Cs4PbBr6. The fast Fourier transformation (FFT) images inset at the bottom of the figure confirm that the cubic CsPbBr3 and hexagonal Cs4PbBr6 exist together. The d-spacing for two red fringes was measured to be 2.9 Å, which belongs to (233) of hexagonal Cs4PbBr6 (JCPDS 74-2378). The d-spacing for brown fringes was measured to be 5.6 Å, belonging to (100) of cubic CsPbBr3 (JCPDS 75-0412). It can be clearly seen that CsPbBr3 is embedded in Cs4PbBr6, which indicates that the small molecules of water enter into the Cs4PbBr6 lattice and react to form CsPbBr3. The existence of the –OH peak at 3460 cm−1 in FTIR spectra also proved the existence of small molecules of water, indirectly. The origin of Cs4PbBr6 green emission has always been controversial. As shown by Fig. 5, the mosaic structure between Cs4PbBr6 and CsPbBr3 is more likely to be the cause of Cs4PbBr6 green emission, and lattice matching between the Cs4PbBr6 and CsPbBr3 contributes to improved luminescent properties,29 which show that the green emission of Cs4PbBr6 comes from the inside CsPbBr3.
 | ||
| Fig. 5 The HRTEM of CsPbBr3 embedded in Cs4PbBr6 in sample D. | ||
X-ray photoelectron spectroscopy (XPS) analysis was performed to investigate the surface chemical elements and valence states of Cs4PbBr6, as shown in Fig. 6. The full survey scan confirms the existence of Cs, Pb, O, and Br in Cs4PbBr6 (Fig. 6a). O may have contributed to –COO–, CO or –OH groups in OA, OM, or water. High-resolution XPS spectra for the core level of Cs 3d, Pb 4f are shown in Fig. 6b and c. As shown in Fig. 6b, peaks at 723.9 and 737.9 eV can be attributed to Cs 3d5/2 and Cs 3d3/2. There was no change in the position of the peak and the distance between the two peaks, indicating the chemical valence state of Cs did not change. The Pb 4f XPS spectrum also showed two peaks at 138.3 eV and 143.1 eV (Fig. 6c), which are assigned to Pb 4f7/2 and 4f5/2. It can be seen that the full width at half maxima (FWHM) of the two peaks is first reduced and then increased with the increase of solvent polarity. The change of FWHM indicates that there are other lead-containing impurity phases on the surface of as-synthesized perovskite Cs4PbBr6 nanocrystals, which can be removed by high polar solvents. Pb 4f7/2 and Pb 4f5/2 peaks of Cs4PbBr6 in sample A can be fitted into two peaks with binding energies of 138.38 eV and 143.08 eV, 139.48 eV and 144.28 eV, respectively, suggesting that Pb in Cs4PbBr6 nanocrystals exists in two chemical environments. The spin–orbit splitting energy is 4.7 eV and 4.8 eV, referring to the Web database (http://srdata.nist.gov/xps/), which can be assigned to Pb2+ ion in PbBr2 and Pb1+ ion in PbBr, respectively, as shown in Fig. 6d, implying that there are many VBr defects existing in sample A. From Fig. 6c, we found that the signal of Pb 4f in PbBr significantly reduced after purification using high polar solvents, suggesting that the VBr defect reduced. The core-level Br 3d XPS scan was fitted into two peaks at the binding energies of 67.9 eV and 68.7 eV, as shown in Fig. 6e–h, which is very similar to findings from the previous studies.30 The observed peak at about 67.9 eV can be attributed to the Br atoms in isolated [PbBr6]4− of Cs4PbBr6,23 and redshifts first and then blueshifts with the increase in polarity. The peak at 68.7 eV correlates with the existence of Br atoms of CsBr impurity or corner-sharing [PbBr6]4− of CsPbBr3.23 The results suggest that the CsPbBr3 phase coexist in the synthesized Cs4PbBr6 phase. It can be seen that the intensity of peak at 68.7 eV increases first and is then reduced with the increase in the polarity of the solvent, indicating that the amount of CsBr impurity or corner-sharing [PbBr6]4− of CsPbBr3 reduces during purification with a high polarity solvent. In addition, we have noticed another strong peak at 75 eV, which can be attributed to Cs 4d5 in CsBr.
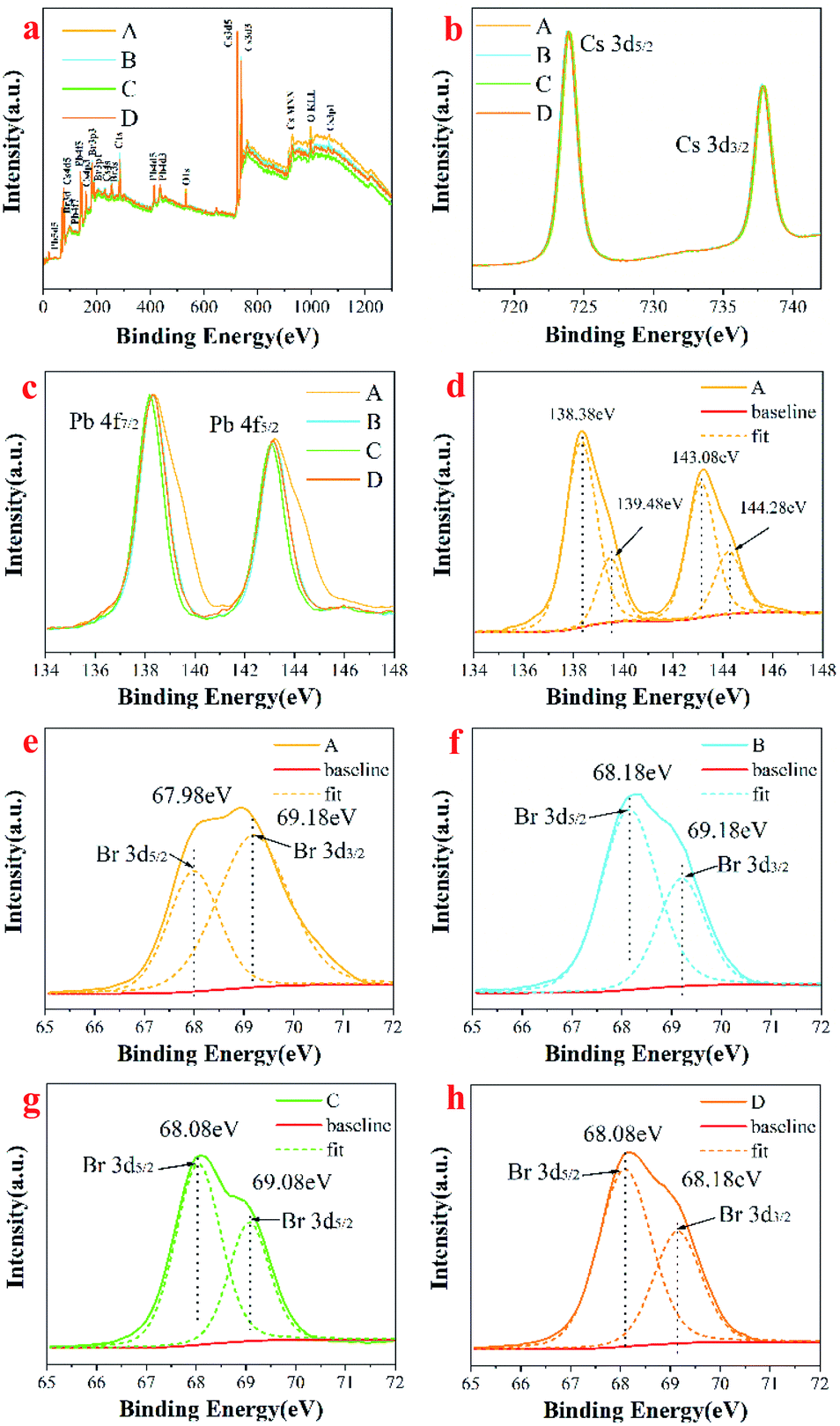 | ||
| Fig. 6 Full survey scan of Cs4PbBr6 nanocrystals (a); high resolution XPS of (b) Cs 3d; (c) Pb 4f; the fitted high resolution XPS of (d) Pb 4f, (e–h) Br 3d. | ||
Raman spectra of the Cs4PbBr6 nanocrystals provided the vibrational modes of the metal-halide sublattice, as shown in Fig. 7. Two Raman-active modes for Cs4PbBr6 nanocrystals were observed, with a peak at 86 cm−1 and a broad peak at 121 cm−1. The peak at 86 cm−1 arises from the vibrational mode of the isolated [PbBr6]4− octahedron in Cs4PbBr6 nanocrystals.31 The broad peak comes from the Cs+ cation surrounding the [PbBr6]4− octahedron. Except for a little broadening, the Raman spectrum is similar to that previously reported for Cs4PbBr6 nanocrystals.32 According to a previous Raman study of CsPbBr3 (ref. 16) and CsPbCl3 crystals with a Pnma phase,33 the peaks assigned to the vibrational mode of [PbBr6]4− octahedron and motion of Cs+ cation are located at 72 cm−1 and 127 cm−1, respectively. It can be seen that the two peaks that originated from CsPbBr3 are submerged in the peaks of Cs4PbBr6 nanocrystals, which may be due to the small amount of CsPbBr3 in Cs4PbBr6 nanocrystals, being consistent with the results of TEM and XRD.
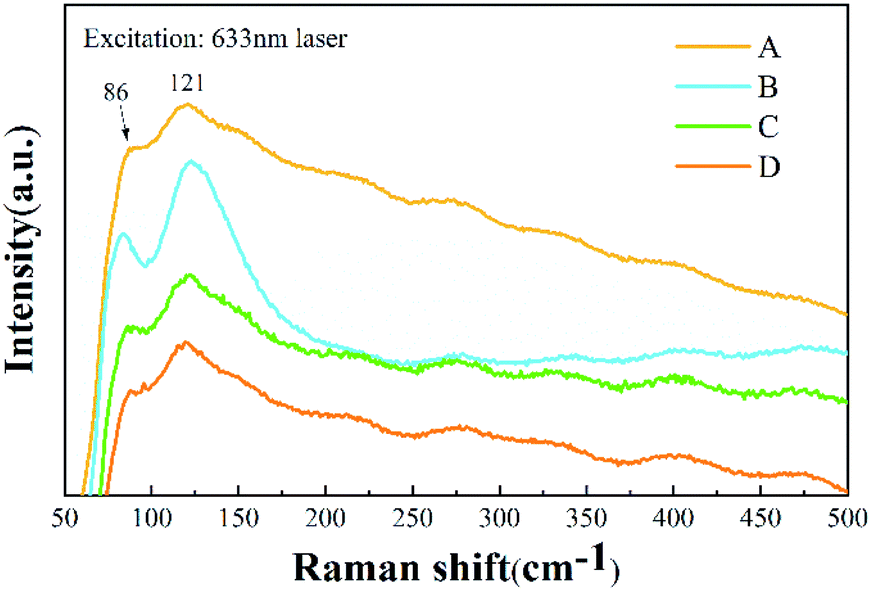 | ||
| Fig. 7 Raman spectra of samples A, B, C and D. | ||
As is known, pure Cs4PbBr6 is an insulator with a large band gap of ∼3.9 eV,14,34 and it itself does not show green emission,19,35–38 whereas CsPbBr3 is a semiconductor with a band gap of about 2.4 eV,39 showing a strong green emission. In order to study the influential mechanism of purification on luminescence properties of the as-synthesized Cs4PbBr6 nanoparticles, a diffuse reflectance absorption spectrum (DRS) was recorded and shown in Fig. 8a, and the corresponding Tauc plot was also measured as shown in Fig. 8b. It can be seen that the DRS was composed of a sharp absorption peak at about 315 nm and a broad absorption band (320–550 nm), while the Tauc plot showed two absorption band edges at about 2.3 eV and 3.7 eV, assigned as Eg1 and Eg2, respectively. The strong absorption peak around 315 nm is related to Cs4PbBr6, originating from the optical transitions between localized states within isolated PbBr64− octahedra.14,34 It has been reported that the absorption peak of pure CsPbBr3 is located at around 505 nm.40 Notably, there is a sharp dip at ∼315 nm in the PLE spectrum (Fig. 1c) and a sharp peak in the absorption spectrum at the same wavelength. This is due to the strong absorption of Cs4PbBr6 at 315 nm,14 and the absorption of CsPbBr3 is comparably weak, which leads to a dip in the PL excitation spectrum. For comparison, band gaps (Eg1) of samples were determined and found to be approximately 2.31 eV (A), 2.33 eV (B), 2.32 eV (C) and 2.29 eV (D), as shown in Fig. 8c. Band gaps (Eg2) of samples were also determined and found to be about 3.69 eV (A), 3.7 eV (B), 3.7 eV (C) and 3.7 eV (D) (Fig. 8d). It can be seen that band gaps (Eg1) and (Eg2) are obviously smaller than those of Cs4PbBr6 nanoparticles. There are three factors that affect the change in the band gap. The first is that a small amount of CsPbBr3 is embedded in Cs4PbBr6 nanocrystals, which leads to lattice distortion and eventual changes in the band gap of Cs4PbBr6 nanocrystals. The formation of CsPbBr3 is triggered by water, which enters the lattice of Cs4PbBr6 nanocrystals according to the following transformation process during the synthesis: Cs4PbBr6 → CsPbBr3 + CsBr. Due to the high solubility of CsBr in water, water drives the reaction to the right. This phenomenon is consistent with the results of XRD and HRTEM, as shown in Fig. 3 and 5, respectively. Secondly, previous researches has show that green fluorescence emission is caused by defects in the Cs4PbBr6 crystal41 and surface ligands can act as passivation agents to reduce crystal defects.42 Considering that structural defects tend to be generated at the surfaces of particles, the emission behaviors of the products might be well expected to be influenced when using polar solutions for the purification, if the emission stems from the structural defects. The defects formed during the synthesis process or the purification mediated mid-band-gap states. This is in agreement with the long Urbach tail shown in Fig. 8a, which is common for disordered or poorly-crystalline semiconductors or defects such as VBr. The third is that a small amount of water enters into Cs4PbBr6 nanocrystals, which leads to lattice distortion and reduces the band gap to about 3.7 eV.28 At the same time, a small amount of water was ionized to form hydroxyl groups, which react with Cs4PbBr6 nanocrystals to form a sub band gap of the hydroxyl group. This speculation is confirmed by the band gap (Eg2), which also shows the change in band gap, as shown in Fig. 8d.
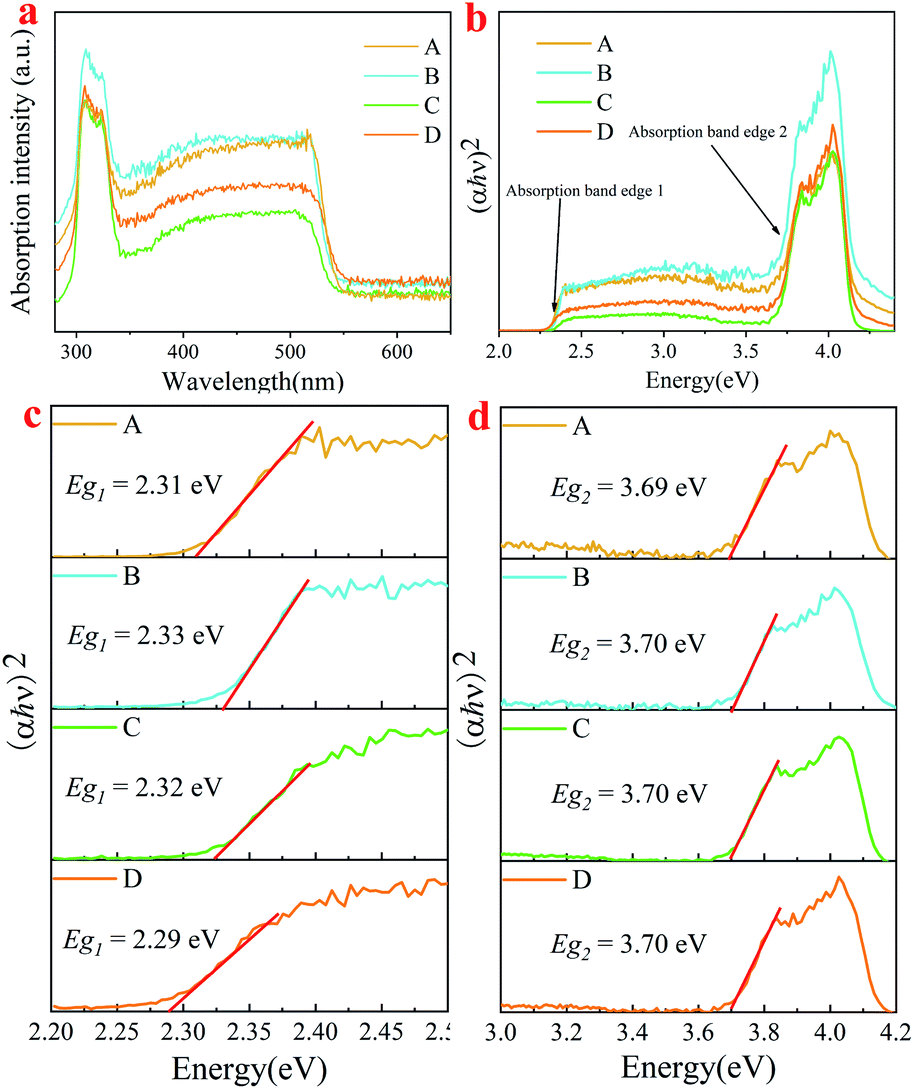 | ||
| Fig. 8 (a) absorption spectra; (b) Tauc plot; local Tauc plot of absorption band edge 1 (c) and edge 2 (d) for absorption of samples A, B, C and D. | ||
The PL decay curve can be fitted well with a biexponential function (S1), where A1 and A2 are constants, t is time. τ1, τ2 in eqn (S1) represent the short-lived and long-lived lifetimes relating to the trap-assisted recombination and carrier radiative recombination, separately.43 Besides, the average lifetime (τav) can be obtained according to eqn (S2) for different nanocrystals.44
The fluorescence intensity I(t) of the sample is fitted with a two exponential decay model described as:
I(t) = A1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) exp(−t/τ1) + A2exp(−t/τ2) exp(−t/τ1) + A2exp(−t/τ2)
| (S1) |
Equation used for the average lifetime calculation:
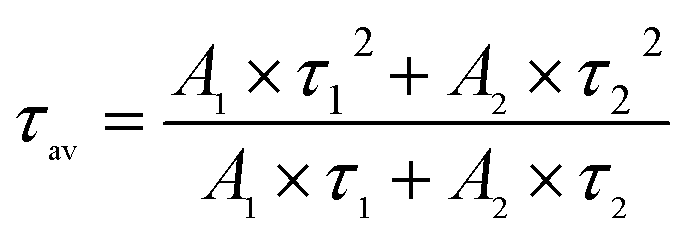 | (S2) |
It can be easily found that the trend of a quantum lifetime is first increasing and then decreasing, and there is a peak for sample C. The reagents used to remove excessive ligands (oleic acid and oleic amine in this synthesis), which may also remove some ligands on the nanocrystal surface. Once the surface ligands are removed, the defect-trapping state will increase and the trapping time will be shortened.27 Then, the electron–hole recombination time has also changed as shown in Fig. 9. To a certain extent, the longer lifetime of photoluminescence transition indicates the decrease of the defect concentration and the increase of crystallinity.45,46
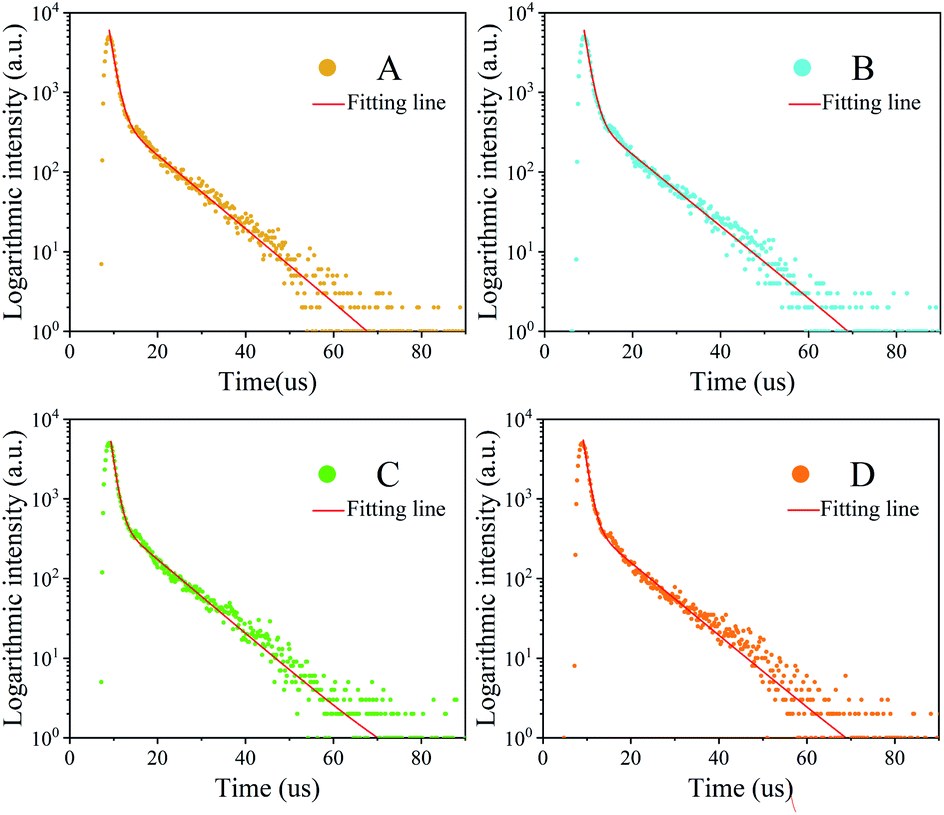 | ||
| Fig. 9 The PL decays spectra of A, B, C and D. | ||
The average PL decay lifetime of the synthesized Cs4PbBr6 nanocrystals from 4.437 μs to 4.912 μs was derived (Table 2). The long decay lifetime reveals high ratios of radiative recombination to nonradiative recombination and less transition to detect states. The results (long lifetime) can be attributed to their less nonradiative energy transfer to the trap-defect and surface state of the Cs4PbBr6 nanocrystals. The small value of τ1 indicates good intrinsic excitonic feature with fewer defect trappers. From Table 2, it can be seen that the average PL decay lifetime is improved by the aprotic polar solvent.
| Sample | τ1 | A1 | τ2 | A2 | τav |
|---|---|---|---|---|---|
| A | 1.18 ± 0.01 | 9.44 ± 0.10 | 6507.48 ± 12 | 529.59 ± 2.0 | 4.437 ± 0.06 |
| B | 1.18 ± 0.01 | 9.67 ± 0.10 | 6498.67 ± 12 | 527.68 ± 2.0 | 4.572 ± 0.06 |
| C | 1.02 ± 0.01 | 9.34 ± 0.10 | 5732.49 ± 13 | 549.59 ± 2.0 | 4.912 ± 0.05 |
| D | 1.09 ± 0.01 | 9.34 ± 0.10 | 5957.84 ± 12 | 501.55 ± 2.0 | 4.668 ± 0.05 |
Conclusions
In summary, we demonstrate the influence mechanism on the optical properties of Cs4PbBr6 during purification with solutions of different protonation capabilities and polarity. During the purification process, the organic groups originating from oleic acid (OA) and the PbBr impurity on the surface of Cs4PbBr6 nanocrystals can be removed by high polarity aprotic and proton solvents, and the concentration of Br vacancy (VBr) can be reduced. The protonic polar solvent can not only etch the organic groups from the surface of nanocrystals, causing the surface reconstruction and particle growth of nanocrystals, but also enter into the lattice of Cs4PbBr6, and react with the embedded CsPbBr3. However, aprotic polar solvent decreases the particle size of Cs4PbBr6 nanocrystals with increasing solvent polarity. The optical properties of Cs4PbBr6 can be effectively improved by using aprotic polar solvents as purification solvents, which is very significant to improve the luminescence efficiency of perovskites.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are thankful for the financial support from the National Natural Science Foundation of China (Grant No. 31660266) and the Natural Science Foundation of Hunan Province, China (Grant No. 2018JJ2509).Notes and references
- T. C. Sum and N. Mathews, Energy Environ. Sci., 2014, 7, 2518–2534 RSC.
- T. M. Brenner, D. A. Egger, A. M. Rappe, L. Kronik, G. Hodes and D. Cahen, J. Phys. Chem. Lett., 2015, 6, 4754–4757 CrossRef CAS PubMed.
- P. C. Harikesh, B. Wu, B. Ghosh, R. A. John, S. Lie, K. Thirumal, L. H. Wong, T. C. Sum, S. Mhaisalkar and N. Mathews, Adv. Mater., 2018, 30, 1802080 CrossRef PubMed.
- R. Mayengbam, S. K. Tripathy and G. Palai, J. Phys. Chem. C, 2018, 122, 28245–28255 CrossRef CAS.
- N. M. Rahman, M. Adnaan, D. Adhikary, M. Islam and M. K. Alam, Comput. Mater. Sci., 2018, 150, 439–447 CrossRef CAS.
- N. Li, Z. Zhu, J. Li, A. K. Y. Jen and L. Wang, Adv. Energy Mater., 2018, 8, 1800525 CrossRef.
- F. Zhang, B. Yang, X. Mao, R. Yang, L. Jiang, Y. Li, J. Xiong, Y. Yang, R. He, W. Deng and K. Han, ACS Appl. Mater. Interfaces, 2017, 9, 14827–14832 CrossRef CAS PubMed.
- J. Li, L. Xu, T. Wang, J. Song, J. Chen, J. Xue, Y. Dong, B. Cai, Q. Shan, B. Han and H. Zeng, Adv. Mater., 2017, 29, 1603885 CrossRef PubMed.
- Q. Zhong, M. Cao, H. Hu, D. Yang, M. Chen, P. Li, L. Wu and Q. Zhang, ACS Nano, 2018, 12, 8579–8587 CrossRef CAS PubMed.
- B. Qiao, P. Song, J. Cao, S. Zhao, Z. Shen, D. Gao, Z. Liang, Z. Xu, D. Song and X. Xu, Nanotechnology, 2017, 28, 445602 CrossRef PubMed.
- A. Dutta, R. K. Behera and N. Pradhan, ACS Energy Lett., 2019, 4, 926–932 CrossRef CAS.
- M. Wu, D. Zhao, Z. Wang and J. Yu, Nanoscale Res. Lett., 2018, 13, 128 CrossRef PubMed.
- G. Li, H. Wang, T. Zhang, L. Mi, Y. Zhang, Z. Zhang, W. Zhang and Y. Jiang, Adv. Funct. Mater., 2016, 26, 8478–8486 CrossRef CAS.
- Q. A. Akkerman, S. Park, E. Radicchi, F. Nunzi, E. Mosconi, F. De Angelis, R. Brescia, P. Rastogi, M. Prato and L. Manna, Nano Lett., 2017, 17, 1924–1930 CrossRef CAS PubMed.
- J. Yin, Y. Zhang, A. Bruno, C. Soci, O. M. Bakr, J.-L. Bredas and O. F. Mohammed, ACS Energy Lett., 2017, 2, 2805–2811 CrossRef CAS.
- J.-H. Cha, J. H. Han, W. Yin, C. Park, Y. Park, T. K. Ahn, J. H. Cho and D.-Y. Jung, J. Phys. Chem. Lett., 2017, 8, 565–570 CrossRef CAS PubMed.
- M. I. Saidaminov, J. Almutlaq, S. Sarmah, I. Dursun, A. A. Zhumekenov, R. Begum, J. Pan, N. Cho, O. F. Mohammed and O. M. Bakr, ACS Energy Lett., 2016, 1, 840–845 CrossRef CAS.
- Y. Zhang, L. Sinatra, E. Alarousu, J. Yin, A. M. El-Zohry, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. C, 2018, 122, 6493–6498 CrossRef CAS.
- D. Chen, Z. Wan, X. Chen, Y. Yuan and J. Zhong, J. Mater. Chem. C, 2016, 4, 10646–10653 RSC.
- C. Reichardt, Chem. Rev., 1994, 94, 2319 CrossRef CAS.
- S. Li, G. Liu, Q. Liu, L. Nie, G. Yao, F. Zeng, Y. He and W. Xiang, ACS Sustainable Chem. Eng., 2020, 8, 10646–10652 CAS.
- S. Yan, S. Yang, L. He, C. Ye, X. Song and F. Liao, Synth. Met., 2014, 198, 142–149 CrossRef CAS.
- X. Chen, F. Zhang, Y. Ge, L. Shi, S. Huang, J. Tang, Z. Lv, L. Zhang, B. Zou and H. Zhong, Adv. Funct. Mater., 2018, 28, 1706567 CrossRef.
- M. He, W. Chunyun, J. Li, J. Wu, S. Zhang, H.-C. Kuo, L.-Y. Shao, S. Zhao, J. Zhang, F. Kang and G. Wei, Nanoscale, 2019, 11, 22899–22906 RSC.
- M. De Bastiani, I. Dursun, Y. Zhang, B. A. Alshankiti, X.-H. Miao, J. Yin, E. Yengel, E. Alarousu, B. Turedi, J. M. Almutlaq, M. I. Saidaminov, S. Mitra, I. Gereige, A. AlSaggaf, Y. Zhu, Y. Han, I. S. Rogan, J.-L. Bredas, O. F. Mohammed and O. M. Bakr, Chem. Mater., 2017, 29, 7108–7113 CrossRef CAS.
- Y. Zhang, M. I. Saidaminov, I. Dursun, H. Yang, B. Murali, E. Alarousu, E. Yengel, B. A. Alshankiti, O. M. Bakr and O. F. Mohammed, J. Phys. Chem. Lett., 2017, 8, 961–965 CrossRef CAS PubMed.
- L. Yuan, R. Patterson, X. Wen, Z. Zhang, G. Conibeer and S. Huang, J. Colloid Interface Sci., 2017, 504, 586–592 CrossRef CAS PubMed.
- X. Wang, J. Yu, M. Hu, Y. Wu, L. Yang, W. Ye and X. Yu, J. Lumin., 2020, 221, 116986 CrossRef CAS.
- L. N. Quan, R. Quintero-Bermudez, O. Voznyy, G. Walters, A. Jain, J. Z. Fan, X. Zheng, Z. Yang and E. H. Sargent, Adv. Mater., 2017, 29, 1605945 CrossRef PubMed.
- P. Uthirakumar, H. Yun, M. Devendiran, W. W. Lee and I.-H. Lee, J. Lumin., 2019, 209, 163–169 CrossRef CAS.
- V. Dracopoulos, D. T. Kastrissios and G. N. Papatheodorou, Polyhedron, 2005, 24, 619–625 CrossRef CAS.
- M. Velazquez, A. Ferrier, S. Pechev, P. Gravereau, J.-P. Chaminade, X. Portier and R. Moncorge, J. Cryst. Growth, 2008, 310, 5458–5463 CrossRef CAS.
- D. M. Calistru, L. Mihut, S. Lefrant and I. Baltog, J. Appl. Phys., 1997, 82, 5391–5395 CrossRef CAS.
- M. Nikl, E. Mihokova, K. Nitsch, F. Somma, C. Giampaolo, G. P. Pazzi, P. Fabeni and S. Zazubovich, Chem. Phys. Lett., 1999, 306, 280–284 CrossRef CAS.
- J. Almutlaq, J. Yin, O. F. Mohammed and O. M. Bakr, J. Phys. Chem. Lett., 2018, 9, 4131–4138 CrossRef CAS PubMed.
- J. Xu, W. Huang, P. Li, D. R. Onken, C. Dun, Y. Guo, K. B. Ucer, C. Lu, H. Wang, S. M. Geyer, R. T. Williams and D. L. Carroll, Adv. Mater., 2017, 29, 1703703 CrossRef PubMed.
- F. Palazon, C. Urso, L. De Trizio, Q. Akkerman, S. Marras, F. Locardi, I. Nelli, M. Ferretti, M. Prato and L. Manna, ACS Energy Lett., 2017, 2, 2445–2448 CrossRef CAS PubMed.
- M. Shin, S.-W. Nam, A. Sadhanala, R. Shivanna, M. Anaya, A. Jimenez-Solano, H. Yoon, S. Jeon, S. D. Stranks, R. L. Z. Hoye and B. Shin, ACS Appl. Energy Mater., 2020, 3, 192–199 CrossRef CAS.
- S. Ananthakumar, J. R. Kumar and S. M. Babu, J. Photon. Energy, 2016, 6, 042001 CrossRef.
- X. Li, Y. Wu, S. Zhang, B. Cai, Y. Gu, J. Song and H. Zeng, Adv. Funct. Mater., 2016, 26, 2435–2445 CrossRef CAS.
- R.-T. Liu, X.-P. Zhai, Z.-Y. Zhu, B. Sun, D.-W. Liu, B. Ma, Z.-Q. Zhang, C.-L. Sun, B.-L. Zhu, X.-D. Zhang, Q. Wang and H.-L. Zhang, J. Phys. Chem. Lett., 2019, 10, 6572–6577 CrossRef CAS PubMed.
- F. Gao, Y. Zhao, X. Zhang and J. You, Adv. Energy Mater., 2020, 10, 1902650 CrossRef CAS.
- S. Sun, D. Yuan, Y. Xu, A. Wang and Z. Deng, ACS Nano, 2016, 10, 3648–3657 CrossRef CAS PubMed.
- Z. Liang, S. Zhao, Z. Xu, B. Qiao, P. Song, D. Gao and X. Xu, ACS Appl. Mater. Interfaces, 2016, 8, 28824–28830 CrossRef CAS PubMed.
- X. Yang, X. Zhang, J. Deng, Z. Chu, Q. Jiang, J. Meng, P. Wang, L. Zhang, Z. Yin and J. You, Nat. Commun., 2018, 9, 570 CrossRef PubMed.
- L. Zhang, X. Yang, Q. Jiang, P. Wang, Z. Yin, X. Zhang, H. Tan, Y. M. Yang, M. Wei, B. R. Sutherland, E. H. Sargent and J. You, Nat. Commun., 2017, 8, 15640 CrossRef CAS PubMed.
Footnote |
| † Co first authors: Zikuan Shi, Yu Yang. |
| This journal is © The Royal Society of Chemistry 2021 |