DOI:
10.1039/D1RA01444G
(Review Article)
RSC Adv., 2021,
11, 15639-15655
Control of hydrogen release during borohydride electrooxidation with porous carbon materials
Received
22nd February 2021
, Accepted 22nd April 2021
First published on 27th April 2021
Abstract
Due to their highly tunable electrical and structural properties, carbon materials are widely used in fuel cells. This study reviews the latest modifications carried out in order to improve the electrochemical properties of carbon-based anodes in Direct Borohydride Fuel Cell (DBFC). However, in this type of fuel cell, various types of carbon (e.g. carbon black, activated carbons, carbon nanotubes, graphene and heteroatom-doped carbons and MOF-derived carbon materials) can provide not only catalyst support, but also hydrogen storage due to the extremely complex process of borohydride electrooxidation. Accurate control of porosity and carbon morphology is therefore necessary for high fuel cell efficiency. Finally, some prospects for the future development of carbon materials for DBFC design are presented. It should be emphasized, that the storage of hydrogen in solid form is a possible breakthrough for the future use of hydrogen as an ecological fuel, which is why scientific research in this topic is so important.
1. Introduction
The primary goal for the modern energy sector should be security of electricity supply, its efficiency and sustainable development based on renewable sources. It should be emphasized that achieving this effect requires not only global environmental protection, but also solidarity with rich and poor countries as well as future generations in the context of economic, social and political factors.1 Although fossil fuels have allowed a spectacular civilization progress, they are also accompanied by negative effects such as climate change and air pollution. Current technological advances in the energy sector offer the prospect of maintaining as well as improving the quality of life without adversely affecting the health of people and the planet.2 The analyses predict a significant share of photovoltaic and wind energy in terms of modern energy.3,4 The existing methods used to produce and utilize energy are not sustainable. Ensuring the energy needs of present and future generations is one of the most difficult political challenges. Competition for fossil fuel resources can be a source of international unease and potential conflicts. Although solar and wind energy can potentially provide an unlimited amount of energy in an efficient manner, this approach is limited by the ability of the electricity grid to supply it.5–7 The grid infrastructure can only manage a limited number of these variable sources. It is very important to ensure that grids maintain electrical voltage and steady frequency to avoid power surges. They also require the ability to meet peak demand. In a situation where supply exceeds the demand, the excess of produced energy could be used to charge car batteries or produce hydrogen fuel.8,9 Hydrogen fuel presents a number of benefits. If hydrogen was generated from renewable sources, it would also act as a carrier of renewable energy, i.e. store electricity generated by energy sources fed from the grid during the overproduction period.10 It is well known that hydrogen-based transport could contribute to climate mitigation. As shown in Fig. 1, there are predictions that in the mid-2040s hydrogen will satisfy one tenth of the global demand for transport energy.11
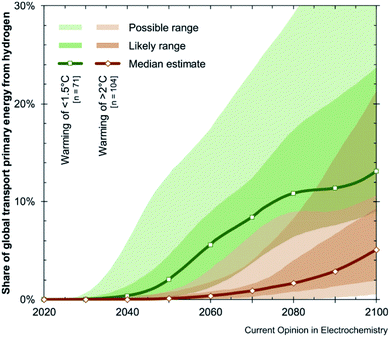 |
| | Fig. 1 Participation in global transport provided by hydrogen. This figure has been adapted from ref. 11 with permission from ELSEVIER, copyright 2019. | |
2. Hydrogen management
Fuel cells (FCs) are electrochemical cells that generate electricity from chemical energy and are often used as alternative energy sources. FCs are associated with modernity and innovation, while in fact these devices have almost 200 years of history. In 1800, two researchers independently proved that water could be decomposed into hydrogen and oxygen using electricity. For this reason, scientists became interested in the reverse process that would enable energy production.12 In 1838 this concept was presented by Christian Friedrich Schönbein, while a year later Sir William Robert Grove became the first inventor of the “gas battery”, now called the fuel cell. L. Mond and C. Langer in the following years improved the Grove's cell using coal as a source of hydrogen. It was believed that the 20th century would be the “Era of Electrochemical Combustion”. Unfortunately, this plan could not be implemented even in the 21st century (Fig. 2).13
 |
| | Fig. 2 Christian Friedrich Schönbein (1799–1868) (A), Sir William Robert Grove (1811–1896) (B), “Gas battery” presentation (C). This figure has been adapted from ref. 12 with permission from WILEY-VCH, copyright 2021. | |
Hydrogen fuel cells use pure hydrogen as fuel, and most importantly, only water is waste.14 Sometimes they are compared to batteries, but due to the fact that FCs require continuous fuel supplies and are not able to store energy, such a comparison is incorrect.15 Under ambient conditions, 1 kg of hydrogen gas equals approx. 11 m3.16 It is therefore important to develop cost-effective storage methods. Storage of hydrogen in high-pressure composite containers is the most common technique.17 They can withstand pressures up to 80 MPa, achieving a hydrogen density of approx. 36 kg m−3.18 Hydrogen can also be stored in the liquid form at −252 °C at ambient pressure. Conversion of hydrogen from a gaseous to a liquid state significantly increases the hydrogen energy density.19 However, liquefaction is much more expensive than compression, because it involves significant energy inputs. The most common liquefaction cycle is the Joule–Thomson cycle.20 Cryoadsorption on high surface area (HSA) materials may be another potential reservoir of hydrogen.21 Porous carbon materials offer a wide range of gas storage options due to the variety of forms and adjustable surface functionality.22 Hydrogen storage in carbon materials is carried out by means of physical sorption (adsorption) on the surface of solids occurring mainly as a result of the influence of Van der Waals capillary effect.23 It would be ideal if the pore diameter was below 1 nm.24 Recently, the maximum measured hydrogen storage capacity (HSC), achieved at −196 °C and 1.2 MPa by activated carbon (AC) with a specific surface area of 3969 m2 g−1, was equal to 7.7 wt%.25 The notable advantages of this method include low operating pressure and relatively small cost of materials. On the other hand, the low volumetric and gravimetric density of hydrogen on carbon materials as well as low temperatures are significant disadvantages.26 Metal hydrides are also interesting materials for hydrogen storage due to such properties as: low process pressure (usually 0.25–10 MPa), reversibility of the hydrogen storage process, low process temperature (hydrogen absorption/desorption can occur even at room temperature) and safety of use (no risk associated with the explosion and flammability of the system).27 In this case, hydrogen atoms occupy interstitial sites in host lattices. Metal hydrides can achieve high volumetric hydrogen density e.g. 115 kg m−3 for LaNi5. However, due to the high atomic weight of transition metals, the gravimetric density of hydrogen is limited to 3 wt%.28 The use of complex hydrides, including borohydrides, is another way to store hydrogen. The amount of hydrogen accumulated in this way is greater than in metal hydrides. An example is LiBH4, which is characterized by the highest gravimetric hydrogen density (18 wt%) at room temperature29 but the most commonly used compound is sodium borohydride. NaBH4 decomposes slowly at temperatures above 673 K, therefore the thermal activation process would be economically inefficient. Interestingly, sodium borohydride is able to release hydrogen by reaction with water, as described in eqn (1):30
| | |
NaBH4 + 2H2O → NaBO2 + 4H2
| (1) |
In 1953, Pecsok first proposed the borohydride oxidation reaction (BOR) according to eqn (2). Over time, NaBH4 was used not only as a safe hydrogen carrier, but also for energy production:31
| | |
BH4− + 8OH− → BO2− + 6H2O + 8e−
| (2) |
It is believed that the first Direct Borohydride Fuel Cell (DBFC) concept was introduced by Indig and Snyder in 1960. Environmental problems as well as the oil crisis in 1973 led to a renewed interest in NaBH4 as an energy/hydrogen carrier.32 In 2000, Amendola et al.33 described the use of this ultra-safe generator for hydrogen production, and in 2001, Daimler–Chrysler presented the Chrysler Natrium prototype powered by a fuel cell using hydrogen produced from a sodium borohydride reformer.34 Although DBFC is more attractive in terms of size and fuel consumption than Direct Methanol Fuel Cell (DMFC), its operating costs are still more expensive. Additionally, DBFC has some serious issues that need to be overcome in order to achieve the theoretical assumptions.35
3. Borohydride electrooxidation
Indirect Borohydride Fuel Cells (IBFCs) utilize metal borohydride as hydrogen carrier, which means that the main goal is to increase the intensification of borohydride hydrolysis to produce as much gas as possible. The hydrogen gas produced as a result of this reaction is then collected and stored for use as fuel.36 On the other hand, Direct Borohydride Fuel Cell (DBFC) can be constantly powered by an alkaline borohydride solution as anodic fuel, because borohydrides exhibit high stability in a strong alkaline aqueous solution. Interestingly, direct borohydride oxidation provides a much lower anode potential, compared to the hydrogen oxidation reaction, which determines the higher cell voltage.37 Due to the low operating temperature and easily recyclable non-toxic products, DBFC attracts much attention.38 In practice, it is impossible to achieve a cell voltage of 1.64 V, not only because of the slow kinetics on most BOR catalysts, but also because the oxygen reduction reaction (ORR) on the cathode is very sluggish (as in PEMFC). Moreover, many other factors, such as reagents concentration, pH of electrolyte as well as competitive reactions affect the reduced performance.39 In addition to direct borohydride electrooxidation, borohydride hydrolysis and hydrogen electrooxidation also occur, resulting in a mixed BOR potential.40 Furthermore, the competition of these reactions leads to the formation of many intermediate boron species.41,42 In situ techniques, such as Fourier transformed infrared (FTIR)43 analysis, rotating ring-disc electrode (RRDE)44 and on-line electrochemical mass spectrometry (OLEMS)45 have enabled the identification of BH3-based species and molecular hydrogen, as BOR intermediates on a gold electrode, according to eqn (3)–(5):| | |
BH4− + s + OH− → BH3,ads + H2O + 2e−
| (3) |
| | |
BH3,ads + OH− → BH3(OH)ads−
| (4) |
| | |
BH3(OH)ads− + 6OH− → BO2− + 5H2O + 6e−
| (5) |
The main problem of using borohydride is its hydrolysis to hydrogen gas. The release of hydrogen not only reduces the effectiveness of borohydride, but also causes the slug flow in anode channel, which increases the resistance of liquid reagent transport from the channel to active sites (Fig. 3a). Moreover, as shown in Fig. 3b, hydrogen bubbles create a “dead zone” between the membrane and the anode during utilization. Despite continuous operation of the fuel pump, bubbles are not released. Consequently, entrapped hydrogen bubbles act as resistance to charge carrier migration. The solution to this problem can be a wavy anode project, which provides a suitable gap between the anode and the membrane for the desired removal of hydrogen bubbles (Fig. 3c).46
 |
| | Fig. 3 Anode channel when hydrogen is generated (a); anode with “dead zones” marked (b); corrugated anode (c). This figure has been adapted from ref. 46 with permission from ELSEVIER, copyright 2017. | |
4. Role of carbon materials in DBFC
The current cost of FCs is a major obstacle to their commercialization and use in automotive applications. Due to this reason, it is so important to find inexpensive anode electrocatalysts, which will promote BH4− electrooxidation, while inhibiting the undesirable hydrolysis reaction. Antolini reported that the catalyst activity significantly depends on its reaction surface area.47 Generally, reducing the size of catalyst particles improves their activity. However, it should be remembered, that there is a so-called “particle size effect”. It means that metal nanoparticles activity can also decline with reduced particle size.48 With regard to DBFC, this effect was investigated only by Olu et al.49 and there is little evidence that particle size effect plays an important role (instead, inter-particle distance has an effect). Accordingly, catalysts are deposited on a HSA support. It is important to ensure that this substrate has a high electrical conductivity in order to allow the electrons to flow, as well as suitable porosity to facilitate the flow of reagents. It should be emphasized, that in addition to the dispersion effect of the support material, there is also a strong interaction between the support material and the catalysts, which includes the modification of electronic and spatial properties of the catalyst particles.50 Yang et al.51 deposited Pd on various carbon substrates (carbon black, multi-walled carbon nanotubes, activated carbon) in order to study the influence of the electrocatalyst carrier on its catalytic activity. Using XRD and HRTEM analyzes, the authors noticed that for Pd/AC sample, the catalyst particles tend to aggregate causing a heterogeneous dispersion, unlike Pd/MWCNTS and Pd/Vulcan XC-72. Also, Hosseini and Mahmoodi52 presented significant evidence for the existence of strong metal-carbon nanotubes interactions due to the presence of the π-electron system. The authors claim that such electronic interaction improves not only the catalytic activity of electrocatalysts, but also their utilization and stability. Carbon materials, due to their high electrical and thermal conductivity as well as their porous structure seem to be excellent candidates for catalyst support in fuel cells.53 Moreover, in DBFC, a porous anode could additionally determine the release of unconsumed gaseous hydrogen that blocks catalytic sites.54 During the borohydride electrooxidation, the catalyst would be separated from the anolyte by gas bubbles in the pores, acting as a physical trap for hydrogen. In the next step, due to the consumption of hydrogen during electrooxidation, the pores would be refilled with anolyte and the BOR reaction would take place again (Fig. 4).55
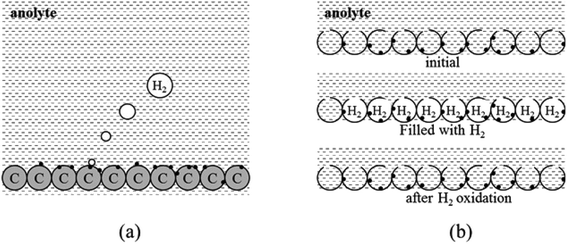 |
| | Fig. 4 Hydrogen release during borohydride electrooxidation on an anode supported with solid carbon (a) and porous carbon (b). This figure has been adapted from ref. 55 with permission from WILEY-VCH, copyright 2015. | |
This article focuses on reviewing various carbon materials as substrates for DBFC anode catalyst to provide better insight into factors affecting catalyst performance as well as hydrogen evolution.
4.1 Support for catalyst
4.1.1 Carbon black. Carbon blacks (CB) are spheroidal particles with a pronounced arrangement of graphitic layers.56 This type of materials is usually produced by the pyrolysis of hydrocarbons.57 Due to their high conductivity and availability as well as low cost, commercially available carbon blacks (e.g. Black Pearls 2000, Vulcan XC-72) are very popular carriers of metal catalysts in low-temperature FCs.58 Kim et al.59 studied the effect of catalyst loading, by comparing unsupported Pt (5.92 mg cm−2) and carbon-supported Pt catalyst (1.50 mg cm−2). According to the authors, modification of anode microstructure determines the fuel utilization. The addition of carbon reduced the average Pt particle size from 8.0 to 4.7 nm. Interestingly, the performance of both catalysts was comparable, which allowed to reduce the cost of anode production. Recently, Lafforgue et al.60 tested four carbon-supported Pd electrocatalysts with different amounts of Pd (22–53%). The authors noted no significant differences in the size of Pd crystallites (all were equal to approx. 5 nm). However, as the metal fraction increased, the kinetics of borohydride oxidation reaction increased. Interestingly, this trend was only relevant for low NaBH4 concentrations (50 mM NaBH4 + 1 M NaOH). Olu et al.49 presented very interesting reports about the influence of Pt particles deposited on a smooth and volumic surface, indicating the superiority of the latter. Compared to glassy carbon, thick active layers ensure a longer residence time of BOR intermediates near certain sites of Pt, which affects the completion of the reaction, and thus increases the faradic efficiency. According to the authors, the concern related to the heterogeneous supply of electrolyte to internal activity of the electroactive material could be easily solved by controlling mass transport. Li et al.55 decided to synthesize carbon materials with different pore size using the template method and compare them with commercial Vulcan XC-72. Pt supported on carbon aerogel with an average pore size of 6.5 nm improved the coulombic efficiency but deteriorated the DBFC performance (Fig. 5a). Macroporous carbon with an average pore size (APS) of 11.2 nm exhibited significantly better properties. As mentioned before, small pores in carbon material enable the capture of hydrogen bubbles which limit H2 escape during borohydride electrooxidation. On the other hand, hydrogen entrapped in pores can decline the charge carrier transport resulting in increased anode polarization (Fig. 5b).55
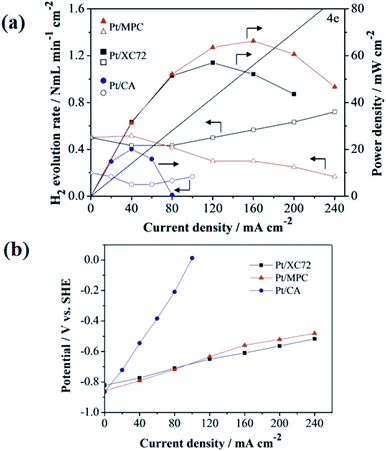 |
| | Fig. 5 The rate of hydrogen evolution and power density response (a); anode polarization curves in borohydride solution (b). This figure has been adapted from ref. 55 with permission from WILEY-VCH, copyright 2015. | |
Nevertheless, it should be emphasized, that a large number of micropores in a carbon support can expand the active area of catalyst. Bai et al.61 noted that as the Pt loading increases, the BET surface area decreases. They explained this phenomenon by Pt intercalation in Vulcan XC-72 micropores. Olu et al.62 pointed out that the texture of the anode also strongly affects the DBFC performance. Pt pasted on carbon cloth (CC) provided a cracked electrode morphology and therefore easy mass transfer of reagents to active sites, while Pt sprayed on a flat Nafion® membrane significantly prevented anolyte circulation (Fig. 6). Additionally, it is concluded that the cracked surface offers better management of hydrogen, due to the more open texture of active anode layer.62
 |
| | Fig. 6 SEM images of Pt pasted on CC (A and B); and Pt sprayed on Nafion® surface (C and D). This figure has been adapted from ref. 62 with permission from ELSEVIER, copyright 2015. | |
4.1.2 Activated carbons. It is well known, that all organic substances consisting of elemental carbon can be used to produce activated carbons (AC).63 Natural residues due to the pyrolysis in high temperature conditions (500–1000 °C) and an inert atmosphere, can be easily transformed into carbonaceous materials. In addition, depending on specific needs, carbonaceous materials can be modified by means of physical or chemical activation.64,65 Physical activation relies on the use of steam or CO2 at the same temperatures as for pyrolysis. This process is characterized by high weight loss of raw material and low microporosity of AC (Fig. 7a).66 Chemical activation allows to get activated carbons with a larger specific surface area (SSA) and porosity at relatively lower conversion temperatures. The most popular activating agent is KOH. The activation mechanism is based on the chemical reaction of potassium compounds with carbon, followed by several solid–liquid reactions, leading to an expansion of carbon network, as shown in Fig. 7b. On the other hand, it is observed that this process reduces the conductivity and density of carbon electrodes and there is a need to remove residual reagents.67
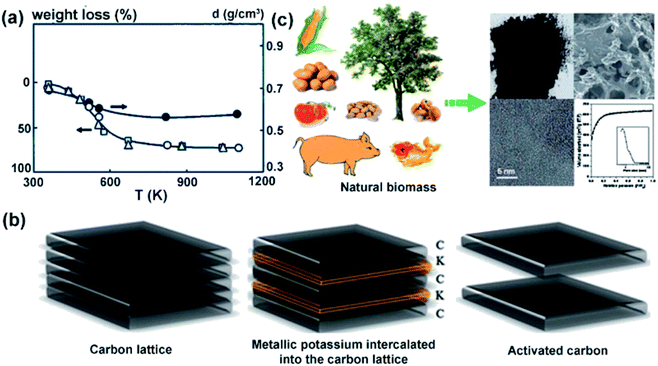 |
| | Fig. 7 Change in weight loss for (Δ) almond shells, (□) olive stones and (○) peach stones during pyrolysis (a); activation mechanism using KOH as activating agent (b); biomass as a source of ACs (c). This figure has been adapted from ref. 67 with permission from TAYLOR & FRANCIS, copyright 2017. | |
Bamboo species,68 waste tea leaves,69 nut shells,70 grape seeds,71 coffee grounds72 or banana peels73 aroused great interest because of its high availability and low cost. Numerous reports68–82 focused on the impact of activation conditions on the microstructure and electrochemical properties of ACs. The diversity of waste products allows to obtain different structures and properties of activated carbons (Table 1).
Table 1 Physical properties of ACs
| Waste products |
Agent |
Temp. (°C) |
SSA (m2 g−1) |
Vtotal (cm3 g−1) |
Vmicropore (cm3 g−1) |
Vmesopore (cm3 g−1) |
Saverage (nm) |
Ref. |
| Bamboo species |
KOH |
700 |
2555 |
1.25 |
0.94 |
— |
1.8 |
68 |
| Grape seeds |
KOH |
600 |
654 |
— |
0.19 |
0.051 |
1.8 |
71 |
| 800 |
1222 |
— |
0.47 |
0.050 |
1.7 |
| K2CO3 |
600 |
33 |
— |
0.11 |
0.0015 |
2.4 |
| 800 |
918 |
— |
0.35 |
0.0254 |
1.6 |
| Apricot shell |
NaOH |
800 |
1342 |
0.559 |
0.496 |
0.063 |
— |
74 |
| Fungi |
KOH |
650 |
2188 |
1.04 |
0.88 |
— |
0.83 |
75 |
| 750 |
2526 |
1.56 |
0.91 |
— |
0.89 |
| Pine cone |
KOH |
750 |
3950 |
2.395 |
1.72 |
— |
2.90 |
76 |
| Coffee grounds |
ZnCl2 |
900 |
1021 |
1.30 |
0.35 |
0.95 |
— |
77 |
| 550 |
1522 |
0.60 |
0.75 |
— |
0.90 |
78 |
| FeCl3 |
280 |
965 |
0.40 |
0.51 |
— |
0.63 |
| Oil palm stone |
CO2 |
650 |
441 |
0.27 |
0.15 |
— |
— |
79 |
| 850 |
1375 |
0.64 |
0.39 |
— |
— |
| Orange peel |
H3PO4 |
700 |
2210 |
1.24 |
1.18 |
— |
— |
80 |
| ZnCl2 |
700 |
1802 |
0.88 |
0.87 |
— |
— |
| KOH |
700 |
1355 |
0.81 |
0.64 |
— |
— |
| Fish scale |
KOH |
— |
2273 |
2.74 |
— |
— |
4.47 |
81 |
| Waste tires |
H3PO4 |
900 |
563 |
0.201 |
0.167 |
0.034 |
0.57 |
82 |
Martins et al.83 obtained good results testing bio-based carbon-supported Pd electrocatalysts towards BOR. Activated carbons were obtained with a SSA of 1411 m2 g−1 and 1689 m2 g−1 using grape stalk and vine shoots, respectively. A larger electrochemical surface area compared to commercial Vulcan XC-72 provided higher current densities and greater number of exchanged electrons (n = 5.6). In turn, Ari et al.84 synthesized carbon materials based on car tire rubbers using modifiers (ethylenediamine – EDA, diethylenetriamine – DETA, triethylenetetramine – TETA, polyethyleneimine – PEI) containing a different number of amine groups (Fig. 8A–C). In general, chemical modification reduced the surface area of this sustainable and cost-effective carbon material. As shown in Fig. 8D, the ability to catalytically produce hydrogen increased as the number of amino groups increased. This specific surface functionality caused the release of the same amount of hydrogen three times faster (at 100% conversion),84 which would mean that the efficiency of borohydride oxidation in DBFC was reduced.
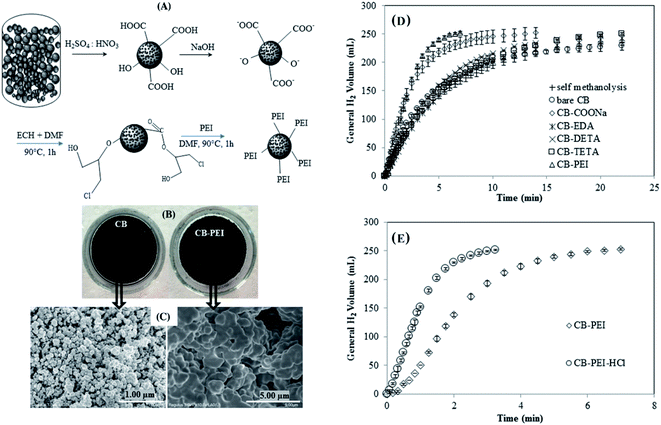 |
| | Fig. 8 The process of car tire rubber (CB) modification with PEI (A); images from a digital camera and scanning electron microscopy (B and C); hydrogen release rate from NaBH4 alcoholysis of unmodified and modified carbons (D and E). This figure has been adapted from ref. 84 with permission from WILEY-VCH, copyright 2019. | |
4.1.3 Carbon nanotubes. Since the first report by Sumio Iijima in 1991,85 there has been a sharp increase in the interest in these fascinating forms of carbon.86 Carbon nanotubes (CNTs) are rolled sheets of graphene with higher conductivity than activated carbons, low mass density and high chemical and electrochemical stability.87 Tubes formed from only one graphite layer are called single-walled carbon nanotubes (SWCNTs), while tubes consisting of more than one layer are called multi-walled carbon nanotubes (MWCNTs). Although, almost 30 years have passed since the report of Iijima was published, the development of cheap and selective methods of CNTs synthesis on an industrial scale still remains an important research direction. Among many synthesis methods, such as arc-discharge deposition88,89 or laser ablation method,90,91 chemical vapour deposition (catalytic pyrolysis, CVD method) definitely arouses the most interest, because the diameter, length, density and purity of produced nanotubes can be precisely controlled.92,93 Deshmukh and Santhanam94 were the first scientists to recommend carbon nanotubes instead of activated carbon as anode support in DBFC. Due to the platinized functionalized multiwalled carbon nanotubes, they obtained a 5% increase in output voltage and a 200% more power density, compared to the Au–Pt catalyst supported on a carbon cloth produced by Amendola et al.95 However, as Demirci rightly pointed out,96 the proper comparison of anode materials performance makes sense only if the experimental system is carried out under identical operating conditions. Nevertheless, Yang et al.97 also described the positive impact of MWCNTs as support for Pd electrode. Compared to carbon black and activated carbon, the anode polarization based on carbon nanotubes has been significantly improved due to a better dispersion of catalyst particles. Moreover, in the case of Pd/MWCNTs and Pd/AC, the open circuit potential (OCP) was more negative, which confirms the dominance of BH4− electrooxidation, because it is well known that OCP is the mixing potential of reaction (6) and (7):97| | |
BH4− + 4OH− = BO2− + 2H2O + 4Had + 4e−
| (6) |
The authors also pointed out that in order to obtain high fuel efficiency, it is necessary to oxidize the adsorbed hydrogen simultaneously. The results confirmed that the support structure is very important.97 The number of available active sites of the catalyst also plays an important role in evaluation of its catalytic activity. Hosseini and Mahmoodi98,99 used a two-sequence reduction method to produce Ni@Pt nanoparticles supported by MWCNTs and Vulcan XC-72. Core@shell nanoparticles provide a large contact area of core and shell metal, which can lead to a change of their catalytic activity. It is well known that interaction between metals is much stronger than the interaction of metal and carbon. Test results indicated that Ni@Pt/MWCNTs provides a larger electrochemically active surface area (EASA) for BOR, which allowed to obtain a power density higher by over 20% (162.2 mW cm−2 for Ni@Pt/MWCNTs and 133.4 mW cm−2 for Ni@Pt/Vulcan XC-72).98 Oh et al.100 also clearly indicated the superiority of carbon nanotubes over commercial Vulcan XC-72 as a carbon support, mainly due to over 40 times higher electrical conductivity. Maximum power density of DBFC using Pd/MWCNTs as anode material was equal to 170.9 mW cm−2, while for Pd/Vulcan XC-72 it was only at 128.9 mW cm−2. Zhang et al.101 decided to combine hydrogen storage alloys with 2 wt% of MWCNTs with an outer diameter of 10–20 nm. The authors believe that the increased efficiency of BOR is a consequence of easier reagents diffusion, higher electrical conductivity of composite electrode and a reduction of hydrogen release. According to the authors, MWCNTs will significantly facilitate the transport of hydrogen to the alloy, enabling the storage of larger amounts of hydrogen. Electrooxidation of this extra hydrogen increases not only the catalytic activity of the anode but also the utilization of NaBH4 (Fig. 9).
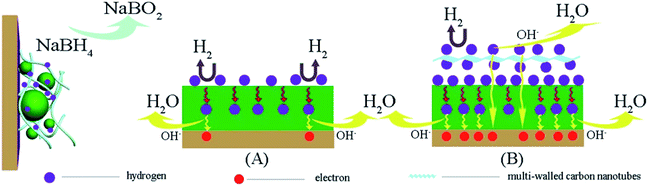 |
| | Fig. 9 Improvement of hydrogen storage properties with carbon nanotubes during borohydride electrooxidation. This figure has been adapted from ref. 101 with permission from ELSEVIER, copyright 2014. | |
In our previous studies,102 we also used MWCNTs as a useful additive to develop the electrochemical properties of anode material. The network formed by MWCNTs created an electron conductive pathway between the hydrogen storage alloy particles. The Nyquist spectrum showed that the composite electrode modified with the addition of 3 wt% of MWCNTs was characterized by the lowest value of charge transfer resistance (CTR). It should be noted that this value is described by the second time constant, referring to the hydrogen adsorption (HA) on the electrode surface and the rate of electrochemical reaction.103 Recently, very interesting works have been presented by Zhang et al.104,105 They created a 3D electrode structure by electrodeposition of Co particles on MWCNTs and supporting body which consisted of cotton cosmetic or waste plastic bag. The preparation process using commercial double-faced adhesive tape is presented in Fig. 10a. Importantly, MWCNTs serve not only as a current collector, but also as an adsorbate for hydrogen released during NaBH4 electrooxidation. In addition to the ecological aspect, the authors emphasized the significant corrosion resistance of the electrodes they produced (Fig. 10b).105
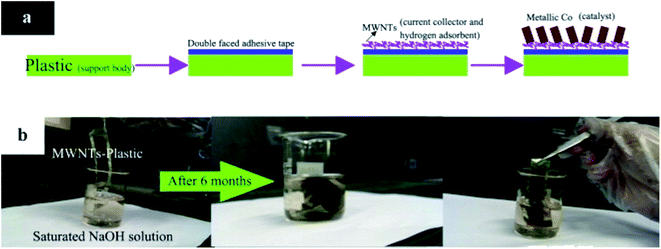 |
| | Fig. 10 Preparation process (a); and stability test in saturated alkaline solution (b). This figure has been adapted from ref. 105 with permission from ELSEVIER, copyright 2015. | |
4.1.4 Graphene. Graphene is composed of a single layer of carbon atoms forming a flat 2D sheet. It can be produced by physical and chemical exfoliation methods106,107 or by epitaxial growth by chemical vapor deposition.108,109 As catalyst support, graphene provides better performance in terms of higher electrical conductivity, greater SSA and flexibility. The presence of oxygen-containing groups on the graphene edge or surface increases the electron transfer rate, while its open structure facilitates fuel transfer.110 Chemically synthesized graphene is widely used in various composites.111 For instance, Hosseini et al.112 used hydrazine as a graphite oxide reducing agent to produce graphene support for Pt and Pt/Ni particles. The sheet-like structure caused uniform dispersion of catalyst nanoparticles, which allowed to obtain a maximum power density of 64.9 mW cm−2 at 60 °C. Liu et al.113 also obtained a homogeneous dispersion of Pt nanoparticles using graphene as anode support. The authors claim that high power density of 42 mW cm−2 at 25 °C was achieved mainly due to the high conductivity, high oxidation stability and corrosion resistance of graphene in an alkaline electrolyte. Zhang et al.114 used porous Ni foam to create a unique reduced 3D graphene - Au network. Importantly, no binder was used, therefore the electrode conductivity and catalyst performance were not reduced. It turned out that good hydrophilicity and three-dimensional structure ensure better contact between the electrode and fuel.114 Several other advanced graphene-based nanomaterials such as Pd/rGO-Fe3O4,115 AuNCage/G,116 PdM (M = Fe, Ag, Au)/rGO117 were also investigated to solve the challenges of DBFC. In a recent work, Li et al.118 created a new self-supporting CoAu electrode based on a reduced graphene oxide (rGO). The authors used the chronoamperometry technique to evaluate the electrocatalytic activity of their materials (Fig. 11a). As shown in Fig. 11b, the amount of released hydrogen was measured during the test and then the final utilization efficiency was calculated (Fig. 11c–e). The authors noted, that efficiency of CoAu/rGO foam is higher than CoAu/Ni foam due to the layered and porous structure of rGO, which is beneficial for the capture of hydrogen produced by hydrolysis (Fig. 11f). During the capture, electrooxidation of entrapped hydrogen occurs, which improved the use of sodium borohydride.118
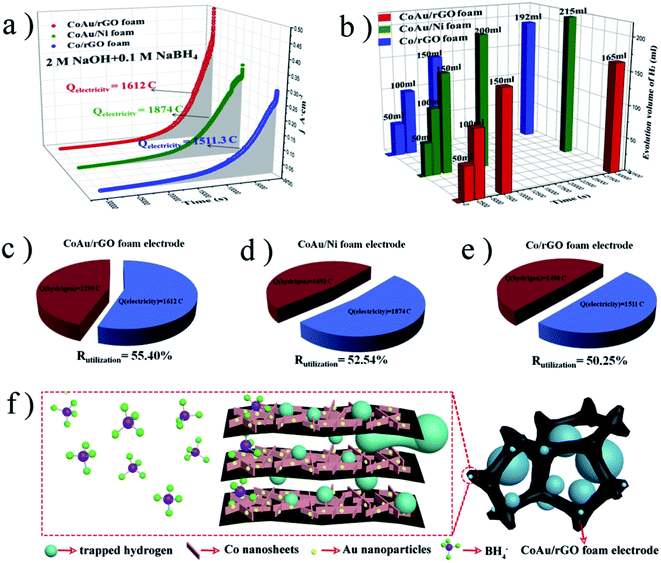 |
| | Fig. 11 Chronoamperometric curves in borohydride solution (a); volume of hydrogen collected during the chronoamperometric test (b); utilization efficiency (c–e) and the role of rGO foam skeleton (f). This figure has been adapted from ref. 118 with permission from ELSEVIER, copyright 2019. | |
4.1.5 Heteroatom-doped carbons. Conductive polymers (electroactive conjugated polymers) are gaining attention due to the possibility of using them as catalytic supports in fuel cell electrodes. Their carbonization in an inert atmosphere leads to the formation of specific carbon materials, as in case of polyaniline or polypyrrole, namely nitrogen-containing carbons.119,120 Rozlívková et al.121 reported, that the conductivity of carbonized polyaniline increases with increasing temperature, reaching a maximum value of 8.6 × 10−4 S cm−1 at 800 °C, while the nitrogen content did not change significantly. Sombatmankhong122 produced a multi-layered macro/mesoporous polypyrrole by electropolymerization and compared its performance with bulk polypyrol as well as commercial carbon black Vulcan XC-72, maintaining the same reaction conditions. The power density reached 56 mW cm−2 due to an almost 3-fold increase of SSA and Vtotal. According to the author, the macroporous structure significantly increased the number of reaction sites, which consequently increased the rate of electrochemical reaction.122 Oliveira et al.123 tested a Pt/polypyrrole-carbon composite (Pt-PPy-C) with different Vulcan XC-72 content (5–35 wt%) and established that the one with the highest carbon content generated the highest current density of 28 mA cm−2 among other composites (Fig. 12A). In addition, as shown in Fig. 12B, based on the mass of Pt catalyst, this value (446 mA mgPt−1) was 30 times greater than the result obtained for a commercial Pt/C catalyst (15 mA mgPt−1). It was also noted that the increase in fuel concentration leads to an increase in the resulting current densities (Fig. 12C) and Pt/PPy-C35% is characterized by the highest stability during BOR chronoamperometric studies (Fig. 12D). This report suggests, that the carbon-based Pt-PPy catalyst creates a good charge transfer channel and thus improves DBFC performance, accomplishing a power density of 83.7 mW cm−2.
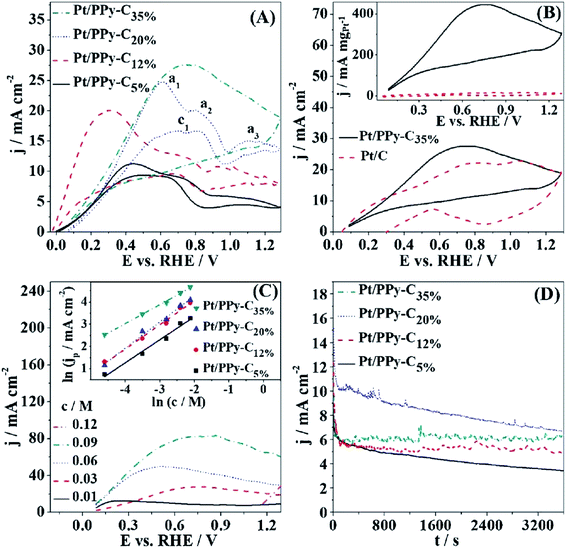 |
| | Fig. 12 Voltammetric curves recorded for all electrocatalysts (A); voltammetric curves for Pt/PPy-C35% catalyst and commercial Pt/C catalyst with corresponding mass-specific current density plots (B); anodic scans for Pt/PPy-C35% at various concentrations of sodium borohydride (C); chronoamperometric test (D). This figure has been adapted from ref. 123 with permission from ELSEVIER, copyright 2018. | |
Lota et al.124 produced composites based on a hydrogen storage alloy with a small amount of carbonized polyaniline, polypyrrole or polyphurfuryl alcohol. The addition of polyaniline caused an almost 10-fold increase of BET (from 0.29 to 2.68 m2 g−1), however, due to its low activity in an alkaline solution, low BOR yield was achieved. Milikić et al.125 also selected carbonized polyaniline as a carbon support for DBFC anode. In this work, the authors doped this conductive polymer with 3,5-dinitrosalicylic acid (PANI-DNSA). The number of electrons exchanged during BOR for Pd/PANI-DNSA was lower (n = 3.6) than for Pd/Vulcan-XC 72 (n = 4.8), mainly due to its lower electrical conductivity. Despite this, the chronoamperometric test confirmed very good catalytic stability of the new electrocatalyst.
4.1.6 MOF-derived carbon nanomaterials. Metal–organic frameworks (MOFs) are relatively new class of crystalline porous materials consisting of transition metal clusters as nodes and organic ligands as spacers. Due to their high carbon content, MOFs can be alternative precursors for the preparation of carbon, metal/carbon composites and metal oxide/carbon composites. Nanoporous carbons are characterized by uniform pores and a high specific surface area (3040 m2 g−1).126 Their structural properties are easily controlled during cost-effective thermal decomposition. It is well known that the encapsulation of metal nanoparticles in carbon structures provides not only a synergistic effect but also corrosion resistance.127 In addition, organic ligands can also contain heteroatoms such as S, N, O, P, making MOFs useful for the production of doped porous materials with uniformly distributed catalytic centers. Due to these outstanding properties, MOF-derived nanocatalysts are widely used in the construction of electrode materials for supercapacitors or catalysts for energy storage and conversion.128,129 Liu et al.126 investigated carbonized metal–organic framework-5 (MOF-5) as the anode catalyst support in DBFC. Furfuryl alcohol was used as an additional carbon source. Compared to the carbon black Vulcan XC-72, the synthesized porous carrier provided a more homogeneous dispersion of Pt, which made it possible to achieve higher power values of 54.34 mW cm−2 at 25 °C. On the other hand, Luo et al.130 presented an environmentally friendly method of synthesizing metal/carbon nanocomposites. The authors noted that Co@ZIF-8 nanocatalyst is highly active for hydrogen release from aqueous NaBH4 hydrolysis (Fig. 13). Similar conclusions were achieved by Zhang et al.131 who tested MOF-derived Co@C composites. The authors noted that after 5 cycles of operation, the catalyst can maintain 93% of its initial catalytic activity. These results present interesting solutions for IBFC. However, the direct electrooxidation process of borohydride would have a reduced coulombic efficiency.
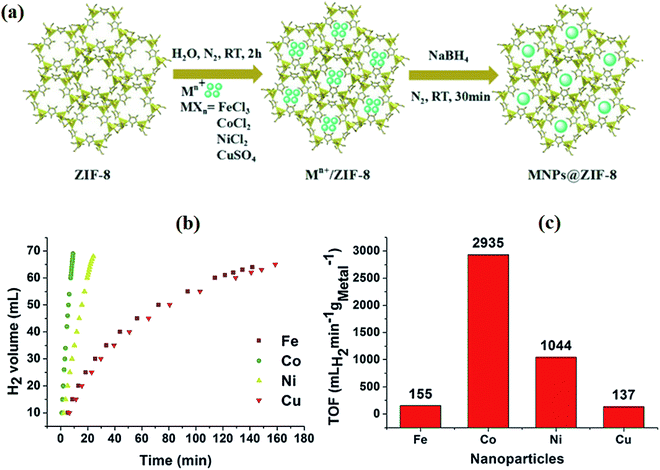 |
| | Fig. 13 Synthesis scheme of metal/ZIF-8 nanocatalysts (a), hydrogen release rate from NaBH4 hydrolysis for various metal NPs (b), comparison of turnover frequency (TOF) values (c). This figure has been adapted from ref. 130 with permission from WILEY-VCH, copyright 2019. | |
4.1.7 Review of selected studies using DBFC configuration. In order to reliably compare the carbon substrates and their influence on the performance of DBFC, only one type of BOR catalyst metal should be examined. Otherwise, due to the strong influence of the nature of the catalyst metal on BOR, a comparison would be very inconvenient and erroneous. For this reason, Table 2 shows only selected studies (different carbons, same catalyst nanoparticles).
Table 2 DBFC performance overview with platinum anode catalyst based on various types of carbon materials
| Anode catalyst |
Loading (mg cm−2) |
Anolyte |
Electrolyte |
Cathode |
Loading (mg cm−2) |
Catholyte |
T (°C) |
Power density (mW cm2) |
Ref. |
| Pt |
5.90 |
0.5 M NaBH4 + 6 M KOH |
6 M KOH |
Pt black |
6 |
Air (natural convection) |
25 |
44.2 |
59 |
| Pt/CB (60%) |
1.50 |
42.0 |
| Pt/CA |
5 |
0.75 M NaBH4 + 1.5 M NaOH |
Nafion-117 membrane |
Commercial Pt/C slurry |
5 |
O2 (150 ml min−1) |
25 |
20.0 |
55 |
| Pt/CB |
55.0 |
| Pt/MPC |
65.0 |
| Pt pasted on carbon cloth |
0.5 |
1 M NaBH4 + 5 M NaOH |
Nafion® NRE-212 |
Pt/C on Toray paper |
2 |
O2 (300 ml min−1) |
25 |
158.0 |
62 |
| Pt sprayed on Nafion |
46.5 |
| Pt/MWCNTs |
0.09 |
10 wt% NaBH4 + 4 M NaOH |
— |
Commercial air electrode |
— |
Air |
25 |
16.0 |
94 |
| Functionalized Pt/MWCNTs |
44.4 |
| Pt/CB |
4 |
1 M NaBH4 + 3 M NaOH |
Nafion-117 membrane |
Au/C |
4 |
2 M H2O2 + 0.5 M H2SO4 |
25 |
34.1 |
113 |
| Pt/Graphene |
41.8 |
126 |
| Pt/NPC |
54.3 |
| Pt/PPY/C5% |
0.4 |
1 M NaBH4 + 4 M NaOH |
Nafion-117 membrane |
Pt mesh |
— |
5 M H2O2 + 1.5 M HCl |
25 |
33.6 |
123 |
| Pt/PPY/C12% |
45.7 |
| Pt/PPY/C20% |
57.0 |
| Pt/PPY/C35% |
83.7 |
| Pt/C (40%) |
0.4 |
10 wt% NaBH4 + 10 wt% NaOH |
Nafion-117 membrane |
Pt/C (40%) |
0.4 |
O2 (200 ml min−1) |
70 |
152 |
132 |
| Pt/C (50%) |
0.3 |
0.01 M NaBH4 + 1 M NaOH |
Nafion-117 membrane |
Commercial Pt/C (50%) |
0.3 |
0.04 M H2O2 + 1 M H2SO4 |
25 |
47–250 |
133 |
| 0.1 M NaBH4 + 1 M NaOH |
0.4 M H2O2 + 1 M H2SO4 |
Despite the fact, that at first glance the results do not seem to differ significantly, it should be remembered that the conditions of the experiment also influence the obtained results. When these are respected, the influence of the porosity of the carbon is clearly visible. The open and fractured structure not only represents a physical trap for hydrogen, but also increases the contact area at the electrode/electrolyte interface, resulting in an increase in maximum DBFC power density.
4.2 Possibility of storing hydrogen
The main limitation in the use of carbon materials for hydrogen storage is their low heat of adsorption. Weak van der Waals forces require very high pressures and low temperatures.134 However, HSA and porosity of nanostructures provide additional binding sites that act as physical traps for hydrogen, which could ultimately improve storage density.135 Therefore, narrow pores are most desirable to ensure strong hydrogen interactions with the surface.136 It should be emphasized that there are highly overestimated reports regarding hydrogen adsorption, especially in the case of carbon nanotubes137,138 which were later adjusted to much lower values.139 Recently, Liu et al.140 reported that under a pressure of 120 bar, the hydrogen storage capacity (HSC) for carbon nanotubes can reach a maximum of 1.7 wt% at room temperature. It is worth noting that the authors considered not only pure SWCNTs and MWCNTs, but also those that were subjected to additional post-treatment in order to enrich structural defects and improve their surface area. Increased HSC, due to the presence of structural defects which ensure more reactive sites for hydrogen adsorption have also been noticed by Rajaura et al.141 It is also well known that the heteroatoms such as N, S, B or P can become hydrogen activation centers mainly due to the lower standard free energy of hydride formation.142 Incorporation of dopants into carbonaceous materials results in increased binding energy.143,144 Ariharan et al.145 used polystyrene and polypyrrole to produce N-doped carbons at various carbonization temperatures. The highest hydrogen storage capacity (2.0 wt% at 100 bar pressure) was demonstrated by N-doped carbon nanotubes carbonized at 900 °C due to the highest surface area (870 m2 g−1) and micropore volume (0.287 cm3 g−1). Although graphene can reach 2630 m2 g−1,146 a single graphene layer does not show superior hydrogen storage parameters compared to other nanostuctured carbons with similar specific surface area.147 Klechikov et al.148 indicated that the storage of hydrogen in graphene materials does not exceed 1 wt% at 20 °C and 120 bar, despite the use of various graphite oxide precursors with a wide range of surface areas (100–2300 m2 g−1). In other experimental studies,149 the authors showed that GO displays better hydrogen uptake capacity (1.90 wt%) compared to rGO (1.34 wt%) at room temperature, due to the presence of oxygen functional groups acting as spacers between layers of graphene. The effect of interlayer spacing was also confirmed by Aboutalebi et al.150 They reported that HSC of GO-MWCNTs hybrid material is equal to 2.6 wt% due to the synergistic effect between components (Fig. 14). Growing the interlayer distance to 8 Å ensures storage of two H2 layers, which gives a capacity of 5.0–6.5 wt%.135 Microporous MOF was first used as a hydrogen adsorbent in 2003.151 It should be emphasized that significant progress has been made since then. It has been proved that the amount of adsorbed hydrogen correlates with the MOF surface area. Interestingly, hydrogen adsorption is higher for MOFs with a surface area below ∼3000 m2 g−1, and when the surface area is higher, the trend is decreasing. This is due to the reduced pore surface occupancy with an increase in surface area and hence an increase in pore volume.152 In addition, the geometry and size of the pores play an important role.153 Zhang et al.152 proved that NPF-200 (cage-type MOF) has the highest volumetric and gravimetric hydrogen capacity (8.7 wt% at 77 K) among the reported MOFs. Nevertheless, under ambient conditions the reported values are much lower, respectively 0.45 wt% at 2 MPa for MIL-101 and 4 wt% at 10 MPa for IRMOF-8.154
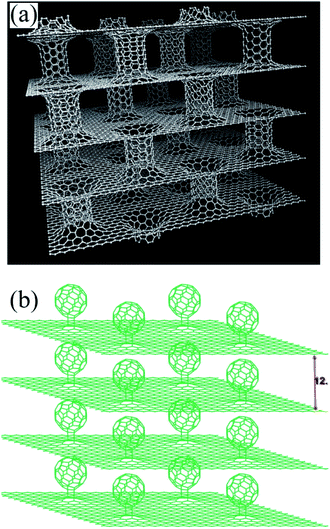 |
| | Fig. 14 Structure of multilayer graphene separated by carbon nanotubes (a) and fullerenes (b). This figure has been adapted from ref. 135 with permission from ELSEVIER, copyright 2017. | |
As mentioned earlier, chemical activation is an effective method for improving the sorption properties. Sun and Webley155 used various activators and activation strategies. They found that corncob-based activated microporous carbon (obtained by two-step KOH activation) was characterized the highest hydrogen capacity of 0.44 wt% at 25 °C and 50 bar pressure. Bader and Ouederni156 optimized hydrogen storage properties by using various KOH weight ratios. When the ratio of KOH to carbonized olive stones was 4, the maximum amount of adsorbed hydrogen reached 1.22 wt% (25 °C, 200 bar). Ramesh et al.157 also used chemical activation. Activated carbon based on jute fibers with a SSA of 1224 m2 g−1 exhibited a gravimetric hydrogen storage capacity of 1.2 wt% (30 °C, 40 bar).
Cathodic polarization during electrode composition of an alkaline solution is an alternative method of storing hydrogen in carbon micropores at ambient conditions. The increased reversible capacity by reducing water is the result of the formation of hydrogen nascent, which easily penetrates the carbon nanostructure, instead of being trapped by surface functional groups.158 The hydrogen electrosorption capacity is estimated by means of an anode charge. Storage of hydrogen in CNTs can be carried out either by physisorption or chemisorption.159 Raman studies showed that electrochemical storage of hydrogen in SWCNTs occurs via physisorption and reaches a value of 0.22 wt%.160 In turn, in the case of MWCNTs, the mechanism of chemisorption dominates. Wang et al.161 investigated the impact of MWCNTs morphology and ball milling treatment on their electrochemical HSC. TEM micrographs display that 12 h ball milling resulted in the formation of shorter carbon nanotubes with open ends. Interestingly, acid treatment did not exhibit such a destructive effect on their structure, as evidenced by the lower ID/IG ratio. According to the authors, an increase of discharge capacity is precisely related to the creation of defects, which ensure better access of hydrogen to hollow interiors of MWCNTs. It should be noted that excessive defects can result in the formation of amorphous carbon, which would result in reduced interaction between H2 and carbon atoms.161 In turn, Reyhani et al.162 investigated the effect of purified MWCNTs by HCl, HNO3, H2SO4 and HF on HSC. The results showed that the purified MWCNTs with HF had the largest HSA and Vmicropore compared to other acids, which allowed to obtain a charge of 496 mA h g−1 (1.83 wt% of stored hydrogen). Babel et al.163 achieved an electric charge of 625 mA h g−1, which corresponds to 2.31 wt% of hydrogen storage. This result was obtained for blackthorn stones activated with KOH at a weight ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 and a temperature of 950 °C. Bleda-Martínez et al.164 noted that HSC also strongly depends on the surface chemistry of the material. The higher the number of oxygen groups, the lower the hydrogen uptake.
:5 and a temperature of 950 °C. Bleda-Martínez et al.164 noted that HSC also strongly depends on the surface chemistry of the material. The higher the number of oxygen groups, the lower the hydrogen uptake.
5. Conclusion and future perspectives
Direct borohydride fuel cell provides high power even at ambient conditions, and the safety of reagents and products makes it desirable for portable applications. However, there are still a number of disadvantages which hinder the commercialization of this type of FC. One of them is a high loss of fuel efficiency due to hydrolysis. The key to success can be the development of electrocatalytic activity of anode catalysts without reducing the overpotential for hydrogen evolution. This is very important because catalysts characterized by high overpotential for hydrogen evolution usually exhibit a big anodic polarization drop during borohydride electrooxidation. In addition, most metals with high electrocatalytic activity towards borohydride electrooxidation reaction are also susceptible to borohydride hydrolysis. Metal particle size and the distance between the particles are the key parameters determining the catalytic activity of supported catalysts. In addition, the metal loading that affects the thickness of the catalyst layer also influences the performance of FC. Therefore, research on anode catalysts for DBFC must also take into account the behavior of the catalyst for the production/oxidation of hydrogen during BOR. As catalysis is a surface effect, the catalyst should be characterized by the largest possible surface area. Surface energy as well as surface chemistry determine the physical and/or chemical interactions which occur at the interface. Firstly, porous carbon materials ensure a longer residence time of BOR intermediates near catalytic sites, which contributes to the completion of the reaction and thus increases the faradic efficiency. Second, porous carbon materials entrap hydrogen bubbles, which suppress gas evolution, but sometimes cause obstructions of ion movement. However, the appropriate pore size can improve the coulombic efficiency of BOR without compromising the anode performance. The consumption of trapped hydrogen means that the anolyte can fill the pores again, and therefore the borohydride electrooxidation reaction occurs again.55 Hydrogen storage is closely related to the adsorption process, hence the specific capacity is dependent on the SSA. However, this SSA should not always be treated as an electrochemically accessible area. It turns out that the pore size and reactivity of carbon atoms are more important. Although graphene sheet provides a large surface area, sp2 carbon atoms are not sufficiently reactive towards dissociation and subsequent hydrogen adsorption, unless there are some structural defects on its surface.160 Among the possible methods of hydrogen storage, electrochemical hydrogen storage is very promising because it can be carried out under ambient conditions. However, there is a notable disproportion between physical and electrochemical storage described in the literature. We are convinced that electrochemical hydrogen storage will be an essential part of future energy systems, especially in DBFC. The development of appropriate carbon materials will allow DBFC to be considered as a promising and alternative energy source when it overcomes the challenges summarized in this review. Activated carbons are widely tested primarily in the context of hydrogen storage, but due to the simple production and full control of porosity during activation, we believe that more attention should be paid to these materials in terms of their use as an anode support in DBFC. Finally, the durability of carbon materials is one of the most important factors determining the performance of FC. Nevertheless, the carbon corrosion rate, which is influenced by the structure and composition of the catalyst support layer, as well as the operating conditions, is often neglected by the authors when characterizing DBFC. The use of selected biomass materials will help diminish the problem of solid waste, the amount of which is constantly increasing. Finally, simulation and modeling will be very useful to understand the diffusion and transfer of a two-phase solution during an extremely complex anode reaction. We hope that our review article will contribute to the stimulation of the development of research on sustainable carbon materials as anode support in DBFC.
Abbreviations
| 2D | Two-dimensional |
| 3D | Three-dimensional |
| AC | Activated carbon |
| APS | Average pore size |
| BOR | Borohydride oxidation reaction |
| CA | Carbon aerogel |
| CB | Carbon black |
| CC | Carbon cloth |
| CNTs | Carbon nanotubes |
| CTR | Charge transfer resistance |
| DBFC | Direct borohydride fuel cell |
| EASA | Electrochemically active surface area |
| FCs | Fuel cells |
| FTIR | Fourier transform infrared |
| HA | Hydrogen adsorption |
| HRTEM | High-resolution transmission electron microscopy |
| HSA | High surface area |
| HSC | Hydrogen storage capacity |
| IBFC | Indirect borohydride fuel cell |
| MPC | Macroporous carbon |
| MWCNTs | Multi-walled carbon nanotubes |
| OCP | Open circuit potential |
| OLEMS | On-line electrochemical mass spectrometry |
| ORR | Oxygen reduction reaction |
| rGO | Reduced graphene oxide |
| RRDE | Rotating ring-disc electrode |
| SSA | Specific surface area |
| SWCNTs | Single-walled carbon nanotubes |
| Saverage | Average pore size |
| Vtotal | Total pore volume |
| Vmicropore | Micropore volume |
| Vmesopore | Mesopore volume |
Conflicts of interest
There is no conflict of interest.
Acknowledgements
The authors would like to acknowledge for the financial support of the National Science Centre (Poland), grant no. DEC-2017/27/N/ST8/02916 and the Ministry of Education and Science, 2021 (Poland).
References
- B. Purvis, Y. Mao and D. Robinson, Sustain. Sci., 2019, 14, 681–695 CrossRef.
- L. de Oliveira Felipe, A. M. de Oliveira and J. L. Bicas, Trends Food Sci. Technol., 2017, 62, 141–153 CrossRef.
- A. Evans, V. Strezov and T. J. Evans, Renewable Sustainable Energy Rev., 2009, 13, 1082–1088 CrossRef.
- A. Hussain, S. M. Arif and M. Aslam, Renewable Sustainable Energy Rev., 2017, 71, 12–28 CrossRef.
- B. S. Silvestre and D. M. Ţîrcă, J. Cleaner Prod., 2019, 208, 325–332 CrossRef.
- V. Khare, S. Nema and P. Baredar, Renewable Sustainable Energy Rev., 2016, 58, 23–33 CrossRef.
- S. Twaha and M. A. M. Ramli, Sustain. Cities Soc., 2018, 41, 320–331 CrossRef.
- B. G. Pollet, I. Staffell and J. L. Shang, Electrochim. Acta, 2012, 84, 235–249 CrossRef CAS.
- D. B. Richardson, Renewable Sustainable Energy Rev., 2013, 19, 247–254 CrossRef.
- E. L. V. Eriksson and E. M. A. Gray, Appl. Energy, 2017, 202, 348–364 CrossRef.
- B. G. Pollet, S. S. Kocha and I. Staffell, Curr. Opin. Electrochem., 2019, 16, 90–95 CrossRef CAS.
- E. Katz and P. Bollella, Isr. J. Chem., 2021, 61, 68–84 CrossRef CAS.
- J. M. Andújar and F. Segura, Renewable Sustainable Energy Rev., 2009, 13, 2309–2322 CrossRef.
- L. Carrette, K. A. Friedrich and U. Stimming, Fuel Cells, 2001, 1, 5–39 CrossRef CAS.
- I. Staffell, D. Scamman, A. Velazquez Abad, P. Balcombe, P. E. Dodds, P. Ekins, N. Shah and K. R. Ward, Energy Environ. Sci., 2019, 12, 463–491 RSC.
- A. Züttel, A. Remhof, A. Borgschulte and O. Friedrichs, Philos. Trans. R. Soc. A, 2010, 368, 3329–3342 CrossRef PubMed.
- A. Alaswad, A. Baroutaji, H. Achour, J. Carton, A. Al Makky and A. G. Olabi, Int. J. Hydrogen Energy, 2016, 41, 16499–16508 CrossRef CAS.
- A. Züttel, Mater. Today, 2003, 6, 24–33 CrossRef.
- A. Züttel, Mitig. Adapt. Strateg. Glob. Chang., 2007, 12, 343–365 CrossRef.
- S. Krasae-, J. H. Stang and P. Neksa, Int. J. Hydrogen Energy, 2010, 35, 4524–4533 CrossRef.
- R. Ströbel, J. Garche, P. T. Moseley, L. Jörissen and G. Wolf, J. Power Sources, 2006, 159, 781–801 CrossRef.
- C. Vix-Guterl, E. Frackowiak, K. Jurewicz, M. Friebe, J. Parmentier and F. Béguin, Carbon, 2005, 43, 1293–1302 CrossRef CAS.
- A. Züttel, P. Sudan, P. Mauron, T. Kiyobayashi, C. Emmenegger and L. Schlapbach, Int. J. Hydrogen Energy, 2002, 27, 203–212 CrossRef.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- H. Chen, H. Wang, Z. Xue, L. Yang, Y. Xiao, M. Zheng, B. Lei, Y. Liu and L. Sun, Int.
J. Hydrogen Energy, 2012, 37, 18888–18894 CrossRef CAS.
- L. Zubizarreta, A. Arenillas and J. J. Pis, Int. J. Hydrogen Energy, 2009, 34, 4575–4581 CrossRef CAS.
- J. O. Abe, A. P. I. Popoola, E. Ajenifuja and O. M. Popoola, Int. J. Hydrogen Energy, 2019, 44, 15072–15086 CrossRef CAS.
- A. Züttel, Naturwissenschaften, 2004, 91, 157–172 CrossRef PubMed.
- L. Schlapbach and A. Züttel, Mater. Sustainable Energy, 2011, 414, 265–270 Search PubMed.
- D. M. F. Santos and C. A. C. Sequeira, Renewable Sustainable Energy Rev., 2011, 15, 3980–4001 CrossRef CAS.
- R. Pecsok, J. Am. Chem. Soc., 1953, 75, 2862–2864 CrossRef CAS.
- U. B. Demirci and P. Miele, C. R. Chim., 2009, 12, 943–950 CrossRef CAS.
- S. C. Amendola, S. L. Sharp-Goldman, M. S. Janjua, M. T. Kelly, P. J. Petillo and M. Binder, J. Power Sources, 2000, 85, 186–189 CrossRef CAS.
- Y. Sakamoto, N. Hoshi, S. Murooka, M. Cao, A. Yoshizaki and K. Hirata, in The 2010 International Power Electronics Conference ECCE ASIA, 2010, pp. 814–819 Search PubMed.
- J. Wee, J. Power Sources, 2006, 161, 1–10 CrossRef CAS.
- C. P. de Leon, F. C. Walsh, D. Pletcher, D. J. Browning and J. B. Lakeman, J. Power Sources, 2006, 155, 172–181 CrossRef.
- Z. P. Li, B. H. Liu, K. Arai and S. Suda, J. Alloys Compd., 2005, 406, 648–652 CrossRef.
- T. H. Oh, Aerosp. Sci. Technol., 2016, 58, 511–517 CrossRef.
- I. Merino-Jiménez, C. Ponce De León, A. A. Shah and F. C. Walsh, J. Power Sources, 2012, 219, 339–357 CrossRef.
- Z. P. Li, B. H. Liu, J. K. Zhu and S. Suda, J. Power Sources, 2006, 163, 555–559 CrossRef CAS.
- J. P. Elder and A. Hickling, Trans. Faraday Soc., 1962, 58, 1852–1864 RSC.
- J. H. Morris, H. J. Gysling and D. Reed, Chem. Rev., 1985, 85, 51–76 CrossRef CAS.
- B. M. Concha, M. Chatenet, F. Maillard, E. A. Ticianelli, F. H. B. Lima and R. B. de Lima, Phys. Chem. Chem. Phys., 2010, 12, 11507–11516 RSC.
- P. Krishnan, T. H. Yang, S. G. Advani and A. K. Prasad, J. Power Sources, 2008, 182, 106–111 CrossRef CAS.
- M. Chatenet, F. H. Lima and E. A. Ticianelli, ECS Trans., 2010, 25, 39–48 CrossRef CAS.
- L. An and C. Y. Jung, Appl. Energy, 2017, 205, 1270–1282 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2016, 181, 298–313 CrossRef CAS.
- T. Frelink, W. Visscher and J. A. R. van Veen, J. Electroanal. Chem., 1995, 382, 65–72 CrossRef.
- P. Y. Olu, C. R. Barros, N. Job and M. Chatenet, Electrocatalysis, 2014, 5, 288–300 CrossRef CAS.
- E. Antolini, Appl. Catal., B, 2009, 88, 1–24 CrossRef CAS.
- J. Q. Yang, B. H. Liu and S. Wu, J. Power Sources, 2009, 194, 824–829 CrossRef CAS.
- M. G. Hosseini and R. Mahmoodi, J. Power Sources, 2017, 370, 87–97 CrossRef CAS.
- J. Tang, J. Liu, N. L. Torad, T. Kimura and Y. Yamauchi, Nano Today, 2014, 9, 305–323 CrossRef CAS.
- H. Cheng and K. Scott, J. Appl. Electrochem., 2006, 36, 1361–1366 CrossRef CAS.
- G. R. Li, Q. Q. Wang, B. H. Liu and Z. P. Li, Fuel Cells, 2015, 15, 270–277 CrossRef CAS.
- A. L. Dicks, J. Power Sources, 2006, 156, 128–141 CrossRef CAS.
- H. P. Boehm, Carbon, 1994, 32, 759–769 CrossRef CAS.
- A. D. Taylor, E. Y. Kim, V. P. Humes, J. Kizuka and L. T. Thompson, J. Power Sources, 2007, 171, 101–106 CrossRef CAS.
- J. H. Kim, H. S. Kim, Y. M. Kang, M. S. Song, S. Rajendran, S. C. Han, D. H. Jung and J. Y. Lee, J. Electrochem. Soc., 2004, 151, A1039–A1043 CrossRef CAS.
- C. Lafforgue, R. W. Atkinson, K. Swider-Lyons and M. Chatenet, Electrochim. Acta, 2020, 341, 135971 CrossRef CAS.
- Y. Bai, C. Wu, F. Wu and B. Yi, Mater. Lett., 2006, 60, 2236–2239 CrossRef CAS.
- P. Y. Olu, F. Deschamps, G. Caldarella, M. Chatenet and N. Job, J. Power Sources, 2015, 297, 492–503 CrossRef CAS.
- D. N. Olowoyo and E. E. Orere, Int. J. Res. Chem. Environ., 2012, 2, 32–35 CAS.
- G. Lota, T. A. Centeno, E. Frackowiak and F. Stoeckli, Electrochim. Acta, 2008, 53, 2210–2216 CrossRef CAS.
- Ł. Kolanowski, M. Graś, M. Bartkowiak, B. Doczekalska and G. Lota, Waste Biomass Valorization, 2020, 11, 3863–3871 CrossRef.
- C. Bouchelta, M. S. Medjram, O. Bertrand and J. P. Bellat, J. Anal. Appl. Pyrolysis, 2008, 82, 70–77 CrossRef CAS.
- Z. Gao, Y. Zhang, N. Song and X. Li, Mater. Res. Lett., 2017, 5, 69–88 CrossRef CAS.
- P. González-García, T. A. Centeno, E. Urones-Garrote, D. Ávila-Brande and L. C. Otero-Díaz, Appl. Surf. Sci., 2013, 265, 731–737 CrossRef.
- C. Peng, X. Bin Yan, R. T. Wang, J. W. Lang, Y. J. Ou and Q. J. Xue, Electrochim. Acta, 2013, 87, 401–408 CrossRef CAS.
- J. Hayashi, T. Horikawa, I. Takeda, K. Muroyama and F. Nasir Ani, Carbon, 2002, 40, 2381–2386 CrossRef CAS.
- I. Okman, S. Karagöz, T. Tay and M. Erdem, Appl. Surf. Sci., 2014, 293, 138–142 CrossRef CAS.
- T. E. Rufford, D. Hulicova-Jurcakova, Z. Zhu and G. Q. Lu, Electrochem. Commun., 2008, 10, 1594–1597 CrossRef CAS.
- K. Wasinski, P. Nowicki, P. Pólrolniczak, M. Walkowiak and R. Pietrzak, Int. J. Electrochem. Sci., 2017, 12, 128–143 CrossRef CAS.
- B. Xu, Y. Chen, G. Wei, G. Cao, H. Zhang and Y. Yang, Mater. Chem. Phys., 2010, 124, 504–509 CrossRef CAS.
- J. Wang, I. Senkovska, S. Kaskel and Q. Liu, Carbon, 2014, 75, 372–380 CrossRef CAS.
- K. Karthikeyan, S. Amaresh, S. N. Lee, X. Sun, V. Aravindan, Y. G. Lee and Y. S. Lee, ChemSusChem, 2014, 7, 1435–1442 CrossRef CAS PubMed.
- T. E. Rufford, D. Hulicova-Jurcakova, E. Fiset, Z. Zhu and G. Q. Lu, Electrochem. Commun., 2009, 11, 974–977 CrossRef CAS.
- L. C. A. Oliveira, E. Pereira, I. R. Guimaraes, A. Vallone, M. Pereira, J. P. Mesquita and K. Sapag, J. Hazard. Mater., 2009, 165, 87–94 CrossRef CAS PubMed.
- A. C. Lua and J. Guo, Carbon, 2000, 38, 1089–1097 CrossRef CAS.
- Q. Wei, Z. Chen, Y. Cheng, X. Wang, X. Yang and Z. Wang, Colloids Surf., A, 2019, 574, 221–227 CrossRef CAS.
- W. Chen, H. Zhang, Y. Huang and W. Wang, J. Mater. Chem., 2010, 20, 4773–4775 RSC.
- M. Zhi, F. Yang, F. Meng, M. Li, A. Manivannan and N. Wu, ACS Sustain. Chem. Eng., 2014, 2, 1592–1598 CrossRef CAS.
- M. Martins, B. Šljukić, C. A. C. Sequeira, Ö. Metin, M. Erdem, T. Sener and D. M. F. Santos, Int. J. Hydrogen Energy, 2016, 41, 10914–10922 CrossRef CAS.
- B. Ari, M. Ay, A. K. Sunol and N. Sahiner, Int. J. Energy Res., 2019, 43, 7159–7172 CAS.
- S. Iijima, Nature, 1991, 354, 737–740 CrossRef.
- R. B. Rakhi, K. Sethupathi and S. Ramaprabhu, Int. J. Hydrogen Energy, 2008, 33, 381–386 CrossRef CAS.
- M. F. L. De Volder, S. H. Tawfick, R. H. Baughman and A. J. Hart, Science, 2013, 339, 535–539 CrossRef CAS PubMed.
- N. D. Hoa, N. Van Quy, Y. Cho and D. Kim, Sens. Actuators, B, 2009, 135, 656–663 CrossRef CAS.
- Y. Su, Z. Yang, H. Wei, E. S. W. Kong and Y. Zhang, Appl. Surf. Sci., 2011, 257, 3123–3127 CrossRef CAS.
- J. Chrzanowska, J. Hoffman, A. Małolepszy, M. Mazurkiewicz, T. A. Kowalewski, Z. Szymanski and L. Stobinski, Phys. Status Solidi, 2015, 252, 1860–1867 CrossRef CAS.
- C. D. Scott, S. Arepalli, P. Nikolaev and R. E. Smalley, Appl. Phys. A: Mater. Sci. Process., 2001, 72, 573–580 CrossRef CAS.
- K. Lota, A. Sierczyńska and G. Lota, J. Solid State Electrochem., 2010, 14, 2209–2212 CrossRef CAS.
- J. Prasek, J. Drbohlavova, J. Chomoucka, J. Hubalek, O. Jasek, V. Adam and R. Kizek, J. Mater. Chem., 2011, 21, 15872–15884 RSC.
- K. Deshmukh and K. S. V. Santhanam, J. Power Sources, 2006, 159, 1084–1088 CrossRef CAS.
- S. C. Amendola, P. Onnerud, M. T. Kelly, P. J. Petillo, S. L. Sharp-Goldman and M. Binder, J. Power Sources, 1999, 84, 130–133 CrossRef CAS.
- U. B. Demirci, J. Power Sources, 2007, 172, 676–687 CrossRef CAS.
- J. Q. Yang, B. H. Liu and S. Wu, J. Power Sources, 2009, 194, 824–829 CrossRef CAS.
- M. G. Hosseini and R. Mahmoodi, J. Power Sources, 2017, 370, 87–97 CrossRef CAS.
- M. G. Hosseini and R. Mahmoodi, J. Colloid Interface Sci., 2017, 500, 264–275 CrossRef CAS PubMed.
- T. H. Oh, B. Jang and S. Kwon, Int. J. Hydrogen Energy, 2014, 39, 6977–6986 CrossRef CAS.
- D. Zhang, G. Wang, K. Cheng, J. Huang, P. Yan and D. Cao, J. Power Sources, 2014, 245, 482–486 CrossRef CAS.
- M. Graś, A. Sierczyńska, K. Lota, I. Acznik and G. Lota, Ionics, 2016, 22, 2539–2544 CrossRef.
- M. Graś, J. Wojciechowski, K. Lota, T. Buchwald, J. Ryl and G. Lota, J. Alloys Compd., 2020, 829, 154553 CrossRef.
- D. Zhang, K. Ye, K. Cheng, D. Cao, J. Yin, Y. Xu and G. Wang, Int. J. Hydrogen Energy, 2014, 39, 9651–9657 CrossRef CAS.
- D. Zhang, K. Ye, D. Cao, B. Wang, K. Cheng, Y. Li, G. Wang and Y. Xu, Electrochim. Acta, 2015, 156, 102–107 CrossRef CAS.
- L. Zhang, X. Li, Y. Huang, Y. Ma, X. Wan and Y. Chen, Carbon, 2010, 48, 2361–2380 CrossRef.
- H. Saleem, M. Haneef and H. Y. Abbasi, Mater. Chem. Phys., 2018, 204, 1–7 CrossRef CAS.
- J. Hwang, M. Kim, D. Campbell, H. A. Alsalman, J. Y. Kwak, S. Shivaraman, A. R. Woll, A. K. Singh, R. G. Hennig, S. Gorantla, M. H. Rümmeli and M. G. Spencer, ACS Nano, 2013, 7, 385–395 CrossRef CAS PubMed.
- L. Banszerus, M. Schmitz, S. Engels, J. Dauber, M. Oellers, F. Haupt, K. Watanabe, T. Taniguchi, B. Beschoten and C. Stampfer, Sci. Adv., 2015, 1, 1500222 CrossRef PubMed.
- N. Shaari and S. K. Kamarudin, Renewable Sustainable Energy Rev., 2017, 69, 862–870 CrossRef CAS.
- C. Wang and D. Astruc, Prog. Mater. Sci., 2018, 94, 306–383 CrossRef CAS.
- M. G. Hosseini, N. Rashidi, R. Mahmoodi and M. Omer, Mater. Chem. Phys., 2018, 208, 207–219 CrossRef CAS.
- X. Liu, L. Yi, X. Wang, J. Su, Y. Song and J. Liu, Int. J. Hydrogen Energy, 2012, 37, 17984–17991 CrossRef CAS.
- D. Zhang, G. Wang, Y. Yuan, Y. Li, S. Jiang, Y. Wang, K. Ye, D. Cao and P. Yan, Int. J. Hydrogen Energy, 2016, 41, 11593–11598 CrossRef CAS.
- M. Martins, Ö. Metin, M. Sevim, B. Šljukić, C. A. C. Sequeira, T. Sener and D. M. F. Santos, Int. J. Hydrogen Energy, 2018, 43, 10686–10697 CrossRef CAS.
- R. Valiollahi, R. Ojani and J.
B. Raoof, Electrochim. Acta, 2016, 191, 230–236 CrossRef CAS.
- M. Martins, B. Šljukić, Ö. Metin, M. Sevim, C. A. C. Sequeira, T. Şener and D. M. F. Santos, J. Alloys Compd., 2017, 718, 204–214 CrossRef CAS.
- B. Li, C. Song, D. Zhang, K. Ye, K. Cheng, K. Zhu, J. Yan, D. Cao and G. Wang, Carbon, 2019, 152, 77–88 CrossRef CAS.
- S. Mentus, G. Ćirić-Marjanović, M. Trchová and J. Stejskal, Nanotechnology, 2009, 20, 245601 CrossRef PubMed.
- I. Sapurina, J. Stejskal, Š. Ivana, M. Trchová, J. Kovárová, J. Hromádková, J. Kopecká, M. Cieslar, A. A. El-Nasr and M. M. Ayad, Synth. Met., 2016, 214, 14–22 CrossRef CAS.
- Z. Rozlívková, M. Trchová, M. Exnerová and J. Stejskal, Synth. Met., 2011, 161, 1122–1129 CrossRef.
- K. Sombatmankhong, J. Porous Mater., 2015, 22, 675–687 CrossRef CAS.
- R. C. P. Oliveira, J. Milikić, E. Daş, A. B. Yurtcan, D. M. F. Santos and B. Šljukić, Appl. Catal., B, 2018, 238, 454–464 CrossRef CAS.
- G. Lota, A. Sierczynska, I. Acznik and K. Lota, Int. J. Electrochem. Sci., 2014, 9, 659–669 Search PubMed.
- J. Milikić, G. Cirić-Marjanović, S. Mentus, D. M. F. Santos, C. A. C. Sequeira and B. Šljukić, Electrochim. Acta, 2016, 213, 298–305 CrossRef.
- J. Liu, H. Wang, C. Wu, Q. Zhao, X. Wang and L. Yi, Int. J. Hydrogen Energy, 2014, 39, 6729–6736 CrossRef CAS.
- Q. Huang, Y. Guo, X. Wang, L. Chai, J. Ding, L. Zhong, T. T. Li, Y. Hu, J. Qian and S. Huang, Nanoscale, 2020, 12, 10019–10025 RSC.
- L. Chai, Z. Hu, X. Wang, Y. Xu, L. Zhang, T. T. Li, Y. Hu, J. Qian and S. Huang, Adv. Sci., 2020, 7, 1903195 CrossRef CAS PubMed.
- X. Wang, Z. Zhu, L. Chai, J. Ding, L. Zhong, A. Dong, T. T. Li, Y. Hu, J. Qian and S. Huang, J. Power Sources, 2019, 440, 227158 CrossRef CAS.
- C. Luo, F. Fu, X. Yang, J. Wei, C. Wang, J. Zhu, D. Huang, D. Astruc and P. Zhao, ChemCatChem, 2019, 11, 1643–1649 CrossRef CAS.
- X. Zhang, X. Sun, D. Xu, X. Tao, P. Dai, Q. Guo and X. Liu, Appl. Surf. Sci., 2019, 469, 764–769 CrossRef CAS.
- K. T. Park, U. H. Jung, S. U. Jeong and S. H. Kim, J. Power Sources, 2006, 162, 192–197 CrossRef CAS.
- R. M. E. Hjelm, C. Lafforgue, R. W. Atkinson, Y. Garsany, R. O. Stroman, M. Chatenet and K. Swider-Lyons, J. Electrochem. Soc., 2019, 166, F1218–F1228 CrossRef CAS.
- G. Sethia and A. Sayari, Carbon, 2016, 99, 289–294 CrossRef CAS.
- X. Yu, Z. Tang, D. Sun, L. Ouyang and M. Zhu, Prog. Mater. Sci., 2017, 88, 1–48 CrossRef.
- R. Pedicini, S. Maisano, V. Chiodo, G. Conte, A. Policicchio and R. G. Agostino, Int. J. Hydrogen Energy, 2020, 45, 14038–14047 CrossRef CAS.
- C. Liu, Y. Y. Fan, M. Liu, H. T. Cong, H. M. Cheng and M. S. Dresselhaus, Science, 1999, 286, 1127–1129 CrossRef CAS PubMed.
- A. C. Dillon, K. M. Jones, T. A. Bekkedahl, C. H. Kiang, D. S. Bethune and M. J. Heben, Nature, 1997, 386, 377–379 CrossRef CAS.
- A. Züttel, P. Sudan, P. Mauron, T. Kiyobayashi, C. Emmenegger and L. Schlapbach, Int. J. Hydrogen Energy, 2002, 27, 203–212 CrossRef.
- C. Liu, Y. Chen, C. Z. Wu, S. T. Xu and H. M. Cheng, Carbon, 2010, 48, 452–455 CrossRef CAS.
- R. S. Rajaura, S. Srivastava, P. K. Sharma, S. Mathur, R. Shrivastava, S. S. Sharma and Y. K. Vijay, Nano-Struct. Nano-Objects, 2018, 14, 57–65 CrossRef CAS.
- B. Viswanathan and M. Sankaran, Diamond Relat. Mater., 2009, 18, 429–432 CrossRef CAS.
- M. Sankaran and B. Viswanathan, Carbon, 2006, 44, 2816–2821 CrossRef CAS.
- A. Ariharan, B. Viswanathan and V. Nandhakumar, Int. J. Hydrogen Energy, 2016, 41, 3527–3536 CrossRef CAS.
- A. Ariharan, B. Viswanathan and V. Nandhakumar, Int. J. Hydrogen Energy, 2018, 43, 5077–5088 CrossRef CAS.
- M. J. McAllister, J. L. Li, D. H. Adamson, H. C. Schniepp, A. A. Abdala, J. Liu, M. Herrera-Alonso, D. L. Milius, R. Car, R. K. Prud’homme and I. A. Aksay, Chem. Mater., 2007, 19, 4396–4404 CrossRef CAS.
- M. Jorda-Beneyto, F. Suarez-Garcıa, D. Lozano-Castello, D. Cazorla-Amoros and A. Linares-Solano, Carbon, 2007, 45, 293–303 CrossRef CAS.
- A. G. Klechikov, G. Mercier, P. Merino, S. Blanco, C. Merino and A. V. Talyzin, Microporous Mesoporous Mater., 2015, 210, 46–51 CrossRef CAS.
- R. S. Rajaura, S. Srivastava, V. Sharma, P. K. Sharma, C. Lal, M. Singh, H. S. Palsania and Y. K. Vijay, Int. J. Hydrogen Energy, 2016, 41, 9454–9461 CrossRef CAS.
- S. H. Aboutalebi, S. Aminorroaya-Yamini, I. Nevirkovets, K. Konstantinov and H. K. Liu, Adv. Energy Mater., 2012, 2, 1439–1446 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, T. Vodak, J. Kim, M. O. Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- X. Zhang, R. Lin, J. Wang, B. Wang, B. Liang, T. Yildirim, J. Zhang, W. Zhou and B. Chen, Adv. Mater., 2020, 32, 1907995 CrossRef CAS PubMed.
- H. S. Cho, J. Yang, X. Gong, Y. Zhang, K. Momma, B. Weckhuysen, H. Deng, J. Kang, O. M. Yaghi and O. Terasaki, Nat. Chem., 2019, 11, 562–570 CrossRef CAS PubMed.
- Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 8136–8137 CrossRef CAS PubMed.
- Y. Sun and P. A. Webley, Chem. Eng. J., 2010, 162, 883–892 CrossRef CAS.
- N. Bader and A. Ouederni, J. Energy Storage, 2016, 5, 77–84 CrossRef.
- T. Ramesh, N. Rajalakshmi and K. S. Dhathathreyan, Renew. Energy Environ. Sustain., 2017, 2, 1–4 CrossRef.
- E. Frackowiak and F. Béguin, Carbon, 2002, 40, 1775–1787 CrossRef CAS.
- A. Eftekhari and B. Fang, Int. J. Hydrogen Energy, 2017, 42, 25143–25165 CrossRef CAS.
- J. B. Martin, I. A. Kinloch and R. A. W. Dryfe, J. Phys. Chem. C, 2010, 114, 4693–4703 CrossRef CAS.
- Y. Wang, W. Deng, X. Liu and X. Wang, Int. J. Hydrogen Energy, 2009, 34, 1437–1443 CrossRef CAS.
- A. Reyhani, S. Z. Mortazavi, A. N. Golikand, A. Z. Moshfegh and S. Mirershadi, J. Power Sources, 2008, 183, 539–543 CrossRef CAS.
- K. Babel, D. Janasiak and K. Jurewicz, Carbon, 2012, 50, 5017–5026 CrossRef CAS.
- M. Bleda-Martinez, J. M. Perez, A. Linares-Solano, E. Morallón and D. Cazorla-Amorós, Carbon, 2008, 46, 1053–1059 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab













