
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTBAI-assisted direct C–H activation of indoles with β-E-styrene sulfonyl hydrazides: a stereoselective access to 3-styryl thioindoles†
Saira Hafeez a and
Aamer Saeed*b
a and
Aamer Saeed*b
aDepartment of Chemistry, University of Science and Technology of China, Hefei, Anhui 230026, China
bDepartment of Chemistry, Quaid-I-Azam University, 45320, Islamabad, Pakistan. E-mail: aamersaeed@yahoo.com; asaeed@aiou.edu.pk
First published on 27th April 2021
Abstract
The current work describes the challenging introduction of a vinyl sulfide group by simple C–H activation on a variety of substrates. The direct C–H activation of indoles with β-(E)-styrene sulfonyl hydrazides under the sulfenylation conditions, assisted by the iodic catalyst tert-butyl ammonium iodide (TBAI), afforded a series of (E)-styrylthioindoles. Accordingly, β-(E)-styrene sulfonyl hydrazides undergo radical cross-coupling reactions with a variety of substituted indoles to afford structurally diverse indole vinyl thioethers in moderate to high yields with E-stereoselectivity. This method is metal-catalyst-free and is valuable not only because of its novelty, but also for providing a convenient synthetic pathway to a variety of (E)-styrylthioindoles with retention of the configuration. The current study paves the way for the use of β-(E)-styrene sulfonyl hydrazides as a unique styryl mercaptan source in chemical synthesis.
1. Introduction
One of the essential goals of modern organic synthesis is to develop efficient and environmentally friendly approaches towards the construction of valuable molecular frameworks. Vinyl sulfides constitute a ubiquitous molecular framework in natural products, pharmaceuticals, and important synthetic intermediates in chemical transformations.1 These compounds find extensive applications in chemical and materials science, and can be used as versatile building blocks for carbonyl compounds2 and as Michael acceptors.3 The most popular synthetic procedures to construct such molecules involve the addition of thiols to alkynes either using transition metal catalysts,4–12 free radicals,13–15 Wittig olefination16–18 or nucleophilic substitution of vinyl halides.19,20 Nevertheless, harsh reaction conditions, expensive starting substrates and a lack of stereo-control are limitations of these methods. Consequently, a milder, more convenient, and more stereoselective protocol to obtain these significant scaffolds would be highly desirable.The stereoselective synthesis of (E)-vinyl sulfides has been achieved by several methods. Banerjee and co-workers reported an indium-iodide-promoted one-pot synthesis of (E)-vinyl sulfides via the palladium-catalyzed condensation of vinyl bromides using disulfides as a sulfur source.21 One of the most significant catalytic processes for the synthesis of (E)-vinyl sulfides was reported by the Venkataraman group, but employs a moisture-sensitive Cu(I) catalyst, and requires a high reaction temperature.22 Later, Cook and co-workers reported a milder protocol for the synthesis of vinyl sulfides.23 The iron-catalyzed cross-coupling reaction of alkyl vinyl halides with thiols has also been reported.24 The triethyl-bromide-catalyzed radical addition of thiols to acetylenes results in alkenyl sulfides with good stereocontrol.25 However, all these methods require the use of either metal catalysts, base, or additives to afford the corresponding vinyl sulfides. Furthermore, all these reports describe the nucleophilic addition of thiols to an unsaturated system, and none describes direct C–H activation via the addition of a vinyl sulfide moiety from a single source. Fig. 1 depicts a comparison of the various synthetic methods towards E-vinyl thioethers with the current work.
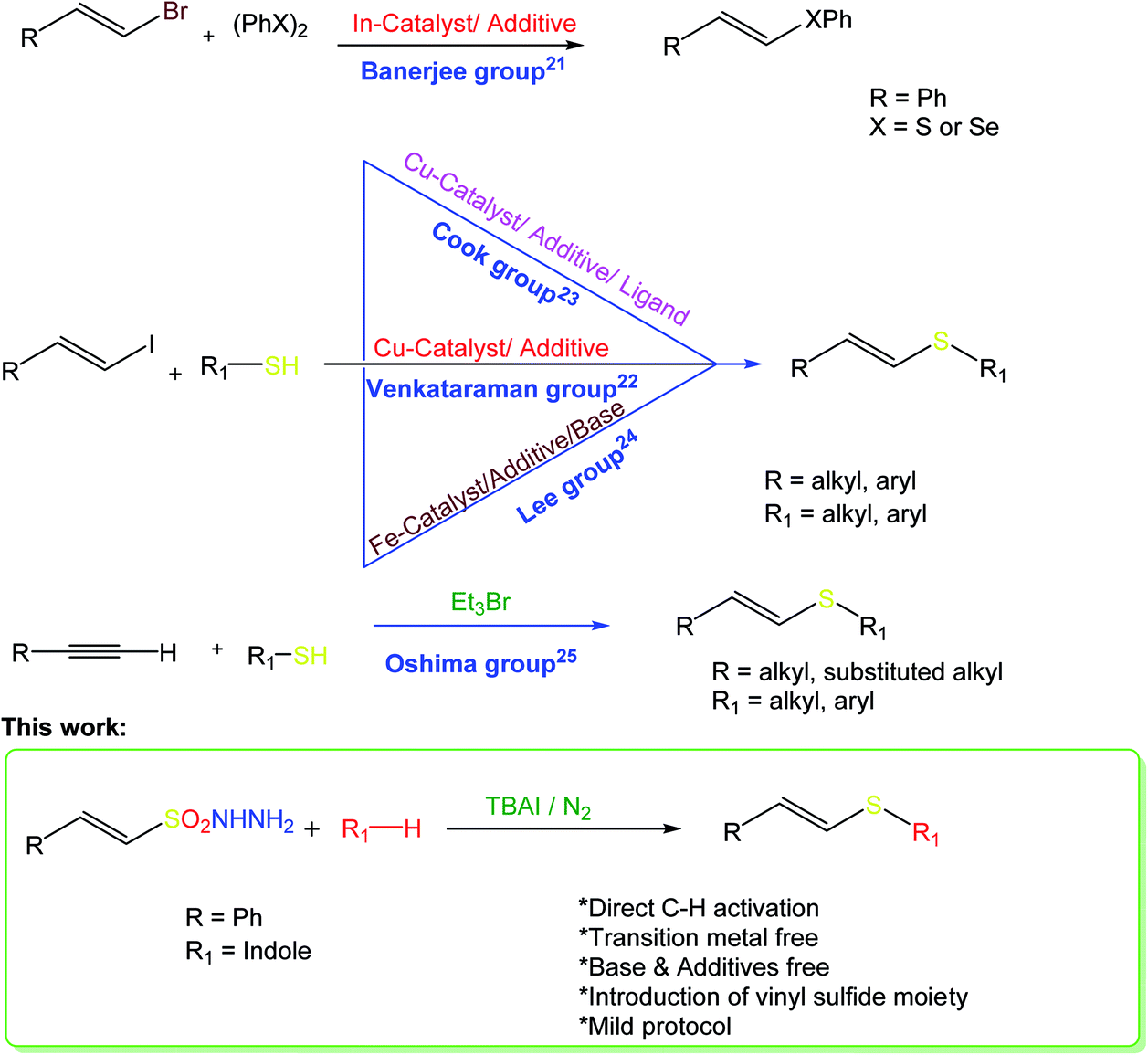 | ||
| Fig. 1 Comparison of synthetic studies of E-vinyl thioethers with the current work. | ||
The indole ring is the key structural unit of many bioactive molecules26,27 and the development of efficient synthetic methods to access differently substituted indoles is a topic of the utmost interest in organic synthesis.28,29 Substituted indoles, particularly thiolated indoles, constitute a significant class of heterocyclic compounds because of their wide range of applications in medicines and biologically active molecules.30–32 Consequently, considerable effort has been made to develop new synthetic routes to indole-based heterocycles.33 3-Thiolated indoles represent one of the most important classes of bioactive molecules used in the treatment of cancer and allergies.34 The various synthetic strategies that have been reported for the 3-arylthiolation of indoles include the use of sulfonyl chlorides,35 sodium sulfinates,36 Bunte salts,37 sulfonyl hydrazides38 and N-hydroxy sulfonamides39 as thiolating agents. The only method to synthesize radio-protective vinyl thioindoles was reported by Skvortsova and co-workers and involves the nucleophilic addition of thioindoles to acetylenes.40 The present work describes the challenging introduction of a vinyl sulfide moiety by simple C–H activation on a variety of substituted indoles using β-(E)-styrene sulfonyl hydrazides (Fig. 2). Sulfonyl hydrazides are readily available, inexpensive, and stable solids that can act as sulfenylation agents.41–43 Consequently, herein, we wish to report the use of β-(E)-styrene sulfonyl hydrazides as unique and direct sulfenylation agents for the synthesis of vinyl thioindoles with excellent stereocontrol under simple reaction conditions.
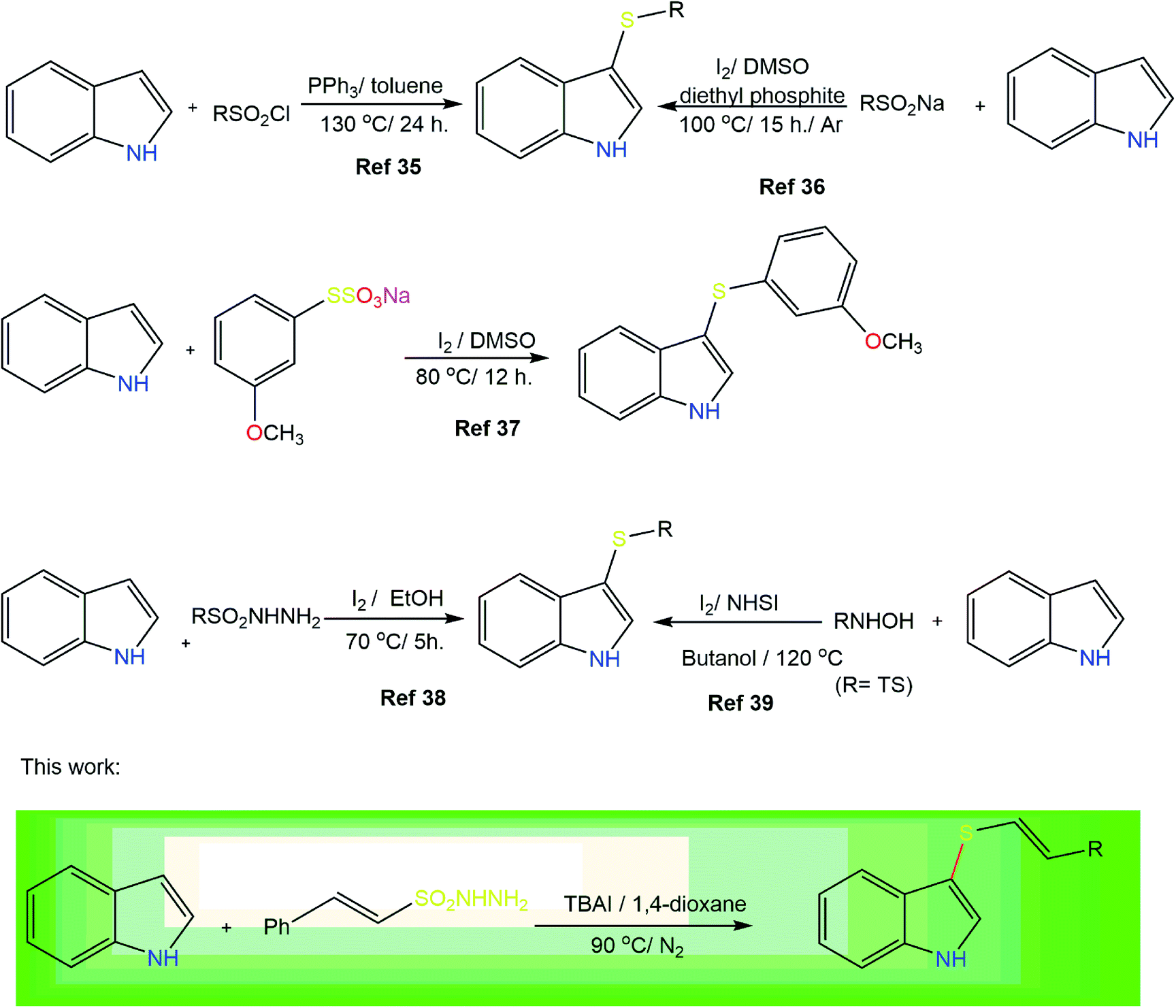 | ||
| Fig. 2 Previous reported work on thioindole synthesis and the current strategy. | ||
2. Results and discussion
2.1 Optimization of the reaction conditions
Scheme 1 outlines the synthetic pathway and conditions adopted for the preparation of the target molecules. We started with the reaction of β-(E)-styrene sulfonyl hydrazide (1a) with indole (2a) in the presence of 10 mol% of iodine at 70 °C in a sealed tube for 12 hours, but the desired product was formed in only 25% yield (Table 1, entry 1). To our delight, the yield was increased to 46% by increasing the temperature to 100 °C (entry 2). Further screening of various solvents afforded the product in moderate yields (entries 3–5) and an improved yield of 65% was obtained using 1,4-dioxane (entry 6). Conducting the reaction under an inert atmosphere to avoid the formation of by-products enhanced the yield to 71% (entry 7). Next, the catalytic effect of various iodic reagents in this transformation was studied (entries 8–13), and the best result was obtained by using TBAI in 1,4-dioxane at 100 °C for 12 hours under an N2 atmosphere (entry 10). Repeating the reaction with a decreased temperature of 90 °C resulted in a slight decrease in the yield of the product (entry 14), and was thus found to be optimum temperature for this conversion. In contrast, further increasing the temperature to 120 °C dropped the yield to 69% (entry 15). A decrease or increase in catalyst loading (5 mol% & 20 mol%) also resulted in a decrease in the yield of the product (entries 16 and 17). | ||
| Scheme 1 Synthetic pathway and optimization of the reaction conditions. | ||
| Entry | Catalyst (mol%) | Solvent (mL) | Temperature (°C) | Time (h) | Yieldb (%) |
|---|---|---|---|---|---|
| a Reaction conditions: 1a (0.48 mmol), 2a (0.4 mmol), catalyst (10 mol%), and solvent (1.5 mL) at the indicated temperature under N2 in a sealed tube.b Isolated yields.c 5 mol% of TBAI was used.d 20 mol% of TBAI was used.e Reaction conducted under an N2 atmosphere. | |||||
| 1a | I2 | EtOH | 70 | 12 | 25 |
| 2a | I2 | EtOH | 100 | 12 | 46 |
| 3a | I2 | DMSO | 100 | 12 | 29 |
| 4a | I2 | DCE | 100 | 12 | 51 |
| 5a | I2 | CH3CN | 100 | 12 | 11 |
| 6a | I2 | 1,4-Dioxane | 100 | 12 | 65 |
| 7e | I2 | 1,4-Dioxane | 100 | 12 | 71 |
| 8e | CuI | 1,4-Dioxane | 100 | 12 | 41 |
| 9e | KI | 1,4-Dioxane | 100 | 12 | 59 |
| 10e | TBAI | 1,4-Dioxane | 100 | 12 | 84 |
| 11e | NIS | 1,4-Dioxane | 100 | 12 | 72 |
| 12e | (CH3)3SOI | 1,4-Dioxane | 100 | 12 | 68 |
| 13e | NaI | 1,4-Dioxane | 100 | 12 | 57 |
| 14e | TBAI | 1,4-Dioxane | 90 | 12 | 86 |
| 15e | TBAI | 1,4-Dioxane | 120 | 12 | 68 |
| 16c | TBAI | 1,4-Dioxane | 100 | 12 | 49 |
| 17d | TBAI | 1,4-Dioxane | 100 | 12 | 63 |
Attempts to decrease in reaction time from 8 to 10 h resulted in lower consumption of the starting materials and thus low yields of the product.
2.2 Scope and limitations of substrates
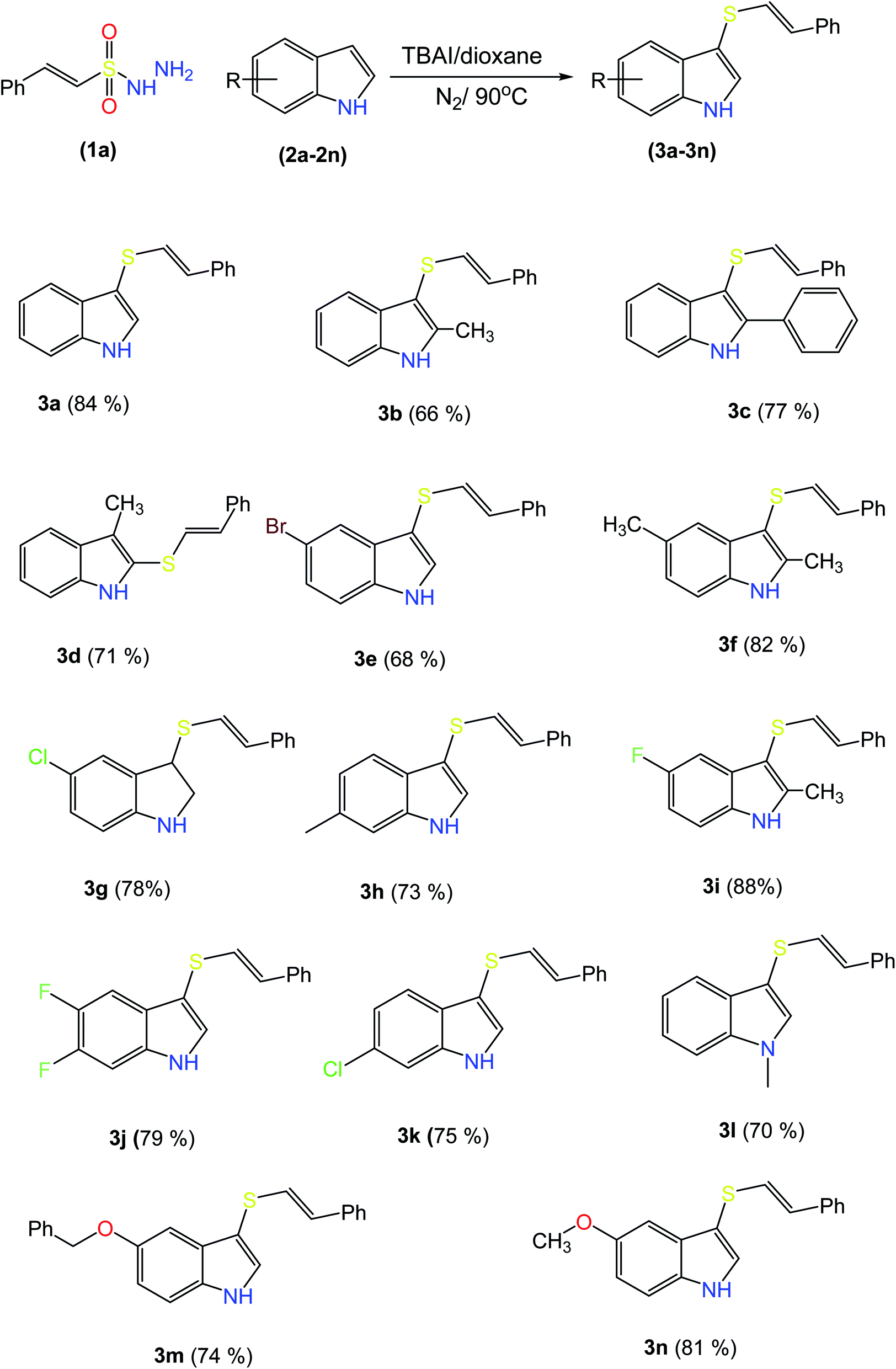 | ||
| Scheme 2 Substrate scope for indoles. a1a (0.48 mmol), 2a (0.4 mmol), TBAI (10 mol%), and 1,4-dioxane (1.5 mL) at 90 °C under an N2 atmosphere for 10 h. | ||
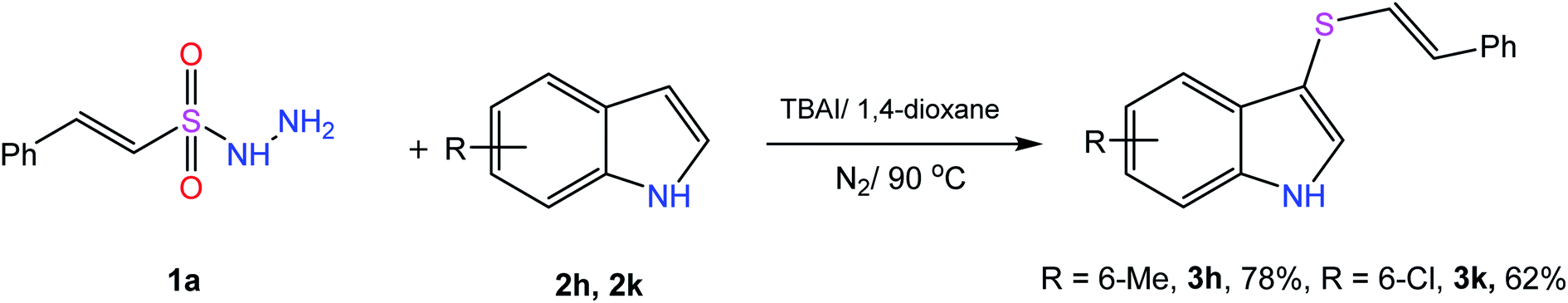
It might be possible to expand this protocol to other activated rings including heteroaromatics by adding some additives and varying the reaction conditions.
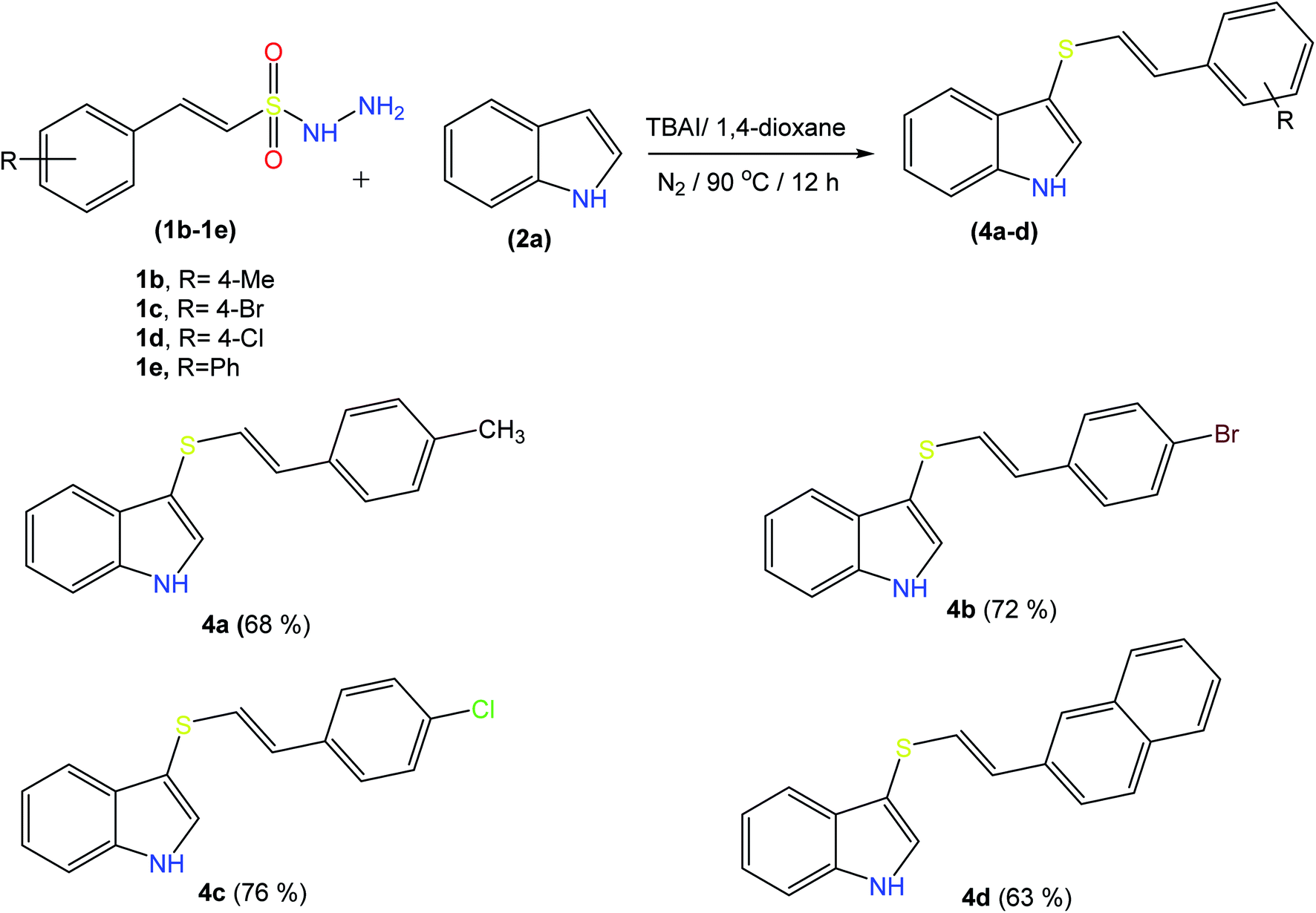 | ||
| Scheme 3 Substrate scope for β-(E)-styrene sulfonyl hydrazides. aReaction conditions: 1a (0.48 mmol), 1b (0.4 mmol), TBAI (10 mol%), and 1,4-dioxane (1.5 mL) at 90 °C under an N2 atmosphere in a sealed tube for 12–14 h. | ||
Examining the 1H-NMR spectra, the broad signal at 8.40 ppm corresponds to the N–H of indole, whilst the sharp singlet at 7.20 ppm is assignable to the C–H of the indole ring; additionally, the two doublets at 6.75 and 6.25 ppm with large coupling constants of 16.0 Hz indicate the presence of mutually coupled protons of the trans double bond, thus confirming the connection of the two partner molecules. The 13C-NMR showed the doubly bonded carbons of the vinyl moiety at 126 and 119 ppm and the C–H of indole at 130 ppm, respectively, whereas the ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–N peak at 138 ppm indicated that the desired transformation had successfully been carried out.
C–N peak at 138 ppm indicated that the desired transformation had successfully been carried out.
2.3 Proposed mechanistic pathway
To elucidate the mechanism of this reaction, we turned our attention to some control experiments. The reaction of indole with β-(E)-styrene sulfonyl hydrazide (1) under the optimal conditions in the presence of the radical inhibitor 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO, 1.2 equiv.) completely inhibited the formation of the desired product, suggesting the involvement of a sulfonyl radical during the reaction (Scheme 4A). The reaction of (1) with indole in the absence of TBAI also results in product formation, but in a very low yield (Scheme 4B). In the absence of indole, a mixture of the products 3s (major) and 3t (minor) was obtained (detected via GC-MS and EI-MS), indicating that (1a) is converted into these species before reacting with indole to give the styryl thioether (Scheme 4C). | ||
| Scheme 4 Control experiments. | ||
In addition, during the course of the reaction, we noticed a change in the color of the reaction mixtures from light brown to purple black, indicating the formation of iodine during this reaction. On the basis of experiment and observations, the following reaction mechanism can be suggested for this radical cross-coupling reaction. Initially, disulfide compound 3s is generated from β-E-styrene sulfonyl hydrazide 1 in the presence of TBAI, and then reacts with iodine to produce PhCHCHSI (II). Nucleophilic attack of the indole on III produces radical intermediate IV and an iodine radical. Extrusion of HI leads to the formation of the final product, thioether 3. The produced HI reacts with TBAOH to remove a water molecule, and the regenerated TBAI can react with sulfonyl hydrazide to produce disulfide again (Scheme 5).
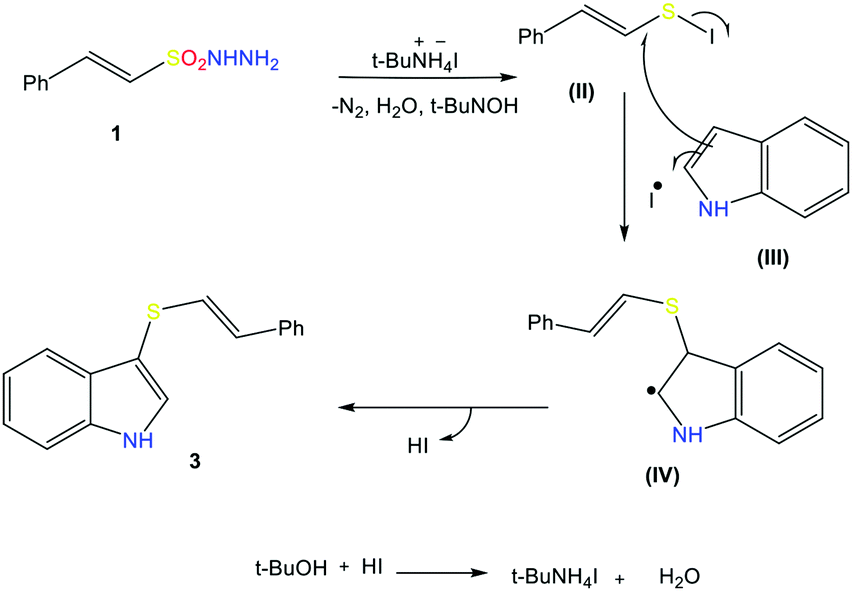 | ||
| Scheme 5 Proposed mechanistic pathway. | ||
The proposed mechanism is based on reports in the literature; the free-radical pathway was confirmed by the control experiment (A) and the formation of the major intermediate 3s was confirmed by EI-MS analysis (ESI S-48†).
3. Experimental section
3.1 Materials and instruments
Indole and its derivatives were purchased from Sinopharm Chemical Reagent Co., Meryer, Acros, Alfa Aesar, and TCI, and were used without further purification. β-(E)-Styrene sulfonyl chlorides and their derivatives were prepared according to literature procedures.9 Melting points were determined in open glass capillaries using a Stuart SMP3 (UK) melting point apparatus and are uncorrected.1H and 13C NMR spectra were recorded using a Bruker AC-400 FT spectrometer (400 MHz and 100 MHz, respectively) using TMS as an internal standard. NMR multiplicities are abbreviated as follows: s = singlet, d = doublet, dd = double doublet, t = triplet, m = multiplet, q = quartet, br = broad signal. Chemical shifts (δ) were recorded using an LC-TOF spectrometer (Micromass). Electron spray ionization (ESI) mass spectrometry data are expressed in ppm, while coupling constants (J) are expressed in Hz. High resolution mass spectra (HRMS) were obtained using a Thermo LTQ Orbitrap XL instrument equipped with an ESI source and controlled by the software Xcalibur. Elemental analyses were conducted using a LECO-183 CHNS analyzer.
3.2 General procedure for the preparation of β-(E)-styrene sulfonyl hydrazides
Hydrazine monohydrate (50 mg, 1 mmol, 2.5 equiv.) was added dropwise to a solution of β-(E)-styrene sulfonyl chloride (0.4 mmol, 1 equiv.) in THF (2 mL) at 0 °C under an N2 atmosphere. The mixture became brown due to the formation of a white precipitate of hydrazine hydrochloride during the addition of hydrazine. The reaction mixture was stirred at 0 °C for 30 to 45 minutes, diluted with ethyl acetate (10 mL), washed with brine (2 × 10 mL), dried over sodium sulfate, and filtered. The filtrate was slowly added to stirred hexane (10 mL) over 5 min. After being stirred for 10 min, the mixture was filtered and then dried under vacuum to obtain the β-(E)-styrene sulfonyl hydrazides as a brown solid.3.3 General procedure for the TBAI-assisted direct C–H activation of indoles with β-(E)-styrene sulfonyl hydrazides
To a mixture of β-(E)-styrene sulfonyl hydrazide 1a (0.48 mmol, 1.2 equiv.), indole 2a (0.4 mmol, 1 equiv.) and TBAI (18.5 mg, 0.05 mmol, 10 mol%) was added 1,4-dioxane (1.5 mL) under an N2 atmosphere in a sealed tube. The resulting mixture was stirred at 100 °C for 12 h and cooled to room temperature, and ethyl acetate (10 mL) was added. The mixture was washed with brine (2 × 10 mL), dried over Na2SO4, and purified using silica gel chromatography eluted with petroleum ether/ethyl acetate (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 1:1) to give the indole styryl thioether 3a.
:1 to 1:1) to give the indole styryl thioether 3a.
4. Conclusion
In conclusion, we have demonstrated an easy and efficient method for the synthesis of various substituted (E)-styryl thioindoles by a TBAI-catalyzed sulfenylation reaction with β-(E)-styrene sulfonyl hydrazides via a radical cross-coupling reaction in moderate to high yields with excellent stereoselectivity. The substituted β-(E)-styrene sulfonyl hydrazide precursors are easily accessible from simple and inexpensive starting materials. The current study paves the way for the use of β-(E)-styrene sulfonyl hydrazides as a unique styryl mercaptan source in chemical synthesis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SH gratefully acknowledges Chinese academy of Sciences and the word Academy of Science CAS-TWAS scholarship for financial support.References
- R. J. Cremlyn, An Introduction to Organosulfur Chemistry, John Wiley & Sons, Chichester, 1996 Search PubMed
.
- R. D. Miller and R. Hassig, Tetrahedron Lett., 1985, 26, 2395 CrossRef CAS
- H. Yamaguchi and Y. Minoura, J. Appl. Polym. Sci., 2003, 15, 1869 CrossRef
- F. Yamashita, H. Kuniyasu, J. Terao and N. Kambe, Org. Lett., 2008, 10, 101 CrossRef CAS PubMed
- A. Sabarre and J. Love, Org. Lett., 2008, 10, 3941 CrossRef CAS PubMed
- C. Cao, L. R. Fraser and J. A. Love, J. Am. Chem. Soc., 2005, 127, 17614 CrossRef CAS
- A. Kondoh, H. Yorimitsu and K. Oshima, Org. Lett., 2007, 9, 1383 CrossRef CAS PubMed
- M. Cai, Y. Wang and W. Hao, Green Chem., 2007, 9, 1180 RSC
- M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368 CrossRef CAS PubMed
- C. J. Weiss and T. J. Marks, J. Am. Chem. Soc., 2010, 132, 10533 CrossRef CAS PubMed
- J. Santandrea, C. Minozzi, C. Cruché and S. K. Collins, Angew. Chem., Int. Ed., 2017, 56, 12255–12259 (Angew. Chem., Int. Ed., 2017, 129, 12423–12427) CrossRef CAS PubMed
- J. Doroszuk, M. Musiejuk, S. Demkowicz, J. Rachon and D. Witt, RSC Adv., 2016, 6, 105449–105453 RSC
- G. Zeni, M. P. Stracke, C. W. Nogueira, A. L. Braga, P. H. Menezes and H. A. Stefani, Org. Lett., 2004, 6, 1135 CrossRef CAS PubMed
- L. Benati, L. Capella, P. C. Montevecchi and P. Spagnolo, J. Org. Chem., 1994, 59, 2818 CrossRef CAS
- Z. Duan, S. Ranjit, P. Zhang and X. Liu, Org. Lett., 2010, 12, 2430 CrossRef CAS PubMed
- V. Aucagne, A. Tatibouet and P. Rollin, Tetrahedron, 2004, 60, 1817 CrossRef CAS
- E. Stephan, A. Olaru and G. Jaouen, Tetrahedron Lett., 1999, 40, 8571 CrossRef CAS
- M. Mikolajczyk, S. Grzejszczak, W. Midura and A. Zatorski, Synthesis, 1975, 278 CrossRef CAS
- H. J. Cristau, B. Chabaud, R. Labaudiniere and H. Christol, J. Org. Chem., 1986, 51, 875 CrossRef CAS
- T. Ogawa, K. Hayami and H. Suzuki, Chem. Lett., 1989, 769 CrossRef CAS
- B. C. Ranu, K. Chattopadhyay and S. Banerjee, J. Org. Chem., 2006, 71, 423 CrossRef CAS
- C. G. Bates, P. Saejueng, M. Q. Doherty and D. Venkataraman, Org. Lett., 2004, 6, 5005 CrossRef CAS PubMed
- M. S. Kabir, M. L. V. Linn, A. Monte and J. M. Cook, Org. Lett., 2008, 10, 3363 CrossRef CAS PubMed
- Y.-Y. Lin, Y.-J. Wang, C.-H. Lin, J.-H. Cheng and C.-F. Lee, J. Org. Chem., 2012, 77, 6100–6106 CrossRef CAS PubMed
- Y. Ichinose, K. Wakamatsu, K. Nozaki, J.-L. Birbaum, K. Oshima and K. Utimoto, Chem. Lett., 1987, 1647 CrossRef CAS
- A. Saeed, P. A. Channar, Q. Iqbal and J. Mahar, Chin. Chem. Lett., 2016, 27, 37–40 CrossRef CAS
- W. Shi, S. L. Marcus and T. L. Lowary, Bioorg. Med. Chem., 2011, 19, 603 CrossRef CAS PubMed
- S. Cacchi, G. Fabrizi and A. Goggiamani, Org. Biomol. Chem., 2011, 9, 641 RSC
- A. R. Katritzky, C. A. Ramsden, J. A. Joule and V. V. Zhdankin, Handbook of Heterocyclic Chemistry, Elsevier, Oxford, 3rd edn, 2010 Search PubMed
- B. Bao, Q. Sun and X. Yao, et al., J. Nat. Prod., 2005, 68, 711–715 CrossRef CAS PubMed
- G. De Martino, G. La Regina and A. Coluccia, et al., J. Med. Chem., 2004, 47, 6120–6123 CrossRef CAS PubMed
- G. La Regina, M. C. Edler and A. Brancale, et al., J Med Chem, 2007, 50, 2865–2874 CrossRef CAS PubMed
- Y. Xi, B. Dong and E. J. McClain, et al., Angew. Chem., Int. Ed., 2014, 53, 4657–4661 CrossRef CAS PubMed
- P. C. Unangst, D. T. Connor and S. R. Stabler, et al., J Med Chem, 1989, 32, 1360–1366 CrossRef CAS PubMed
- Q. Wu, D. Zhao, X. Qin, J. Lan and J. You, Chem. Commun., 2011, 47, 9188–9190 RSC
- F. Xiao, H. Xie, S. Liu and G.-J. Denga, Adv. Synth. Catal., 2014, 356, 364–368 CrossRef CAS
- H. Qi, T. Zhang, K. Wan and M. Luo, J. Org. Chem., 2016, 81, 4262–4268 CrossRef CAS PubMed
- F.-L. Yang and S.-K. Tian, Angew. Chem., Int. Ed., 2013, 52, 4929–4932 CrossRef CAS
- F.-X. Wang, S.-D. Zhou, C. Wang and S.-K. Tian, Org. Biomol. Chem., 2017, 15, 5284–5288 RSC
- G. G. Skvortsova, B. V. Trzhtsinskaya and L. F. Teterina, et al., Khim.-Farm. Zh., 1986, 20, 51 CAS
- R. Rahaman, N. Devi, K. Sarma and P. Barman, RSC Adv., 2016, 6, 10873 RSC
- Y. Yang, S. Zhang, L. Tang, Y. Hu, Z. Zha and Z. Wang, Green Chem., 2016, 18, 2609–2613 RSC
- D. S. Raghuyanshi and N. Verma, RSC Adv., 2017, 7, 22860 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra02444b |
| This journal is © The Royal Society of Chemistry 2021 |