
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCobalt-catalyzed, directed arylation of C–H bonds in N-aryl pyrazoles†
Tung T. Nguyen *ab,
Lam V. Leab,
Hai H. Phamab,
Dung H. Nguyenab,
Nam T. S. Phanab,
Ha V. Leab and
Anh N. Q. Phan*ab
*ab,
Lam V. Leab,
Hai H. Phamab,
Dung H. Nguyenab,
Nam T. S. Phanab,
Ha V. Leab and
Anh N. Q. Phan*ab
aFaculty of Chemical Engineering, Ho Chi Minh City University of Technology (HCMUT), 268 Ly Thuong Kiet, District 10, Ho Chi Minh City, Vietnam. E-mail: tungtn@hcmut.edu.vn; pnqanh@hcmut.edu.vn
bVietnam National University Ho Chi Minh City, Linh Trung Ward, Thu Duc District, Ho Chi Minh City, Vietnam
First published on 1st March 2021
Abstract
We report a method for directed ortho-arylation of N-aryl pyrazoles with arylboronic acids. Reactions proceeded in the presence of a Co(hfacac)2 catalyst, CeSO4 oxidant, and HFIP solvent. Functionalities such as nitro, ester, bromo, and ketone groups were compatible with the reaction conditions. Using heterocycles including thiophene and carbazole was also feasible.
The last few decades have witnessed extensive development of methods for directed functionalization of C–H bonds.1 Since many functionalities are weak coordinating groups toward C–H activation, directing groups are commonly required, thus facilitating the cleavage of C–H bonds.2 Among these, using N-heterocycles which could bring the transition metal proximal to the targeted C–H bonds have attracted substantial attention. It should be noted that most of the general methods have relied on the use of bidentate, strongly coordinating groups. Meanwhile, monodentate, neutral, nitrogen-based directing groups have often suffered from the utilization of second- or third-row transition metals, which are relatively expensive and scarce.3
The pyrazole moiety is ubiquitously found in bio-related molecules (Scheme 1).4 Functionalization of C–H bonds in pyrazoles would offer a convenient route to diversify the structures which are useful for further studies. Using the nitrogen atom in pyrazole for directed C–H activation has been documented.5 Arylation of ortho C–H bonds in N-aryl pyrazoles would afford condensed, synthetically useful anilides.5b Methods for palladium-catalyzed, pyrazole-directed arylation of sp2 C–H bonds with aryl iodides have been developed.5a,5b Aryl (pseudo)halides were competent substrates for ruthenium-catalyzed, ortho-functionalization of N-aryl pyrazoles.5c–h Notably, using complexes of first-row metals for pyrazole-directed arylation is rare.5i Herein we report our attempts for arylation of ortho C–H bonds in N-aryl pyrazoles with arylboronic acids. Successes relied on the use of cobalt(II) hexafluoroacetylacetonate Co(hfacac)2 catalyst, CeSO4 co-oxidant, and HFIP solvent.
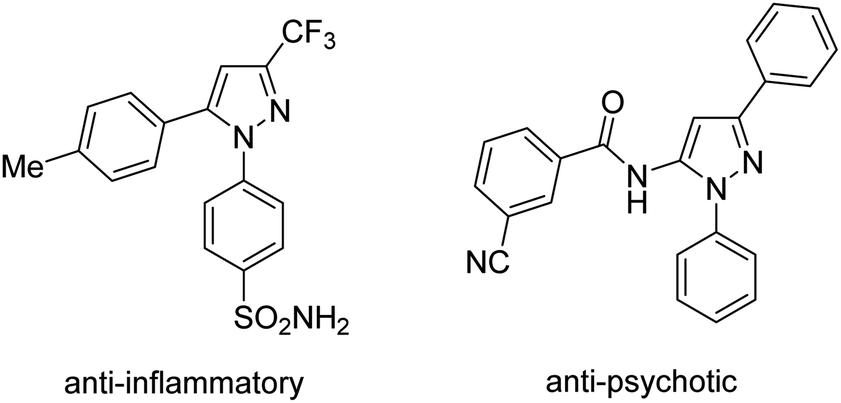 | ||
| Scheme 1 N-Aryl pyrazole based bio-related molecules. | ||
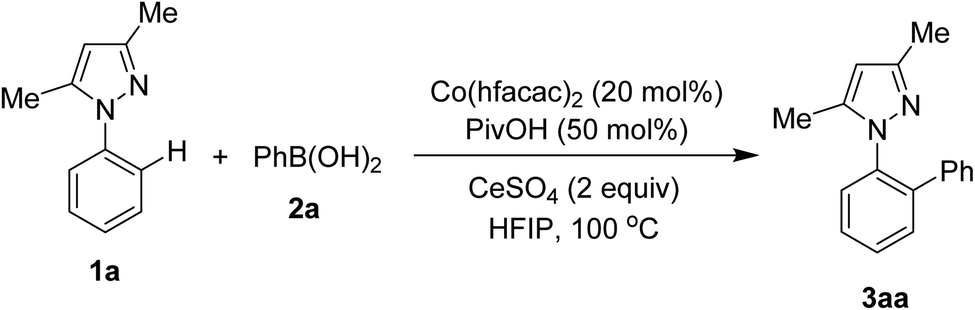
Reaction of N-phenyl pyrazole 1a and phenylboronic acid 2a was firstly discovered. Results and control experiments are shown in Table 1. Using Co(hfacac)2 catalyst afforded the ortho-arylation product 3aa in 82% yield (entry 1). That Co(acac)2 was an inferior catalyst (entry 2) to Co(hfacac)2 somewhat implied the importance of electronic properties of cobalt(II) complexes. The additive pivalic acid PivOH was crucial to obtain an acceptable yield of 3aa (entry 3). Other co-oxidants rather than CeSO4 were not effective for this directed arylation (entries 4 and 5). Trifluoroethanol could be used as solvent for the coupling of 1a and 2a, thus furnishing 3aa in 77% yield (entry 6). The reaction should not be run at the temperature lower than 100 °C (entry 7). Lastly, omitting the presence of Co(hfacac)2 gave no arylation product (entry 8), confirming the crucial role of the cobalt complex.
|
||
|---|---|---|
| Entry | Variation from standard conditions | Yield of 3aa (%) |
| a 1a (0.1 mmol), 2a (0.2 mmol), solvent (1 mL), under air for 24 h. Yields are GC yields using diphenyl ether internal standard. Abbreaviations: hfacac = hexafluoroacetylacetonate, TFE = trifluoroethanol, HFIP = hexafluoroisopropanol. | ||
| 1 | None | 82 |
| 2 | Co(acac)2 instead of Co(hfacac)2 | 65 |
| 3 | No PivOH | 57 |
| 4 | Mn(OAc)3·2H2O instead of CeSO4 | 22 |
| 5 | PhI(OAc)2 instead of CeSO4 | 0 |
| 6 | TFE instead of HFIP | 77 |
| 7 | 80 °C instead of 100 °C | 52 |
| 8 | No Co(hfacac)2 | 0 |
Scope of the arylation with respect to arylboronic acids was next studied. The results are presented in Scheme 2. Electron-neutral and electron-rich arylboronic acids were competent subtrates (3aa, 3ab). In contrast to palladium-catalyzed methods,5a,5b our conditions were tolerant of arylboronic acids containing nitro functionality (3ac, 3ad). Ketone (3af) and ester (3ag) groups were still intact during the course of the arylation. However, the nitrile functional group was hydrolyzed, affording the arylation product in moderate yield (3ah). Heteroaryl boronic acids were also attempted. Arylation of C–H bonds with 2-thienylboronic acid gave only 28% yield of the desired product (3ai). Meanwhile, a 72% yield of a carbazole-derived pyrazole (3aj) was successfully isolated. It should be noted that ortho-substituted arylboronic acids were not competent subtrates.
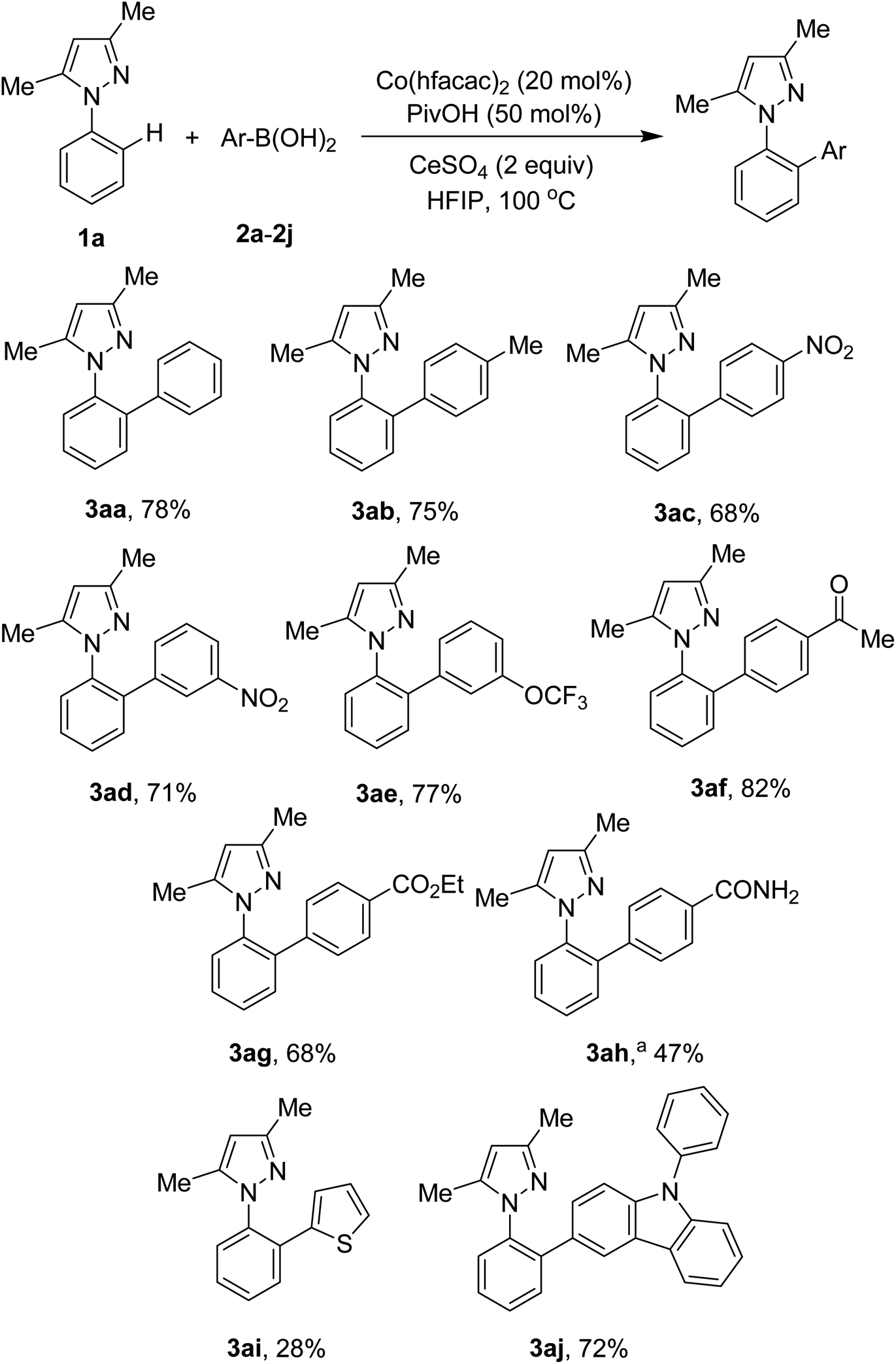 | ||
| Scheme 2 Scope of arylboronic acids. Reaction conditions: 1a (0.5 mmol), 2a–2j (1 mmol), Co(hfacac)2 (0.1 mmol), PivOH (0.25 mmol), CeSO4 (1 mmol), HFIP (2.5 mL), under air, at 100 °C for 24 h. Yields are isolated yields. a4-Cyanophenylboronic acid was used. | ||
Different N-aryl pyrazoles were examined. The results are shown in Scheme 3. Reaction conditions were compatible with electron-rich arenes (3ba, 3ca, 3ef, 3ej). Less hindered C–H bonds were selectively activated if a biased arene was attempted (3dg). N-Aryl pyrazoles containing bromo and trifluoromethoxy groups were successfully arylated (3fd, 3gg). If 2-naphthyl derived pyrazole was used, arylation occurred at the C1–H bond (3ha).
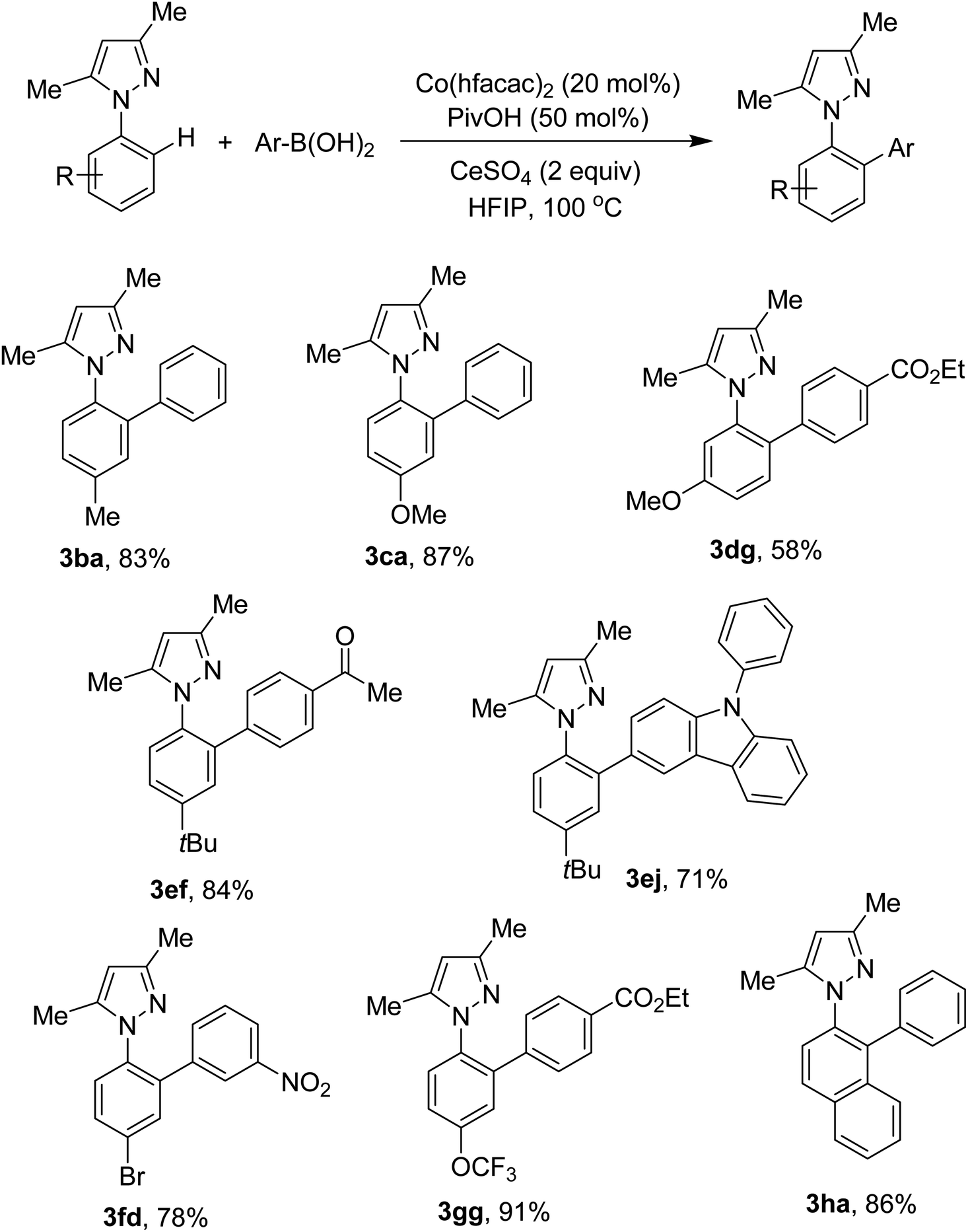 | ||
| Scheme 3 Arylation of C–H bonds in N-aryl pyrazoles. Reaction conditions: N-aryl pyrazole (0.5 mmol), arylboronic acid (1 mmol), Co(hfacac)2 (0.1 mmol), PivOH (0.25 mmol), CeSO4 (1 mmol), HFIP (2.5 mL), under air, at 100 °C for 24 h. Yields are isolated yields. | ||
Based on the results of previous studies,6 our hypothesis on the reaction mechanism may include a Co(II)/Co(III) catalytic cycle. Coordination of nitrogen atom in pyrazole directing group would bring the cobalt(III) center proximal to the ortho C–H bond, thus facilitating the C–H activation. Transmetallation followed by reductive elimination would afford the arylation product.
In conclusion, we have developed a method for cobalt-catalyzed, pyrazole-directed arylation of sp2 C–H bonds. Reaction conditions were tolerant of many functionalities including ester, ketone, nitro, and bromo groups. Ongoing projects will be involving investigation of other pyrazole-directed functionalization of inert C–H bonds.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the National Foundation for Science and Technology Development (NAFOSTED) for financial support (via project no. 104.05-2018.330).References
-
(a) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754 CrossRef CAS
; (b) P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192 CrossRef CAS
-
(a) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788 CrossRef CAS
-
(a) M. Zhang, Y. Zhang, X. Jie, H. Zhao, G. Li and W. Su, Org. Chem. Front., 2014, 1, 843 RSC
-
(a) T. de Paulis, K. Hemstapat, Y. Chen, Y. Zhang, S. Saleh, D. Alagille, R. M. Baldwin, G. D. Tamagnan and P. J. Conn, J. Med. Chem., 2006, 49, 3332 CrossRef CAS
-
(a) D. Shabashov and O. Daugulis, Org. Lett., 2005, 7, 3657 CrossRef CAS
-
(a) R. Mei, U. Dhawa, R. C. Samanta, W. Ma, J. Wencel-Delord and L. Ackermann, ChemSusChem, 2020, 13, 3306 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00975c |
| This journal is © The Royal Society of Chemistry 2021 |