
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA comparison study of sodium ion- and potassium ion-modified graphitic carbon nitride for photocatalytic hydrogen evolution†
Siyu Hu,
Anchi Yu * and
Rong Lu*
* and
Rong Lu*
Department of Chemistry, Renmin University of China, Beijing 100872, P. R. China. E-mail: lurong@ruc.edu.cn; yuac@ruc.edu.cn; Fax: +86-10-6251-6444
First published on 27th April 2021
Abstract
It is well known that modifying graphitic carbon nitride (GCN) is an imperative strategy to improve its photocatalytic activity. In this study, Na-doped and K-doped graphitic carbon nitride (GCN-Na and GCN-K) were prepared via the simple thermal polymerization of a mixture of melamine and NaCl or KCl, respectively. The structure characterization showed that both Na+ and K+ intercalation could reduce the interlayer distance of GCN and introduce cyano defects in GCN, while K+ apparently had a stronger influence on the structure variation of GCN. The chemical composition data showed that both Na+ and K+ could easily interact with GCN, while K-doping caused a greater change in the C/N ratio than Na-doping. Moreover, compared to GCN-Na-5 (5 represents weight ratio of alkali halide to melamine), the conduction and valence bands of GCN-K-5 both shifted upward based on the electronic and optical measurements. Consequently, GCN-K-5 yielded an H2 evolution rate around 4 times higher than that of GCN-Na-5 under visible light irradiation (>420 nm). The cation size effect on GCN was proposed to be mainly responsible for the variation in the structure, optical and electronic properties of ion-doped GCNs, and hence the enhanced photocatalytic H2 evolution. The current work can provide new insight into optimizing photocatalysts for enhanced photocatalytic performances.
1. Introduction
Graphitic carbon nitride (GCN), as a metal-free layered polymeric semiconductor, has become a potential photocatalyst due to its unique optical, electronic and physiochemical properties.1–5 However, to date, the photocatalytic efficiency of pristine GCN is still low and far from its practical application. Thus, it is imperative to modify GCN to improve its photocatalytic activity. Accordingly, elemental doping is considered a facile and effective modification method,4–9 and especially introducing alkali metal ions into GCN has been frequently utilized to modify GCN, resulting in enhanced photocatalytic activity.5–8In non-noble metal ion-doping GCN studies, the most ions used are Na+ and K+ given that Li+ strongly absorbs water and the size of Cs+ is too big to form a stable product.10,11 There are several ways to intercalate Na+ or K+ into GCN including the calcination of a mixture of carbon nitride precursors (cyanamide, melamine, etc.) with alkali halide,11–22 a mixture of the prepared GCN and alkali halide,23–27 a mixture of carbon nitride precursors and alkali metal hydroxide,28–31 and a mixture of the prepared GCN and alkali metal hydroxide.6,32–35 Most of the studies revealed that for one or several factors, alkali metal ion doping in GCN can help increase its surface specific area,6,11,19,20,23,29 promote its crystallinity,15,17,22,27 enhance its light absorption,13,15–17,19,21,22,27 reduce its bandgap,6,13 shift its conduction/valence band,12,17,20 improve its charge carrier transfer and separation efficiency,6,13,15,17–19,21–23,25,27 and finally achieve higher photocatalytic activity compared with pristine GCN. Nevertheless, among these studies, it has been found that some inconsistences exist such as optical bandgap change,6,12,13,17,20 crystallinity,11,12,14,15,18,19 interlayer distance change,12,15,19,27 conduction/valence band shift6,12,13,17,20 and photocatalytic H2 evolution efficiency.14–16,19 Even for the same element ion-doped GCN from different research groups,14,16–18,20 some contradictory experimental data exists. These inconsistences may be correlated with the synthetic conditions employed for doping (precursor type, doping amount, calcination temperature, etc.) by different research groups.
Several mechanisms have been proposed to illustrate the effect of ions on GCN, such as tailoring its layered structure12 and synergistic effects.15–17,19,21,23,29 To the best of our knowledge, only a few reports explored a series of alkali metal element-doped GCN.6,11,15,25 To fully understand the effect of alkali metal ions on GCN, a systematic examination of alkali halide-modified GCN is still needed. In this study, we comparatively investigated Na-doped and K-doped GCN by heating a mixture of melamine and NaCl or KCl, and then washing off the redundant ions, respectively. The morphology, crystalline structure, chemical structure, chemical composition, surface specific area, optical and electronic properties, and photocatalytic H2 evolution were carefully explored for both Na-doped and K-doped GCN. In addition, the characterization of the pristine GCN is shown in all the figures and tables for reference.
2. Experimental section
2.1. Samples
Melamine (>99.0%) was purchased from Sigma-Aldrich. Potassium chloride (KCl, ≥99.0%) and sodium chloride (NaCl, ≥99.0%) were purchased from Alfa Aesar. Pure water (18 MΩ cm) was obtained using a Millipore Milli-Q water purification system. All other reagents used in this work were analytically pure and used as received without further purification.2.2. Synthesis of graphitic carbon nitride (GCN)
Typically, 1.5 g melamine was placed in an alumina crucible covered with aluminum foil in a muffle furnace and heated to 550 °C for 4 h at a ramping rate of 2 °C min−1 in air, and then cooled naturally to room temperature. The as-prepared light yellow sample was washed several times with deionized water and dried at 60 °C in a vacuum oven for 24 h, and then fully ground into a fine powder. The final product was labeled as GCN.2.3. Synthesis of Na-doped and K-doped GCN
The precursor melamine (1.5 g) and a certain amount of NaCl or KCl were dissolved in 80 mL deionized water under constant stirring, and then heated to dry the mixture completely. The resultant mixture was heated to 550 °C for 4 h at a ramping rate of 2 °C min−1 in air and cooled naturally to room temperature. The obtained sample was ground into fine power and dispersed in boiling deionized water, and then washed several times until the conductivity of the supernatant reached a constant value (∼0.2 mS cm−1). The obtained pale yellow precipitate was separated by centrifugation and dried at 60 °C in a vacuum oven for 24 h. The final ion-doped product was fully ground again and denoted as GCN-Na-0.5 and GCN-Na-5 and GCN-K-0.5 and GCN-K-5, where 0.5 and 5 represent the initial weight ratio of NaCl or KCl to melamine, respectively.2.4. Characterization
Powder X-ray diffraction (XRD) was performed on a Shimadzu XRD-7000 diffractometer with Cu radiation (λ = 1.54056 Å). Fourier transform-infrared (FTIR) spectra were measured on a Bruker Tensor 27 spectrometer. Raman spectra were obtained using a Fourier infrared Raman spectrometer (laser source 1064 nm, Vertex 70v&RAM II, Bruker). X-ray photoelectron spectroscopy (XPS) was performed on a Thermo ESCALab250Xi electron spectrometer (Thermo Scientific) with an Al radiation source. The C 1s peak (284.8 eV) was referenced for all binding energies. Scanning electron microscopy (SEM) images were obtained using a Hitachi SU8010 scanning electron microscope to characterize the morphology of the obtained samples, and their element mappings and chemical compositions were measured using a SEM energy dispersive spectrometer (SEM-EDS). The crystalline structures of the as-prepared samples were examined using a Hitachi JEM-2100F transmission electron microscope (TEM). The chemical compositions of the samples were also determined using a Thermo Flash-EA-1112 elemental analyzer (EA). The Na+ and K+ content in the products was measured using an Agilent ICP-OES 715 inductively coupled plasma atomic emission spectrometer (ICP-AES). The UV-Vis diffuse reflectance spectra (UVDRS) of the samples were recorded on a Shimadzu UV-2600 spectrophotometer. Photoluminescence spectra were recorded on a Horiba FluoroMax+ spectrophotometer, and the excitation wavelength was set at 370 nm. The fluorescence lifetime was obtained under 375 nm excitation and 470 nm detection with a UV-Vis near infrared spectrophotometer (FLS980, Edinburgh). The Brunauer–Emmett–Teller (BET) surface areas of the samples were determined using a Belsorp-Mini II equipment (MicrotracBEL) by measuring their N2 adsorption–desorption isotherms at 77 K.Electrochemical measurements (Mott–Schottky curve, transient photocurrent response and electrochemical impedance spectroscopy (EIS)) were performed on an electrochemical workstation (CHI-660D, Shanghai Chenhua) with a conventional three-electrode cell. A Pt wire was used as a counter electrode, and Ag/AgCl (3 M KCl) was used as the reference electrode. To fabricate the working electrode, 10 mg of sample was dispersed in a 200 μL Nafion solution (5%) to obtain a slurry, and the as-prepared slurry was dropped onto the platinum-carbon electrode, and subsequently dried under an infrared lamp. Na2SO4 aqueous solution (0.5 M) was used as the electrolyte solution. The transient photocurrent response was measured under irradiation from a 420 nm LED lamp (∼600 mW cm−2).
The photocatalytic H2 evolution reaction was measured using a Pyrex top-irradiation reaction vessel connected to a closed gas system.36 In detail, 50 mg catalyst powder was dispersed in 100 mL of aqueous solution, which contained 10 vol% triethanolamine. A total of 3 wt% Pt cocatalyst was deposited on the surface of the catalyst powder directly by adding H2PtCl6 in 100 mL of aqueous solution. A 300 W xenon lamp (MICROSOLAR300, PerfectLight, Beijing) with a λ > 420 nm filter was used as the light source. The air was completely removed before the reaction solution was irradiated. A gas chromatography (GC-2014C, Shimadzu) was used to determine the amount of H2 from the photocatalytic reaction.
3. Results and discussion
The morphologies of GCN, GCN-Na-5 and GCN-K-5 were investigated via SEM, as shown in Fig. 1(a–c). The SEM images shown in Fig. 1(a–c) reveal that both GCN-Na-5 and GCN-K-5 possess a sheet-like structure but exhibit a less condensed stacking feature, implying that the ion-doped samples maintain a similar structure to that of GCN. The SEM element mapping images of GCN-Na-5 and GCN-K-5 are shown in Fig. S1 and S2,† indicating that the C, N, O and Na or K elements uniformly coexist in the entire sample region, but the Cl element remained undetected. BET measurements were performed to obtain the specific surface areas and pore structures of the ion-doped GCNs (Fig. 1(d)), and the extracted data is displayed in Table S1.† The specific surface areas of GCN-Na-5 and GCN-K-5 are 10.3 and 7.0 m2 g−1, respectively, and the specific surface area of GCN-Na-5 is close to that of GCN (10.4 m2 g−1), while the specific surface area of GCN-K-5 is slightly less than that of GCN, indicating that the effect of K-doping on the specific surface area is slightly greater than that of Na-doping. The specific surface area of GCN (10.4 m2 g−1) is consistent with the previous report.37 The slight change in the specific surface areas/pore size distributions and the less condensed stacking of GCN-Na-5 and GCN-K-5 compared to that of the pristine GCN suggest that the ion-doped GCNs experienced interlayer exfoliation upon treatment with alkali halide salt.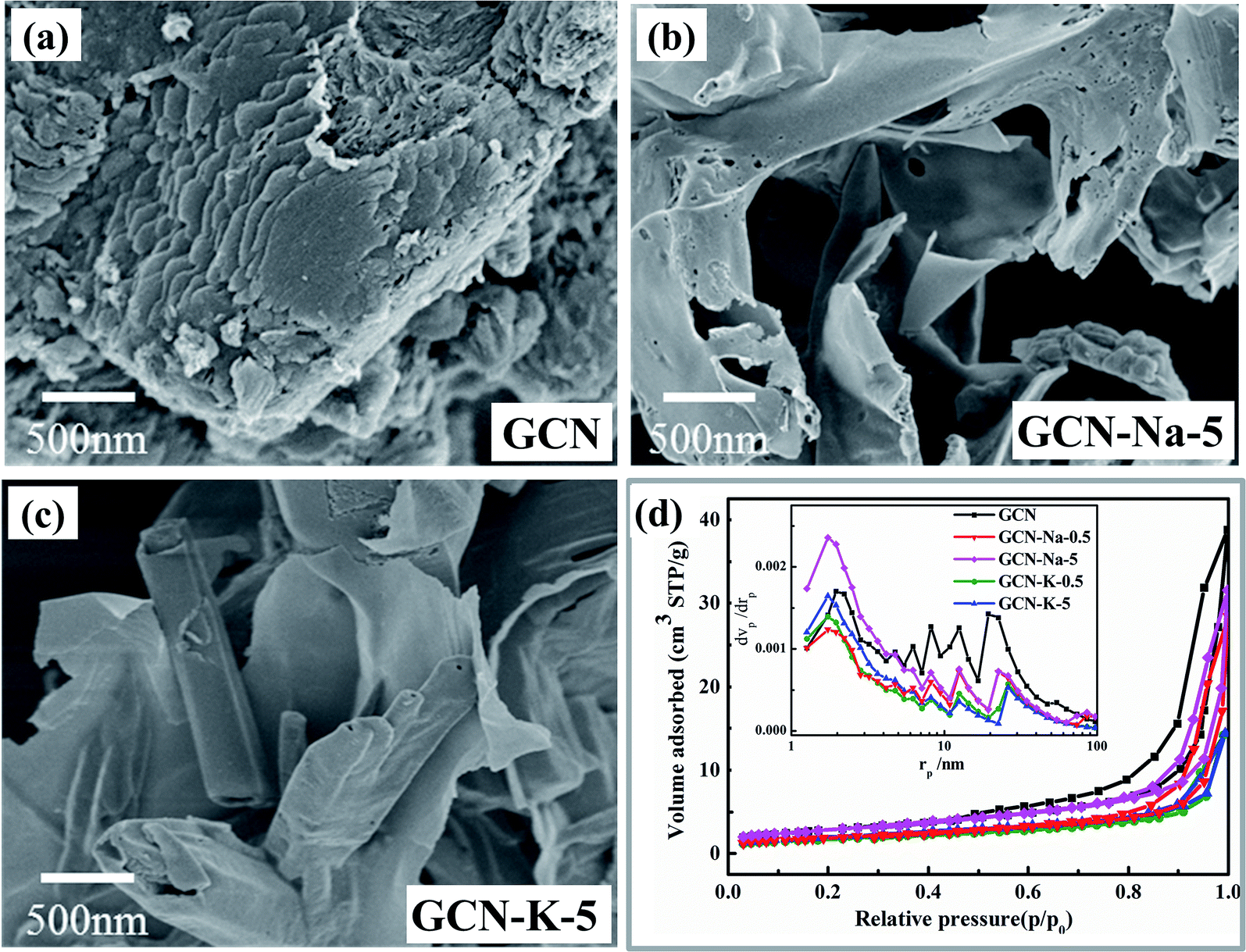 | ||
| Fig. 1 SEM images of GCN (a), GCN-Na-5 (b) and GCN-K-5 (c). BET nitrogen adsorption/desorption isotherms and corresponding pore size distribution curves (inset) of GCN and ion-doped GCNs (d). | ||
The XRD patterns of GCN and ion-doped GCNs are shown in Fig. 2(a). For GCN, there are two diffraction peaks at around 13.0° and 27.53°, which represent the intralayer long-range order packing (100) and interlayer stacking (002), respectively.30 For the ion-doped GCNs, the intensities of both peaks decrease, indicating that the doping of the metal ions resulted in a decrease in crystallinity. Especially for GCN-K-5, the (100) peak almost disappears and the (002) peak presents the lowest intensity and simultaneously shifts to 28.05°, which implies a narrower interlayer distance.18 The high-resolution TEM images of GCN, GCN-Na-5 and GCN-5-K are shown in Fig. 2(b–d), respectively, and the larger-scale TEM and high-resolution TEM images of GCN are presented in Fig. S3.† As shown in Fig. 2, the d-spacing of the lattice fringe for the (002) plane is measured as 0.324 nm for GCN, which is similar to the value in previous reports.38–41 Also, the d-spacing of the lattice fringe for the (002) plane is 0.322 nm for GCN-Na-5 and 0.317 nm for GCN-K-5, which are consistent with their respective XRD data. Both the XRD and TEM data shown in Fig. 2 illustrate that the interlayer distance of the (002) plane becomes narrow upon the doping of the metal ions, and K+ has greater impact on the crystalline structure of GCN than that of Na+.
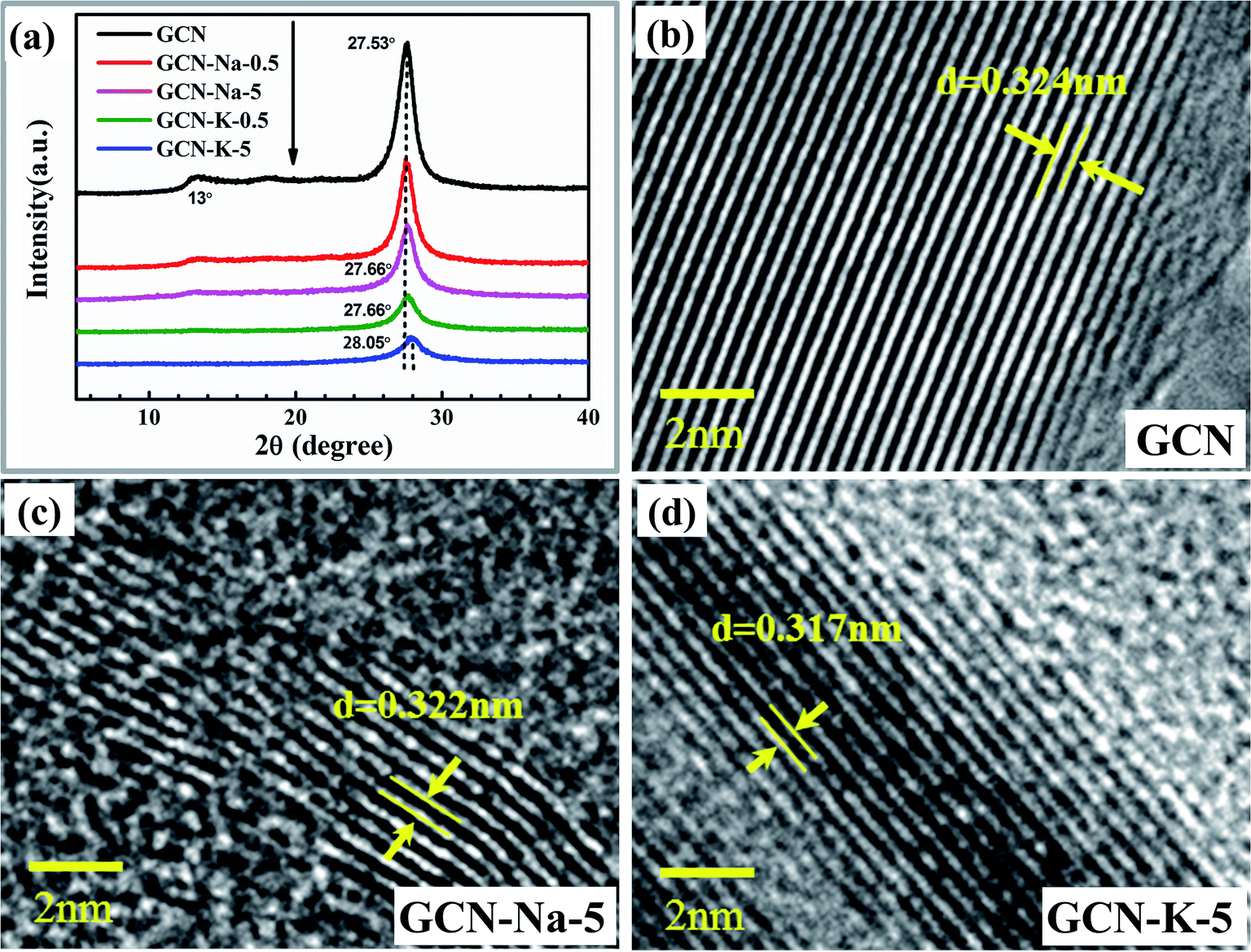 | ||
| Fig. 2 XRD patterns (a) of GCN and ion-doped GCNs. High-resolution TEM images of GCN (b), GCN-Na-5 (c) and GCN-K-5 (d). | ||
The chemical structures of GCN and the ion-doped samples were first examined by FTIR (Fig. 3(a)). In Fig. 3(a), the bands at 808 cm−1 and 1200–1800 cm−1 correspond to the triazine units and aromatic CN heterocycles of GCN, respectively.13,29,42 However, compared to GCN, there are three extra peaks located at 998 cm−1, 1158 cm−1 and 2180 cm−1 in the FTIR spectra of the ion-doped GCNs. The peaks at 998 cm−1 and 1158 cm−1 are assigned to the C–O vibrations and the peak at 2180 cm−1 originates from the cyano group (-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N).12,43 Ye et al. suggested that the appearance of CN and C–O groups after doping is due to the tailoring of the layered structure of GCN by Na+ or K+.12 Besides, the intensity of the N–H peak located at 3000–3300 cm−1 decreases, while the band located at 3300–3600 cm−1 originating from the O–H stretching mode becomes stronger upon Na+ or K+ doping in GCN.12 According to the FTIR spectrum shown in Fig. 3(a), it can be further found that all the peaks for the vibrations of CN, C–O and O–H in the K-doped GCN are stronger than that of Na-doped GCN, implying that K+ has stronger perturbation on the structure of GCN than Na+.
N).12,43 Ye et al. suggested that the appearance of CN and C–O groups after doping is due to the tailoring of the layered structure of GCN by Na+ or K+.12 Besides, the intensity of the N–H peak located at 3000–3300 cm−1 decreases, while the band located at 3300–3600 cm−1 originating from the O–H stretching mode becomes stronger upon Na+ or K+ doping in GCN.12 According to the FTIR spectrum shown in Fig. 3(a), it can be further found that all the peaks for the vibrations of CN, C–O and O–H in the K-doped GCN are stronger than that of Na-doped GCN, implying that K+ has stronger perturbation on the structure of GCN than Na+.
 | ||
| Fig. 3 FTIR (a) and Raman (b) spectra of GCN and ion-doped GCNs. | ||
The chemical structures of GCN and the ion-doped GCNs were further examined by Raman spectroscopy (Fig. 3(b)). As shown in Fig. 3(b), one main difference among the Raman spectra of GCN and the ion-doped GCNs is the change in the relative intensity of the peak at 707 cm−1 to the peak at 725 cm−1. Here, the peaks located at 707 cm−1 and 725 cm−1 are assigned as the breathing modes of the triazine rings.18,25,44 Xu et al. reported the appearance of a peak at 706 cm−1 for pristine CN, whereas a peak at 726 cm−1 appeared for Cs+-modified CN.44 According to Fig. 3(b), it is obvious that the intensity ratio (I707/I725) decreases in the order of GCN, GCN-Na-0.5, GCN-Na-5, GCN-K-0.5 and GCN-K-5, which implies that the effect of K-doping on the chemical structure of GCN is stronger than of Na-doping, and hence the intensity ratio I707/I725 is suggested as an indicator to estimate the structure perturbation degree of GCN. Therefore, both the FTIR and Raman measurements suggest that K-doping has a stronger effect on the chemical structure variation of GCN than Na-doping.
The element chemical states of GCN and the ion-doped GCNs were also analyzed by XPS, as shown in Fig. 4, Fig. S4 and S5.† The high-resolution XPS spectra of Na 1s for NaCl and K 2p for KCl are also displayed for reference. The C 1s spectrum (Fig. 4(a)) for GCN contains three components located at 284.8, 286.4 and 288.3 eV, corresponding to the adventitious carbon, C–NHx (amino group), and N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N coordination in the framework of carbon nitride, respectively.19 It can be seen that the peak at 286.4 eV for the ion-doped GCN is stronger than that of pristine GCN, which is due to the presence of the cyano group14 (Fig. 4(a)) and is consistent with the FTIR and Raman data shown in Fig. 3. The N 1s spectrum (Fig. 4(b)) can be deconvoluted into four peaks at 398.6 eV, 400.1 eV, 401.3 eV and 404.4 eV, which are assigned to the N species in NC–N, N–(C)3, N–H and positive charge localization in the heptazine rings.15 As shown in Fig. 4(a) and (b), both the C 1s and N 1s XPS spectra of the ion-doped GCNs exhibit no obvious change compared to that of GCN, which suggests that the main chemical structure is still kept after salt modification. In addition, Fig. 4(c) presents the O 1s spectrum with peaks at the binding energies of 532.5 eV and 530.8 eV. The peak at 532.5 eV can be ascribed to the adsorbed H2O, and the peak at 530.8 eV may be related to the (N)2C–OH group,12 which is also agreement with the presence of the C–OH vibration in the FTIR spectra of ion-doped GCNs (Fig. 3). Both the Na 1s and K 2p XPS peaks exhibit a negligible shift relative to the Na 1s peak for NaCl and K 2p peak for KCl (Fig. 4(e) and (f)), respectively, which suggests that the chemical state of Na+ or K+ presents little change upon doping in GCN. In addition, the Cl 2p XPS data (Fig. 4(d)) implies that there is a trace amount of Cl element existing in the ion-doped GCNs.
N coordination in the framework of carbon nitride, respectively.19 It can be seen that the peak at 286.4 eV for the ion-doped GCN is stronger than that of pristine GCN, which is due to the presence of the cyano group14 (Fig. 4(a)) and is consistent with the FTIR and Raman data shown in Fig. 3. The N 1s spectrum (Fig. 4(b)) can be deconvoluted into four peaks at 398.6 eV, 400.1 eV, 401.3 eV and 404.4 eV, which are assigned to the N species in NC–N, N–(C)3, N–H and positive charge localization in the heptazine rings.15 As shown in Fig. 4(a) and (b), both the C 1s and N 1s XPS spectra of the ion-doped GCNs exhibit no obvious change compared to that of GCN, which suggests that the main chemical structure is still kept after salt modification. In addition, Fig. 4(c) presents the O 1s spectrum with peaks at the binding energies of 532.5 eV and 530.8 eV. The peak at 532.5 eV can be ascribed to the adsorbed H2O, and the peak at 530.8 eV may be related to the (N)2C–OH group,12 which is also agreement with the presence of the C–OH vibration in the FTIR spectra of ion-doped GCNs (Fig. 3). Both the Na 1s and K 2p XPS peaks exhibit a negligible shift relative to the Na 1s peak for NaCl and K 2p peak for KCl (Fig. 4(e) and (f)), respectively, which suggests that the chemical state of Na+ or K+ presents little change upon doping in GCN. In addition, the Cl 2p XPS data (Fig. 4(d)) implies that there is a trace amount of Cl element existing in the ion-doped GCNs.
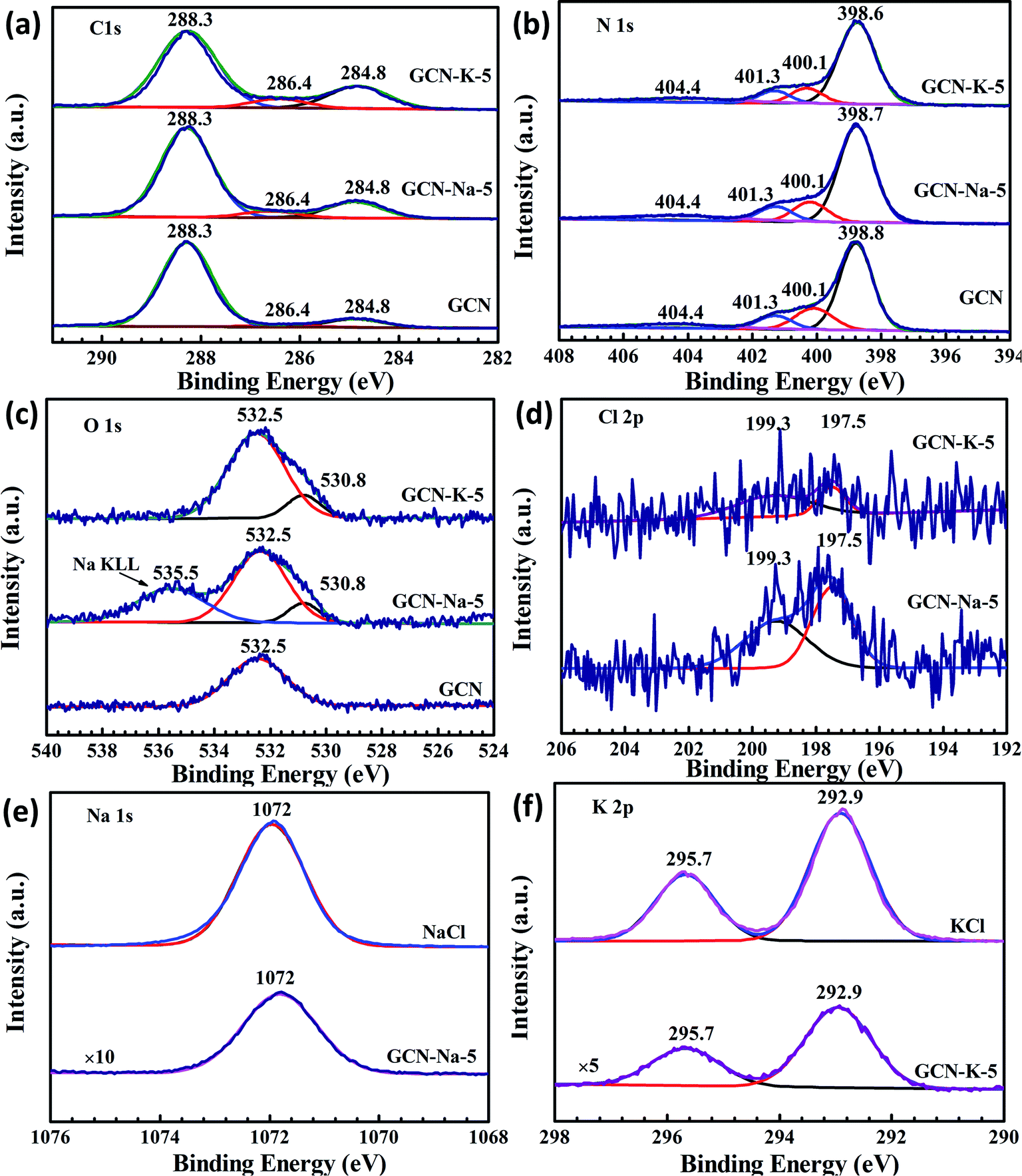 | ||
| Fig. 4 High resolution XPS spectra. C 1s (a), N 1s (b) and O 1s (c) XPS spectra for GCN, GCN-Na-5 and GCN-K-5; Cl 1s (d) XPS spectra for GCN-Na-5 and GCN-K-5; Na 1s (e) XPS spectra for GCN-Na-5 and NaCl; and K 2p (f) XPS spectra for GCN-K-5 and KCl. | ||
Considering that K-doping caused greater variation in both the crystalline structure and chemical structure than Na-doping, the elemental compositions of GCN and the ion-doped GCNs were further explored via the combination of EDS, XPS, EA, and ICP-AES, as listed in Tables S2–S5,† respectively. According to the EDS (Table S2†) and EA (Table S4†) data, the values of the C/N ratio present an increasing trend in the order of GCN, GCN-Na-0.5, GCN-Na-5, GCN-K-0.5 and GCN-K-5, again indicating that K+ has a stronger influence on GCN than Na+. Furthermore, according to the EDS data in Table S2,† it can be found that the content of Na element in GCN-Na-0.5 (2.46 at%) is very close to that of GCN-Na-5 (2.49 at%) and the content of K element in GCN-K-0.5 (2.47 at%) is also close to that of GCN-K-5 (3.01 at%). The XPS (Table S3†) and ICP-AES (Table S5†) measurements further show that the Na element content in GCN-Na-0.5 and GCN-Na-5 is close and the K element content in GCN-K-0.5 and GCN-K-5 is also close, which suggest that both Na+ and K+ have the ability to interact particularly easily with GCN. Besides, the reason why the Na or K element content is not proportional to their respective initial feeding amount is still under investigation.
Based on the above analysis, it can be concluded that the main framework of GCN was still maintained after doping with Na+ or K+, while K-doping has stronger influence on its structure variation. According to the interlayer distances of GCN, GCN-Na-5 and GCN-K-5 derived from the XRD and TEM measurements in Fig. 2 and diameters of Na+ (0.190 nm) and K+ (0.266 nm),29 the proposed structures of GCN, GCN-Na-5 and GCN-K-5 from the side view are presented in Scheme 1. Compared to that of Na+, the diameter of K+ is closer to the interlayer distance of GCN. Thus, this suggests that there may be stronger interaction between K+ and the interlayer lattices of GCN, which will give rise to a narrower interlayer distance in K-doped GCN, as illustrated in Scheme 1, and further have a greater impact on the chemical structure of GCN-K-5.
 | ||
| Scheme 1 Illustration of the proposed structures of GCN, GCN-Na-5 and GCN-K-5 from the side view. | ||
To examine whether the size effect of K+ has an impact on the optical and electronic properties, GCN and the ion-doped GCNs were further investigated by UVDRS, valence band (VB) XPS and photoelectrochemistry measurements. The UVDRS spectra of GCN and ion-doped GCNs are shown in Fig. 5(a). All the ion-doped GCNs exhibit a similar optical absorption envelop to that of GCN, indicating that the intrinsic backbone structure of GCN did not change after doping with Na+ or K+. Furthermore, the optical bandgap widths of all the samples (Table S6†) can be estimated from their Tauc plots, as shown in the inset of Fig. 5(a). It is clear that the optical bandgap change for each ion-doped sample is negligible compared with GCN. Fig. 5(b) presents the VB-XPS data, and the VB values of each sample were derived from the results shown in Fig. 5(b). Fig. 5(c) shows the Mott–Schottky plots of all the samples and the derived flat band potentials are also displayed. The positive slopes of the Mott–Schottky plots shown in Fig. 5(c) indicate that GCN and the ion-doped GCNs are both n-type semiconductors, and thus their conduction band minimum (CBM) should be close to their respective flat band potential. Combining the flat band potentials and VB values of each sample, the corresponding valence band maximum (VBM) was deduced, and further combining the respective optical bandgap width, the CBM of each sample was also derived.45,46 Here, GCN is taken as an example to illustrate how its electron band structure was constructed, as shown in Fig. 5(d). Firstly, using the flat band potential (−0.95 V) of GCN obtained from its Mott–Schottky plot (Fermi energy level) in Fig. 5(c) and the VB value (2.43 eV) of GCN obtained from its VB-XPS spectrum in Fig. 5(b), the VBM (1.48 V) of GCN was derived. Also, further using the optical bandgap (2.80 eV) of GCN obtained from Fig. 5(a), the CBM (−1.32 V) of GCN was derived. Similarly, the electronic band structures for the ion-doped GCNs were obtained, as schematically illustrated in Fig. 5(d). As shown in Fig. 5(d), compared to GCN-Na-5, both the CBM and VBM of GCN-K-5 are upshifted, which may be due to the charge redistribution arising from the stronger interaction between K+ and the interlayer lattices in GCN-K-5, as suggested above. However, the high-resolution XPS data suggests that the chemical state of Na+ or K+ presented little change upon their doping in GCN, perhaps because XPS was insensitive to detect the chemical surrounding change of Na+ or K+ after their doping in GCN.
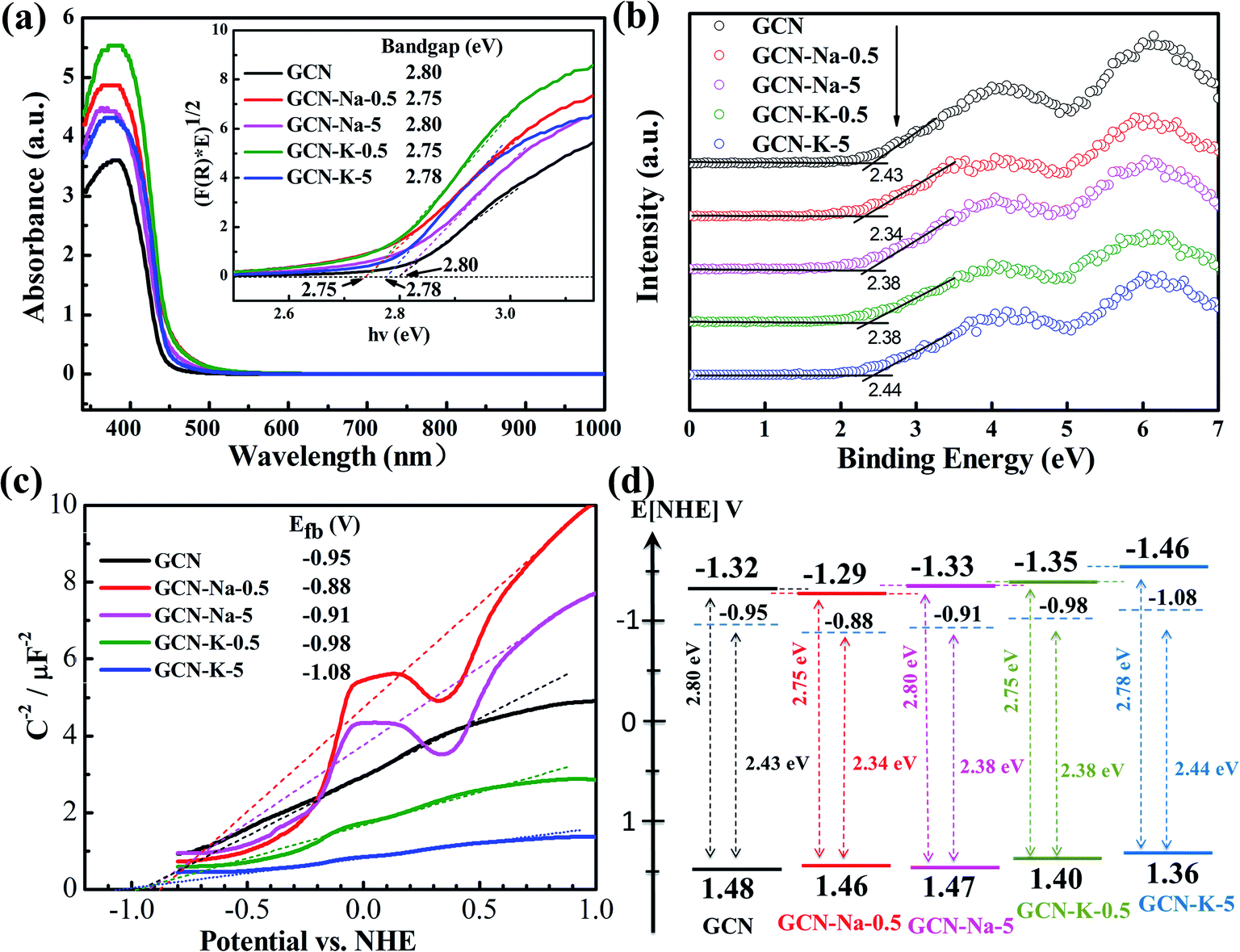 | ||
| Fig. 5 UV-Vis diffuse reflectance spectra of GCN and ion-doped GCNs (a). Inset: Tauc plots transformed from the respective UV-Vis diffuse reflectance spectra. Valence band XPS spectra of GCN and ion-doped GCNs (b). Mott–Schottky plots of GCN and ion-doped GCNs at 1500 Hz (c). Band structure diagram for GCN and ion-doped GCNs (d). | ||
Photoelectrochemical measurements were conducted to investigate the charge transport properties of GCN and the ion-doped GCNs. Fig. 6(a) shows the photocurrent densities of GCN, GCN-Na-5 and GCN-K-5, and it is obvious that GCN-K-5 exhibits a significant promotion in photocurrent density, demonstrating that K-doping is a key factor to improve the electronic conductivity of GCN-K-5. Fig. 6(b) shows that the semiconductor GCN-K-5 possess the smallest arc radius in the Nyquist curve from electrochemical impedance spectroscopy, which is consistent with its photocurrent density data. Therefore, the results again confirmed that K-doping has a greater impact on the optical and electronic properties of GCN than Na-doping.
 | ||
| Fig. 6 Transient photocurrent response of GCN, GCN-Na-5 and GCN-K-5 under visible light (420 nm) illumination (a). Electrochemical impedance spectra (EIS) of GCN, GCN-Na-5 and GCN-K-5 (b). Fluorescence spectra of GCN, GCN-Na-5 and GCN-K-5 under 370 nm light excitation (c). Photocatalytic H2 evolution rate of GCN and ion-doped GCNs under visible light irradiation (d). Inset in (c): fluorescence decays of GCN, GCN-Na-5 and GCN-K-5. Excitation wavelength, 375 nm and detection wavelength, 470 nm. | ||
To further investigate the properties of the photogenerated carriers in GCN and ion-doped GCNs, both the fluorescence emission and fluorescence decay of GCN and ion-doped GCNs were measured, as shown in Fig. 6(c), S6 and S7.† The fluorescence decay lifetimes of all the samples are listed in Table S7,† and the fluorescence decay lifetime of GCN is consistent with previous report.47 As shown in Fig. 6(c), the fluorescence emission intensity of each ion-doped GCN is much smaller than that of GCN, which is consistent with its greatly reduced fluorescence decay lifetime, indicating the more efficient separation of photogenerated electrons and holes in the ion-doped GCNs compared to that of the pristine GCN. Besides, the K-doped samples presented relatively faster fluorescence decay than that of the Na-doped samples, also indicating that K+ has stronger interaction with the GCN interlayer than Na+.
The photocatalytic H2 evolution with GCN and ion-doped GCNs was evaluated by loading 3 wt% Pt as a co-catalyst and using triethanolamine as the sacrificial reagent under visible light irradiation (>420 nm). The average H2 evolution rates of GCN and ion-doped GCNs are displayed in Fig. 6(d) and the measurements are summarized in Table S8.† The H2 evolution rate of GCN (4.8 μmol h−1) is consistent with the previous report (∼5.1 μmol h−1) under similar experimental conditions.12 As show in Fig. 6(d), the K-doped samples resulted in a higher H2 evolution rate than that of the Na-doped samples, and especially GCN-K-5 exhibited the highest photocatalytic activity, and its average H2 evolution rate is about 4 times higher than that of GCN-Na-5. Considering that the structure, optical and electronic properties present a more evident variation in the K-doped GCN than that in the Na-doped GCN, and the diameter of K+ is closer to the interlayer distance of GCN, it is reasonable to propose a stronger interaction between K+ and the interlayer lattices of GCN. Thus, the stronger interaction may give rise to charge redistribution in the K-doped GCN and generate a more negative CBM. Consequently, GCN-K-5 achieved the highest H2 evolution rate among the samples. In addition, the K element content of GCN-K-0.5 is similar to that of GCN-K-5, as revealed by the chemical composition measurement; however, their XRD, optical and electronic properties as well as H2 evolution rates are quite different, which needs further investigation.
4. Conclusion
In summary, Na-doped and K-doped GCNs were prepared via the simple thermal polymerization of melamine and NaCl or KCl, respectively. Compared to the pristine GCN, the ion-doped GCNs presented a less condensed morphology, while the intrinsic structure of GCN was still maintained after doping. The XRD and TEM data indicate that the interlayer distance of GCN after K-doping became narrower than that after Na+ doping. Furthermore, both the FTIR and Raman spectra of the ion-doped GCNs indicate that K+ has a stronger influence on the chemical structural variation in GCN than that of Na+, and cyano defects were introduced in GCN after ion-doping. Also, the ratio of the intensity of the peak at 707 cm−1 to the peak at 726 cm−1 was an indicator of the structural change in GCN according to the Raman measurements. The chemical composition data show that both Na+ and K+ could easily interact with GCN, while K-doping caused a greater change in the C/N ratio than Na-doping. Moreover, the UVDRS data of GCN and ion-doped GCNs reveals that there was a negligible optical bandgap change caused by Na-doping or K-doping, while their derived electronic band structure illustrates that there was a clear up-shift in the CBM and VBM of GCN-K-5. Finally, the most significant promotion in photocurrent density, the most heavily reduced florescence, and the fastest florescence decay were observed for GCN-K-5. Consequently, GCN-K-5 yielded an H2 evolution rate around 4 times higher than that of GCN-Na-5 under visible light irradiation. This work highlights the importance of the K+ size effect on the structure variation and also the optical and electrical properties of GCN to improve the photocatalytic activity of K-doped GCN.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21773306 and 21273280).References
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717–6731 RSC.
- W. J. Ong, L. L. Tan, Y. H. Ng, S. T. Yong and S. P. Chai, Chem. Rev., 2016, 116, 7159–7329 CrossRef CAS PubMed.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS PubMed.
- T. S. Miller, A. B. Jorge, T. M. Suter, A. Sella, F. Cora and P. F. McMillan, Phys. Chem. Chem. Phys., 2017, 19, 15613–15638 RSC.
- J. Jiang, S. Cao, C. Hu and C. Chen, Chin. J. Catal., 2017, 38, 1981–1989 CrossRef CAS.
- V. Hasija, P. Raizada, A. Sudhaik, K. Sharma, A. Kumar, P. Singh, S. B. Jonnalagadda and V. K. Thakur, Appl. Mater. Today, 2019, 15, 494–524 CrossRef.
- X. Liu, R. Ma, L. Zhuang, B. Hu, J. Chen, X. Liu and X. Wang, Crit. Rev. Environ. Sci. Technol., 2020, 51, 751–790 CrossRef.
- S. Patnaik, D. P. Sahoo and K. Parida, Carbon, 2021, 172, 682–711 CrossRef CAS.
- Z. Ge, A. Yu and R. Lu, Mater. Lett., 2019, 250, 9–11 CrossRef CAS.
- Z. Chen, A. Savateev, S. Pronkin, V. Papaefthimiou, C. Wolff, M. G. Willinger, E. Willinger, D. Neher, M. Antonietti and D. Dontsova, Adv. Mater., 2017, 29, 170555 Search PubMed.
- Y. Li, H. Xu, S. Ouyang, D. Lu, X. Wang, D. Wang and J. Ye, J. Mater. Chem. A, 2016, 4, 2943–2950 RSC.
- Y. Wang, S. Zhao, Y. Zhang, J. Fang, Y. Zhou, S. Yuan, C. Zhang and W. Chen, Appl. Surf. Sci., 2018, 440, 258–265 CrossRef CAS.
- Y. Xu, X. He, H. Zhong, D. J. Singh, L. Zhang and R. Wang, Appl. Catal., B, 2019, 246, 349–355 CrossRef CAS.
- W. Wang, Z. Shu, J. Zhou, D. Meng, Z. Zhao and T. Li, J. Mater. Chem. A, 2020, 8, 6785–6794 RSC.
- F. Guo, J. Chen, M. Zhang, B. Gao, B. Lin and Y. Chen, J. Mater. Chem. A, 2016, 4, 10806–10809 RSC.
- F. Yang, D. Liu, Y. Li, L. Cheng and J. Ye, Appl. Catal., B, 2019, 240, 64–71 CrossRef CAS.
- M. Wu, J. M. Yan, X. N. Tang, M. Zhao and Q. Jiang, ChemSusChem, 2014, 7, 2654–2658 CrossRef CAS PubMed.
- X. Liu, X. Wu, D. Long, X. Rao and Y. Zhang, J. Photochem. Photobiol., A, 2020, 391, 112337 CrossRef CAS.
- Z. Fang, D. Li, R. Chen, Y. Huang, B. Luo and W. Shi, ACS Appl. Mater. Interfaces, 2019, 11, 22255–22263 CrossRef CAS PubMed.
- Y. Guo, T. Chen, Q. Liu, Z. Zhang and X. Fang, J. Phys. Chem. C, 2016, 120, 25328–25337 CrossRef CAS.
- P. Deng, L. Shi, H. Wang and W. Qi, Colloids Surf., A, 2020, 601, 125023 CrossRef CAS.
- Z. Shu, Y. Wang, W. Wang, J. Zhou, T. Li, X. Liu, Y. Tan and Z. Zhao, Int. J. Hydrogen Energy, 2019, 44, 748–756 CrossRef CAS.
- A. Yuan, H. Lei, Z. Wang and X. Dong, J. Colloid Interface Sci., 2020, 560, 40–49 CrossRef CAS PubMed.
- W. Xu, X. Zhao, X. An, S. Wang, J. Zhang, Z. Li, W. Wu and M. Wu, ACS Appl. Mater. Interfaces, 2020, 12, 48526–48532 CrossRef CAS PubMed.
- W. Wu, W. Xu, X. An, L. Wang, J. Zhang, Z. Li and M. Wu, Appl. Catal., B, 2018, 229, 204–210 CrossRef CAS.
- Y. Xu, C. Qiu, X. Fan, Y. Xiao, G. Zhang, K. Yu, H. Ju, X. Ling, Y. Zhu and C. Su, Appl. Catal., B, 2020, 268, 118457 CrossRef CAS.
- J. Zhang, S. Hu and Y. Wang, RSC Adv., 2014, 4, 62912–62919 RSC.
- L. Zhang, N. Ding, M. Hashimoto, K. Iwasaki, N. Chikamori, K. Nakata, Y. Xu, J. Shi, H. Wu, Y. Luo, D. Li, A. Fujishima and Q. Meng, Nano Res., 2018, 11, 2295–2309 CrossRef CAS.
- H. Yu, R. Shi, Y. Zhao, T. Bian, Y. Zhao, C. Zhou, G. I. N. Waterhouse, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2017, 29, 1605148 CrossRef PubMed.
- S. Sun, J. Li, J. Cui, X. Gou, Q. Yang, Y. Jiang, S. Liang and Z. Yang, Int. J. Hydrogen Energy, 2019, 44, 778–787 CrossRef CAS.
- X. Li, X. Sun, L. Zhang, S. Sun and W. Wang, J. Mater. Chem. A, 2018, 6, 3005–3011 RSC.
- P. Hu, C. Chen, R. Zeng, J. Xiang, Y. Huang, D. Hou, Q. Li and Y. Huang, Nano Energy, 2018, 50, 376–382 CrossRef CAS.
- W. Fang, J. Liu, L. Yu, Z. Jiang and W. Shangguan, Appl. Catal., B, 2017, 209, 631–636 CrossRef CAS.
- G. Liu, S. Yan, L. Shi and L. Yao, Front. Chem., 2019, 7, 639 CrossRef CAS PubMed.
- J. Wen, R. Li, R. Lu and A. Yu, Chem.–Asian J., 2018, 13, 1060–1066 CrossRef CAS PubMed.
- H. Zhang and A. Yu, J. Phys. Chem. C, 2014, 118, 11628–11635 CrossRef CAS.
- D. Vidyasagar, S. G. Ghugal, S. S. Umare and M. Banavoth, Sci. Rep., 2019, 9, 7186 CrossRef PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Mueller, R. Schloegl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
- Y. Dedkov, H. Y. Xu, L. C. Wu, H. Zhao, L. G. Jin and S. Y. Qi, PLoS One, 2015, 10, e0142616 CrossRef PubMed.
- H. Wang, X. Zhang, J. Xie, J. Zhang, P. Ma, B. Pan and Y. Xie, Nanoscale, 2015, 7, 5152–5156 RSC.
- J. Y. Kang, W. Ha, H. X. Zhang and Y. P. Shi, Talanta, 2020, 215, 120916 CrossRef CAS PubMed.
- X. L. Wang, W. Q. Fang, H. F. Wang, H. Zhang, H. Zhao, Y. Yao and H. G. Yang, J. Mater. Chem. A, 2013, 1, 14089–14096 RSC.
- W. Xu, X. An, Q. Zhang, Z. Li, Q. Zhang, Z. Yao, X. Wang, S. Wang, J. Zheng, J. Zhang, W. Wu and M. Wu, Int. J. Hydrogen Energy, 2019, 7, 12351–12357 CAS.
- W. Wang, P. Xu, M. Chen, G. Zeng, C. Zhang, C. Zhou, Y. Yang, D. Huang, C. Lai, M. Cheng, L. Hu, W. Xiong, H. Guo and M. Zhou, ACS Sustainable Chem. Eng., 2018, 6, 15503–15516 CrossRef CAS.
- H. Huang, K. Xiao, N. Tian, F. Dong, T. Zhang, X. Du and Y. Zhang, J. Mater. Chem. A, 2017, 5, 17452–17463 RSC.
- H. Zhang, S. Li, R. Lu and A. Yu, ACS Appl. Mater. Interfaces, 2015, 7, 21868–21874 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01395e |
| This journal is © The Royal Society of Chemistry 2021 |