
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZeolitic imidazole framework derived N-doped porous carbon/metal cobalt nanoparticles hybrid for oxygen electrocatalysis and rechargeable Zn–air batteries†
Xia Liua,
Yuanyuan Ma b,
Yongliang Caia,
Song Hua,
Jian Chena,
Zhaolin Liu*c and
Zhijuan Wang*a
b,
Yongliang Caia,
Song Hua,
Jian Chena,
Zhaolin Liu*c and
Zhijuan Wang*a
aInstitute of Advanced Synthesis (IAS), School of Chemistry and Molecular Engineering (SCME), Jiangsu National Synergetic Innovation Center for Advanced Materials (SICAM), Nanjing Tech University, 30 South Puzhu Road, Nanjing 211816, PR China. E-mail: ias_zjwang@njtech.edu.cn
bDepartment of Materials Science and Engineering, National University of Singapore, 117574, Singapore
cInstitute of Materials Research and Engineering, Agency for Science, Technology and Research (A*STAR), 2 Fusionopolis Way, #08-03 Innovis 138634, Singapore. E-mail: zl-liu@imre.a-star.edu.sg
First published on 27th April 2021
Abstract
Bifunctional electrocatalysts with high catalytic property for the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER) are vital for high-performance zinc–air batteries (ZnABs). In this study, an efficient bifunctional electrocatalyst with hollow structure (C–N/Co (1/2)) has been successfully prepared through carbonization of ZIF-8@ZIF-67 and evaporation of Zn ions at high temperature. With Co nanoparticles encapsulated by an N-doped porous carbon matrix, the catalyst exhibits excellent stability in aqueous alkaline solution over an extended period and good tolerance to the methanol crossover effect. The integration of an N-doped graphitic carbon outer shell and Co nanoparticles enables high ORR and OER activity, as evidenced by ZnAB using the catalyst C–N/Co (1/2) in an air cathode.
Introduction
Given the rapid growth of the global energy demand and increasingly severe pollution of the environment, the development of highly efficient materials and technologies for energy storage and conversion is of great significance.1–3 Among various technologies developed, Zn–air batteries have proven a promising battery system owing to the high theoretical energy density, the appropriate operating voltage (∼1.65 V), abundant zinc resources, and free supply of oxygen for the air cathode.4–6 However, the inherent sluggish kinetics of the oxygen reduction reaction (ORR) and oxygen evolution reaction (OER), which are the basis of Zn–air batteries,7 have hampered its development and applications.8Noble metals (Pt for ORR, IrO2 for OER, etc.) and their derivatives constitute the state-of-the-art ORR and OER catalysts.7–9 Unfortunately, their scarcity on the earth, insufficient stability, and high cost make their practical commercialization difficult. As an alternative, non-precious metal-based materials with carbon support, which also show potentially good electrocatalytic performances, have recently drawn considerable attention.8,10–18 In this regard, metal–organic framework (MOF)-based catalysts have become the focus of electrocatalyst design, given its remarkable advantages including the porous structure, the existence of abundant transition metal, and the easy modification of architecture and composition.1,15,19–21 Moreover, the evenly dispersed metal species in the MOF-derived materials are able to function as active sites for electrochemical reactions.12,22–24 It has been reported that Co-based MOF electrocatalysts have shown good catalytic activity, while the dissatisfactory bifunctional ORR/OER property,25,26 the poor electrical conductivity, and aggregation of nanoparticles19 still limit their practical application in Zn–air batteries.
In order to address these issues, our group has reported a series of core–shell carbon materials derived from MOF25 and its hybrid26 as bifunctional electrocatalysts. Stability and catalytic activities in these materials were remarkable. However, either OER25 or ORR26 cannot meet the requirements as a bifunctional electrocatalyst.
In this work, we designed a new class of C–N/Co-based MOF that could be derived from core–shell ZIF-8@ZIF-67-based zeolitic imidazolate frameworks (ZIFs)27,28 precursor, wherein ZIF-8 ([Zn(2-MeIm)2]n, 2-MeIm = 2-methylimidazole) serves as the core and self-sacrificed template to formulate and control the morphology and size of the shell of ZIF-67 ([Co(2-MeIm)2]n). As zinc metal has a relatively low boiling temperature (907 °C),15,29 it can be evaporated under high-temperature pyrolysis. Therefore, a porous carbon network was produced, which is beneficial to enhance electrocatalytic activity. During this process, Co ions can be pushed onto the surface, which will provide more catalytically active sites for ORR and OER. Meanwhile, the possibility of Co atoms aggregation has been reduced as the concentration of Co atoms in the shell decreases. As a result, the synthesized C–N/Co-based MOF was found to exhibit good performance as a bifunctional electrocatalyst in the Zn–air battery.
Experimental
Chemicals and materials
Zinc nitrate hexahydrate (Zn(NO3)2·6H2O, 99%, Adamas), cobalt nitrate hexahydrate (Co(NO3)2·6H2O, 97.7%, Alfa Aesar), 2-methylimidazole (2-MeIm, 99%, Acros), methanol (CH3OH, 99.9%, Adamas), potassium hydroxide (KOH, 85%, Nanjing Reagent), Nafion (5 wt%, Alfa Aesar), carbon paper (SGL 10BC, Germany), 20 wt% platinum supported on Vulcan XC-72 carbon (Pt/C, Premetek) and 20 wt% iridium on Vulcan XC-72 carbon (Ir/C, Premetek) were used as received without any further purification. Ultrapure water (≥18 MΩ cm−1), obtained from a Milli Q Plus water purification system (Millipore, USA), was used throughout the experiment.Characterizations
The surface morphologies of the samples were obtained from scanning electron microscopy (SEM, FEI Quanta 250 FEG) at an accelerating voltage of 15 kV. The inner nanostructures were characterized by a HT7700 Transmission Electron Microscopy operated at 100 kV and a JSM-2100F High-Resolution Transmission Electron Microscopy operated at 200 kV. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo ESCALAB 250Xi spectrometer with a monochrome aluminum Kα radiation. Powder X-ray diffraction (XRD) patterns were conducted using a Bruker D8 Advanced diffractometer with copper Kα radiation source. Nitrogen adsorption–desorption isotherm measurements were used to analyze the specific surface area and pore size distribution of the samples.Synthesis of ZIF-8 and nitrogen-doped carbon (NC)
2-MeIm (6.16 g, 0.075 mol) was first dissolved in 150 mL of methanol. Another solution with 150 mL of methanol and zinc nitrate hexahydrate (Zn(NO3)2·6H2O) (5.95 g, 0.02 mol) was subsequently poured into the above solution under stirring. The mixed solution was kept stirring for 24 h at room temperature. Then the white precipitates were collected by centrifugation and washed with methanol four times and dried at 60 °C in a vacuum to obtain ZIF-8. The ZIF-8 was calcined at 920 °C in a tube furnace for 3 h with a ramp rate of 2 °C min−1 in argon to get nitrogen-doped carbon (NC).Synthesis of ZIF-8@ZIF-67 and C–N/Co (x)
In a typical synthesis, cobalt nitrate hexahydrate (Co(NO3)2·6H2O) (5.82 g, 0.02 mol) and 2-MeIm (6.16 g, 0.075 mol) were dissolved in 100 mL of methanol to form clear solutions, respectively. ZIF-8 (0.50 g) was dispersed in 100 mL of methanol by sonication for 5 min. Then, the methanol solution of Co(NO3)2·6H2O was first poured into the suspension of ZIF-8 and the methanol solution of 2-MeIm was subsequently poured into the above mixture solution under stirring. After stirring for 24 h at room temperature, the resulting bright purple precipitates were collected by centrifugation, washed with methanol four times and dried at 60 °C in a vacuum. Finally, the ZIF-8@ZIF-67 was calcined at 920 °C in a tube furnace for 3 h with a ramp rate of 2 °C min−1 in argon to obtain sample C–N/Co. In order to explore the influence of Co content on the properties of catalysts, a series of C–N/Co (x) (x = 1/2, 1/3 and 2/3, which denotes the molar ratio of Co(NO3)2·6H2O between the sample and C–N/Co) catalysts with different cobalt contents were prepared using the similar processes by changing the amount of Co(NO3)2·6H2O. In addition, the sample C–N/Co (1/2) was also pyrolyzed at 820 °C using the corresponding ZIF-8@ZIF-67 in argon.Synthesis of ZIF-67 and graphitic carbon (GC)
ZIF-67 was prepared similar to that of ZIF-8@ZIF-67 except in the absence of ZIF-8. Then, the ZIF-67 was calcined at 920 °C in a tube furnace for 3 h with a ramp rate of 2 °C min−1 in argon to get highly graphitic carbon (GC).Electrochemical measurements
All of the electrochemical measurements were performed on an Autolab AUT302N.FRA32M.V (Metrohm) with a standard three-electrode setup using a glass carbon electrode (GCE, 5 mm in diameter), a graphite rod and a Hg/HgO electrode (1 M KOH) as the working, counter and reference electrode, respectively. To prepare catalyst ink, 5 mg of catalyst and 20 μL of Nafion solution (5 wt%) were dispersed in 980 μL of water–ethanol solution (volume ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) to generate homogeneous ink assisted by ultrasound for 2 h. Then, 10 μL of the suspension was dropped onto a polished glass carbon (GC) rotating disk electrode (RDE) surface (loading 0.254 mg cm−2) and dried in air at room temperature. Linear sweep voltammetry (LSV) measurements were performed at a scan rate of 5 mV s−1. All measured potentials in this research were referenced to reversible hydrogen electrode (RHE) by ERHE = EHg/HgO + 0.059 × pH + 0.098. Before each measurement, high-purity N2 or O2 flow was bubbled through the electrolyte for at least 30 min to obtain a N2- or O2-saturated electrolyte solution. During the experiment, the high-purity N2/O2 flow was passed over the electrolyte in order to maintain the N2/O2 saturation.
:1) to generate homogeneous ink assisted by ultrasound for 2 h. Then, 10 μL of the suspension was dropped onto a polished glass carbon (GC) rotating disk electrode (RDE) surface (loading 0.254 mg cm−2) and dried in air at room temperature. Linear sweep voltammetry (LSV) measurements were performed at a scan rate of 5 mV s−1. All measured potentials in this research were referenced to reversible hydrogen electrode (RHE) by ERHE = EHg/HgO + 0.059 × pH + 0.098. Before each measurement, high-purity N2 or O2 flow was bubbled through the electrolyte for at least 30 min to obtain a N2- or O2-saturated electrolyte solution. During the experiment, the high-purity N2/O2 flow was passed over the electrolyte in order to maintain the N2/O2 saturation.
In ORR measurements, cyclic voltammetry (CV) test was executed in N2- and O2-saturated 0.1 M KOH aqueous solution at a scan rate of 50 mV s−1, respectively. Linear sweep voltammetry (LSV) curves at various rotating speeds from 400 to 2400 rpm and chronoamperometry measurements at 0.6 V (vs. RHE) with a rotation rate of 1600 rpm were recorded in O2-saturated 0.1 M KOH. The number of transferred electrons (n) per oxygen molecule involved in ORR was calculated according to the Koutecky–Levich equations:
| J−1 = JL−1 + JK−1 = B−1ω−1/2 + JK−1 |
| B = 0.2nF(D0)2/3υ−1/6C0 |
485 C mol−1), D0 is the diffusion coefficient of O2 in 0.1 M KOH (1.9 × 10−5 cm2 s−1), υ is the kinetic viscosity of the electrolyte (0.01 cm2 s−1), and C0 is the bulk concentration of O2 (1.2 × 10−6 mol cm−3); ω is the electrode rotating rate. The constant 0.2 is adopted when the rotation speed is expressed in rpm. The generated peroxide ion content during ORR was determined by a rotating ring-disk electrode (RRDE) setup on an Autolab AUT302N.FRA32M.V (Metrohm). RRDE experiment was carried out at a rotating rate of 1600 rpm. The formation of peroxide ions (HO2−) and n during the ORR can be calculated according to following equations:30,31| n = 4ID/(ID + IR/N) |
| HO2−% = 100 × 2IR/(NID + IR)% = 100 × (4 − n)/2% |
For OER, linear sweep voltammograms were recorded in O2-saturated 1.0 M KOH with a rotation rate of 1600 rpm. The resulted polarization curves involved OER were corrected by iR-compensation, where R was the electrolyte ohmic resistance and can be measured from the electrochemical impedance spectroscopy (EIS) in the frequency range from 105 Hz to 0.01 Hz with an excitation amplitude of 5 mV. The electrochemical double-layer capacitance Cdl was determined by measuring CVs with multiple scan rates in a nonfaradaic potential region between 1.2 V and 1.3 V.
Zn–air battery (ZnAB) assembly and test
To evaluate the catalytic performance and durability of the C–N/Co (1/2) catalyst, battery cycling tests were performed using a home-made zinc–air cell device. Commercial Ir/C was chosen as a benchmark. Therefore, two Zn–air batteries will be assembled and tested. In brief, two cathodes were prepared by spreading C–N/Co (1/2) and commercial Ir/C slurries onto carbon paper, respectively, to achieve a loading density of 4.0 mg cm−2, with the working area of the air cathode being 4 cm2. A metallic zinc plate was employed as the anode, with a 6.0 M KOH aqueous solution containing 20.0 g L−1 ZnCl2 as the electrolyte. The battery tests were carried out at 25 °C on a Maccor 4300 battery tester. In each test cycle, the cell was discharged at a constant current of 20 mA over 4 h and then charged at a constant current of 10 mA over 8 h.Results and discussion
In this study, core–shell ZIF-8@ZIF-67 materials with different Co contents have been synthesized by seed epitaxial growth of ZIF-67 on ZIF-8, which was schematically depicted in Scheme 1. The content of Co ions in ZIF-67 was able to be easily changed by modifying the concentration of Co(NO3)2·6H2O in the reactants. C–N/Co and a series of C–N/Co (x) (x = 1/3, 1/2 and 2/3) catalysts have been fabricated by the pyrolysis of the corresponding ZIF-8@ZIF-67 core–shell structures at 920 °C under Ar atmosphere. During the pyrolysis, Co2+ ions were reduced to metal Co nanoparticles (NPs) by the reductive gas (such as H2 and NH3) released from the linkers.4,5,32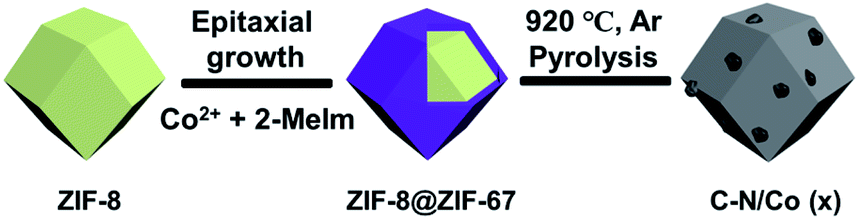 | ||
| Scheme 1 Schematic illustration of the synthesis procedure of C–N/Co (x). | ||
The surface morphology of C–N/Co (1/2) was characterized by SEM. As shown in Fig. 1a, the synthesized C–N/Co (1/2) inherits a uniform rhombic dodecahedral skeleton from the core–shell precursor (Fig. S1a†) with a distorted and rough surface. The magnified SEM image of a single particle with a collapsed shell (inset of Fig. 1a) demonstrates the hollow structure because of the evaporation of Zn and decomposition of 2-MeIm.4,33 From the larger magnification of SEM images (Fig. S1b and c†), it can be seen that Co NPs were pushed onto the surfaces of C–N networks, which would be further verified by TEM images. Moreover, numerous carbon nanotubes (CNTs) derived from the carbon matrix and wrapped the surface, which comes from the thermal decomposition of 2-MeIm under the catalytic effect of Co NPs.32 These CNTs provide an effective and convenient tunnel for mass and electron transfer and thus improve the electrical conductivity of the final product.15,34
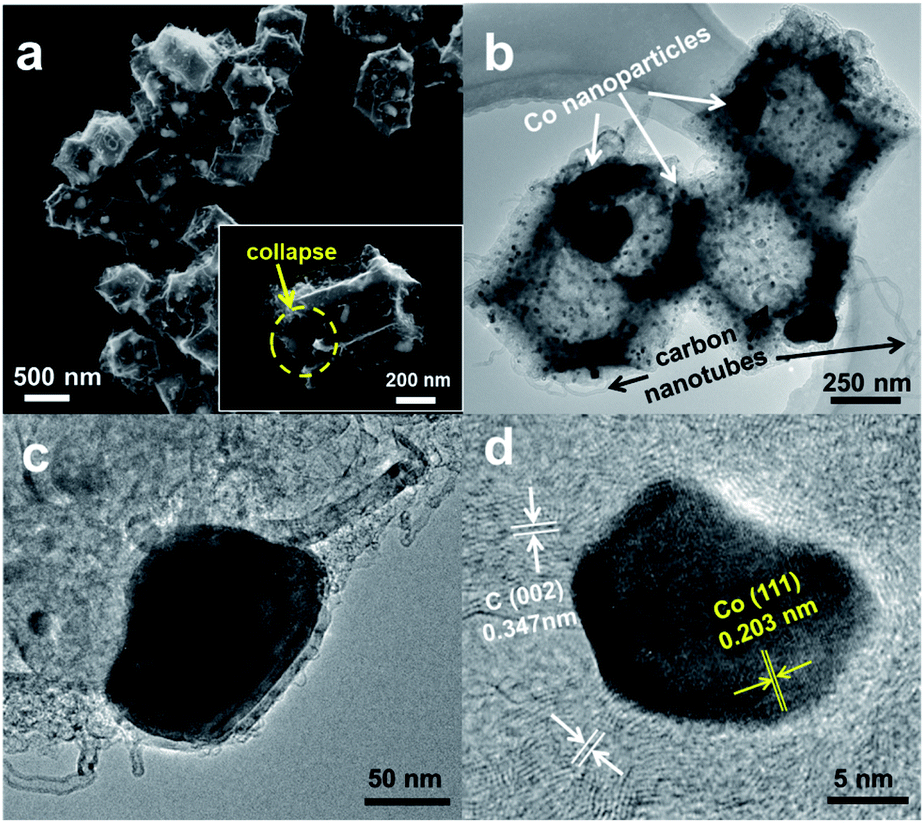 | ||
| Fig. 1 (a) SEM, (b and c) TEM and (d) high-resolution TEM images of C–N/Co (1/2). Inset of (a): the magnification SEM image of single C–N/Co (1/2) particle. | ||
TEM image (Fig. 1b) further reveals the hollow structure of C–N/Co (1/2), which is consistent with the SEM result (inset of Fig. 1a). The magnified TEM image in Fig. 1c demonstrates that few large Co NPs (∼100 nm) (Fig. 1c) and many small Co NPs (∼15 nm) (Fig. 1d) are encapsulated in the carbon matrix, which is consistent with the SEM images. High-resolution TEM (HRTEM) images (Fig. 1d and S1d†) showed that the lattice distances of 0.347 nm and 0.203 nm corresponded to the (002) crystal faces of graphitic carbon and (111) planes of nanocrystalline cobalt, respectively. Importantly, it can be seen clearly that Co NPs are well wrapped by the carbon layers, which could protect these Co NPs from agglomerating and being excessively oxidized by the oxygen in ambient air.
The powder XRD was performed to analyze the crystal structure of C–N/Co (1/2) (Fig. 2). A relatively small peak located at 25.9° can be assigned to the (002) diffraction planes of graphite carbon (JCPDS no. 75-1621), which agrees with the HRTEM result (Fig. 1d). Besides, three characteristic peaks at 44.2°, 51.7° and 75.9° match well with the (111), (200) and (220) crystalline faces of metal Co (JCPDS no. 15-0806). These results verify that Co ions have been successfully reduced to a zero valent state.
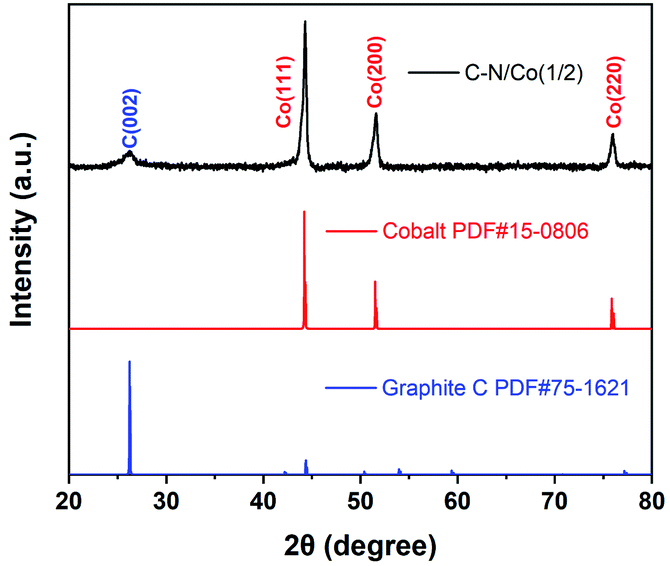 | ||
| Fig. 2 Powder XRD pattern of C–N/Co (1/2). | ||
XPS was employed to investigate the chemical composition and elemental valence state of C–N/Co (1/2) (Fig. 3). The survey spectrum suggests the existence of C, N, Co and O elements in C–N/Co (1/2) (Fig. 3a). Notably, the absence of Zn peak displays complete evaporation of Zn ions during the pyrolysis, which agrees with XRD results (Fig. 2). The high-resolution XPS spectrum of the C 1s peak can be deconvolved into two peaks at 284.7 and 285.7 eV (Fig. 3b), corresponded to C–C and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N species, respectively.20 It confirms that N heteroatoms have been successfully doped into carbon matrix. The majority of C–C bonds are responsible for the excellent electron conductivity,35 which is consistent to the SEM and TEM results (Fig. 1d). The deconvolved N 1s spectrum exhibits five peaks, corresponding to pyridinic N (398.5 eV), Co–N (399.1 eV), pyrrolic N (400.2 eV), graphitic N (401.6 eV), and oxidized N (403.4 eV) (Fig. 3c), respectively.22 It has been reported that pyridinic N, graphitic N and Co–N bonds have a positive effect on OER and ORR performances.20,36 Particularly, Co–N bond can accelerate the charge transfer between carbon and metal/N-dopants, providing additional sites for ORR process.22,37 In the high-resolution Co 2p spectrum (Fig. 3d), two main peaks at 796.2 and 780.6 eV can be attributed to Co 2p1/2 and Co 2p3/2, while two broad peaks located at 803.9 and 785.6 eV are assigned to the satellites of Co 2p1/2 and Co 2p3/2, respectively.38 The resolved Co 2p3/2 reveals the co-existence of Co0 (779.7 eV) and Co2+ (781.5 eV), which indicates that the Co NPs on the surface of the C–N/Co (1/2) composite are slowly oxidized in the air.39,40
N species, respectively.20 It confirms that N heteroatoms have been successfully doped into carbon matrix. The majority of C–C bonds are responsible for the excellent electron conductivity,35 which is consistent to the SEM and TEM results (Fig. 1d). The deconvolved N 1s spectrum exhibits five peaks, corresponding to pyridinic N (398.5 eV), Co–N (399.1 eV), pyrrolic N (400.2 eV), graphitic N (401.6 eV), and oxidized N (403.4 eV) (Fig. 3c), respectively.22 It has been reported that pyridinic N, graphitic N and Co–N bonds have a positive effect on OER and ORR performances.20,36 Particularly, Co–N bond can accelerate the charge transfer between carbon and metal/N-dopants, providing additional sites for ORR process.22,37 In the high-resolution Co 2p spectrum (Fig. 3d), two main peaks at 796.2 and 780.6 eV can be attributed to Co 2p1/2 and Co 2p3/2, while two broad peaks located at 803.9 and 785.6 eV are assigned to the satellites of Co 2p1/2 and Co 2p3/2, respectively.38 The resolved Co 2p3/2 reveals the co-existence of Co0 (779.7 eV) and Co2+ (781.5 eV), which indicates that the Co NPs on the surface of the C–N/Co (1/2) composite are slowly oxidized in the air.39,40
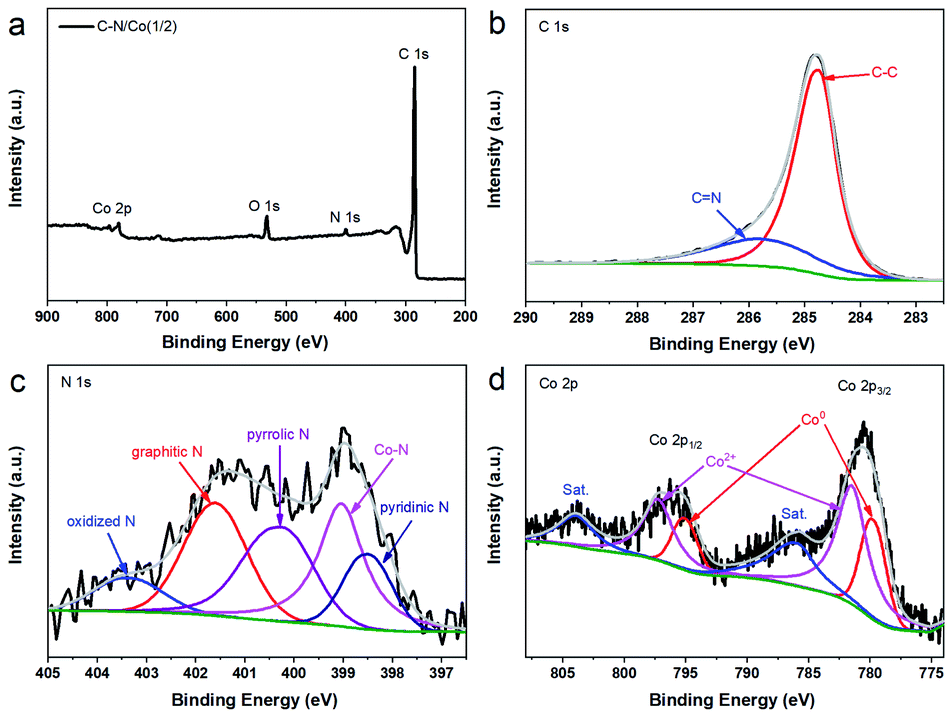 | ||
| Fig. 3 (a) XPS survey spectrum of C–N/Co (1/2), high-resolution and the corresponding deconvoluted spectrum of (b) C 1s, (c) N 1s and (d) Co 2p. | ||
The electrocatalytic ORR performance of C–N/Co (1/2) was first executed by cyclic voltammetry (CV) measurements in N2- and O2-saturated 0.1 M KOH solution at a scan rate of 50 mV s−1. As shown in Fig. 4a, no oxygen reduction peak is observed in N2-saturated solution. However, C–N/Co (1/2) displays an obvious cathodic reduction peak at 0.79 V in O2-saturated solution, which is more positive than C–N/Co (0.78 V), C–N/Co (1/3) (0.77 V) and C–N/Co (2/3) (0.76 V) (Fig. S2†). To further investigate the ORR performance of C–N/Co (x) composites, linear sweep voltammetry (LSV) tests were performed on a rotating disk electrode (RDE) in O2-saturated 0.1 M KOH at a scan rate of 5 mV s−1 with a rotation speed of 1600 rpm (Fig. 4b). Taken a current density of 0.1 mA cm−2 as criterion,41–43 C–N/Co (1/2) exhibits a more positive ORR onset potential (0.89 V) compared to C–N/Co (0.87 V), C–N/Co (1/3) (0.86 V) and C–N/Co (2/3) (0.85 V), which is comparable to the benchmark Pt/C catalyst (20 wt% Pt) at 0.93 V. The results suggest that appropriate content of Co atoms is crucial to the formation of available surface active sites and enhance the ORR activity.22,35,44 The RDE tests were carried out at various rotating speeds to study the ORR diffusion kinetics of C–N/Co (1/2). The number (n) of transferred electrons per O2 molecule in the ORR procedure was evaluated using the slopes from Koutecky–Levich (K–L) plots (J−1 vs. W−1/2) (Fig. S3a†). The slopes at different electrode potentials (between 0.6 and 0.2 V) remain an almost constant of 4 (from 3.97 to 4.08), implying a possible near one-step and four-electron transfer process of the ORR on C–N/Co (1/2). The ORR activity of C–N/Co (1/2) was further evaluated in RRDE experiments conducted in alkaline solution, and a typical sweeping voltammogram at a rotating rate of 1600 rpm is shown in Fig. S3b.† The yield of HO2− was found below 5% over the potential range of 0 V to 0.85 V, giving an n value of 3.91 to 3.99 (inset of Fig. S3b†). This is highly consistent with the results derived from the K–L plots of RDE measurements (inset of Fig. S3a†), confirming a single 4-electron step for the efficient ORR of C–N/Co (1/2). Long-term stability is another vital parameter for evaluating the practical applications of electrocatalysts. The durability test of C–N/Co (1/2) was conducted by chronoamperometry measurements toward ORR, with commercial Pt/C taken as contrast sample (Fig. 4c). The result shows that C–N/Co (1/2) remains the 90% of the initial current density after 34200 s. On the contrary, Pt/C undertakes a rapid decay in activity at the beginning and suffers a total current loss of 26% over the same period. In addition, the tolerance of C–N/Co (1/2) and Pt/C to methanol crossover effect was also investigated using a chronoamperometric method (Fig. S4†). After the addition of 1.0 M methanol, an immediate and sharp decrease is observed in the current density of Pt/C while C–N/Co (1/2) shows a negligible change (99.8% retention). The better durability and tolerance of C–N/Co (1/2) may originate from that carbon layers confine the Co NPs in the matrix to protect the metal Co from leaching and corrosion, and improve the long-term stability.1,45,46
 | ||
| Fig. 4 (a) Cyclic voltammograms of the ORR in O2-saturated and N2-saturated 0.1 M KOH solution on C–N/Co (1/2); (b) LSV curves for the ORR in O2-saturated 0.1 M KOH solution on C–N/Co (1/2), C–N/Co, C–N/Co (1/3), C–N/Co (2/3), and Pt/C; (c) chronoamperometry test of C–N/Co (1/2) and Pt/C in O2-saturated 0.1 M KOH solution at 0.6 V; (d) LSV curves for the OER in O2-saturated 1.0 M KOH solution on C–N/Co (1/2), C–N/Co, C–N/Co (1/3), C–N/Co (2/3), Ir/C and Pt/C; (e) the corresponding Tafel plots; (f) LSV curves of C–N/Co (1/2) before and after 1000 CV cycles in the stability test over 1.0 M KOH. | ||
The electrocatalytic activity of the C–N/Co (1/2) hybrid towards OER was also investigated in O2-saturated KOH solution (Fig. 4d). All C–N/Co (x) composites manifest better OER performance compared with the commercial Ir/C and Pt/C. Clearly, C–N/Co (1/2) exhibits the highest current density and lowest onset potential for OER among all hybrids. Taken a current density of 10 mA cm−2 as criterion, C–N/Co (1/2) electrocatalyst has a lower overpotential of 0.29 V in contrast to that of C–N/Co (0.37 V), C–N/Co (1/3) (0.34 V), C–N/Co (2/3) (0.35 V). Co NPs encapsulated in N-doped graphitized carbon networks are supposed to improve electron conductivity of the catalyst and act as active sites for OER, which result in effective oxygen electrocatalysis.1,39,47–49 In addition, the ORR and OER performances of NC, GC and NC + GC (the obtained NC (11 mg) and GC (11 mg) powder were mixed physically) and C–N/Co (1/2) were compared (Fig. S5†). Clearly, the ORR and OER results of C–N/Co (1/2) is much better than that of other samples, which proves that the superior catalytic activities of the catalyst were due to the unique core–shell structure and synergic effects between C–N matrices and Co NPs. Moreover, the LSV curves of the sample with Zn element (C–N/Co (1/2, 820 °C)) have also been characterized (Fig. S6†). After evaporation of Zn ions, a distinguished improvement is observed for the OER while the ORR property is kept constant. This result confirmed that the evaporation of Zn ions is vital to obtain the excellent bifunctional electrocatalyst in this study.
In order to further explore the electrocatalytic performance for OER, the Tafel plots (Fig. 4e) of the prepared materials and the commercial Ir/C were obtained from the corresponding LSV curves in Fig. 4d. The Tafel slopes of C–N/Co (1/2), C–N/Co, C–N/Co (1/3), C–N/Co (2/3) and Ir/C were found to be 66 mV dec−1, 88 mV dec−1, 85 mV dec−1, 78 mV dec−1 and 126 mV dec−1. These results distinctly reveal that C–N/Co (1/2) exhibits the smallest Tafel slope among the above electrocatalysts under the same kinetic current density.
In addition, the electrochemical active surface area (ECSA) is estimated by double-layer capacitance (Cdl).1,35 It can be seen that the C–N/Co (1/2) composite manifests larger Cdl (22.9 mF cm−2) than C–N/Co (14.2 mF cm−2), C–N/Co (1/3) (16.5 mF cm−2) and C–N/Co (2/3) (18.2 mF cm−2) (Fig. S7†). The increase of ECSA leads to the enhancement of catalytic activity,50 which is one of the reasons for C–N/Co (1/2) composite to show the best catalytic activity. In addition, the exposure of the electrochemically active sites is another important factor considering the similar BET specific surface areas and pore size distribution of the as-prepared composites (Fig. S8†). In this respect, the best catalytic activity of C–N/Co (1/2) among the above comes from the more active sites that have been exposed to its surface.
The durability and stability of the C–N/Co (1/2) were also assessed by continuous CV measurements with a scan rate of 100 mV s−1 (Fig. 4f). After 1000 cycles of continuous potential cycling, negligible current loss can be observed by comparing the first and the 1000th LSV curves, implying the excellent stability of C–N/Co (1/2) in OER process. The marvelous stability comes from the protection system built by outer carbon layers, which protects the OER active sites from impurities and oxidation products.49
Based on the above results, the C–N/Co (1/2) demonstrated advanced electrocatalysis for both ORR and OER, which is comparable to the reported catalysts (Tables S1 and S2†). Therefore, it is expected as a bifunctional candidate for Zn–air batteries application. A home-built Zn–air cell device was assembled using C–N/Co (1/2) as electrocatalyst. As shown in Fig. 5, the rechargeability of the assembled battery was executed by repeating discharging at a constant current of 5 mA cm−2 over 4 h and charging at a constant current of 2.5 mA cm−2 over 8 h for 28 cycles. The C–N/Co (1/2) displays close charge and discharge voltage compared with Ir/C at first, but lower than Ir/C after 50 h. Here the cycling stability and durability of our catalyst are much better than Ir/C with negligible potential attenuations. This is attributed to carbon layers around Co NPs, which protect them from aggregation during reactions, and supply good electron conductivity and large amount of catalytic active sites. All of these results prove that the Zn–air batteries with C–N/Co (1/2) catalyst as an air cathode are promising secondary cell devices in the future.
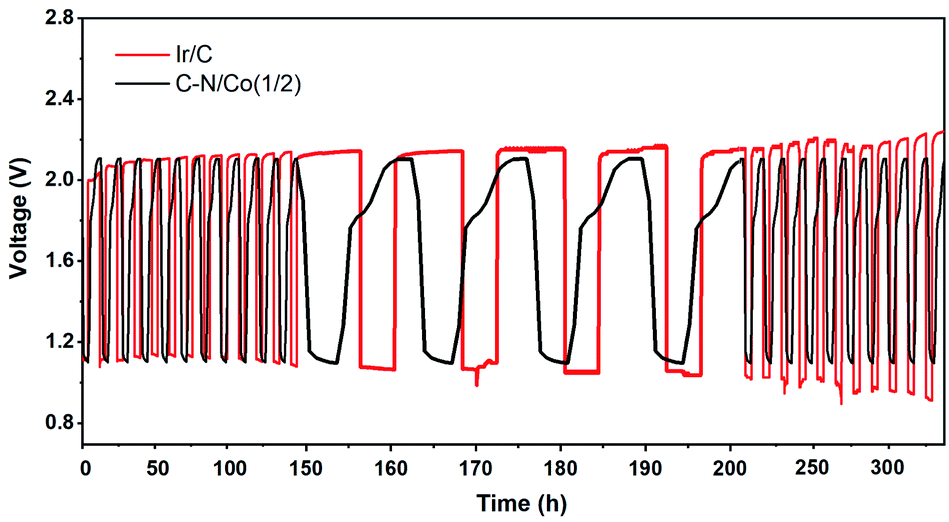 | ||
| Fig. 5 Galvanostatic charge/discharge cycling curves of rechargeable Zn–air batteries with C–N/Co (1/2) and Ir/C catalysts as air cathodes. | ||
Conclusions
In summary, we have successfully synthesized a series of C–N/Co (x) catalysts with different content of Co via direct pyrolysis of the core–shell ZIF-8@ZIF-67 structures. ZIF-8 was used as self-sacrificed template to restrict the morphology and size of external ZIF-67 shell. The final product of C–N/Co (1/2) showed the good electrochemical performances owing to its special content and structure. During the pyrolysis process, the Zn ions have been evaporated and the metal Co NPs were pushed onto the surface to expose more active sites, which is the main reason for the good ORR and OER performances. In addition, the synergistic influences between Co NPs and N-doped porous carbon matrix endow the C–N/Co (1/2) remarkable electrocatalysis with good stability. Based on the above results, C–N/Co (1/2) was used as a bifunctional electrocatalyst in the home-built Zn–air cell and showed good performance. This work not only provides a promising carbon-based electrocatalytic material for energy conversion and utilization, but also offers a new strategy to investigate the relationships between the MOF precursors and electrochemical properties of the products.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a Start-up Grant from Nanjing Tech University (39837119), the National Science Foundation for Young Scientists of China (21705078), and the Natural Science Foundation for Youth of Jiangsu Province (BK20170972).Notes and references
- C. Feng, Y. Guo, Y. H. Xie, X. L. Cao, S. Li, L. G. Zhang, W. Wang and J. Wang, Nanoscale, 2020, 12, 5942–5952 RSC.
- M. S. Faber and S. Jin, Energy Environ. Sci., 2014, 7, 3519–3542 RSC.
- Y.-J. Wang, B. Fang, X. Wang, A. Ignaszak, Y. Liu, A. Li, L. Zhang and J. Zhang, Prog. Mater. Sci., 2018, 98, 108–167 CrossRef CAS.
- S. Agarwal, X. W. Yu and A. Manthiram, Mater. Today Energy, 2020, 16, 100405 CrossRef.
- N. Y. Cheng, L. Ren, X. Xu, Y. Du and S. X. Dou, Adv. Energy Mater., 2018, 8, 1801257 CrossRef.
- J. Fu, Z. P. Cano, M. G. Park, A. Yu, M. Fowler and Z. Chen, Adv. Mater., 2017, 29, 1604685 CrossRef PubMed.
- C. Tang, H. F. Wang and Q. Zhang, Acc. Chem. Res., 2018, 51, 881–889 CrossRef CAS PubMed.
- L. B. Zong, W. C. Wu, S. L. Liu, H. J. Yin, Y. N. Chen, C. Liu, K. C. Fan, X. X. Zhao, X. Chen, F. M. Wang, Y. Yang, L. Wang and S. H. Feng, Energy Storage Materials, 2020, 27, 514–521 CrossRef.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 146–158 CrossRef PubMed.
- Y. Zhao, R. Nakamura, K. Kamiya, S. Nakanishi and K. Hashimoto, Nat. Commun., 2013, 4, 2390 CrossRef PubMed.
- L. M. Dai, Y. H. Xue, L. T. Qu, H.-J. Choi and J.-B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- H. Xu, Z. X. Shi, Y. X. Tong and G. R. Li, Adv. Mater., 2018, 30, e1705442 CrossRef PubMed.
- K. Z. Jiang, P. T. Wang, S. J. Guo, X. Zhang, X. Shen, G. Lu, D. Su and X. Q. Huang, Angew. Chem., Int. Ed., 2016, 128, 9176–9181 CrossRef.
- K. Shen, X. Chen, J. Chen and Y. Li, ACS Catal., 2016, 6, 5887–5903 CrossRef CAS.
- H. F. Wang, L. Y. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- Y. J. Wu, S. L. Zhao, K. Zhao, T. X. Tu, J. Z. Zheng, J. Chen, H. F. Zhou, D. J. Chen and S. X. Li, J. Power Sources, 2016, 311, 137–143 CrossRef CAS.
- S. W. Liu, H. M. Zhang, Q. Zhao, X. Zhang, R. R. Liu, X. Ge, G. Z. Wang, H. J. Zhao and W. P. Cai, Carbon, 2016, 106, 74–83 CrossRef CAS.
- Y. J. Wang, B. Fang, D. Zhang, A. Li, D. P. Wilkinson, A. Ignaszak, L. Zhang and J. Zhang, Electrochem. Energy Rev., 2018, 1, 1–34 CrossRef CAS.
- S. Ren, X. Duan, S. Liang, M. Zhang and H. Zheng, J. Mater. Chem. A, 2020, 8, 6144–6182 RSC.
- I. S. Amiinu, Z. H. Pu, X. B. Liu, K. A. Owusu, H. G. R. Monestel, F. O. Boakye, H. N. Zhang and S. C. Mu, Adv. Funct. Mater., 2017, 27, 1702300 CrossRef.
- D. Liu, X. M. Zhang, Y. J. Wang, S. Y. Song, L. F. Cui, H. B. Fan, X. C. Qiao and B. Z. Fang, Nanoscale, 2020, 12, 9524–9532 RSC.
- Y. Hou, Z. H. Wen, S. M. Cui, S. Q. Ci, S. Mao and J. H. Chen, Adv. Funct. Mater., 2015, 25, 872–882 CrossRef CAS.
- H. Y. Qiao, Y. Yang, X. P. Dai, H. H. Zhao, J. X. Yong, L. Yu, X. B. Luan, M. L. Cui, X. Zhang and X. L. Huang, Electrochim. Acta, 2019, 318, 430–439 CrossRef CAS.
- L. Du, L. X. Xing, G. X. Zhang and S. H. Sun, Carbon, 2020, 156, 77–92 CrossRef CAS.
- Z. J. Wang, Y. Z. Lu, Y. Yan, T. Y. P. Larissa, X. Zhang, D. Wuu, H. Zhang, Y. H. Yang and X. Wang, Nano Energy, 2016, 30, 368–378 CrossRef CAS.
- C. Bao, X. Liu, M. Li, J. Meng, Y. L. Cai, X. Huang, T.-P. Loh and Z. J. Wang, Electrochem. Commun., 2019, 106, 106515 CrossRef CAS.
- D. K. Panchariya, R. K. Rai, E. Anil Kumar and S. K. Singh, ACS Omega, 2018, 3, 167–175 CrossRef CAS PubMed.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed.
- Y. X. Zhao, Q. X. Lai, J. J. Zhu, J. Zhong, Z. M. Tang, Y. Luo and Y. Y. Liang, Small, 2018, 14, 1704207 CrossRef PubMed.
- J. Liu, L. H. Jiang, B. S. Zhang, J. T. Jin, D. S. Su, S. L. Wang and G. Q. Sun, ACS Catal., 2014, 4, 2998–3001 CrossRef CAS.
- H. H. Liu, S. Y. Wang, L. Long, J. B. Jia and M. C. Liu, Nanotechnology, 2021, 32, 205402 CrossRef CAS PubMed.
- J. S. Meng, C. J. Niu, L. H. Xu, J. T. Li, X. Liu, X. P. Wang, Y. Z. Wu, X. M. Xu, W. Y. Chen, Q. Li, Z. Z. Zhu, D. Y. Zhao and L. Q. Mai, J. Am. Chem. Soc., 2017, 139, 8212–8221 CrossRef CAS PubMed.
- H. B. Wu, S. Y. Wei, L. Zhang, R. Xu, H. H. Hng and X. W. D. Lou, Chem.–Eur. J., 2013, 19, 10804–10808 CrossRef CAS PubMed.
- Y. Yan, X. M. Ge, Z. L. Liu, J. Y. Wang, J. M. Lee and X. Wang, Nanoscale, 2013, 5, 7768–7771 RSC.
- L. Yan, L. Cao, P. Dai, X. Gu, D. Liu, L. Li, Y. Wang and X. Zhao, Adv. Funct. Mater., 2017, 27, 1703455 CrossRef.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- K. X. Niu, B. P. Yang, J. F. Cui, J. T. Jin, X. G. Fu, Q. P. Zhao and J. Y. Zhang, J. Power Sources, 2013, 243, 65–71 CrossRef CAS.
- Y. N. Guo, J. Tang, H. Y. Qian, Z. L. Wang and Y. Yamauchi, Chem. Mater., 2017, 29, 5566–5573 CrossRef CAS.
- S. Dou, X. Y. Li, L. Tao, J. Huo and S. Y. Wang, Chem. Commun., 2016, 52, 9727–9730 RSC.
- P. He, X. Y. Yu and X. W. Lou, Angew. Chem., Int. Ed., 2017, 56, 3897–3900 CrossRef CAS PubMed.
- N. Daems, X. Sheng, I. F. J. Vankelecom and P. P. Pescarmona, J. Mater. Chem. A, 2014, 2, 4085–4110 RSC.
- W. Xia, A. Mahmood, Z. B. Liang, R. Q. Zou and S. J. Guo, Angew. Chem., Int. Ed., 2016, 55, 2650–2676 CrossRef CAS PubMed.
- J. L. Long, R. Li and X. L. Gou, Catal. Commun., 2017, 95, 31–35 CrossRef CAS.
- Z.-F. Huang, J. J. Song, Y. H. Du, S. B. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. C. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- D. H. Deng, L. Yu, X. Q. Chen, G. X. Wang, L. Jin, X. L. Pan, J. Deng, G. Q. Sun and X. H. Bao, Angew. Chem., Int. Ed., 2013, 52, 371–375 CrossRef CAS PubMed.
- K. Shen, L. Chen, J. L. Long, W. Zhong and Y. W. Li, ACS Catal., 2015, 5, 5264–5271 CrossRef CAS.
- Y. H. Su, Y. H. Zhu, H. L. Jiang, J. H. Shen, X. L. Yang, W. J. Zou, J. D. Chen and C. Z. Li, Nanoscale, 2014, 6, 15080–15089 RSC.
- S. A. Shah, Z. Y. Ji, X. P. Shen, X. Y. Yue, G. X. Zhu, K. Q. Xu, A. Yuan, N. Ullah, J. Zhu, P. Song and X. Y. Li, ACS Appl. Energy Mater., 2019, 2, 4075–4083 CrossRef CAS.
- V. K. Abdelkader-Fernández, D. M. Fernandes, S. S. Balula, L. Cunha-Silva, M. J. Pérez-Mendoza, F. J. López-Garzón, M. F. Pereira and C. Freire, ACS Appl. Energy Mater., 2019, 2, 1854–1867 CrossRef.
- T. Zhang, J. Du, P. X. Xi and C. L. Xu, ACS Appl. Mater. Interfaces, 2017, 9, 362–370 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra01350e |
| This journal is © The Royal Society of Chemistry 2021 |