DOI:
10.1039/D1RA01102B
(Paper)
RSC Adv., 2021,
11, 14899-14904
Copper(I) pyrimidine-2-thiolate cluster-based polymers as bifunctional visible-light-photocatalysts for chemoselective transfer hydrogenation of α,β-unsaturated carbonyls†
Received
9th February 2021
, Accepted 14th April 2021
First published on 21st April 2021
Abstract
The photoinduced chemoselective transfer hydrogenation of unsaturated carbonyls to allylic alcohols has been accomplished using cluster-based MOFs as bifunctional visible photocatalysts. Assemblies of hexanuclear clusters [Cu6(dmpymt)6] (1, Hdmpymt = 4,6-dimethylpyrimidine-2-thione) as metalloligands with CuI or (Ph3P)CuI yielded cluster-based metal organic frameworks (MOFs) {[Cu6(dmpymt)6]2[Cu2(μ-I)2]4(CuI)2}n (2), {[Cu6(dmpymt)6]2[Cu2(μ-I)2]4}n (3), respectively. Nanoparticles (NPs) of 2 and 3 served both as photosensitizers and photocatalysts for the highly chemoselective reduction of unsaturated carbonyl compounds to unsaturated alcohols with high catalytic activity under blue LED irradiation. The photocatalytic system could be reused for several cycles without any obvious loss of efficiency.
Introduction
The chemoselective hydrogenation of unsaturated carbonyl compounds is a preferred method for the synthesis of allylic alcohols, a class of important chemical, pharmaceutical and agrochemical intermediates.1–4 However, reduction of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is both thermodynamically and kinetically favored over the reaction of the CO bond,5–9 thereby creating a synthetic challenge.10–17 Extensive effort has been devoted to developing catalytic systems that enable to efficiently and selectively hydrogenate unsaturated carbonyls to the corresponding unsaturated alcohols. However, current methods often use expensive transition metal catalysts, or toxic and hard to separate phosphine organic ligands at elevated reaction temperatures (Table 1).18–20 The design of a catalytic system that efficiently promotes chemoselective transfer hydrogenation of unsaturated carbonyls with alcohol activated by visible light at room temperature is highly desired.21–26 We are aware of only one report that is a visible light photoactive noble metal Au/SiC catalyst, achieving selective hydrogenation of cinnamaldehyde.27 To the end, the utilization of efficient and robust earth-abundant catalysts with visible-light activity for this transformation is a long-term target. Metal–organic frameworks (MOFs) offer an exceptional opportunity as potential scaffolds for non-noble photocatalytic materials. There are two main strategies to achieve photocatalytically active MOFs. One approach is to introduce both the photon absorption and catalytic center into the coordinated frameworks of the MOF.28 These photoactive MOFs based on a ligand-to-metal charge transfer (LMCT) mechanism are not readily tunable to visible light absorption. The second approach is the incorporation of the desired catalytic species (molecular catalysts, metal nanoparticle etc.) as a co-catalyst into a visible-light photoactive MOF with visible-light absorbent components (e.g. porphyrins, 2-aminoterephthalates, perylene diimides, organic dyes, metal-complexes), yielding a dual-catalyst system.29,30 These dual-catalysts mostly require cumbersome preparation methods. An alternative strategy, single-step construction of visible-light photoactive MOFs using a bifunctional photocatalyst as a building block/linker serving both as a photosensitizer and as a photocatalyst, is a promising approach because it is simpler and easier-to-use. Only a few MOFs have been doped with a bifunctional visible light photocatalyst based on (bpy)Re(CO)3 (bpy = 2,2′-bipyridine),31 (bpy)Ru(CO)2Cl2,32 Ir(bpy)3,33 or (bpy)PtCl2 (ref. 34) functional groups for CO2 reduction and hydrogen production. Interestingly, the resulting solid MOF materials displayed higher photocatalytic activity than the corresponding homogeneous catalysts. We postulated that a bifunctional photocatalyst as a building block, using as both visible-light photosensitizer and transfer-hydrogenation catalyst, could be developed to heterogeneously catalyze reduction of unsaturated carbonyls to allylic alcohols under visible light.
C bond is both thermodynamically and kinetically favored over the reaction of the CO bond,5–9 thereby creating a synthetic challenge.10–17 Extensive effort has been devoted to developing catalytic systems that enable to efficiently and selectively hydrogenate unsaturated carbonyls to the corresponding unsaturated alcohols. However, current methods often use expensive transition metal catalysts, or toxic and hard to separate phosphine organic ligands at elevated reaction temperatures (Table 1).18–20 The design of a catalytic system that efficiently promotes chemoselective transfer hydrogenation of unsaturated carbonyls with alcohol activated by visible light at room temperature is highly desired.21–26 We are aware of only one report that is a visible light photoactive noble metal Au/SiC catalyst, achieving selective hydrogenation of cinnamaldehyde.27 To the end, the utilization of efficient and robust earth-abundant catalysts with visible-light activity for this transformation is a long-term target. Metal–organic frameworks (MOFs) offer an exceptional opportunity as potential scaffolds for non-noble photocatalytic materials. There are two main strategies to achieve photocatalytically active MOFs. One approach is to introduce both the photon absorption and catalytic center into the coordinated frameworks of the MOF.28 These photoactive MOFs based on a ligand-to-metal charge transfer (LMCT) mechanism are not readily tunable to visible light absorption. The second approach is the incorporation of the desired catalytic species (molecular catalysts, metal nanoparticle etc.) as a co-catalyst into a visible-light photoactive MOF with visible-light absorbent components (e.g. porphyrins, 2-aminoterephthalates, perylene diimides, organic dyes, metal-complexes), yielding a dual-catalyst system.29,30 These dual-catalysts mostly require cumbersome preparation methods. An alternative strategy, single-step construction of visible-light photoactive MOFs using a bifunctional photocatalyst as a building block/linker serving both as a photosensitizer and as a photocatalyst, is a promising approach because it is simpler and easier-to-use. Only a few MOFs have been doped with a bifunctional visible light photocatalyst based on (bpy)Re(CO)3 (bpy = 2,2′-bipyridine),31 (bpy)Ru(CO)2Cl2,32 Ir(bpy)3,33 or (bpy)PtCl2 (ref. 34) functional groups for CO2 reduction and hydrogen production. Interestingly, the resulting solid MOF materials displayed higher photocatalytic activity than the corresponding homogeneous catalysts. We postulated that a bifunctional photocatalyst as a building block, using as both visible-light photosensitizer and transfer-hydrogenation catalyst, could be developed to heterogeneously catalyze reduction of unsaturated carbonyls to allylic alcohols under visible light.
Table 1 Examples for the selective hydrogenation CO bond of cinnamaldehyde
| Entry |
Catalyst |
Reaction conditions |
Yield (%) |
Selec. (%) |
Ref. |
| T (°C) |
PH2 (bar) |
t (h) |
| Reactions were promoted under visible light irradiation. |
| 1 |
Pt/YCo0.3Fe0.7O3 |
90 |
20 |
0.5 |
93.8 |
94.9 |
18 |
| 2 |
Ir/H–MoOx |
100 |
20 |
2 |
93 |
93 |
19 |
| 3 |
Co@CN-900 |
80 |
1 |
48 |
98 |
99 |
20 |
| 4 |
Cu6(dmpymt)6 |
100 |
0 |
10 |
98 |
99 |
38 |
| 5a |
Au/SiC |
20 |
0 |
4 |
100 |
100 |
27 |
| 6a |
3 |
20 |
0 |
24 |
94 |
100 |
— |
Typical metal–ligand bifunctional H–M–NH catalysis have provided an efficient method for this reaction.35–37 Very recently, we have found that Cu(I) pyrimidine-2-thiolate clusters can efficiently catalyze transfer hydrogenation reactions via a cooperative metal hydride and ligand proton transfer mechanism.38 Cu(I) N-heterocycle thiolate clusters are potentially attractive precursors for the construction of bifunctional photocatalyst MOFs based on the following considerations: (i) metal thiolate complexes can be used as co-catalysts for the production of hydrogen compatible with a photosensitizer,39–45 (ii) Cu(I) N-heterocycle thiolates absorb visible light and are intensely luminescent with long fluorescent lifetimes,46–48 (iii) they can smoothly achieve such conversion at 100 °C,38 (iv) some clusters such as [Cu6(dmpymt)6] with uncoordinated N atoms can be used as metalloligands for the construction of MOFs. We have investigated this possibility using the hexanuclear Cu(I) pyrimidine-2-thiolate cluster [Cu6(dmpymt)6] (1) as a precursor to construct cluster-based coordination complexes {[Cu6(dmpymt)6]2[Cu2(μ-I)2]4(CuI)2}n (2) and {[Cu6(dmpymt)6]2[Cu2(μ-I)2]4}n (3) from CuI and (Ph3P)CuI, respectively. Nanoparticles of 2 and 3 can efficiently catalyse transfer hydrogenation of unsaturated carbonyl compounds to the corresponding unsaturated alcohols under the irradiation of blue LEDs using 2-propanol as the hydrogen source.
Results and discussion
Syntheses and characterization
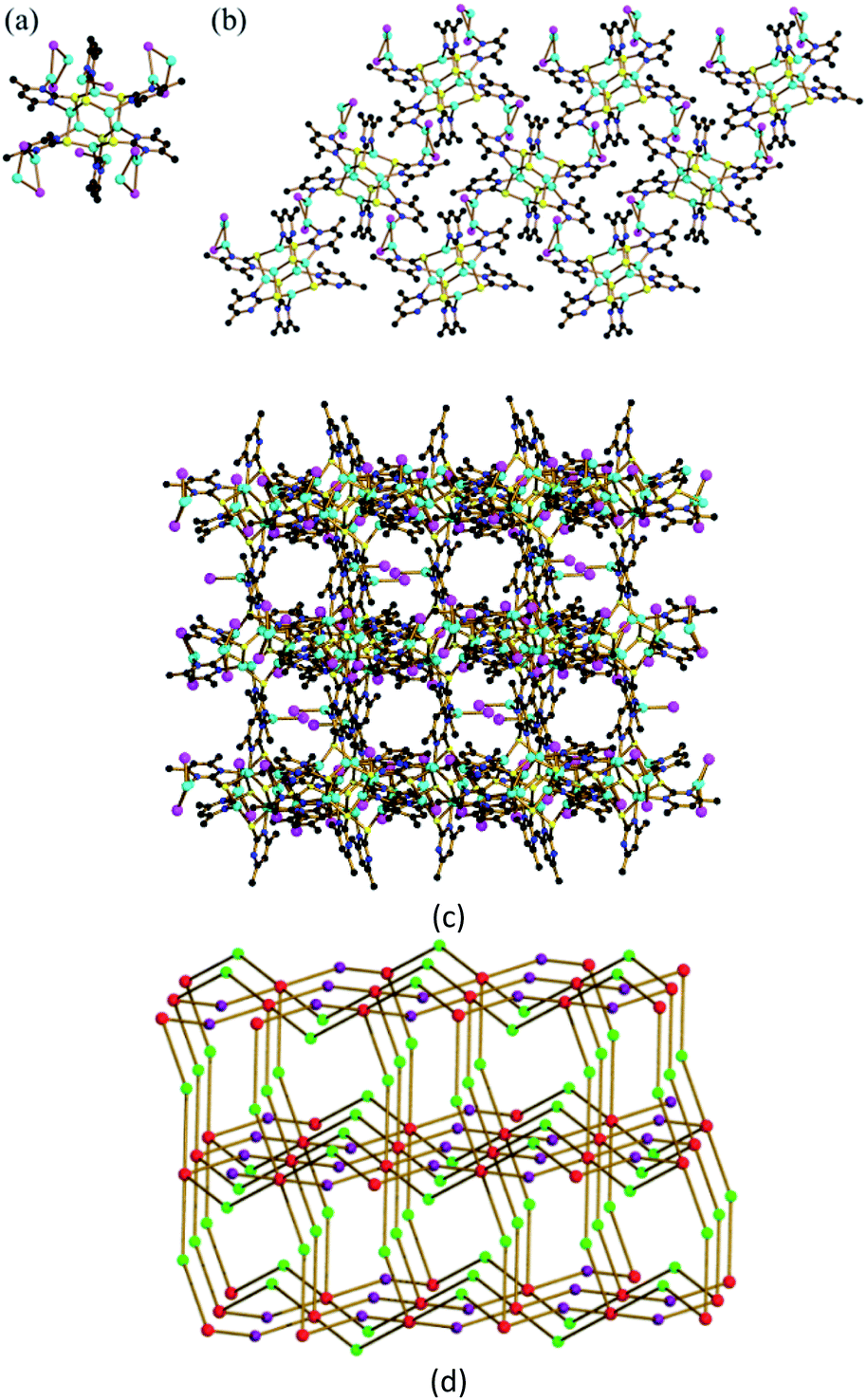
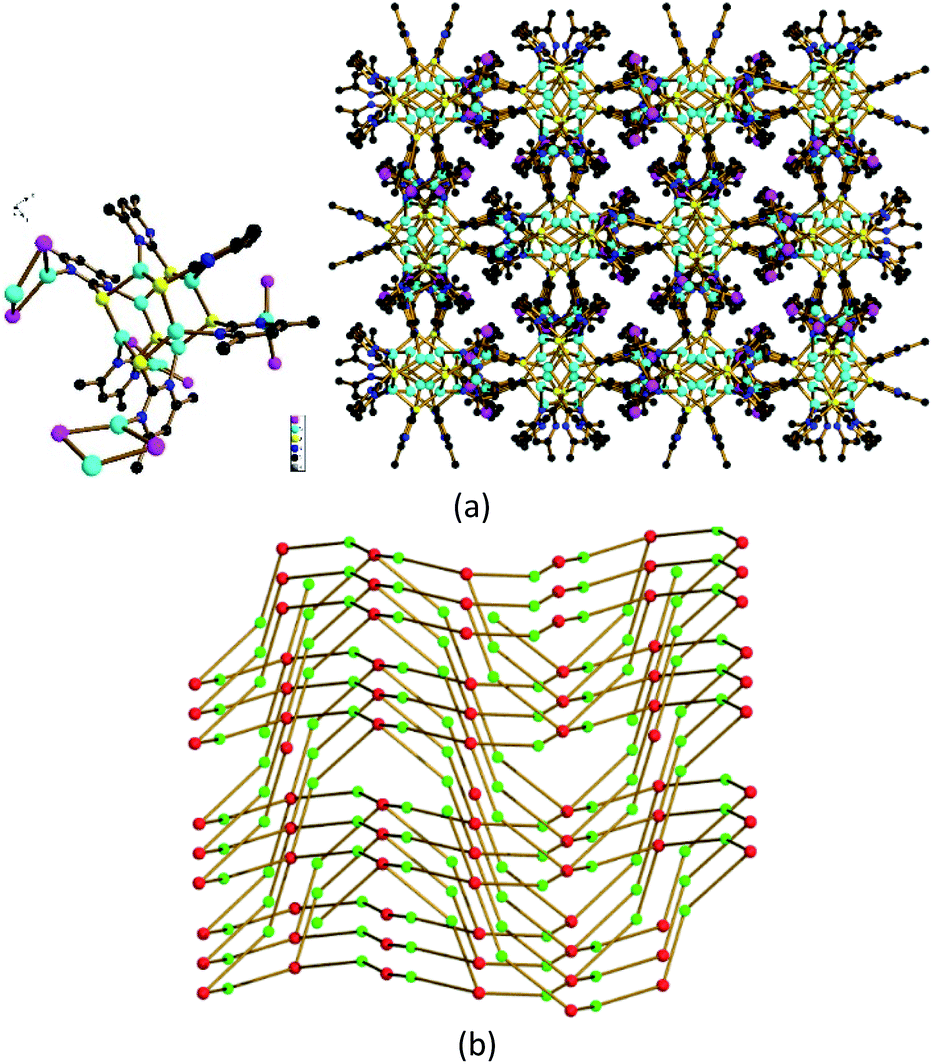
As remarked above, the coordinative N atom in [Cu6(dmpymt)6] (1) can further coordinate with metal ion. At the outset, we carried out reaction of 1 with CuI in MeCN and CHCl3 at room temperature to result in the formation of one 3D cluster-based polymer {[Cu6(dmpymt)6]2[Cu2(μ-I)2]4(CuI)2}n (2) in 75% yield. Solvothermal reaction of [Cu6(dmpymt)6] with (Ph3P)CuI in MeCN/toluene at 120 °C yielded several crystals of a 3D complex {[Cu6(dmpymt)6]2[Cu2(μ-I)2]4}n (3) in 89% yield. Single-crystal analysis revealed that 2 crystallized in the tetragonal space group P41212, 3 crystallizes in the monoclinic space group C2/c. In 2, each “Cu6(dmpymt)6” unit is interconnected with the other four through “Cu2(μ-I)2” linkages forming a 2D layer, which is further connected to equivalent layers via N–Cu–N bonds to form a 3D net (Fig. 1). Compound 3 also consists of a single 3D network in which each “Cu6(dmpymt)6” unit, serving as a tetrahedral node, interconnects with the other four nearby units through N-{Cu(μ-I)2Cu}-N bridges (Fig. 2a). All six (in 2) or four (in 3) of six dmpymt ligands in the [Cu6(dmpymt)6] units take a μ-(κ1-N,κ1-N′,κ3-S) mode to bridge four Cu(I) atoms. Other dmpymt ligands bridge three Cu(I) atoms via μ-(κ1-N,κ3-S) coordination fashion. If each “Cu2(μ-I)2” or “CuI” is considered as a 2-connecting node, then each “Cu6(dmpymt)6” is simplified as 6- or 4-connecting node and the 3D topologies can be described using the {(8)(8)(812·123)} (2) (Fig. 1d) and {(8)(84·122)} (3) (Fig. 2b) Schläfli symbols. In 2, Cu⋯Cu of contacts of 2.5460(19) Å in “Cu2(μ-I)2” unit is longer than the corresponding one in 3 (2.501(2) Å). For “Cu6(dmpymt)6” unit, Cu⋯Cu of distances in 2 are 2.7767(18) Å, 2.7957(18) Å, 2.998(2) Å, which are comparable with those in 3 (2.663(3) Å, 2.7825(19) Å, 2.963(3) Å). The measured X-ray powder patterns of 2 and 3 show a good agreement with those of the corresponding simulated ones (Fig. S1 and S2†). Complexes 2 and 3 are air and moisture stable and insoluble in common organic solvents such as CHCl3, MeOH, MeCN, DMF and DMSO. Complexes 2 and 3 are thermally stable up to 270 °C, and 3 has the better thermostability (Fig. S3†).
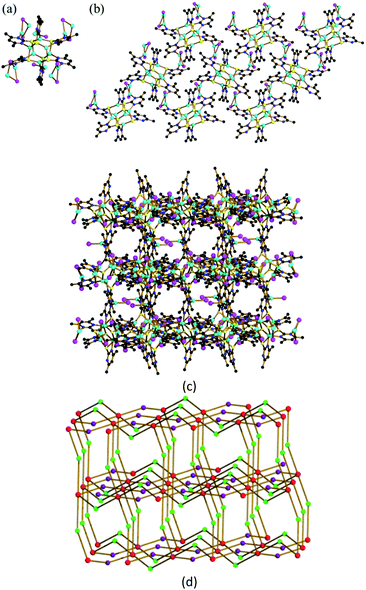 |
| | Fig. 1 (a) The minimum structure unit in 2; (b) [Cu6(dmpymt)6] and [Cu(μ-I)]2 form a two-dimensional network structure extending along the bc surface in 2; color codes: Cu (cyan), I (pink), S (yellow), N (blue), C (black); (c) view of the 3D network of 2. (d) Schematic view of the topological net of 2. Red represents 4-connected Cu6(dmpymt)6; green represents Cu2I2 linking; violet represents [CuI] units. | |
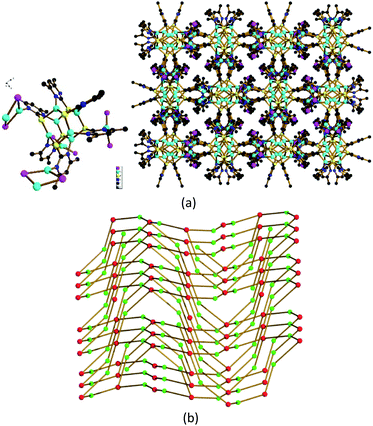 |
| | Fig. 2 (a) View of the 3D network of 3. (b) Schematic view of the topological net of 3. See Fig. 1 for color codes. | |
Nanoparticles of 2 were generated by mixing a solution of [Cu6(dmpymt)6] with CuI in MeCN and CHCl3 for several minutes. Nanowires of 3 were prepared by mixing a solution of [Cu6(dmpymt)6] with (Ph3P)CuI and PVP in MeCN and toluene, followed by heating to 120 °C for 24 h in a glass pressure tube. Their XRD patterns of as-synthesized nanoparticles 2 and 3 match their crystallographic data well, respectively. Scanning electron microscopy (SEM) images (Fig. 3a and b) show that nanoparticles of 2 consist of an approximate spherical structure with the diameter about 50–100 nm. NPs 3 grow into nanowires (≈50–100 nm in width; tens of micrometers in length). As shown in Fig. S4,† the peaks at 931.9 eV (2) or 932.2 eV (3) and 951.8 eV (2)/952.1 eV (3), can be attributed to Cu 2p3/2 and Cu 2p1/2 of Cu+, respectively. The observed type III isotherm in the N2 adsorption–desorption measurements supported this hypothesis (Fig. S5†).
 |
| | Fig. 3 SEM images of nanostructures obtained from 2 (a) and 3 (b). | |
Optical properties
Complex 1 exhibits an ultraviolet and visible light absorption behavior (Fig. S6†). Complexes 2 and 3 show similar absorption bands, but wider and red shifted relative to that of 1. Upon the excitation at λ = 420 nm, 1 exhibits largely enhanced photoluminescence (quantum yield QY = 36.23%, emission centered at λ = 742 nm), whereas the CHCl3 solution of [Cu6(dmpymt)6] is only weakly luminescent (QY = 4.37%). These emissions are most probably arising from a ligand-to-metal charge-transfer (LMCT) and/or metal-to-ligand charge-transfer (MLCT), possibly involving metal-centered (ds/dp) states.49,50 The solid-state emission spectra of 2 and 3 were investigated at room temperature with excitation at 420 nm (Fig. S7†). The room temperature emission of 2 is similar to that of [Cu6(dmpymt)6] (λex = 742 nm) while complex 3 exhibited a broad emission over the range 550–850 nm with a maxima at ca. 677 nm. The emission of 3 is blue-shifted compared to those of 1 and 2. Their quantum yields of 2 and 3 are 42.47% (2) and 55.28% (3), respectively, which are much higher than that of 1. The photoluminescence lifetimes of 1, 2 and 3 in the solid state were measured to be 3.6, 4.1 and 7.1 μs, respectively. These results indicate that the assemblies of cluster could expand its absorption region and improve its luminescent properties. What's more, the transient photocurrent curves for NPs 2 and 3 were also investigated (Fig. S8†), which shows that the stable photocurrent value for NPs 3 was higher, suggesting its fastest charge separation efficiency.
Catalytic properties
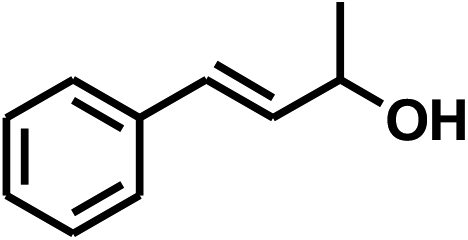
The band gap energies (Eg) of 1–3 were determined using the Kubelka–Munk method based on their diffuse reflectance spectra (Fig. S9†). The energy band gaps obtained by extrapolation of the linear portion of the absorption edges are 2.20 eV (1 and 2) and 2.02 eV (3). These narrow band gaps encouraged us to investigate their visible light photocatalytic activity, which can be evaluated to catalyze the selective transfer hydrogenation of unsaturated carbonyls. We chose cinnamaldehyde (4a) as our model substrate to optimize the reaction conditions. As shown in Table S3,† incubating a mixture of 4a, NaOH as a base and NPs of 3 as a catalyst in isopropanol under visible light afforded full conversion of 4a to the target product cinnamyl alcohol (5a) in 80% yield, the saturated aldehyde of 3-phenylpropanal (6a) in 6% yield and saturated alcohol of 3-phenylpropan-1-ol (7a) in 14% yield after 24 h (Table S3,† entry 1). When this reaction was carried out in MeOH or EtOH, the conversion and selectivity of 4a became lower (entries 2 and 3). When the reaction was performed in i-PrOH–MeCN mixture, the selectivity of 5a was increased significantly with high activity (entries 4–7). The i-PrOH/MeCN volume ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 provided highest selectivity to 5a with highly catalytic activity (entry 6). Control experiments showed that reaction hardly proceeded in the absence of catalyst, base or light (entries 8–10). Other bases KOH, t-BuOK, NaOMe, Cs2CO3, Na2CO3, K2CO3, K3PO4 exhibited reduced selectivity (entries 11–19) relative to NaOH. The catalytic performances of other two compounds were also investigated. NPs 3 exhibited higher selectivity and activity toward reduction of 4a to 5a in comparison with 1 and 2. It is notable that the finely ground synthesized crystals of 2 or 3 as photocatalyst gave the conversion in 84% or 87% under the same conditions. As such, the visible-light-catalyzed chemoselective reduction of 4a to 5a was able to efficiently proceed under the optimized conditions, i.e., 3 as a both photosensitizer and photocatalyst and 20 mol% NaOH as a base, in i-PrOH–MeCN mixture solution, irradiated by blue LEDs for 24 h at room temperature.
:1 provided highest selectivity to 5a with highly catalytic activity (entry 6). Control experiments showed that reaction hardly proceeded in the absence of catalyst, base or light (entries 8–10). Other bases KOH, t-BuOK, NaOMe, Cs2CO3, Na2CO3, K2CO3, K3PO4 exhibited reduced selectivity (entries 11–19) relative to NaOH. The catalytic performances of other two compounds were also investigated. NPs 3 exhibited higher selectivity and activity toward reduction of 4a to 5a in comparison with 1 and 2. It is notable that the finely ground synthesized crystals of 2 or 3 as photocatalyst gave the conversion in 84% or 87% under the same conditions. As such, the visible-light-catalyzed chemoselective reduction of 4a to 5a was able to efficiently proceed under the optimized conditions, i.e., 3 as a both photosensitizer and photocatalyst and 20 mol% NaOH as a base, in i-PrOH–MeCN mixture solution, irradiated by blue LEDs for 24 h at room temperature.
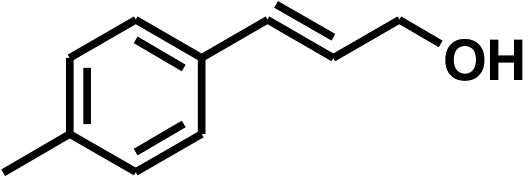
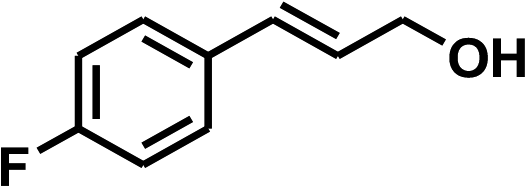
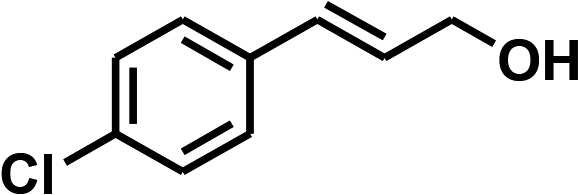
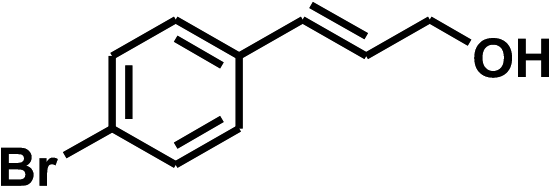
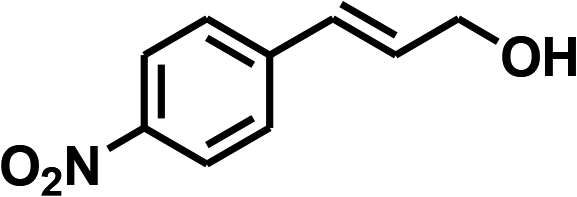
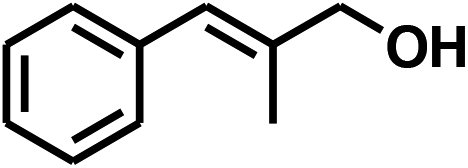
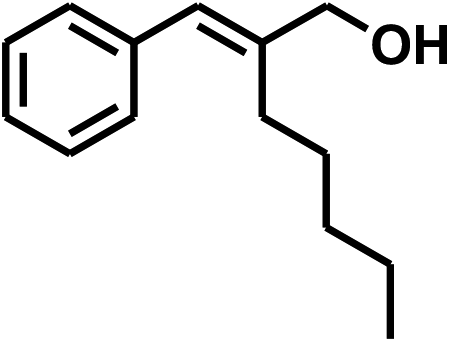
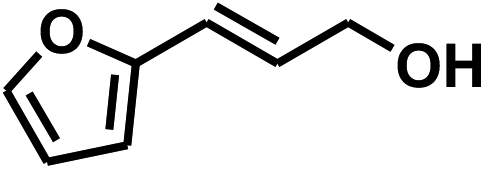
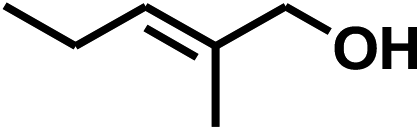
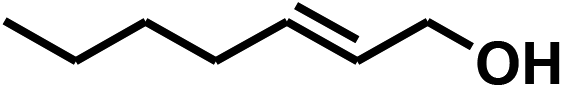
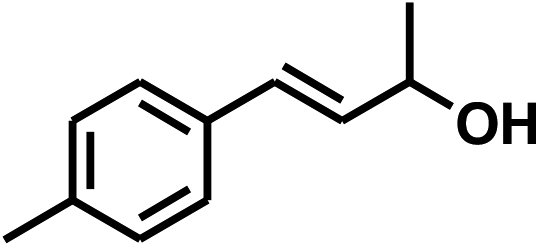
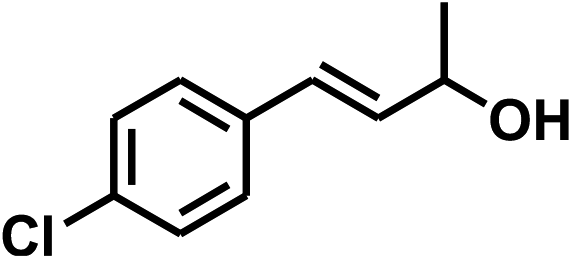
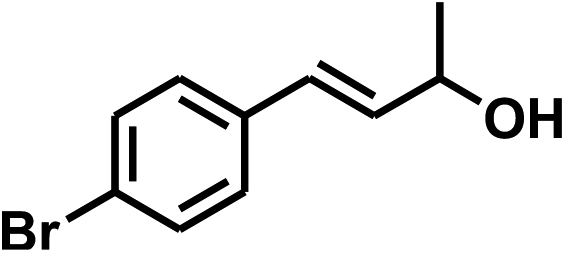
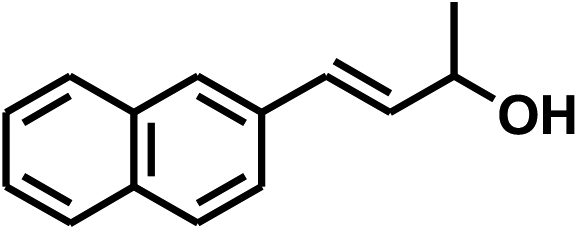
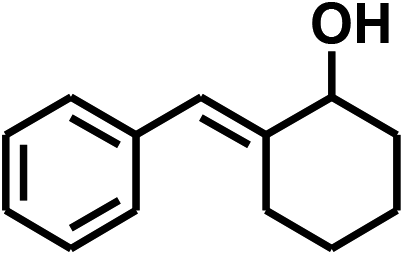
With the optimized conditions in hand, our initial study focused on the chemoselective reduction of α,β-unsaturated aldehydes (Table 2). Pleasedly, the transfer hydrogenation of cinnamaldehyde type substrates showed photoinduced selective hydrogenation with high selectivity toward the production of corresponding unsaturated alcohol (entries 1–10). The electron-withdrawing substituent slightly reduced the conversion of cinnamaldehydes (entries 4–7 and 10), but had little influence on the selectivity to the unsaturated alcohols. What's more, the reduction of heteroatom-containing α,β-unsaturated aldehyde resulted in the CO bond-selective hydrogenation to α,β-unsaturated alcohol in good yield (85%). The aliphatic unsaturated aldehyde exhibited lower activity and selectivity than the aromatic unsaturated aldehydes (entries 12–13). The catalyst system can also be applied to selective hydrogenation of α,β-unsaturated ketones. NPs 3 can catalyse hydrogenation of benzylideneacetone with high activity, obtaining the corresponding α,β-unsaturated alcohols in excellent yields. p-substituted benzylideneacetones were selectively transformed to the corresponding unsaturated alcohols, providing good yields (entries 14–19). α,β-unsaturated ketone without a conjugated aromatic ring was also converted, but the selectivities was lower (entry 20) than those of α,β-unsaturated ketones with a conjugated aromatic ring.
Table 2 Selective photocatalytic hydrogenation of unsaturated carbonylsa
| Entry |
Main product |
Conv.b (%) |
Select.b (%) |
Yieldc (%) |
| General conditions: unsaturated carbonyls (0.2 mmol), 3 (6 mg), NaOH (20 mol%), i-PrOH/MeCN (8 mL, 2/1), room temperature, blue LEDs (455 nm, 26 W), 24 h, N2. Yields determined by HPLC with biphenyl as an internal standard. Isolated Yields. Reaction time = 36 h in parentheses. |
| 1 |
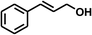 |
100 |
94 |
94 |
| 2 |
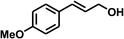 |
100 |
91 |
91 |
| 3 |
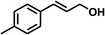 |
100 |
94 |
93 |
| 4 |
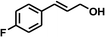 |
83(90) |
92(91) |
75(79) |
| 5 |
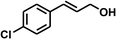 |
95 |
93 |
88 |
| 6 |
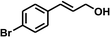 |
95 |
95 |
89 |
| 7 |
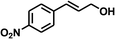 |
86(92) |
93(92) |
78(82) |
| 8 |
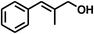 |
93 |
91 |
83 |
| 9 |
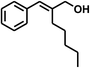 |
88 |
91 |
82 |
| 10 |
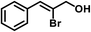 |
73(85) |
90(91) |
65(74) |
| 11 |
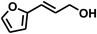 |
90 |
91 |
85 |
| 12 |
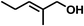 |
84(92) |
86(83) |
67(72) |
| 13 |
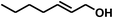 |
80(90) |
84(82) |
62(70) |
| 14 |
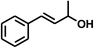 |
87 |
84 |
75 |
| 15 |
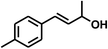 |
92 |
87 |
80 |
| 16 |
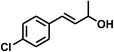 |
85 |
86 |
72 |
| 17 |
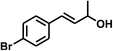 |
89 |
84 |
75 |
| 18 |
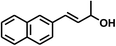 |
82 |
86 |
70 |
| 19 |
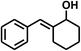 |
75 |
92 |
68 |
| 20 |
 |
72(85)d |
85(83)d |
61(68)d |
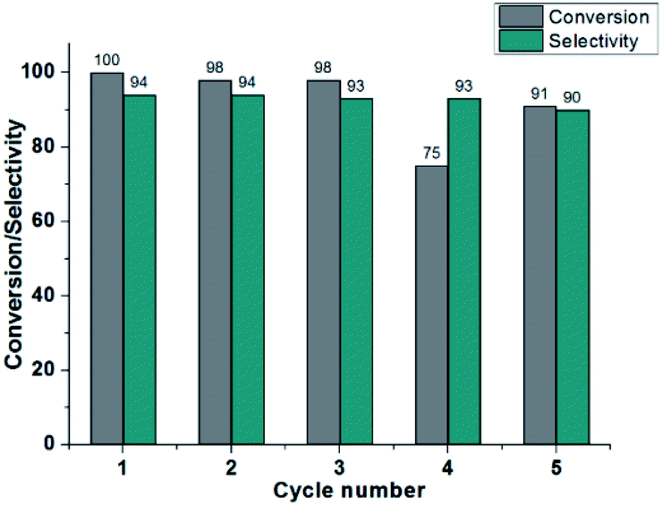
The reusability of the catalyst 3 is also investigated for the selectivity hydrogenation of cinnamaldehyde to cinnamyl alcohol. After the first reaction cycle, the catalyst is directly separated from the reaction mixture by centrifugation and washed with MeCN, H2O and Et2O. The isolated catalyst is used for the second cycle by adding 4a and NaOH in MeCN/i-PrOH under the blue LEDs irradiation. After the catalytic reaction, catalyst 3 was collected by filtration, washed with MeCN, H2O and Et2O, dried in air and then characterized by PXRD. The PXRD patterns were in good agreement with the simulated ones generated from the single crystal data of 3, suggesting that the original framework structure of 3 was retained during the catalysis (Fig. S10†). After the hydrogenation reaction was performed for the fourth cycle, the SEM images of the resulting catalytic species revealed the obvious shape changes of NPs 3 (Fig. S11†), resulting in poor hydrogenation activity, but still with high selectivity. When the fifth cycle was performed for longer time (36 h), about 82% of 4a was converted to 5a. The catalyst deactivation was witnessed after each run (Fig. 4). This result also suggested that photocatalysis is very likely to be limited to the surface layers of NPs 3. We propose the mechanism involving a metal–ligand bifunctional mechanism.51–56 As shown in scheme S1,† the proton from i-PrOH is delivered to the dmpymt ligand, yielding Cat II. Meanwhile, the β-elimination of acetone from Cu isopropoxide intermediate (I) produces Cat I, which is subsequently protonated by alcohol to obtain a hydride/proton complex Cat II. In order to investigate the role of the NH or SH group, we modified Hdmpymt by methylation, obtaining a ligand 4,6-dimethyl-2-(methylthio)pyrimidine (dmmtpym), which doesn't contain a basic nitrogen for protonation. The resulting Cu(I) complexes [Cu6(L)6] (4, L = dmmtpym) exhibited poorer activity and selectivity than that of anologue 1 (Table S3,† entries 18 and 20). The result supports the idea of an NH proton transfer concomitant with carbonyl reduction by Cu–H.
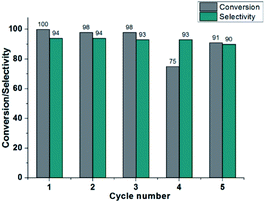 |
| | Fig. 4 Recycling tests for photoinduced chemoselective transfer hydrogenation of cinnamaldehyde using 3 under visible light irradiation. | |
Conclusions
In summary, we developed a straightforward synthesis of bifunctional cluster-based MOFs 2 and 3 through the reaction of a preformed cluster [Cu6(dmpymt)6] as a metalloligand with CuI and (Ph3P)CuI. 2 and 3 serve both as a photosensitizer and a co-catalyst. Under the irradiation of blue LEDs, they display high photocatalytic activity toward the transfer hydrogenation of unsaturated carbonyl compounds to unsaturated alcohols with high selectivity at room temperature. This work also clearly demonstrates that the synthetic methodology may be applied to other bifunctional clusters to yield recyclable heterogeneous photocatalysts with enhanced catalytic activities. Further studies are in progress to design robust and efficient MOFs-based photocatalysts for visible light-driven transfer hydrogenation reactions.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by Bozhou University and Natural Science Research Key Projects of Anhui Province Universities (KJ2020A0774).
Notes and references
- R. H. Morris, Acc. Chem. Res., 2015, 48, 1494–1502 CrossRef CAS PubMed.
- J.-i. Ito and H. Nishiyama, Tetrahedron Lett., 2014, 55, 3133–3146 CrossRef.
- S. G. Ouellet, A. M. Walji and D. W. C. Macmillan, Acc. Chem. Res., 2007, 40, 1327–1339 CrossRef CAS PubMed.
- S. Rösler, J. Obenauf and R. Kempe, J. Am. Chem. Soc., 2015, 137, 7998–8001 CrossRef PubMed.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS PubMed.
- D. Guijarro, Ó. Pablo and M. Yus, J. Org. Chem., 2013, 78, 3647–3654 CrossRef CAS PubMed.
- M. Tamura, K. Tokonami, Y. Nakagawa and K. Tomishige, ACS Catal., 2016, 6, 3600–3609 CrossRef CAS.
- R. Moser, Ž. V. Bošković, C. S. Crowe and B. H. Lipshutz, J. Am. Chem. Soc., 2010, 132, 7852–7853 CrossRef CAS PubMed.
- T. Mitsudome and K. Kaneda, Green Chem., 2013, 15, 2636–2654 RSC.
- R. H. Crabtree, Chem. Rev., 2017, 117, 9228–9246 CrossRef CAS PubMed.
- Z. Y. Guo, C. X. Xiao, R. V. Maligal-Ganesh, L. Zhou, T. W. Goh, X. L. Li, D. Tesfagaber, A. Thiel and W. Y. Huang, ACS Catal., 2014, 4, 1340–1348 CrossRef CAS.
- C. J. Stephenson, C. L. Whitford, P. C. Stair, O. K. Farha and J. T. Hupp, ChemCatChem, 2016, 8, 855–860 CrossRef CAS.
- D. Loffreda, F. Delbecq, F. Vigné and P. Sautet, J. Am. Chem. Soc., 2006, 128, 1316–1323 CrossRef CAS PubMed.
- G. H. Wang, X. H. Deng, D. Gu, K. Chen, H. Tüysüz, B. Spliethoff, H. J. Bongard, C. Weidenthaler, W. Schmidt and F. Schüth, Angew. Chem., Int. Ed., 2016, 55, 11101–11105 CrossRef CAS PubMed.
- S. Tuokko, P. M. Pihko and K. Honkala, Angew. Chem., Int. Ed., 2016, 55, 1670–1674 CrossRef CAS PubMed.
- E. Plessers, D. E. De Vos and M. B. J. Roeffaers, J. Catal., 2016, 340, 136–143 CrossRef CAS.
- C. M. Moore and N. K. Szymczak, Chem. Commun., 2013, 49, 400–402 RSC.
- Y. J. Xue, H. Y. Xin, W. H. Xie, P. Wu and X. H. Li, Chem. Commun., 2019, 55, 3363–3366 RSC.
- S. He, L. F. Xie, M. W. Che, H. C. Chan, L. C. Yang, Z. P. Shi, Y. Tang and Q. S. Gao, J. Mol. Catal. A: Chem., 2016, 425, 248–254 CrossRef CAS.
- X. M. Liu, S. J. Cheng, J. L. Long, W. Zhang, X. H. Liu and D. P. Wei, Mater. Chem. Front., 2017, 1, 2005–2012 RSC.
- S. Choudhury, J.-O. Baeg, N.-J. Park and R. K. Yadav, Angew. Chem., Int. Ed., 2012, 51, 11624–11628 CrossRef CAS PubMed.
- A. Call, C. Casadevall, F. Acuña-Parés, A. Casitas and J. Lloret-Fillol, Chem. Sci., 2017, 8, 4739–4749 RSC.
- J. Li, J. H. Yang, F. Y. Wen and C. Li, Chem. Commun., 2011, 47, 7080–7082 RSC.
- T. Ghosh, T. Slanina and B. König, Chem. Sci., 2015, 6, 2027–2034 RSC.
- X. B. Ke, X. G. Zhang, J. Zhao, S. Sarina, J. Barry and H. Y. Zhu, Green Chem., 2013, 15, 236–244 RSC.
- X. B. Liu, D. R. Sun, R. S. Yuan, X. Z. Fu and Z. H. Li, J. Catal., 2013, 304, 1–6 CrossRef CAS.
- C. H. Hao, X. N. Guo, Y. T. Pan, S. Chen, Z. F. Jiao, H. Yang and X. Y. Guo, J. Am. Chem. Soc., 2016, 138, 9361–9364 CrossRef CAS PubMed.
- H. Xiao, W. Y. Zhang, G. S. Yao, L. L. Hang, L. H. Chen, B. Boury and Z. W. Chen, Appl. Catal., B, 2019, 244, 719–731 CrossRef CAS.
- C. G. Silva, A. Corma and H. García, J. Mater. Chem., 2010, 20, 3141 RSC.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Férey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- C. Wang, Z. Xie, K. E. deKrafft and W. Lin, J. Am. Chem. Soc., 2011, 133, 13445–13454 CrossRef CAS PubMed.
- A. G. Condie, J. C. González-Gómez and C. R. J. Stephenson, J. Am. Chem. Soc., 2010, 132, 1464–1465 CrossRef CAS PubMed.
- C. Wang, K. E. deKrafft and W. B. Lin, J. Am. Chem. Soc., 2012, 134, 7211–7214 CrossRef CAS PubMed.
- D. Shi, R. Zheng and M. J. Sun, Angew. Chem., Int. Ed., 2017, 56, 14637–14641 CrossRef CAS PubMed.
- J. A. Fuentes, I. Carpenter, N. Kann and M. L. Clarke, Chem. Commun., 2013, 49, 10245–10247 RSC.
- N. Arai, K. Azuma, N. Nii and T. Ohkuma, Angew. Chem., Int. Ed., 2008, 47, 7457–7460 CrossRef CAS PubMed.
- H. G. Nedden, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2623–2643 CrossRef PubMed.
- M. J. Zhang, D. W. Tang, H. X. Li, D. J. Young, H. F. Wang, H. Y. Li and J. P. Lang, J. Org. Chem., 2018, 83, 1204–1215 CrossRef CAS PubMed.
- C. Zhang, G. Y. Li and X. L. Cai, Int. J. Energy Res., 2017, 1–8 Search PubMed.
- F. Kamatsos and C. A. Mitsopoulou, Int. J. Hydrogen Energy DOI:10.1016/j.ijhydene.2021.02.005.
- A. Das, Z. J. Han, W. W. Brennessel, P. L. Holland and R. Eisenberg, ACS Catal., 2015, 5, 1397–1406 CrossRef CAS.
- Z. J. Han, L. X. Shen, W. W. Brennessel, P. L. Holland and R. Eisenberg, J. Am. Chem. Soc., 2013, 135, 14659–14669 CrossRef CAS PubMed.
- X. L. Cai, G. Y. Li, Y. Yang and C. Zhang, Russ. J. Appl. Chem., 2016, 89, 1506–1511 CrossRef CAS.
- S. Gao, W. Y. Zhang, Q. Duan, Q. C. Liang, D. Y. Jiang, J. X. Zhao and J. H. Hou, Chem. Pap., 2017, 71, 617–625 CrossRef CAS.
- H. Tang, E. N. Brothers, C. A. Grapperhaus and M. B. Hall, ACS Catal., 2020, 10, 3778–3789 CrossRef CAS.
- H. N. Kagalwala, A. B. Maurer, I. N. Mills and S. Bernhard, ChemCatChem, 2014, 6, 3018–3026 CrossRef CAS.
- D. Li, W. J. Shi and L. Hou, Inorg. Chem., 2005, 44, 3907–3913 CrossRef CAS PubMed.
- A. Gallego, O. Castillo, C. J. Gómez-García, F. Zamora and S. Delgado, Inorg. Chem., 2012, 51, 718–727 CrossRef CAS PubMed.
- Y. X. Weng, K. C. Chan, B. C. Tzeng and C. M. Che, J. Chem. Phys., 1998, 109, 5948–5955 CrossRef CAS.
- L. Han, M. C. Hong, R. H. Wang, B. L. Wu, Y. Xu, B. Y. Lou and Z. Z. Lin, Chem. Commun., 2004, 2578–2579 RSC.
- T. Ohkuma, H. Ooka, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1995, 117, 10417–10418 CrossRef CAS.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- B. L. Conley, M. K. Pennington-Boggio, E. Boz and T. J. Williams, Chem. Rev., 2010, 110, 2294–2312 CrossRef CAS PubMed.
- H. G. Nedden, A. Zanotti-Gerosa and M. Wills, Chem. Rec., 2016, 16, 2623–2643 CrossRef PubMed.
- J. A. Fuentes, I. Carpenter, N. Kann and M. L. Clarke, Chem. Commun., 2013, 49, 10245–10247 RSC.
- X. F. Wu, J. K. Liu, X. H. Li, A. Zanotti-Gerosa, F. Hancock, D. Vinci, J. Ruan and J. L. Xiao, Angew. Chem., Int. Ed., 2006, 45, 6718–6722 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, 1H and 13C NMR of products, crystallographic data, PXRD patterns, fluorescence spectra, etc. CCDC 2061814 and 2061807. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01102b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
David James Youngb,
Ji Long Maa and
Guo Quan Shaoa
*a,
David James Youngb,
Ji Long Maa and
Guo Quan Shaoa