
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDual utility of a single diphosphine–ruthenium complex: a precursor for new complexes and, a pre-catalyst for transfer-hydrogenation and Oppenauer oxidation†
Aparajita Mukherjee and
Samaresh Bhattacharya *
*
Department of Chemistry, Inorganic Chemistry Section, Jadavpur University, Kolkata–700 032, India. E-mail: samaresh_b@yahoo.com; Fax: +91-33-24146223
First published on 27th April 2021
Abstract
The diphosphine–ruthenium complex, [Ru(dppbz)(CO)2Cl2] (dppbz = 1,2-bis(diphenylphosphino)benzene), where the two carbonyls are mutually cis and the two chlorides are trans, has been found to serve as an efficient precursor for the synthesis of new complexes. In [Ru(dppbz)(CO)2Cl2] one of the two carbonyls undergoes facile displacement by neutral monodentate ligands (L) to afford complexes of the type [Ru(dppbz)(CO)(L)Cl2] (L = acetonitrile, 4-picoline and dimethyl sulfoxide). Both the carbonyls in [Ru(dppbz)(CO)2Cl2] are displaced on reaction with another equivalent of dppbz to afford [Ru(dppbz)2Cl2]. The two carbonyls and the two chlorides in [Ru(dppbz)(CO)2Cl2] could be displaced together by chelating mono-anionic bidentate ligands, viz. anions derived from 8-hydroxyquinoline (Hq) and 2-picolinic acid (Hpic) via loss of a proton, to afford the mixed-tris complexes [Ru(dppbz)(q)2] and [Ru(dppbz)(pic)2], respectively. The molecular structures of four selected complexes, viz. [Ru(dppbz)(CO)(dmso)Cl2], [Ru(dppbz)2Cl2], [Ru(dppbz)(q)2] and [Ru(dppbz)(pic)2], have been determined by X-ray crystallography. In dichloromethane solution, all the complexes show intense absorptions in the visible and ultraviolet regions. Cyclic voltammetry on the complexes shows redox responses within 0.71 to −1.24 V vs. SCE. [Ru(dppbz)(CO)2Cl2] has been found to serve as an excellent pre-catalyst for catalytic transfer-hydrogenation and Oppenauer oxidation.
Introduction
There has been considerable current interest in the chemistry of ruthenium complexes largely because of their versatile catalytic applications.1 Complexes of ruthenium are also finding application in biology,2 several ruthenium-species, such as RAPTA, NAMI-A, KP1019, etc., have already shown promise as anti-tumor agents.3 A third application of ruthenium complexes, viz. synthetic application, is also there, which is relatively less talked about. Synthesis of any new ruthenium complex is essentially achieved via displacement of pre-coordinated ligand(s) from an appropriate starting ruthenium precursor. Predictable displaceability of ligand(s) in any ruthenium complex is thus of significant importance, and is a much sought after property. In the present study, which has emerged from a recently reported work of us involving a mixed-ligand ruthenium(II) diphosphine complex, [Ru(dppbz)(CO)2Cl2], where dppbz depicts 1,2-bis(diphenylphosphino)benzene (Chart 1),4 our aim was to explore potential of this complex as a precursor for (i) synthesis of new complexes, and (ii) catalysis. This [Ru(dppbz)(CO)2Cl2] complex was found to serve as an useful precursor for the in situ generation of a ruthenium(0) species, viz. [Ru(dppbz)(CO)2], which could efficiently catalyze C–C and C–N coupling reactions. Herein we wish to describe two types of utility of the same [Ru(dppbz)(CO)2Cl2] complex, viz. synthetic utility and catalytic utility, which are not associated with any change of oxidation state of the metal center. There are four monodentate ligands in this complex, two carbonyls that are mutually cis and two chlorides that are trans. We came up with experimental methods whereby one, two or all four of these monodentate ligands can be predictably displaced by ligand(s) of appropriate nature leading to generation of new molecules – a phenomenon demonstrating synthetic utility of the parent complex. | ||
| Chart 1 | ||
Ruthenium complexes with a pre-existing Ru–H bond or with the capability of formation of such a fragment in situ, have the potential to serve as catalyst/pre-catalyst in transfer-hydrogenation of suitable substrates.5–11 Complexes having Ru–Cl bond(s) are particularly useful in this respect, as they can easily give rise to a Ru–H fragment.7f,g,i We have also successfully explored this possibility in the [Ru(dppbz)(CO)2Cl2] complex. Herein we report our observations on exploration of dual utility of the [Ru(dppbz)(CO)2Cl2] complex, with particular reference to (i) formation and characterization of the new complexes, and (ii) efficiency of the [Ru(dppbz)(CO)2Cl2] complex in catalyzing transfer-hydrogenation reactions.
Results and discussion
Syntheses and structures
As outlined in the introduction, this study was intended to explore two types of reactivity in [Ru(dppbz)(CO)2Cl2] without bringing about any change in the oxidation state of ruthenium. The reactivity that we describe first has its origin in our previous study involving the same ruthenium complex. During our attempts to grow single crystals of [Ru(dppbz)(CO)2Cl2] from its solutions in different solvents, we noticed that whenever a coordinating solvent was used for such crystallization, the crystalline solid obtained back was a mixture of [Ru(dppbz)(CO)2Cl2] and another species that seemed to have only one carbonyl in it, as indicated by IR.12 Prompted by this indication that a carbonyl in [Ru(dppbz)(CO)2Cl2] probably undergoes facile displacement by a monodentate ligand (L) leading to formation of a new complex of type [Ru(dppbz)(CO)(L)Cl2], herein we have carried out reactions of [Ru(dppbz)(CO)2Cl2] with three selected monodentate ligands, viz. dimethyl sulfoxide (dmso), acetonitrile and 4-picoline. Each of these ligands is found to readily react with [Ru(dppbz)(CO)2Cl2] and afford the expected monosubstituted product of type [Ru(dppbz)(CO)(L)Cl2] (L = dmso, CH3CN, 4-picoline) in quantitative yields. Bulk characterization data on these three complexes were found to be consistent with their compositions. The structure of one member of this family, viz. [Ru(dppbz)(CO)(dmso)Cl2], was determined by X-ray crystallography. The structure is shown in Fig. 1 and selected bond parameters are presented in Table S1 (ESI†). The structure confirms the loss of one CO group. The ancillary dmso ligand is S-bonded to the ruthenium center. The Ru–S distance is normal,13 and the other bond distances around ruthenium compare well with those in the parent [Ru(dppbz)(CO)2Cl2] complex.4 It was interesting to note that unlike the disposition of the CO and chloride ligands in the parent complex, the remaining carbonyl and a chloride have undergone a positional interchange that results in the formal trans-to-cis isomerization of the two chlorides in [Ru(dppbz)(CO)(dmso)Cl2]. In order to help rationalize this change in geometry, the structures of both the trans and cis isomers of [Ru(dppbz)(CO)(dmso)Cl2] were optimized by DFT. The computed energies of the optimized isomers (Fig. 2) indicate that the cis isomer is 8.40 kcal mol−1 more stable than the trans isomer. We speculate that the initial ligand substitution occurs stereoselectively with the displacement of a carbonyl from the parent complex by dmso to furnish the trans form of [Ru(dppbz)(CO)(dmso)Cl2] as the kinetic product of substitution; conversion of this isomer into the thermodynamically more stable cis isomer results via a subsequent sequence of events involving the mutual permutation of the carbonyl and one chloride from the trans isomer. The isomerization seems to be facilitated by the elevated temperature of refluxing dimethyl sulfoxide.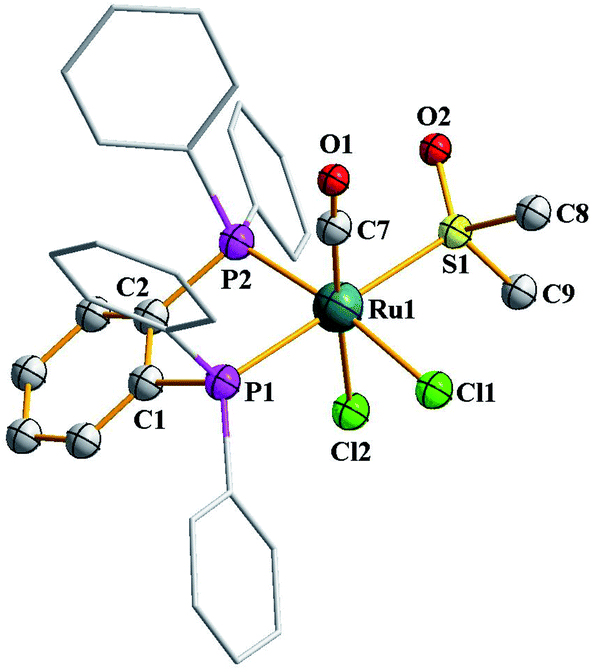 | ||
| Fig. 1 Crystal structure of [Ru(dppbz)(CO)(dmso)Cl2]. | ||
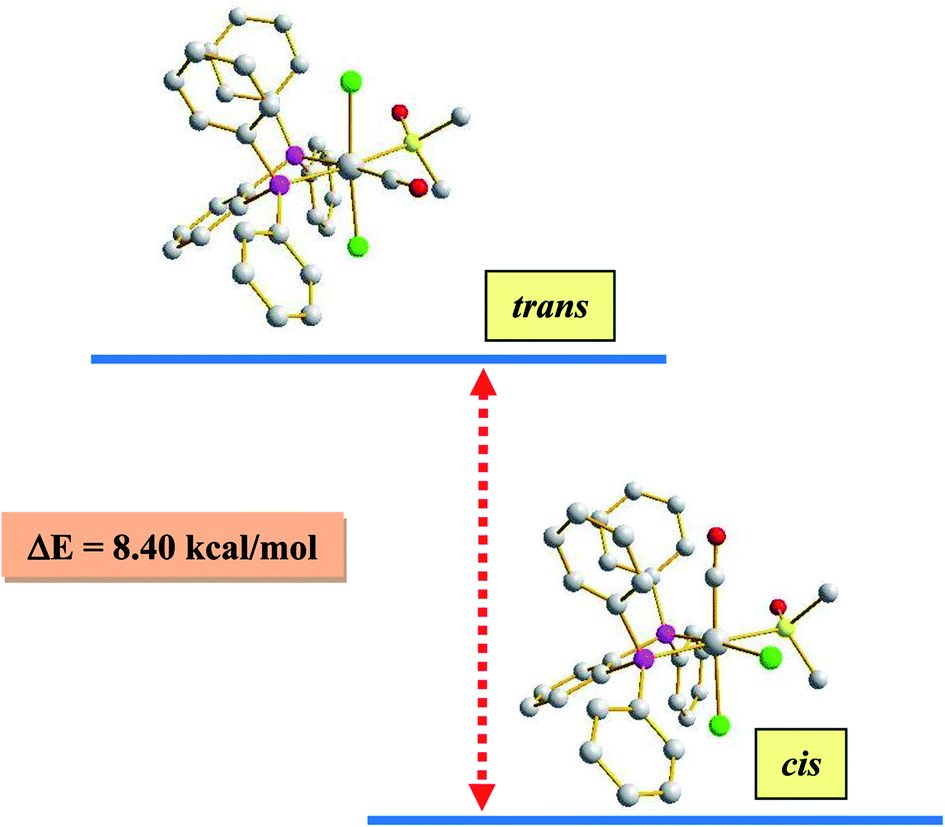 | ||
| Fig. 2 DFT-optimized structures of the cis and trans isomers of the [Ru(dppbz)(CO)(dmso)Cl2], and the energy difference (ΔE) between them. | ||
As crystals of the other two [Ru(dppbz)(CO)(L)Cl2] (L = CH3CN and 4-picoline) complexes could not be grown, structures of both the trans and cis isomers of these two complexes were examined by DFT method, and the computed energies of the optimized isomers (Fig. S1 and S2; ESI†) indicate that the cis isomer is thermodynamically more stable than the trans isomer in both cases. However, in view of the relatively milder experimental conditions used for the synthesis of these two complexes, viz. [Ru(dppbz)(CO)(CH3CN)Cl2] and [Ru(dppbz)(CO)(4-picoline)Cl2], they are believed to have the trans structures, which are shown in Fig. S3 and S4 (ESI†). Some computed bond parameters of these DFT-optimized structures are listed in Table S2 (ESI†), which are found to compare well with each other, and also with those in the crystal structure of [Ru(dppbz)(CO)(dmso)Cl2].
Formation of the [Ru(dppbz)(CO)(L)Cl2] (L = dmso, CH3CN, 4-picoline) complexes demonstrates the labile nature of one carbonyl in the parent [Ru(dppbz)(CO)2Cl2] complex. It is also noteworthy in this context that use of an excess quantity of the monodentate ligand (L) does not lead to the displacement of both carbonyls from [Ru(dppbz)(CO)2Cl2]. However, treatment of [Ru(dppbz)(CO)2Cl2] with a second equivalent of dppbz was found to successfully displace both carbonyls and furnish the bis-dppbz complex, [Ru(dppbz)2Cl2], in good yield. It is relevant to mention here that presence of acetonitrile during the synthesis was found to be crucial, as the bis-dppbz complex is not formed at all when the same synthesis was attempted in dichloromethane alone. This indicates that displacement of one carbonyl by acetonitrile probably takes place first, which then allows the added dppbz to bind itself to the metal center and eventually furnish the bis-dppbz complex. This hypothesis is further supported by the fact that reaction of [Ru(dppbz)(CO)(CH3CN)Cl2] with dppbz ligand in dichloromethane under ambient conditions readily affords the same bis-dppbz complex. The molecular structure of [Ru(dppbz)2Cl2], which was established by X-ray crystallography, is shown in Fig. 3, and some relevant bond parameters are presented in Table S3 (ESI†). The structure of [Ru(dppbz)2Cl2] reveals the presence of two chelating dppbz ligands that share a common equatorial plane with the ruthenium center, and the two chlorides are mutually trans as in the parent complex. Formation of [Ru(dppbz)2Cl2] thus exemplifies stereoretentive displacement of the two mutually cis carbonyls from [Ru(dppbz)(CO)2Cl2] by a neutral chelating ligand.
 | ||
| Fig. 3 Crystal structure of [Ru(dppbz)2Cl2]. | ||
Encouraged by the facile displacement of carbonyl(s) from [Ru(dppbz)(CO)2Cl2] by a variety of neutral ligands, we next investigated the lability of the coordinated chloride(s) in the same complex. Here we selected two bidentate chelating ligands, viz. quinolin-8-ol (Hq) and 2-picolinic acid (Hpic), both of which are known to bind to ruthenium, and also other metal ions, as monoanionic N,O-donors, via loss of the acidic proton, to form five-membered chelate rings (I and II).14,15 The pyridine-type nitrogen was expected to displace carbonyl and the phenolate or carboxylate oxygen was expected to displace chloride. The reaction of quinolin-8-ol with [Ru(dppbz)(CO)2Cl2] was first carried out in equimolar ratio with the intention of obtaining a complex of type [Ru(dppbz)(q)(CO)Cl], but we only obtained a complex formulated as [Ru(dppbz)(q)2] in low yield. This result indicates that, compared to [Ru(dppb)(CO)2Cl2], the initial product from the reaction, [Ru(dppbz)(q)(CO)Cl], is kinetically more reactive towards the deprotonated quinolin-8-ol. And thus it rapidly undergoes an additional substitution with the deprotonated quinolin-8-ol to afford [Ru(dppbz)(q)2]. Realizing the requirement of two moles of Hq per mole of [Ru(dppbz)(CO)2Cl2] for the unstoppable production of [Ru(dppbz)(q)2], it was synthesized in good yield from a reaction between these two ingredients in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mole ratio. Depending on the relative disposition of the two N,O-ligands, three geometrical isomers, viz. ct, tc and cc, may be envisioned for [Ru(dppbz)(q)2].16 In order to sort out this isomer assignment problem, crystal structure of the isolated [Ru(dppbz)(q)2] complex was determined. The structure (Fig. 4 and Table S4 (ESI†)) reveals that both the quinolin-8-olate ligands are chelated to ruthenium in the usual fashion (I), with the nitrogens exhibiting a cis disposition and contained in the plane defined by the ruthenium center and the two phosphorus atoms of the dppbz ligand. The two oxygens atoms adopt a trans orientation relative to the plane containing the RuP2N2 atoms. Therefore the isolated [Ru(dppbz)(q)2] complex has the ct-geometry. Comparison of the stereochemistry around ruthenium in the parent [Ru(dppbz)(CO)2Cl2] complex and the derived [Ru(dppbz)(q)2] complex reveals that, interestingly, the anionic halide positions and the neutral carbonyl sites in parent complex are taken up respectively by the anionic phenolate-oxygens and neutral pyridine-nitrogens in the derived bis-quinolinolate complex. Bond parameters within the Ru(q) fragments are similar to those distances and angles reported in related structures having the same quinolinolate auxiliary.14 The mean Ru–P distance of 2.2489 Å in [Ru(dppbz)(q)2] is 0.1443 Å shorter than the mean Ru–P distance in [Ru(dppbz)(CO)2Cl2], a feature we attribute to the relatively poor π-acid character of the heterocyclic platform of the quinolin-8-olate ligands compared to the two carbonyls in the parent complex.
:1 mole ratio. Depending on the relative disposition of the two N,O-ligands, three geometrical isomers, viz. ct, tc and cc, may be envisioned for [Ru(dppbz)(q)2].16 In order to sort out this isomer assignment problem, crystal structure of the isolated [Ru(dppbz)(q)2] complex was determined. The structure (Fig. 4 and Table S4 (ESI†)) reveals that both the quinolin-8-olate ligands are chelated to ruthenium in the usual fashion (I), with the nitrogens exhibiting a cis disposition and contained in the plane defined by the ruthenium center and the two phosphorus atoms of the dppbz ligand. The two oxygens atoms adopt a trans orientation relative to the plane containing the RuP2N2 atoms. Therefore the isolated [Ru(dppbz)(q)2] complex has the ct-geometry. Comparison of the stereochemistry around ruthenium in the parent [Ru(dppbz)(CO)2Cl2] complex and the derived [Ru(dppbz)(q)2] complex reveals that, interestingly, the anionic halide positions and the neutral carbonyl sites in parent complex are taken up respectively by the anionic phenolate-oxygens and neutral pyridine-nitrogens in the derived bis-quinolinolate complex. Bond parameters within the Ru(q) fragments are similar to those distances and angles reported in related structures having the same quinolinolate auxiliary.14 The mean Ru–P distance of 2.2489 Å in [Ru(dppbz)(q)2] is 0.1443 Å shorter than the mean Ru–P distance in [Ru(dppbz)(CO)2Cl2], a feature we attribute to the relatively poor π-acid character of the heterocyclic platform of the quinolin-8-olate ligands compared to the two carbonyls in the parent complex.

 | ||
| Fig. 4 Crystal structure of [Ru(dppbz)(q)2]. | ||
The reaction of [Ru(dppbz)(CO)2Cl2] was next carried out with 2-picolinic acid (Hpic) in 1:2 mole ratio,17 and the bis-picolinate complex, [Ru(dppbz)(pic)2], was isolated in good yield. The molecular structure of this complex was determined by X-ray crystallography (Fig. 5 and Table S5 (ESI†)), which shows that the picolinate ligands are chelated to the ruthenium in the usual fashion (II). The complex has a similar stereochemical disposition of the two N,O-donor ligands as observed in [Ru(dppbz)(q)2]. The Ru–P distances (2.2553 Å mean distance) are again found to be significantly shorter in [Ru(dppbz)(pic)2] than in the parent complex. The observed bond parameters in the Ru(pic) fragments compare well with those data reported in other pic-chelated compounds.15 Formation of these two [Ru(dppbz)(N–O)2] complexes (where N–O = quinolin-8-olate, 2-picolinate) demonstrates that all of the monodentate ligands in [Ru(dppbz)(CO)2Cl2], viz. the two neutral carbonyls and the two anionic chlorides, are easily displaced by appropriate chelating bidentate ligands. And in view of the electronic nature of the displacing as well as displaced donor sites, the observed displacement reactions are stereoretentive in nature.
 | ||
| Fig. 5 Crystal structure of [Ru(dppbz)(pic)2]. | ||
[Ru(dppbz)(CO)2Cl2] thus serves as a useful precursor for the synthesis of new and unique dppbz-chelated ruthenium(II) complexes, as summarized in Scheme 1. Neutral monodentate ligands (L) are found to displace only one carbonyl from the parent complex, independent of the amount of ligand used, to furnish complexes with the formula [Ru(dppbz)(CO)(L)Cl2]. Whereas, use of a rigid diphosphine ligand promotes the facile displacement of both carbonyls to give[Ru(dppbz)2Cl2]. The loss of the mutually cis carbonyls in [Ru(dppbz)(CO)2Cl2] is attributable to the chelate effect imposed by the dppbz ligands. Mono-anionic chelating ligands (N–O) are found to bring about stereoretentive displacement of the monodentate CO and chloride ligands in [Ru(dppbz)(CO)2Cl2] to yield complexes of the type [Ru(dppbz)(N–O)2]. The reactions illustrated in Scheme 1 demonstrate that, by proper choice of ligands, new octahedral ruthenium(II) complexes based on [Ru(dppbz)(CO)2Cl2] may be synthesized via selective displacement of monodentate ligand(s).
 | ||
| Scheme 1 Formation of different types of complexes from [Ru(dppbz)(CO)2Cl2]. | ||
Spectral properties
Magnetic susceptibility measurements confirm that all six of the new complexes are diamagnetic, consistent with their formulated structures and 2+ oxidation state for ruthenium (low-spin d6, S = 0). The 1H NMR spectra of all six complexes show broad signals within 6.36–8.10 ppm for the dppbz ligand. The 1H chemical shift for the methyl group(s) in [Ru(dppbz)(CO)(L)Cl2] (L = dmso, CH3CN, 4-picoline) is observed at 3.37, 2.16 and 2.31 ppm, respectively. In both [Ru(dppbz)(N–O)2] (N–O = q, pic) complexes, appropriate signals for the N–O ligands, consistent with C2 symmetry existing in these molecules, are observed.The IR spectra of the complexes show many bands of varying intensities within 450–2100 cm−1, among which several bands observed from 492–1584 cm−1 are attributed, by comparison with the spectrum of the parent [Ru(dppbz)(CO)2Cl2] complex, to the Ru(dppbz) unit. The [Ru(dppbz)(CO)(L)Cl2] (L = dmso, CH3CN, 4-picoline) complexes show a strong ν(CO) stretch within 1973–1980 cm−1, while no such band was found in the spectra of the [Ru(dppbz)2Cl2] and [Ru(dppbz)(N–O)2] (N–O = q, pic) complexes consistent with the absence of a terminal CO ligand. In [Ru(dppbz)(pic)2] a broad and strong band was observed at 1703 cm−1, which is due to the carboxylate fragment in the coordinated picolinates.
The mixed-ligand ruthenium dppbz complexes are readily soluble in dichloromethane and chloroform, producing yellow solutions for [Ru(dppbz)(CO)(L)Cl2] and [Ru(dppbz)2Cl2], red solution for [Ru(dppbz)(q)2], and orange solution for [Ru(dppbz)(pic)2]. The electronic spectra of the complexes were recorded in dichloromethane solutions, and the spectral data are given in Table 1. Each complex shows several absorptions in the visible and ultraviolet region. To gain insight into the nature of the observed absorptions TDDFT calculations, which include dichloromethane as solvent, have been performed on all six complexes using the Gaussian 09 program package.18 The main calculated transitions and compositions of the molecular orbitals associated with these transitions for all the six complexes are presented in Tables S6–S17 (ESI†), and contour plots of the same molecular orbitals are shown in Fig. S5–S10 (ESI†). Each of the [Ru(dppbz)(CO)(L)Cl2] (L = dmso, CH3CN, 4-picoline) complexes show three absorptions. The lowest energy absorption, observed within 340–363 nm, is found to result from a combination of transitions involving several filled and vacant orbitals, and based on the composition of these participating orbitals this absorption is attributable to a combination of metal-to-ligand charge-transfer (MLCT), ligand-to-ligand charge-transfer (LLCT), ligand-to-metal charge-transfer (LMCT), and intra-ligand charge-transfer (ILCT) transitions.19 The second absorption observed within 302–316 nm, and the third one near 270 nm, are also found to be of qualitatively similar nature. In the [Ru(dppbz)2Cl2] complex the lowest energy absorption at 370 nm is found to result due to a transition from the HOMO to LUMO+2. As the HOMO has dominant (61%) ruthenium character and the LUMO+2 is localized almost entirely (99%) on the dppbz ligand, this absorption is assignable to a predominantly MLCT transition with minor LLCT character. The next two absorptions at 313 nm and 276 nm are found to be primarily due to ILCT transition, with much less LLCT and LMCT character. In the [Ru(dppbz)(q)2] complex the absorptions at 686 nm and 475 nm are found to be due mainly to ILCT transition with some MLCT character. The third absorption at 366 nm has major MLCT character, while the fourth one at 248 nm has major ILCT character. In the [Ru(dppbz)(pic)2] complex, the band at 433 nm has dominant MLCT nature with much less ILCT component. The next band at 343 nm also has dominant MLCT nature, while the third band at 259 has major LLCT character.
| Complex | Electronic spectral dataa λmax, nm (ε, M−1 cm−1) | Cyclic voltammetric datab E/V vs. SCE |
|---|---|---|
| a In dichloromethane.b Solvent, dichloromethane; supporting electrolyte, TBHP; scan rate, 50 mV s−1.c Shoulder.d Epc value.e E1/2 value, where E1/2 = 0.5(Epa + Epc).f ΔEp value, where ΔEp = Epa − Epc. | ||
| [Ru(dppbz)(CO)(dmso)Cl2] | 340 (2418), 302c (2181), 270c (13200) |
−1.20d |
| [Ru(dppbz)(CO)(CH3CN)Cl2] | 356c (800), 316c (2000), 268c (14000) |
−1.16d |
| [Ru(dppbz)(CO)(4-picoline)Cl2] | 363 (3100), 307c (6100), 269c (15100) |
−1.17d |
| [Ru(dppbz)2Cl2] | 370 (1800), 313c (1500), 276c (3900) | 0.91e (99)f, −1.03d |
| [Ru(dppbz)(q)2] | 686 (620), 475 (5000), 366 (6300), 248 (19000) |
0.99e (263)f, 0.14e (83)f, −1.16d |
| [Ru(dppbz)(pic)2] | 432c (1200), 343 (8100), 259c (16200) |
0.71e (94)f, −1.24d |
Electrochemical properties
The redox properties of the mixed-ligand ruthenium dppbz complexes have been examined in dichloromethane solution (0.1 M TBHP) by cyclic voltammetry. Voltammetric data are presented in Table 1. The three [Ru(dppbz)(CO)(L)Cl2] (L = dmso, CH3CN, 4-picoline) complexes show an irreversible reduction near −1.2 V.20 In [Ru(dppbz)2Cl2], a similar reduction is observed at −1.03 V, along with a reversible oxidation recorded at 0.91 V that is tentatively assigned to the Ru(II)/Ru(III) redox couple. It is interesting to note that this ruthenium-based oxidation is not observed in [Ru(dppbz)(CO)2Cl2] within the positive limit of the voltage window, indicating that the two carbonyls in the parent complex more effectively stabilize ruthenium(II) than the dppbz ligand. [Ru(dppbz)(q)2] shows a reversible Ru(II)/Ru(III) oxidation at 0.14 V and an irreversible reduction at −1.16 V. A second oxidative wave is observed at 0.99 V, which is believed to be due to oxidation of the quinolin-8-olate ligand.21 In [Ru(dppbz)(pic)2], the Ru(II)/Ru(III) oxidation is observed at 0.71 V and the reduction of dppbz at −1.24 V. The anodic shift in the Ru(II)/Ru(III) oxidation couple in [Ru(dppbz)(pic)2] compared to [Ru(dppbz)(q)2] reflects a lower efficiency of the carboxylate moiety, compared to phenolate moiety, to stabilize the ruthenium(III) oxidation product.Catalytic transfer-hydrogenation
As indicated in the introduction, the second objective of this study was to explore catalytic activity of the parent [Ru(dppbz)(CO)2Cl2] complex. The presence of Ru–Cl bond in this complex, together with the labile nature of one carbonyl as observed during exploration of its synthetic utility, tempted us to assess its catalytic potential towards transfer-hydrogenation reaction. We began our study by examining the transfer-hydrogenation of 4-chlorobenzaldehyde to 4-chlorobenzyl alcohol. After extensive optimization it was found that 0.02 mol% catalyst, 0.2 mol% KOtBu, 1-propanol as solvent, a reaction temperature of 95 °C, and a reaction time of 6 h furnished an excellent (99%) yield of the product (Table S18, entry 1; ESI†). Using the optimized reaction conditions, transfer-hydrogenation of twelve different aldehydes has been performed (entries 1–12, Table 2). Benzaldehyde and, para-substituted benzaldehydes having both electron-donating and electron-withdrawing substituent at the para-position, furnished the corresponding alcohols in excellent (97–99%) yields (entries 1–5). However, reduction of bulkier aldehydes could be achieved with less efficiency as reflected in lower yields of the product alcohols (entries 6–8). Both the aldehyde groups in 1,3-diformyl benzene are found to be reduced similarly and smoothly (entry 9). In cinnamaldehyde the aldehyde function underwent selective reduction in presence of the olefinic fragment (entry 10). Aldehydes having another strong donor atom, such as salicylaldehyde or pyridine-2-aldehyde, are found hard or impossible to hydrogenate (entries 11 and 12 respectively), presumably due to catalyst inhibition caused through coordination. After successful transfer-hydrogenation of aryl aldehydes, we attempted reduction of aryl ketones under the similar experimental condition.22 Several aryl ketones were used as substrate, and majority of them could be reduced to their corresponding alcohols with good (78–97%) yields (entries 13–19). 2-Cyclohexen-1-one was found to undergo selective reduction of the keto-group in presence of the olefinic fragment (entry 19). 2-Hydroxyacetophenone gave the corresponding alcohol in poor (32%) yield (entry 20), while 2-acetylpyridine did not afford the expected alcohol at all (entry 21); a manifestation of catalyst inhibition via coordination also observed in case of aldehyde reduction.
|
||
|---|---|---|
| Entry | Substrate | Yieldb (%) |
| a Reaction conditions: aldehydes and ketones (1.0 mmol), KOtBu (0.2 mol%), Ru catalyst (0.02 mol%), 1-propanol (5 mL).b Yields are determined by GCMS based on the quantity of substrate remaining after the reaction. Besides the substrate and the expected product, no other species was detected in any of the reactions.c For ketones Ru catalyst 0.2 mol% and 10 h time are required, instead of Ru catalyst 0.02 mol% and 6 h time respectively. | ||
| 1 |  |
99 |
| 2 |  |
98 |
| 3 |  |
99 |
| 4 |  |
99 |
| 5 |  |
97 |
| 6 | 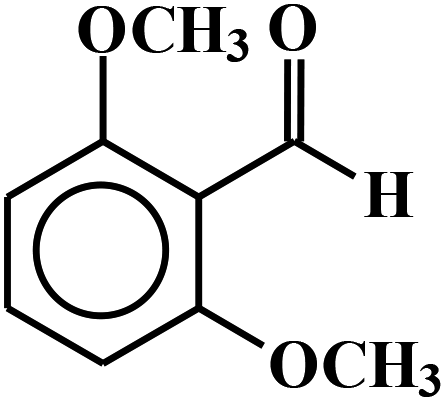 |
61 |
| 7 | 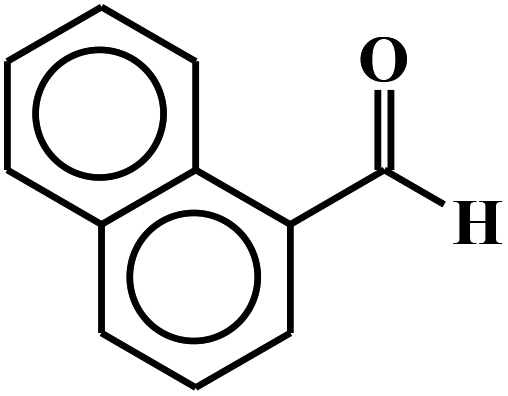 |
91 |
| 8 | 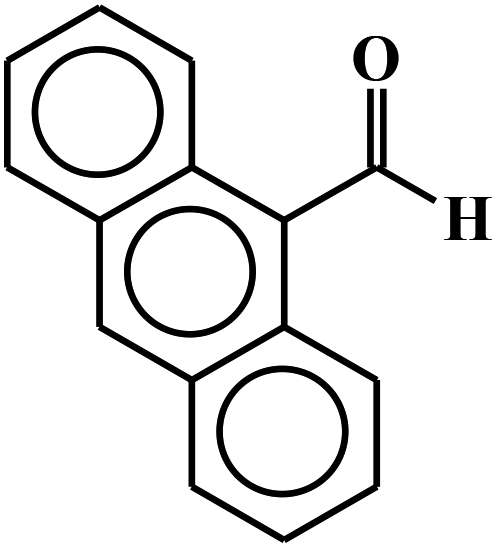 |
88 |
| 9 |  |
|
| 10 |  |
|
| 11 | 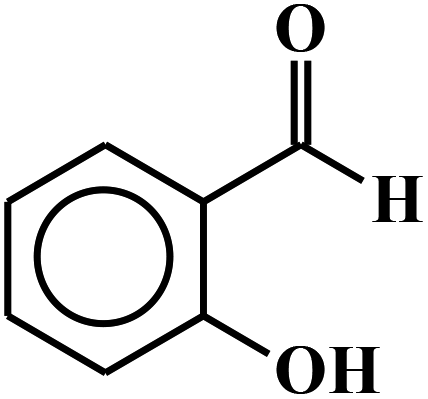 |
25 |
| 12 |  |
0 |
| 13c | 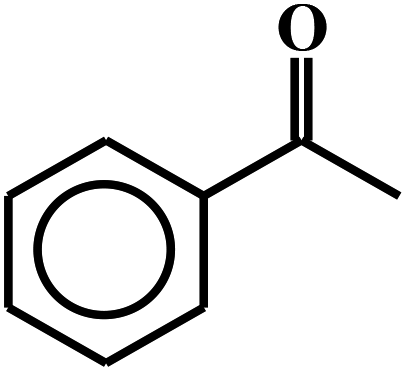 |
97 |
| 14c | 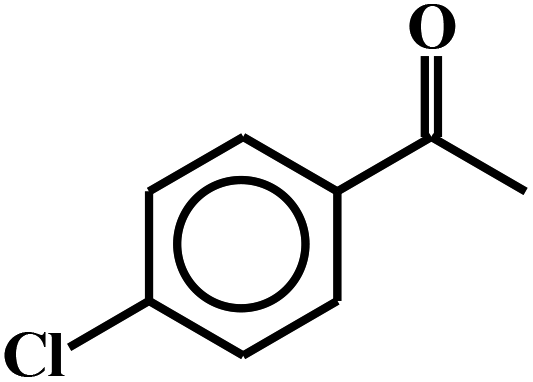 |
99 |
| 15c |  |
90 |
| 16c | 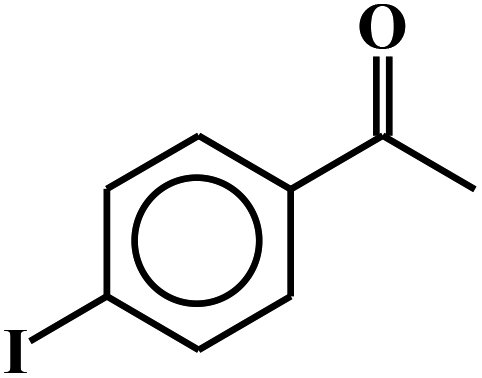 |
92 |
| 17c |  |
82 |
| 18c |  |
81 |
| 19c |  |
78 |
| 20c |  |
32 |
| 21c |  |
0 |
The observed catalysis is believed to be initiated by the formation of a Ru–H bonded species, which is formed in situ via interaction of the Ru–Cl moiety in the catalyst-precursor with 1-propanol. The other steps are envisaged to be similar as described by us and others.23,7f,7g,7i Facile dissociation of one carbonyl from the parent [Ru(dppbz)(CO)2Cl2] complex in presence of monodentate neutral ligands (vide supra) seems to be an added advantage for substrate approach to the catalyst. The observed catalytic activity of [Ru(dppbz)(CO)2Cl2] is found to be better than that of many other reported complexes,6,7a–d,g including our own reports.23b–e However, catalytic efficiency of [Ru(dppbz)(CO)2Cl2] is comparable to that of some reported Ru-catalysts,7f,i,j and less than few others.7h,23a
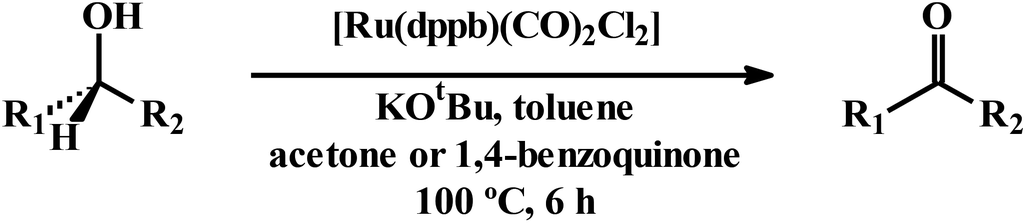
The observed efficiency in transfer-hydrogenation of aldehydes and ketones using [Ru(dppbz)(CO)2Cl2] as the pre-catalyst, reflects successful intermediacy of the in situ generated ruthenium-hydrido species. This encouraged us to examine whether a reverse reaction, viz. catalytic dehydrogenation of alcohols to corresponding oxidation product(s), known as Oppenauer oxidation, is also achievable using the same catalyst precursor. Ru-catalyzed Oppenauer oxidation is well precedent in the literature.24 Some of these reactions are known to proceed via the intermediacy of ruthenium-hydrido species,24a,c,f,j that is also derivable in situ from [Ru(dppbz)(CO)2Cl2]. We planned to explore catalytic potential of [Ru(dppbz)(CO)2Cl2] towards Oppenauer-type oxidation of secondary alcohols, a green oxidation protocol that utilizes acetone as hydrogen acceptor. We first examined the oxidation of cyclohexanol to cyclohexanone and, after several trials for optimization, we have observed that the best result is obtained with 3:2 toluene-acetone as solvent, 0.1 mol% of catalyst, KOtBu as base, 100 °C temperature and 6 h reaction time (Table S19, entry 1; ESI†). Presence of acetone in this oxidation was found to be a must, as manifested in reaction in toluene alone that did not yield any oxidized product (entry 12). However, acetone alone was not found to be the best solvent either, as it afforded the product in significantly lower yield (entry 13). And a 3:2 toluene-acetone mixture was found to be the best choice. Scope of the reaction is illustrated in Table 3. When 2-propanol was used as the substrate, acetone was the expected product, and hence acetone could not be utilized as a reagent. We therefore tried oxidizing 2-propanol using 1,4-benzoquinone as the alternative hydrogen acceptor, which furnished the expected product in excellent yield (entry 1).25 Also in the oxidation of 2-butanol or 3-methyl-2-butanol, 1,4-benzoquinone was found to be a better reagent than acetone (entries 2 and 3). For the oxidation of the other three secondary alcohols, both acetone and 1,4-benzoquinone were found to show comparable efficiency (entries 4–6).
|
|||
|---|---|---|---|
| Entry | Reactant | Oxidant | Yieldb, % |
| a Reaction conditions: secondary alcohol (1.0 mmol), KOtBu (0.25 mol%), Ru catalyst (0.1 mol%).b Yields are determined by GCMS based on the quantity of substrate remaining after the reaction. Besides the substrate and the expected product, no other species was detected in any of the reactions.c toluene (3 mL), acetone (2 mL).d toluene (5 mL), benzoquinone (1.0 mmol). | |||
| 1 | 2-Propanol | Acetonec | — |
| 1,4-Benzoquinoned | 95 | ||
| 2 | 2-Butanol | Acetonec | 57 |
| 1,4-Benzoquinoned | 95 | ||
| 3 | 3-Methyl-2-butanol | Acetonec | 66 |
| 1,4-Benzoquinoned | 97 | ||
| 4 | Cyclopentanol | Acetonec | 96 |
| 1,4-Benzoquinoned | 91 | ||
| 5 | Cyclohexanol | Acetonec | 91 |
| 1,4-Benzoquinoned | 89 | ||
| 6 | Diphenylmethanol | Acetonec | 98 |
| 1,4-Benzoquinoned | 99 | ||
We have also attempted dehydrogenation of primary alcohols using the same [Ru(dppbz)(CO)2Cl2] complex as the catalyst-precursor. We investigated benzyl alcohol as the first substrate and, after screening the experimental parameters, found that the same experimental condition used for oxidation of secondary alcohols was most effective, which afforded benzyl benzoate as the major product along with benzaldehyde in much lower yield (Table S20, entry1; ESI†). The scope of this dehydrogenation reaction is presented in Table 4. Six primary alcohols were tried as substrate, and with acetone as the acceptor of hydrogen, the corresponding ester was obtained as the major dehydrogenation product from each reaction. While 1,4-benzoquinone was utilized as hydrogen acceptor, yield of the ester decreased and that of the aldehyde was found to improve significantly (entries 1–6). We also tried 1,5-pentanediol as a substrate with two alcoholic groups in a single molecule, and, with acetone as the hydrogen acceptor, the corresponding cyclic ester or lactone, viz. 1-oxacyclohexan-2-one, was obtained in excellent yield along with the mono-aldehyde as a minor product (entry 7). Such oxidation of primary alcohol to ester in presence of acceptor appears to be unprecedented. The catalytic efficiency of our complex towards oxidation of secondary alcohol to ketone is better than that of many other Ru-catalysts,24b–h and comparable to that of few.24a,j It is interesting to mention here that Oppenauer type oxidation using 1,4-benzoquinone as acceptor of hydrogen appears to be unprecedented.
|
||||
|---|---|---|---|---|
| Entry | Reactant | Oxidant | Yieldb,% (Ester) | Yieldb,% (Aldehyde) |
| a Reaction conditions: secondary alcohol (1.0 mmol), KOtBu (0.25 mol%), Ru catalyst (0.1 mol%).b Yields are determined by GCMS based on the quantity of substrate remaining after the reaction. Besides the substrate and the reported product(s), no other species was detected in any of the reactions.c Toluene (3 mL), acetone (2 mL).d Toluene (5 mL), benzoquinone (1.0 mmol). | ||||
| 1 | Benzyl alcohol | Acetonec | 75 | 10 |
| 1,4-Benzoquinoned | 59 | 32 | ||
| 2 | 4-Methoxy benzyl alcohol | Acetonec | 41 | 43 |
| 1,4-Benzoquinoned | 48 | 40 | ||
| 3 | 1-Butanol | Acetonec | 70 | 12 |
| 1,4-Benzoquinoned | 33 | 56 | ||
| 4 | Ethanol | Acetonec | 65 | 9 |
| 1,4-Benzoquinoned | 18 | 51 | ||
| 5 | 1-Propanol | Acetonec | 68 | 8 |
| 1,4-Benzoquinoned | 29 | 62 | ||
| 6 | Isoamyl alcohol | Acetonec | 72 | 11 |
| 1,4-Benzoquinoned | 59 | 32 | ||
| 7 |  |
Acetonec |  |
 |
| 1,4-Benzoquinoned |  |
 |
||

Conclusions
The present study shows that the [Ru(dppbz)(CO)2Cl2] complex can be utilized for two purposes, as a precursor for synthesis of new complexes and as a catalyst precursor for transfer-hydrogenation and Oppenauer type oxidation. The coordinated carbonyls and chlorides in this complex could be displaced under relatively mild condition, and the nature of displacement can be predicted based on the nature of displacing ligands. Moreover, [Ru(dppbz)(CO)2Cl2] is found to be an excellent pre-catalyst for transfer-hydrogenation of aldehydes and ketones, as well as Oppenauer type oxidation of alcohols. While secondary alcohols yield ketones as expected, primary alcohols are found to furnish esters, which is quite unusual. Besides, the application of 1,4-benzoquinone as an alternative to acetone in Oppenauer type oxidation is another interesting finding of this study.Experimental
Materials
Ruthenium trichloride was purchased from Arora Matthey, Kolkata, India. 1,2-Bis(diphenylphosphino)benzene (dppbz) was procured from Aldrich. [{Ru(CO)2Cl2}n] was prepared by following a reported method.26 [Ru(dppbz)(CO)2Cl2] was synthesized starting from [{Ru(CO)2Cl2}n] as reported earlier by us.4 Tetrabutylammonium hexafluorophosphate (TBHP) used in the electrochemical studies was purchased from Aldrich. All other chemicals and solvents were reagent grade commercial materials and were used as received.Synthesis of the complexes
:1 dichloromethane–acetonitrile (40 mL) was added 1,2-bis(diphenylphosphino)benzene (35 mg, 0.080 mmol), and the solution was refluxed for 4 h. Upon partial (∼50%) evaporation of the solvents under ambient condition, [Ru(dppbz)2Cl2] separated as a crystalline yellow solid, which was collected by filtration, washed thoroughly with diethyl ether, and dried in air. Yield: (57 mg) 72%. Anal. calc. for C60H48P4Cl2Ru: C, 67.67; H, 4.51. Found: C, 67.72; H, 4.48. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.26–7.70 (48H)*. IR (KBr, cm−1): 468, 492, 511, 521, 531, 551, 618, 667, 697, 727, 748, 758, 924, 998, 1027, 1092, 1109, 1156, 1187, 1251, 1431, 1451, 1482, and 1569.:7 acetonitrile–benzene as the eluant a slow moving reddish-orange band separated, which was extracted with acetonitrile. Evaporation of the acetonitrile extract afforded [Ru(dppbz)(q)2] as a deep red solid. Yield: (46 mg) 74%. Anal. calc. for C48H36N2O2P2Ru: C, 68.95; H, 4.31; N, 3.35. Found: C, 69.34; H, 4.29; N, 3.32. 1H NMR (300 MHz, CDCl3): δ (ppm) = 6.51 (d, H, J = 7.6), 6.58 (d, H, J = 7.9), 6.74 (s, H), 6.77 (s, H), 6.80 (d, H, J = 3.3), 6.82 (d, H, J = 3.6), 6.97 (t, H, J = 7.1), 7.09 (t, H, J = 7.9), 7.24–7.42 (7H)*, 7.65 (s, H), 7.67 (s, H), 8.14 (d, H, J = 4.3). IR (KBr, cm−1): 490, 508, 531, 548, 595, 628, 671, 694, 740, 780, 801, 818, 915, 998, 1028, 1096, 1109, 1170, 1186, 1217, 1288, 1321, 1366, 1383, 1432, 1456, 1483, 1496, 1563, and 1592.:5 acetonitrile–benzene as the eluant an orange band separated, which was extracted with acetonitrile. Evaporation of the acetonitrile extract afforded [Ru(dppbz)(pic)2] as a crystalline orange solid. Yield: (42 mg) 71%. Anal. calc. for C42H32N2O4P2Ru: C, 63.71; H, 4.05; N, 3.54. Found: C, 63.67; H, 4.11; N, 3.56. 1H NMR (300 MHz, CDCl3): δ (ppm) = 7.05–7.35 (6H)*, 7.58 (t, H, J = 7.6), 7.62–7.79 (8H)*, 8.32 (d, H, J = 7.1). IR (KBr, cm−1): 507, 530, 551, 671, 695, 761, 851, 998, 1026, 1047, 1095, 1111, 1161, 1186, 1283, 1348, 1384, 1433, 1482, 1564, 1595, 1629, and 1703.Physical measurements
Microanalyses (C, H, N) were performed using a Heraeus Carlo Erba 1108 elemental analyzer. IR spectra were obtained on a Perkin Elmer Spectrum Two IR spectrometer as KBr pellets. Magnetic susceptibilities were measured using a Sherwood MK-1 balance. 1H NMR spectra were recorded in CDCl3 solution on a Bruker Avance DPX 300 NMR spectrometer using TMS as the internal standard. Electronic spectra were recorded on a JASCO V-630 spectrophotometer. Electrochemical measurements were made using a CH Instruments model 600A electrochemical analyzer. A platinum disc working electrode, a platinum wire auxiliary electrode and an aqueous saturated calomel reference electrode (SCE) were used in the cyclic voltammetry experiments. All electrochemical experiments were performed under a dinitrogen atmosphere. The reported electrochemical data were collected at 298 K and are uncorrected for junction potentials. Geometry optimization by density functional theory (DFT) method and electronic spectral analysis by time-dependent density-functional theory (TDDFT) calculation were performed using the Gaussian 09 (B3LYP/GEN) package.18 GC-MS analyses were performed using a Perkin Elmer CLARUS 680 instrument.Crystallography
Single crystals of the four complexes were obtained by: (i) [Ru(dppbz)(CO)(dmso)Cl2]: slow diffusion of hexane into a dichloromethane solution of the complex; (ii) [Ru(dppbz)2Cl2]: slow diffusion of acetonitrile into a dichloromethane solution of the complex; (iii) [Ru(dppbz)(q)2]: slow diffusion of toluene into a dichloromethane solution of the complex; and (iv) [Ru(dppbz)(pic)2]: slow evaporation of solvent from a solution of the complex in acetonitrile. Table S21 (ESI†) shows the X-ray data processing and collection parameters for the four complexes. Data on all the crystals were collected on a Bruker SMART CCD diffractometer. X-ray data reduction, structure solution, and refinement were done using the SHELXS-97 and SHELXL-97 packages.28 The structures were solved by the direct methods.Computational modeling details
All calculations were done using density functional theory (DFT) with the B3LYP exchange correlation functional,29 as implemented in Gaussian 09 program.18 The lanl2dz basis set was used for Ru,30 and 6-31G(d) was employed for the other elements.31 Vertical electronic excitations were computed based on optimized geometries using time-dependent density functional theory (TD-DFT) in dichloromethane using the conductor-like polarizable continuum model (CPCM).32 GaussSum was used to calculate the fractional contributions from groups or atoms to each molecular orbital.33Application as catalysts
:1) mixture of diethyl ether and hexane, and the filtrate was analyzed by GC-MS.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the reviewers for their constructive comments, which have been very helpful in preparing the revised manuscript. Financial assistance received from the Council of Scientific and Industrial Research (CSIR), New Delhi [Sanction no. 01(2994)/19/EMR-II] is gratefully acknowledged. The DST-FIST and DST-PURSE programs of the Department of Chemistry, Jadavpur University, are also gratefully acknowledged for providing financial and infrastructural supports. AM thanks CSIR for her fellowship [grant number 09/096(0962)/2019-EMR-I]. The authors thank Prof. Saurabh Das (Department of Chemistry, Jadavpur University) for his help in recording the UV-vis spectra of the complexes.References
- (a) M. Hirano and S. Komiya, Oxidative coupling reactions at ruthenium(0) and their applications to catalytic homo-and cross-dimerizations, Coord. Chem. Rev., 2016, 314, 182–200 CrossRef CAS; (b) G. Chelucci, S. Baldino and W. Baratta, Ruthenium and osmium complexes containing 2-(aminomethyl)pyridine (Ampy)-based ligands in catalysis, Coord. Chem. Rev., 2015, 300, 29–85 CrossRef CAS; (c) C. Liu, J. Yuan, M. Gao, S. Tang, W. Li, R. Shi and A. Lei, Oxidative coupling between two hydrocarbons: An update of recent C-H functionalizations, Chem. Rev., 2015, 115, 12138–12204 CrossRef CAS PubMed; (d) L. Souillart and N. Cramer, Catalytic C–C bond activations via oxidative addition to transition metals, Chem. Rev., 2015, 115, 9410–9464 CrossRef CAS PubMed; (e) W. H. Wang, Y. Himeda, J. T. Muckerman, G. F. Manbeck and E. Fujita, CO2 hydrogenation to formate and methanol as an alternative to photo- and electrochemical CO2 reduction, Chem. Rev., 2015, 115, 12936–12973 CrossRef CAS PubMed; (f) Q. Zeng, F. W. Lewis, L. M. Harwood and F. Hartl, Role of ligands in catalytic water oxidation by mononuclear ruthenium complexes, Coord. Chem. Rev., 2015, 304–305, 88–101 CrossRef CAS; (g) L. Duan, L. Wang, F. Li, F. Li and L. Sun, Highly efficient bioinspired molecular Ru water oxidation catalysts with negatively charged backbone ligands, Acc. Chem. Res., 2015, 48, 2084–2096 CrossRef CAS PubMed; (h) C. Gunanathan and D. Milstein, Bond activation and catalysis by ruthenium pincer complexes, Chem. Rev., 2014, 114, 12024–12087 CrossRef CAS PubMed; (i) M. D. Kärkäs, O. Verho, E. V. Johnston and B. Åkermark, Artificial photosynthesis: molecular systems for catalytic water oxidation, Chem. Rev., 2014, 114, 11863–12001 CrossRef PubMed; (j) D. J. Nelson, S. Manzini, C. A. Urbina-Blanco and S. P. Nolan, Key processes in ruthenium-catalysed olefin metathesis, Chem. Commun., 2014, 50, 10355–10375 RSC; (k) A. M. Smit and R. Whyman, Review of methods for the catalytic hydrogenation of carboxamides, Chem. Rev., 2014, 114, 5477–5510 CrossRef PubMed; (l) J. J. Verendel, O. Pàmies, M. Diéguez and P. G. Andersson, Asymmetric hydrogenation of olefins using chiral crabtree-type catalysts: scope and limitations, Chem. Rev., 2014, 114, 2130–2169 CrossRef CAS PubMed; (m) L. Ackermann, Carboxylate-assisted ruthenium-catalyzed alkyne annulations by C–H/Het–H bond functionalizations, Acc. Chem. Res., 2014, 47, 281–295 CrossRef CAS PubMed; (n) F. B. Hamad, T. Sun, S. Xiao and F. Verpoort, Olefin metathesis ruthenium catalysts bearing unsymmetrical heterocylic carbenes, Coord. Chem. Rev., 2013, 257, 2274–2292 CrossRef CAS; (o) D. S. Wang, Q. A. Chen, S. M. Lu and Y. G. Zhou, Asymmetric hydrogenation of heteroarenes and arenes, Chem. Rev., 2012, 112, 2557–2590 CrossRef CAS PubMed; (p) P. B. Arockiam, C. Bruneau and P. H. Dixneuf, Ruthenium (II)-catalyzed C–H bond activation and functionalization, Chem. Rev., 2012, 112, 5879–5918 CrossRef CAS PubMed; (q) C. Gunanathan and D. Milstein, Metal–ligand cooperation by aromatization–dearomatization: a new paradigm in bond activation and “Green” catalysis, Acc. Chem. Res., 2011, 44, 588–602 CrossRef CAS PubMed; (r) S. Lai-Fung Chan, Y. H. Kan, K. L. Yip, J. S. Huang and C. M. Che, Ruthenium complexes of 1, 4, 7-trimethyl-1, 4, 7-triazacyclononane for atom and group transfer reactions, Coord. Chem. Rev., 2011, 255, 899–919 CrossRef; (s) J. H. Xie, S. F. Zhu and Q. L. Zhou, Transition metal-catalyzed enantioselective hydrogenation of enamines and imines, Chem. Rev., 2011, 111, 1713–1760 CrossRef CAS PubMed; (t) L. Ackermann, Carboxylate-assisted transition-metal-catalyzed C-H bond functionalizations: mechanism and scope, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- (a) A. R. Simović, R. Masnikosa, I. Bratsos and E. Alessio, Chemistry and reactivity of ruthenium(II) complexes: DNA/protein binding mode and anticancer activity are related to the complex structure, Coord. Chem. Rev., 2019, 398, 113011 CrossRef; (b) M. Mital and Z. Ziora, Biological applications of Ru(II) polypyridyl complexes, Coord. Chem. Rev., 2018, 375, 434–458 CrossRef CAS; (c) V. Brabec and J. Kasparkova, Ruthenium coordination compounds of biological and biomedical significance. DNA binding agents, Coord. Chem. Rev., 2018, 376, 75–94 CrossRef CAS; (d) F. Heinemann, J. Karges and G. Gasser, Critical overview of the use of Ru(II) polypyridyl complexes as photosensitizers in one-photon and two-photon photodynamic therapy, Acc. Chem. Res., 2017, 50, 2727–2736 CrossRef CAS PubMed; (e) H.-K. Liu and P. J. Sadler, Metal complexes as DNA intercalators, Acc. Chem. Res., 2011, 44, 349–359 CrossRef CAS PubMed.
- (a) C. K. Rono, W. K. Chu, J. Darkwa, D. Meyer and B. C. E. Makhubela, Triazolyl RuII, RhIII, OsII, and IrIII Complexes as Potential Anticancer Agents: Synthesis, Structure Elucidation, Cytotoxicity, and DNA Model Interaction Studies, Organometallics, 2019, 38, 3197–3211 CrossRef CAS; (b) F. Pelletier, V. Comte, A. Massard, M. Wenzel, S. Toulot, P. Richard, M. Picquet, P. L. Gendre, O. Zava, F. Edafe, A. Casini and P. J. Dyson, Development of bimetallic titanocene− ruthenium− arene complexes as anticancer agents: relationships between structural and biological properties, J. Med. Chem., 2010, 53, 6923–6933 CrossRef CAS PubMed; (c) A. Levina, A. Mitra and P. A Lay, Recent developments in ruthenium anticancer drugs, Metallomics, 2009, 1, 458–470 CrossRef CAS PubMed; (d) L. Leyva, C. Sirlin, L. Rubio, C. Franco, R. L. Lagadec, J. Spencer, P. Bischoff, C. Gaiddon, J. -P. Loeffler and M. Pfeffer, Synthesis of cycloruthenated compounds as potential anticancer agents, Eur. J. Inorg. Chem., 2007, 3055–3066 CrossRef CAS; (e) Y. K. Yan, M. Melchart, A. Habtemariam and P. J. Sadler, Organometallic chemistry, biology and medicine: ruthenium arene anticancer complexes, Chem. Commun., 2005, 4764–4776 RSC.
- A. Mukherjee, D. A. Hrovat, M. G. Richmond and S. Bhattacharya, A new diphosphine-carbonyl complex of ruthenium: an efficient precursor for C-C and C-N bond coupling catalysis, Dalton Trans., 2018, 47, 10264–10272 RSC.
- Reviews on Ru-catalyzed transfer-hydrogenation: (a) D. A. Hey, R. M. Reich, W. Baratta and F. E. Kühn, Current advances on ruthenium(II) N-heterocyclic carbenes in hydrogenation reactions, Coord. Chem. Rev., 2018, 374, 114–132 CrossRef CAS; (b) W. P. Hems, M. Groarke, A. Zanotti-Gerosa and G. A. Grasa, [(Bisphosphine) Ru(II) diamine] complexes in asymmetric hydrogenation: expanding the scope of the diamine ligand, Acc. Chem. Res., 2007, 40, 1340–1347 CrossRef CAS PubMed.
- Ru-catalyzed transfer-hydrogenation reaction of aldehydes: (a) S. Baldino, S. Facchetti, A. Zanotti-Gerosa, H. G. Nedden and W. Baratta, Transfer hydrogenation and hydrogenation of hommercial-grade aldehydes to primary alcohols catalyzed by 2-(aminomethyl)pyridine and pincer benzo[h]quinoline ruthenium complexes, ChemCatChem, 2016, 8, 2279–2288 CrossRef CAS; (b) W. Baratta, K. Siega and P. Rigo, Fast and chemoselective transfer hydrogenation of aldehydes catalyzed by a terdentate CNN ruthenium complex [RuCl (CNN)(dppb)], Adv. Synth. Catal., 2007, 349, 1633–1636 CrossRef CAS.
- Ru-catalyzed transfer-hydrogenation reaction of ketones:
(a) J. P. Byrne, P. Musembi and M. Albrecht, Carbohydrate-functionalized N-heterocyclic carbene Ru(II) complexes: synthesis, characterization and catalytic transfer hydrogenation activity, Dalton Trans., 2019, 48, 11838–11847 RSC;
(b) C. M. Moore, B. Bark and N. K. Szymczak, Simple Ligand Modifications with Pendent OH Groups Dramatically Impact the Activity and Selectivity of Ruthenium Catalysts for Transfer
Hydrogenation: The Importance of Alkali Metals, ACS Catal., 2016, 6, 1981–1990 CrossRef CAS;
(c) T. Touge, H. Nara, M. Fujiwhara, Y. Kayaki and T. Ikariya, Efficient Access to Chiral Benzhydrols via Asymmetric Transfer Hydrogenation of Unsymmetrical Benzophenones with Bifunctional Oxo-Tethered Ruthenium Catalysts, J. Am. Chem. Soc., 2016, 138, 10084–10087 CrossRef CAS PubMed;
(d) C. M. Moore and N. K. Szymczak, 6,6’-Dihydroxy terpyridine: a proton-responsive bifunctional ligand and its application in catalytic transfer hydrogenation of ketones, Chem. Commun., 2013, 49, 400–402 RSC;
(e) F. Hasanayn, A. Baroudi, A. A. Bengali and A. S. Goldman, Hydrogenation of dimethyl carbonate to methanol by trans-[Ru(H)2(PNN)(CO)] catalysts: DFT evidence for ion-pair-mediated metathesis paths for C–OMe bond cleavage, Organometallics, 2013, 32, 6969–6985 CrossRef CAS;
(f) M. J. Page, J. Wagler and B. A. Messerle, Pyridine-2, 6-bis (thioether)(SNS) complexes of ruthenium as catalysts for transfer hydrogenation, Organometallics, 2010, 29, 3790–3798 CrossRef CAS;
(g) B. J. Sarmah and D. K. Dutta, Chlorocarbonyl ruthenium(II) complexes of tripodal triphos {MeC(CH2PPh2)3}: Synthesis, characterization and catalytic applications in transfer hydrogenation of carbonyl compounds, J. Organomet. Chem., 2010, 695, 781–785 CrossRef CAS;
(h) W. Baratta, M. Bosco, G. Chelucci, A. D. Zotto, K. Siega, M. Toniutti, E. Zangrando and P. Rigo, Terdentate RuX (CNN)(PP)(X= Cl, H, OR) complexes: synthesis, properties, and catalytic activity in fast transfer hydrogenation, Organometallics, 2006, 25, 4611–4620 CrossRef CAS;
(i) V. Cadierno, P. Crochet, J. Díez, S. E. García-Garrido and J. Gimeno, Efficient transfer hydrogenation of ketones catalyzed by the bis(isocyanide)-ruthenium(II) complexes trans,cis,cis-[RuX2(CNR)2(dppf)] (X = Cl, Br; dppf = 1,1ʹ-bis(diphenylphosphino)ferrocene): isolation of active mono- and dihydride intermediates, Organometallics, 2004, 23, 4836–4845 CrossRef CAS;
(j) K. Abdur-Rashid, M. Faatz, A. J. Lough and R. H. Morris, Catalytic cycle for the asymmetric hydrogenation of prochiral ketones to chiral alcohols: direct hydride and proton transfer from chiral catalysts trans-Ru(H)2(diphosphine)(diamine) to ketones and direct addition of dihydrogen to the resulting hydridoamido complexes, J. Am. Chem. Soc., 2001, 123, 7473–7474 CrossRef CAS PubMed.
- Ru-catalyzed transfer-hydrogenation reaction of alkynes: (a) R. Kusy and K. Grela, E- and Z-Selective transfer semihydrogenation of alkynes catalyzed by standard ruthenium olefin metathesis catalysts, Org. Lett., 2016, 18, 6196–6199 CrossRef CAS PubMed; (b) B. Y. Park, K. D. Nguyen, M. R. Chaulagain, V. Komanduri and M. J. Krische, Alkynes as allylmetal equivalents in redox-triggered C-C couplings to primary alcohols: (Z)-homoallylic alcohols via ruthenium-catalyzed propargyl C-H oxidative addition, J. Am. Chem. Soc., 2014, 136, 11902–11905 CrossRef CAS PubMed.
- Ru-catalyzed transfer-hydrogenation reaction of nitriles: (a) I. D. Alshakova, B. Gabidullin and G. I. Nikonov, Ru-catalyzed transfer hydrogenation of nitriles, aromatics, olefins, alkynes and esters, ChemCatChem, 2018, 10, 4860–4869 CrossRef; (b) V. H. Mai and G. I. Nikonov, Transfer hydrogenation of nitriles, olefins, and N-heterocycles catalyzed by an N-heterocyclic carbene-supported half-sandwich complex of ruthenium, Organometallics, 2016, 35, 943–949 CrossRef CAS; (c) B. Paul, K. Chakrabarti and S. Kundu, Optimum bifunctionality in a 2-(2-pyridyl-2-ol)-1,10-phenanthroline based ruthenium complex for transfer hydrogenation of ketones and nitriles: impact of the number of 2-hydroxypyridine fragments, Dalton Trans., 2016, 45, 11162–11171 RSC; (d) S.-H. Lee and G. I. Nikonov, Transfer hydrogenation of ketones, nitriles, and esters catalyzed by a half-sandwich complex of ruthenium, ChemCatChem, 2015, 7, 107–113 CrossRef CAS.
- Ru-catalyzed transfer-hydrogenation reaction of imines: (a) S. Ibáñez, M. Poyatos and E. Peris, A ferrocenyl-benzo-fused imidazolylidene complex of ruthenium as redox-switchable catalyst for the transfer hydrogenation of ketones and imines, ChemCatChem, 2016, 8, 3790–3795 CrossRef; (b) N. Pannetier, J. B. Sortais, J. T. Issenhuth, L. Barloy, C. Sirlin, A. Holuigue, L. Lefort, L. Panella, J. G. de Vries and M. Pfeffer, Cyclometalated complexes of ruthenium, rhodium and iridium as catalysts for transfer hydrogenation of ketones and imines, Adv. Synth. Catal., 2011, 353, 2844–2852 CrossRef CAS.
- Ru-catalyzed transfer-hydrogenation reaction of nitroarenes: (a) A. Bolje, S. Hohloch, J. Košmrlj and B. Sarkar, RuII, IrIII and OsII mesoionic carbene complexes: efficient catalysts for transfer hydrogenation of selected functionalities, Dalton Trans., 2016, 45, 15983–15993 RSC; (b) B. Paul, K. Chakrabarti, S. Shee, M. Maji, A. Mishra and S. Kundu, A simple and efficient in situ generated ruthenium catalyst for chemoselective transfer hydrogenation of nitroarenes: kinetic and mechanistic studies and comparison with iridium systems, RSC Adv., 2016, 6, 100532–100545 RSC; (c) S. Hohloch, L. Suntrup and B. Sarkar, Arene-ruthenium(II) and -iridium(III) complexes with “click”-based pyridyl-triazoles, bis-triazoles, and chelating abnormal carbenes: applications in catalytic transfer hydrogenation of nitrobenzene, Organometallics, 2013, 32, 7376–7385 CrossRef CAS; (d) R. V. Jagadeesh, G. Wienhöfer, F. A. Westerhaus, A.-E. Surkus, H. Junge, K. Junge and M. Beller, A convenient and general ruthenium-catalyzed transfer hydrogenation of nitro-and azobenzenes, Chem.–Eur. J., 2011, 17, 14375–14379 CrossRef CAS PubMed.
- Three CO stretches are observed within 1900–2100 cm−1.
- (a) J. S. Jaswal, S. J. Rettig and B. R. James, Ruthenium(III) complexes containing dimethylsulfoxide or dimethylsulfide ligands, and a new route to trans-dichlorotetrakis(dimethylsulfoxide)ruthenium(II), Can. J. Chem., 1990, 68, 1808–1817 CrossRef CAS; (b) J. D. Oliver and D. P. Riley, Synthesis and crystal structure of dibromotetrakis (dimethyl sulfoxide) ruthenium (II). Structural implications for oxygen oxidation catalysis, Inorg. Chem., 1984, 23, 156–158 CrossRef CAS.
- (a) C. F. Leung, S. M. Ng, J. Xiang, W. Y. Wong, M. H. W. Lam, C. C. Ko and T. C. Lau, Synthesis and photophysical properties of ruthenium(II) isocyanide complexes containing 8-quinolinolate ligands, Organometallics, 2009, 28, 5709–5714 CrossRef CAS; (b) J. G. Małecki, M. Jaworska, R. Kruszynski and J. Kłak, Reaction of [(C6H6)RuCl2]2 with 7,8-benzoquinoline and 8-hydroxyquinoline, Polyhedron, 2005, 24, 3012–3021 CrossRef; (c) H. Wang, T. Onozuka, H. Tomizawa, M. Tanaka and E. Miki, The unexpected reactions of [RuCl3(2mqn)NO]− (H2mqn=2-methyl-8-quinolinol) with 2-chloro-8-quinolinol (H2cqn) and of [RuCl(2cqn)(2mqn)NO] on photoirradiation, Inorg. Chim. Acta, 2004, 357, 1303–1308 CrossRef CAS; (d) H. Wang, T. Hagihara, H. Ikezawa, H. Tomizawa and E. Miki, Electronic effects of the substituent group in 8-quinolinolato ligand on geometrical isomerism for nitrosylruthenium(II) complexes, Inorg. Chim. Acta, 2000, 299, 80–90 CrossRef CAS; (e) J. T. Warren, W. Chen, D. H. Johnston and C. Turro, Ground-state properties and excited-state reactivity of 8-quinolate complexes of ruthenium(II), Inorg. Chem., 1999, 38, 6187–6192 CrossRef CAS PubMed.
- (a) I. Bratsos, E. Mitri, F. Ravalico, E. Zangrando, T. Gianferrara, A. Bergamo and E. Alessio, New half sandwich Ru(II) coordination compounds for anticancer activity, Dalton Trans., 2012, 41, 7358–7371 RSC; (b) D. Ooyama, T. Kobayashi, K. Shiren and K. Tanaka, Regulation of electron donating ability to metal center: isolation and characterization of ruthenium carbonyl complexes with N,N- and/or N,O-donor polypyridyl ligands, J. Organomet. Chem., 2003, 665, 107–113 CrossRef CAS; (c) T. Hirano, M. Kuroda, N. Takeda, M. Hayashi, M. Mukaida, T. Oi and H. Nagao, Cis–trans isomerization of {RuNO}6-type nitrosylruthenium complexes containing 2-pyridinecarboxylate and structural characterization of a μ-H3O2 bridged dinuclear nitrosylruthenium complex, Dalton Trans., 2002, 2158–2162 RSC.
- The isomer labels were assigned considering the mutual disposition of the two nitrogens first, and that of the two the oxygens next. For example, ct- means the two nitrogens are cis, and the two oxygens are trans.
- The same [Ru(dppbz)(pic)2] complex is also obtained from reaction of equimolar [Ru(dppbz)(CO)2Cl2] and 2-picolinic acid, but in much less yield.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- In the [Ru(dppbz)(CO)(4-picoline)Cl2] complex the ILCT component was found to be absent.
- All potentials are referenced to saturated calomel electrode (SCE).
- G. K. Lahiri, S. Bhattacharya, B. K. Ghosh and A. Chakravorty, Ruthenium and osmium complexes of N,O chelators: syntheses, oxidation levels, and distortion parameters, Inorg. Chem., 1987, 26, 4324–4331 CrossRef CAS.
- For transfer-hydrogenation of ketones, higher catalyst loading and longer reaction time were necessary to achieve optimum yield of the desired products.
- (a) R. Saha, A. Mukherjee and S. Bhattacharya, Heteroleptic 1,4-diazabutadiene complexes of ruthenium: Synthesis, characterization and utilization in catalytic transfer hydrogenation, Eur. J. Inorg. Chem., 2020, 4539–4548 CrossRef CAS; (b) J. Karmakar and S. Bhattacharya, Arene-ruthenium complexes with 2-(arylazo)phenol as ancillary ligand: Synthesis, characterization, and utilization in catalytic transfer-hydrogenation, Polyhedron, 2019, 172, 39–44 CrossRef CAS; (c) P. Dhibar, P. Paul and S. Bhattacharya, Formation of acetone thiosemicarbazone complex of ruthenium via usual chelation and unexpected fragmentation: Characterization and catalytic application, J. Indian Chem. Soc., 2016, 93, 781–788 CAS; (d) J. Dutta, M. G. Richmond and S. Bhattacharya, Cycloruthenation of N-(naphthyl)salicylaldimine and related ligands: Utilization of the Ru–C bond in catalytic transfer hydrogenation, Eur. J. Inorg. Chem., 2014, 4600–4610 CrossRef CAS; (e) N. Saha Chowdhury, C. GuhaRoy, R. J. Butcher and S. Bhattacharya, Mixed-ligand 1,3-diaryltriazenide complexes of ruthenium: Synthesis, structure and catalytic properties, Inorg. Chim. Acta, 2013, 406, 20–26 CrossRef.
- Ru-catalyzed Oppenauer oxidation: (a) L. Pardatscher, B. J. Hofmann, P. J. Fischer, S. M. Hölzl, R. M. Reich, F. E. Kühn and W. Baratta, Highly Efficient Abnormal NHC Ruthenium Catalyst for Oppenauer-Type Oxidation and Transfer Hydrogenation Reactions, ACS Catal., 2019, 9, 11302–11306 CrossRef CAS; (b) R. Labes, C. Battilocchio, C. Mateos, G. R. Cumming, O. d. Frutos, J. A. Rincón, K. Binder and S. V. Ley, Chemoselective continuous Ru-catalyzed hydrogen-transfer Oppenauer-type oxidation of secondary alcohols, Org. Process Res. Dev., 2017, 21, 1419–1422 CrossRef CAS; (c) Q. Wang, W. Du, T. Liu, H. Chai and Z. Yu, Ruthenium(II)–NNN complex catalyzed Oppenauer-type oxidation of secondary alcohols, Tetrahedron Lett., 2014, 55, 1585–1588 CrossRef CAS; (d) C. M. Nicklaus, P. H. Phua, T. Buntara, S. Noel, H. J. Heeres and J. G. d. Vries, Ruthenium/1,1′-bis-(diphenylphosphino)ferrocene-catalysed Oppenauer oxidation of alcohols and lactonisation of α,ω-diols using methyl isobutyl ketone as oxidant, Adv. Synth. Catal., 2013, 355, 2839–2844 CrossRef CAS; (e) S. Manzini, C. A. Urbina-Blanco and S. P. Nolan, Chemoselective oxidation of secondary alcohols using a ruthenium phenylindenyl complex, Organometallics, 2013, 32, 660–664 CrossRef CAS; (f) W. Du, L. Wang, P. Wu and Z. Yu, A versatile ruthenium (II)–NNC complex catalyst for transfer hydrogenation of ketones and Oppenauer-type oxidation of alcohols, Chem.–Eur. J., 2012, 18, 11550–11554 CrossRef CAS PubMed; (g) R. Mello, J. Martínez-Ferrer, G. Asensio and M. E. González-Núñez, Oppenauer oxidation of secondary alcohols with 1,1,1-trifluoroacetone as hydride acceptor, J. Org. Chem., 2007, 72, 9376–9378 CrossRef CAS PubMed; (h) S. Gauthier, R. Scopelliti and K. Severin, A heterobimetallic rhodium(I)-ruthenium(II) catalyst for the Oppenauer-type oxidation of primary and secondary alcohols under mild conditions, Organometallics, 2004, 23, 3769–3771 CrossRef CAS; (i) Y. R. S. Laxmi and J.-E. Bäckvall, Mechanistic studies on ruthenium-catalyzed hydrogen transfer reactions, Chem. Commun., 2000, 611–612 RSC; (j) M. L. S. Almeida, M. Beller, G.-Z. Wang and J.-E. Bäckvall, Ruthenium(II)-catalyzed Oppenauer-type oxidation of secondary alcohols, Chem.–Eur. J., 1996, 2, 1533–1536 CrossRef CAS.
- Benzene-1,4-diol was detected as the hydrogenated product of 1,4-benzoquinone.
- P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway and A. H. White, Designed synthesis of mononuclear tris(heteroleptic)ruthenium complexes containing bidentate polypyridyl ligands, Inorg. Chem., 1995, 34, 6145–6157 CrossRef CAS.
- Chemical shifts for all NMR data are given as δ (ppm) and the multiplicity of the signals, along with the associated coupling constant(s), is given in parentheses. Overlapping signals are marked with an asterisk (*).
- G. M. Sheldrick, SHELXS-97 and SHELXL-97, Fortran programs for crystal structure solution and refinement, University of Göttingen: Göttingen, Germany 1997 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Development of the Colic-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- L. E. Roy, P. J. Hay and R. L. Martin, Revised basis sets for the LANL effective core potentials, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef CAS PubMed.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. Defrees and J. A. Pople, Self-consistent molecular orbital methods. XXIII. A polarization-type basis set for second-row elements, J. Chem. Phys., 1982, 77, 3654–3665 CrossRef CAS.
- (a) R. Bauernschmitt and R. Ahlrichs, Treatment of electronic excitations within the adiabatic approximation of time dependent density functional theory, Chem. Phys. Lett., 1996, 256, 454–464 CrossRef CAS; (b) G. Scalmani, M. J. Frisch, B. Mennucci, J. Tomasi, R. Cammi and V. Barone, Geometries and properties of excited statesin the gas phase and in solution: Theory and application of a time-dependent density functional theory polarizable continuum model, J. Chem. Phys., 2006, 124, 94107(1)–94107(15) CrossRef.
- E. D. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO Version 3.1, University of Wisconsin, Madison, 1998 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond parameters for [Ru(dppbz)(CO)(dmso)Cl2], (Table S1); energy differences between the cis- and trans-isomers of [Ru(dppbz)(CO)(CH3CN)Cl2] and [Ru(dppbz)(CO)(4-picoline)Cl2], (Fig. S1 and S2); DFT-optimized structures of trans-[Ru(dppbz)(CO)(CH3CN)Cl2] and trans-[Ru(dppbz)(CO)(4-picoline)Cl2], (Fig. S3 and S4); some computed bond parameters of the DFT-optimized structures of [Ru(dppbz)(CO)(CH3CN)Cl2] and [Ru(dppbz)(CO)(4-picoline)Cl2], (Table S2); selected bond parameters for [Ru(dppbz)2Cl2], [Ru(dppbz)()2] and [Ru(dppbz)(pic)2], (Tables S3–S5); parameters and figures from TDDFT calculations, (Tables S6–S17 and Fig. S5–S10); optimization for catalytic transfer-hydrogenation, (Table S18); optimization for catalytic Oppenauer oxidation, (Tables S19 and S20); crystal data (Table S21). CCDC 1489487–1489490. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01594j |
| This journal is © The Royal Society of Chemistry 2021 |