
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEgyptian blue: from pigment to battery electrodes†
J. Landon Tylera,
Robert L. Sacci a,
Jinliang Ningb,
David R. Mullinsa,
Kun Liangb,
Jagjit Nandaa,
Jianwei Sunb and
Michael Naguib*b
a,
Jinliang Ningb,
David R. Mullinsa,
Kun Liangb,
Jagjit Nandaa,
Jianwei Sunb and
Michael Naguib*b
aChemical Sciences Division, Oak Ridge National Laboratory, Oak Ridge, Tennessee 37831, USA
bDepartment of Physics and Engineering Physics, Tulane University, New Orleans, LA 70118, USA. E-mail: naguib@tulane.edu
First published on 2nd June 2021
Abstract
Herein we report on using Egyptian blue as an anode material for Li-ion batteries. A 1st cycle lithiation capacity of 594 mA h g−1 and reversible capacity of 210 mA h g−1 at 20 mA g−1, and at 500 mA g−1 a reversible capacity of 120 mA h g−1 (stable over 1000 cycles) were achieved with coulombic efficiency more than 99.5%. Using X-ray diffraction, and FTIR and X-ray absorption spectroscopies we found that the material goes through a conversion reaction during the 1st cycle that results in the formation of amorphous mixed oxides with copper nanoclusters.
Calcium copper silicate (CaCuSi4O10), known as Egyptian Blue (EB), was first used about 5000 years ago by the ancient Egyptians as a blue pigment for pottery, jewelry and paintings.1 Even under the harsh conditions that some of the artifacts from that era have been in, the EB remained without significant degradation mostly due to its high physical and chemical stability.2 Even though this material has been thoroughly characterized for identification purposes in ancient pottery, Renaissance era paintings and other art pieces, recent studies on EB have yielded newly discovered properties such as near infrared photoluminescence.3–5 Even more recently, the discovery of exfoliating EB into nanosheets has breathed new life into studies with EB as a functional nanomaterial.6 By combining the luminescent properties and the ability to process the nanosheets in ink-jet printing, EB may have a promising future in security inks and near-IR-based biomedical imaging.6,7 With its new momentum, studies into EB may produce results never expected of an ancient pigment.8
As shown in Fig. 1, the crystal structure of EB consists of corner shared SiO4 tetrahedra rings linked together by square-planar coordinated copper, and the copper silicates blocks are interleaved by calcium atoms. Copper silicates with elements other than Ca between the blocks such as BaCuSi4O10 and SrCuSi4O10 were also reported.9
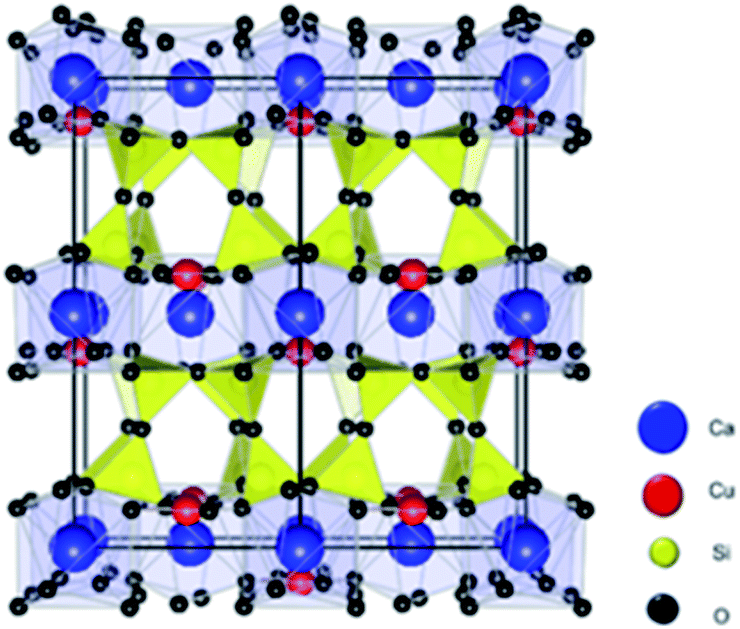 | ||
| Fig. 1 Atomistic model for the CaCuSi4O10 (Egyptian blue) structure. Ca, Cu, Si, and O atoms are represented by blue, red, yellow, and black spheres, respectively. | ||
With the need to find new electrode materials for Li-ion batteries, we decided to explore EB as an electrode material. The structure contains two hypothetical sites for hosting Li by intercalation or insertion. The first site is similar to other silicate structures that can host lithium ions (e.g. Li2MnSiO4).10 In EB, this storage mechanism would be an intercalation of the lithium ions between the silicate layers by replacing Ca or coexisting with it. The coexistence of Ca and Li ions has been reported before by Pruvost et al. for graphite.11 In addition, Li insertion in the silicate rings similar to Li3VO4 is the second hypothetical site.12
Herein we show that, using X-ray and infrared based techniques, none of these hypothetical mechanisms take place but rather that EB goes through a conversion reaction that irreversibly changes the crystalline structure into amorphous composite of oxides decorated with copper nanoclusters. Conversion reactions are of interest to the battery community due to their high capacities and low costs.13
X-ray diffraction (XRD) pattern of the as-received EB powder (Fig. 2a) shows a predominantly single phase of CaCuSi4O10 [PDF#12-0512].1,3 Scanning electron microscopy (SEM) image of the as received EB (Fig. 2b-1) shows large particles (10–100 μm). The galvanostatic cycling (at 100 mA g−1), of the as-received EB (Fig. 2c) shows 1st cycle lithiation and delithiation capacities of 290 and 100 mA h g−1, respectively. Both values are lower than that of commercial graphite anodes (372 mA h g−1). This can be explained by the large bulky particles (>45 μm) of the as-received EB.
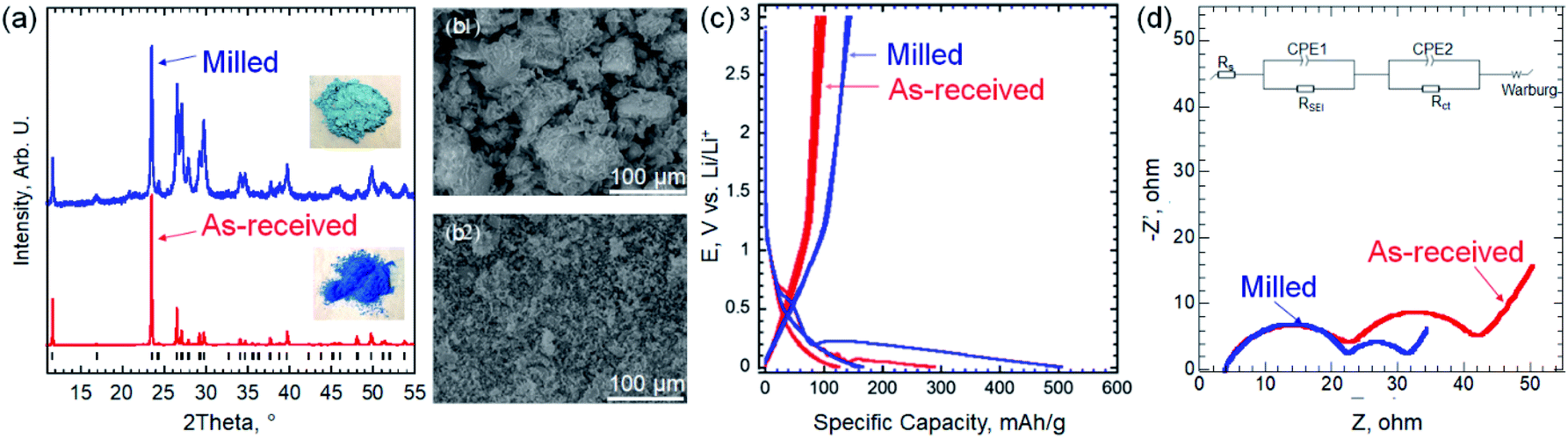 | ||
| Fig. 2 (a) XRD patterns for the as-received EB powder (red) and after milling (blue). The black markers represent the diffraction peaks position of CaCuSi4O10 [PDF#12-0512]. The insets are photographs of the EB powders as received (bottom) and after milling (top) (b) SEM images of EB before (1) and after (2) milling. (c) The voltage profiles of EB at 100 mA g−1 before and after milling. (d) Nyquist plots before and after milling. The inset is the equivalent electrical circuit used to fit EIS data. | ||
To reduce the particle size (PS), the as-received EB was milled as described in the ESI.† XRD pattern of the milled powder showed broader diffraction peaks that can be explained by the smaller PS, which agrees with the SEM image in Fig. 2b-2. Using Scherrer formula,14 the average PS after milling was estimated to be ∼100 nm. It is worth noting that by milling the EB, its color lightened up significantly (insets in Fig. 2a), which can be explained by the PS reduction. Peak broadening, splitting and impure peaks measured after the milling process can be attributed to changes in the lattice structure induced by the stress of mechanical milling.15 As shown in Fig. 2c, this simple milling step resulted in a large increase in the capacity. Thus, the rest of the study was performed using milled EB. At 100 mA g−1, the 1st lithiation capacity increased from 290 to 520 mA h g−1 by milling and the delithiation capacity was 150 mA h g−1 after milling. The smaller PS facilitates ion transport by reducing the diffusion length of the ions and increases specific surface area.16–18
To understand the kinetics before and after milling, electrochemical impedance spectroscopy (EIS) was conducted at 5 mV vs. Li/Li+. As shown in Fig. 2d, Nyquist plots before and after milling exhibit the similar shape, including two semicircles in the high and medium/low frequency regions, and a straight line at the low frequency range. The EIS data were fitted using the inset equivalent electrical circuit, indicating two time constants of solid electrolyte interphase (SEI) and charge transfer processes. The same system resistances of 3.7 Ω and the resistance from SEI of 18.7 Ω are delivered before and after milling. However, the charge transfer resistances are 19.6 and 8.9 Ω before and after milling, respectively. Combining the feature of charger transfer resistance and straight lines, we can clearly see the attribution to the phase transformation or the formation of new phases after intercalating Li+. In addition, the diffusion coefficients before and after milling are calculated to ∼1.3 × 10−12 and 8.8 × 10−12 cm2 s−1, respectively (details for the calculations can be found in the ESI and Fig. S1†). The smaller charge transfer resistance and fast diffusion coefficient facilitate the better electrochemical performance after milling.
The cyclic voltammogram of milled EB is shown in Fig. S2.† Large irreversible lithiation peak below 0.3 V vs. Li/Li+ was observed and it is discussed in more details below. To further investigate the electrochemical performance of milled EB, galvanostatic cycling at 20, 100 and 500 mA g−1 was conducted. A 1st cycle lithiation capacities of 598, 504 and 267 mA h g−1 were measured at 20, 100 and 500 mA g−1, respectively (Fig. 3a). Even though the first cycle irreversibility is high due to irreversible reactions taking place in the 1st cycle (discussed latter), the coulombic efficiencies after the first cycle were over 90% for the length of cycling. At 20 mA g−1, reversible specific capacity of 210 mA h g−1 was delivered. As shown in Fig. 3b, at 500 mA g−1, a stable (over 1000 cycles) reversible capacity of 120 mA h g−1 was achieved. It is worth noting that the coulombic efficiency was larger than 99.5% after 100 cycles. After 100 cycles, the capacity at 100 mA g−1 increased with extended cycling (Fig. 3b and c). One explanation for this initial decrease in capacity can be attributed to extensive structural changes that occur during the first 100 cycles. As these changes begin to form a unique electrode structure, the reactivation of lithium sites is made possible and more active lithium sites over time are developed as observed in other electrode materials.19–22 More work is needed to fully explain this observation.
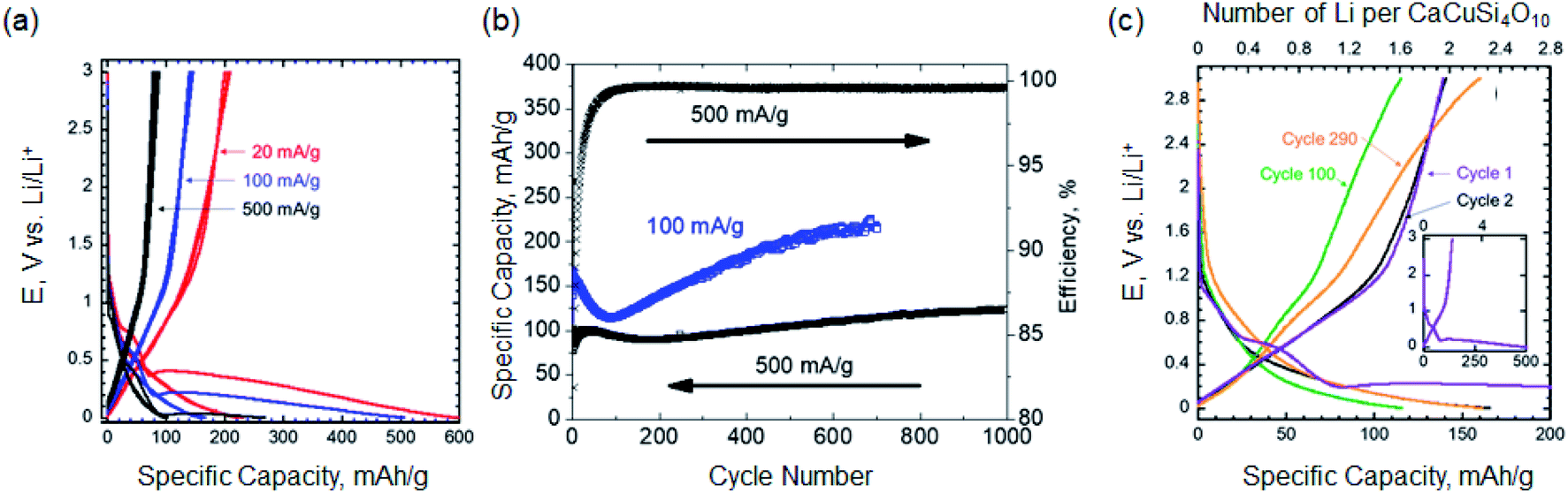 | ||
| Fig. 3 (a) The voltage profiles of the EB at 20 mA g−1 (red), 100 mA g−1 (blue), and 500 mA g−1 (black) (b) cycling performance of 100 mA g−1 and 500 mA g−1 (c) the voltage profile and number of lithium ions participating electrochemically per mole of EB of select cycles at 100 mA g−1. The inset shows the lithiation and delithiation profiles for the first cycle. | ||
As shown in Fig. 4a, XRD pattern after the first lithiation cycle (discharging to 5 mV) showed no new peaks, beside the copper foil current collector peak, but rather a significant drop in the diffraction peaks intensities of the EB, almost vanished, without any measurable shift in peaks position, compared to pristine EB (i.e., before any electrochemical cycling). These results suggest that the material became completely amorphous after lithiation. Thus, we eliminated the possibility of either intercalation or insertion reactions taking place, but rather a conversion reaction that results in the amorphization of the material.
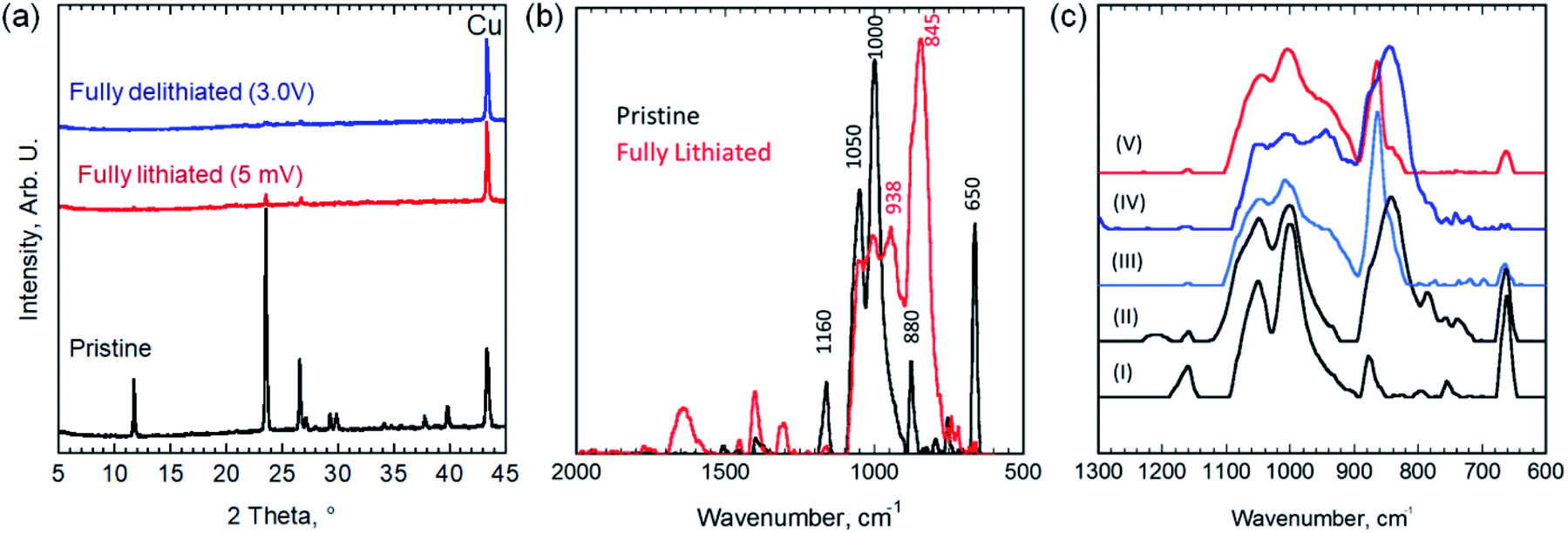 | ||
| Fig. 4 (a) XRD of pristine EB electrode on Cu foil (black) and fully lithiated electrode-discharged to 5 mV (red) and fully delithiated electrode-recharged back to 3 V (blue). (b) The FTIR spectra of pristine (black) and fully lithiated-discharged to 5 mV (red). (c) FTIR spectra of pristine (I), partially discharged to 0.3 V (II) and 0.075 V (III), and fully discharged to 5 mV (IV), and fully discharged then charged to 3 V (V) EB electrodes. | ||
To further understand this conversion reaction, FTIR was performed to characterize changes taking place during lithiation (Fig. 4b and c). Si–O–R bending and stretching vibrations usually appear at around 1000–1100 cm−1 (with a shoulder around 1200 cm−1).23 The absorption peaks at both 1000 and 1050 cm−1 can be attributed to Si–O–Si and Cu–O–Si bonds in the EB and presumably with the peak at 1160 cm−1.24 This peak splitting behavior has been observed in other silicates such as CuMeSiO2.25,26 The absorption peak at 880 cm−1 before lithiation is associated with the C–C–C asymmetric stretching vibration and CF stretching vibration of the polyvinylidene fluoride (PVDF) binder.27 The peaks emerged after lithiation at 845 and 1407 cm−1 indicate the formation of Li2CO3 during SEI formation.28 Normally, absorption peaks around 660 cm−1 represent Cu(I)–O vibrations, but in the case of the EB, the spacing in the crystal structure causes the Cu(II)–O bonds to have a wavenumber indicative of Cu(I)–O bonds (i.e. 650 cm−1).29
In the fully lithiated state, a noticeable loss in the Cu–O bonding peak (at 650 cm−1) has occurred along with the formation of absorption peaks at 845 and 938 cm−1 and smaller peaks after 1300 cm−1. The peaks at 845 cm−1 and at 1300 cm−1 and beyond can be attributed to the formation of organo-lithium compounds and Li2CO3 and SEI formation.30 The disappearance of the Cu–O peak at 650 cm−1 indicates vanishing the Cu–O bonds through an irreversible reaction. The change in the relative ratio between the peaks at 1000 and 1050 cm−1 and the formation of a peak at 938 cm−1 indicates that Si–O–Si and Si–O–Cu bonds are giving way to the formation of Li–Si and Li–Si–O bonds.30
We used X-ray absorption spectroscopy (XAS) to study the changes in the Cu K-edge peak of EB upon lithiation. Fig. 5a shows the XAS spectra of pristine EB compared to CuO and Cu foil. The Cu K-edge spectra features of pristine EB are comparable to what was reported by Pagès-Camagna et al. for archaeological EB.31 As shown in Fig. 5a, oxidation state of Cu in EB is very close to what we measured for copper oxide, CuO, reference material, but after fully lithiation the edge peak was shifted to lower oxidation state and became comparable to copper metal, which agrees with the FTIR results discussed above. After delithiation, the Cu K-edge did not change any further and remained comparable to Cu metal. Fig. 5b shows the Cu K-edge for electrodes lithiated from OCV to 0.3 V and 0.075 V vs. Li/Li+. A gradual shift toward lower oxidation state from pristine EB (Fig. 5a) to lithiated to 0.3 V then 0.075 V (Fig. 5b) and finally fully lithiated to 5 mV (Fig. 5a) was observed. The absence of pronounced peaks around 8994 eV and 9003 eV in the fully lithiated EB compared to what was observed for Cu foil can be explained by formation of atomic clusters of Cu that result in smearing these peaks.32
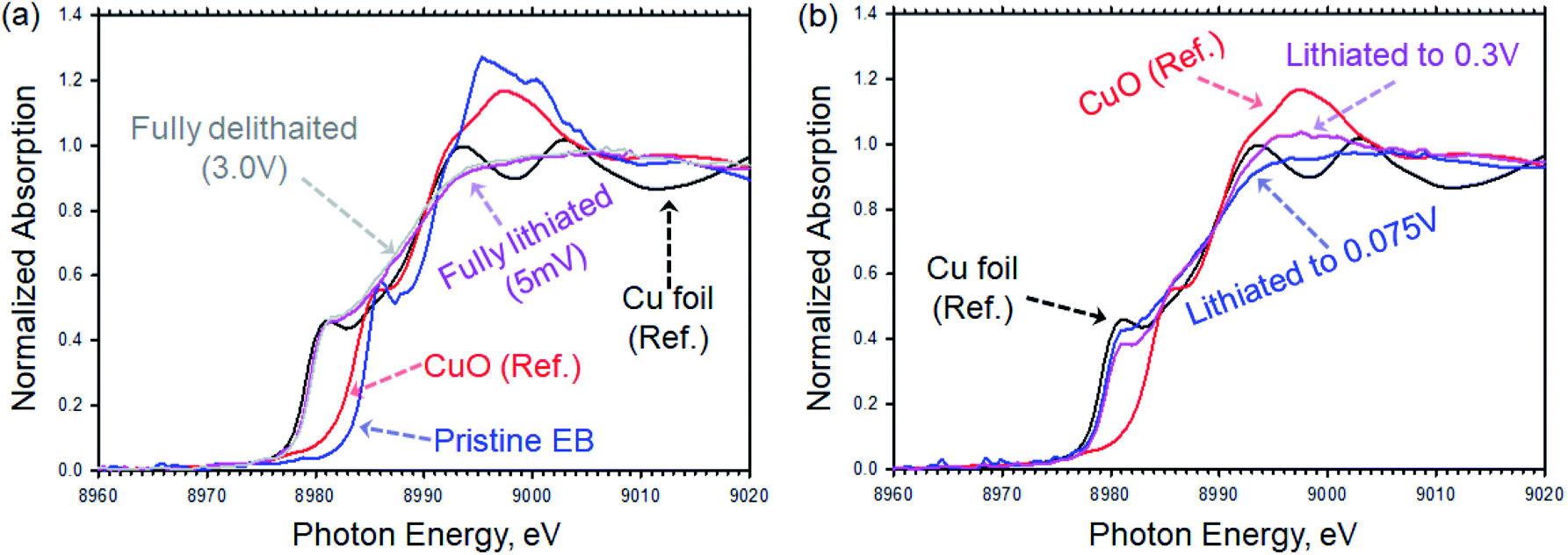 | ||
| Fig. 5 Cu K-edge XAS for EB electrodes compared to reference Cu foil and CuO. (a) Pristine EB electrode (blue), fully lithiated electrode by discharging to 5 mV (purple) and fully delithiated electrode by recharging back to 3 V (gray) compared to Cu foil reference (black) and CuO reference (red). (b) Partially lithiated EB electrodes by discharging to 0.3 V (purple) and 0.075 V (blue) compared to Cu foil reference (black) and CuO reference (red). | ||
There are definitive evidences from the XRD, FTIR and XAS results that the structure of the EB is changing irreversibly during the initial lithiation, and three proposed reactions are taking place to explain the three slopes observed in the voltage profile of the 1st cycles (Fig. 3a and c). First, the SEI is formed by electrolytic reduction of carbonate solvents. This reaction can clearly be seen in FTIR spectra upon lithiation by the presence of newly formed lithium compounds such as LiF and Li2CO3, which make up the SEI layer.33 Moreover, the Cu–O bonds are breaking and forming copper metal that was confirmed using XAS. During this process, a combination of copper nanoclusters and Li2O is formed.34,35 The new-formed copper nanoclusters can act as conductive material enhancing the electrochemical performance. Finally, lithium reacts reversibly with the conversion reaction products (e.g., CaSiO3, SiO2 or SiOx). The exact reversible reaction mechanism of the mixed oxides with Li is not well understood. Even for SiO2, its lithiation mechanism is still debated.36
To find out why does the lithiation of EB is through a conversion reaction, we employed density functional theory (DFT) calculations to determine the energies of the two following reactions:
| 8Li + 2CaCu(Si2O5)2 = Li2Si2O5 + 2CaSiO3 + 3Li2SiO3 + Si + 2Cu, | (1) |
| 2Li4CaCu(Si2O5)2 = Li2Si2O5 + 2CaSiO3 + 3Li2SiO3 + Si + 2Cu. | (2) |
As shown in Table S1,† both reactions are notably exothermic, indicating that both a mixture of Li and EB (with a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mol ratio) or a well relaxed imaginary lithiation compound Li4CaCu(Si2O5)2 (Fig. S3†) are obviously not stable thermodynamically. In both reactions, the reactants tend to decompose into the resultant phases that are all low temperature stable phases determined using Pymatgen and the Materials Project.37,38 This explains why a conversion reaction takes place rather than insertion/intercalation in the EB crystal.
:1 mol ratio) or a well relaxed imaginary lithiation compound Li4CaCu(Si2O5)2 (Fig. S3†) are obviously not stable thermodynamically. In both reactions, the reactants tend to decompose into the resultant phases that are all low temperature stable phases determined using Pymatgen and the Materials Project.37,38 This explains why a conversion reaction takes place rather than insertion/intercalation in the EB crystal.
Confirmed by XRD, FTIR and XAS, as Li-ion electrode material, Egyptian blue hosts Li through a conversion reaction that results in a composite of copper nanoclusters and amorphous oxides. DFT suggest that the lithiated crystal of EB is thermodynamically unstable and therefore conversion reaction takes place. In lieu of the newly formed amorphous composite, EB exhibits a decent reversible capacity (∼210 mA h g−1 at 20 mA g−1), high coulombic efficiencies, and an increase in capacity with cycling time. At 500 mA g−1 a reversible capacity of 120 mA h g−1 was stable over 1000 cycles. Considering that this is the first report on using EB as electrode material, further optimization is inevitable and better electrochemical performance is possible. Also, the presence of Ca ions between the copper silicate layers suggests that EB could be used in Ca-ion batteries.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
JLT and JN are supported by Office of Electricity, Department of Energy through the Energy Storage Research Program, managed by Dr Imre Gyuk. RLS was supported by the U. S. Department of Energy, Office of Science, Basic Energy Sciences, Materials Sciences and Engineering. JN and JS at Tulane University acknowledge the support of the U. S. DOE, Office of Science, Basic Energy Sciences Grant No. DE-SC0014208. MN efforts at Tulane University was partially supported by NASA MIRO Grant Number 80NSSC20M0249.Notes and references
- E. Jope and G. Huse, Nature, 1940, 146, 26 CrossRef CAS.
- D. Johnson-McDaniel, C. A. Barrett, A. Sharafi and T. T. Salguero, J. Am. Chem. Soc., 2013, 135, 1677–1679 CrossRef CAS PubMed.
- W. Chen, Y. Shi, Z. Chen, X. Sang, S. Zheng, X. Liu and J. Qiu, J. Phys. Chem. C, 2015, 119, 20571–20577 CrossRef CAS.
- G. Accorsi, G. Verri, M. Bolognesi, N. Armaroli, C. Clementi, C. Miliani and A. Romani, Chem. Commun., 2009, 3392–3394 RSC.
- Y.-J. Li, S. Ye, C.-H. Wang, X.-M. Wang and Q.-Y. Zhang, J. Mater. Chem. C, 2014, 2, 10395–10402 RSC.
- D. Johnson-McDaniel and T. T. Salguero, J. Visualized Exp., 2014, e51686 Search PubMed.
- B. Errington, G. Lawson, S. W. Lewis and G. D. Smith, Dyes Pigm., 2016, 132, 310–315 CrossRef CAS.
- S. M. Borisov, C. Würth, U. Resch-Genger and I. Klimant, Anal. Chem., 2013, 85, 9371–9377 CrossRef CAS PubMed.
- E. Kendrick, C. Kirk and S. Dann, Dyes Pigm., 2007, 73, 13–18 CrossRef CAS.
- N. Kuganathan and M. Islam, Chem. Mater., 2009, 21, 5196–5202 CrossRef CAS.
- S. Pruvost, C. Hérold, A. Hérold and P. Lagrange, Eur. J. Inorg. Chem., 2004, 2004, 1661–1667 CrossRef.
- Z. Liang, Z. Lin, Y. Zhao, Y. Dong, Q. Kuang, X. Lin, X. Liu and D. Yan, J. Power Sources, 2015, 274, 345–354 CrossRef CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, E170–E192 CrossRef CAS PubMed.
- P. Scherrer, Göttinger Nachrichten Math. Phys., 1918, 2, 98–100 Search PubMed.
- A. E. Mohmoud, H. S. Wasly and M. A. Doheim, J. Eng. Sci., 2014, 42, 1430 Search PubMed.
- A. J. Rennie, V. L. Martins, R. M. Smith and P. J. Hall, Sci. Rep., 2016, 6, 22062 CrossRef CAS PubMed.
- K. Liang, K. Marcus, L. Guo, Z. Li, L. Zhou, Y. Li, S. De Oliveira, N. Orlovskaya, Y.-H. Sohn and Y. Yang, Chem. Commun., 2017, 53, 7608–7611 RSC.
- K. Liang, L. Ju, S. Koul, A. Kushima and Y. Yang, Adv. Energy Mater., 2018, 1802543 Search PubMed.
- Y. Ma, U. Ulissi, D. Bresser, Y. Ma, Y. Ji and S. Passenrini, Electrochim. Acta, 2017, 258, 535–543 CrossRef CAS.
- X. Wei, C. Tang, X. Wang, L. Zhou, Q. Wei, M. Yan, J. Sheng, P. Hu, B. Wang and L. Mai, ACS Appl. Mater. Interfaces, 2015, 7, 26572–26578 CrossRef CAS PubMed.
- H. Sun, G. Xin, T. Hu, M. Yu, D. Shao, X. Sun and J. Lian, Nat. Commun., 2014, 5, 1–8 Search PubMed.
- Z. Cai, L. Xu, M. Yan, C. Han, L. He, K. M. Hercule, C. Niu, Z. Yuan, W. Xu, L. Qu, K. Zhao and L. Mai, Nano Lett., 2015, 15, 738–744 CrossRef CAS PubMed.
- Z. Zhan and H. Zeng, J. Non-Cryst. Solids, 1999, 243, 26–38 CrossRef CAS.
- I. Garofano, A. Duran, J. Perez-Rodriguez and M. Robador, Spectrosc. Lett., 2011, 44, 560–565 CrossRef CAS.
- R. S. d. Cruz, J. M. d. S. Silva, U. Arnold, M. S. Sercheli and U. Schuchardt, J. Braz. Chem. Soc., 2002, 13, 170–176 CrossRef.
- W. Dimassi, I. Haddadi, R. Bousbih, S. Slama, M. A. Kanzari, M. Bouaïcha and H. Ezzaouia, J. Lumin., 2011, 131, 829–833 CrossRef CAS.
- H. Bai, X. Wang, Y. Zhou and L. Zhang, Prog. Nat. Sci.: Mater. Int., 2012, 22, 250–257 CrossRef.
- S. Bhattacharya, A. R. Riahi and A. T. Alpas, Carbon, 2014, 77, 99–112 CrossRef CAS.
- Y. C. Zhang, J. Y. Tang, G. L. Wang, M. Zhang and X. Y. Hu, J. Cryst. Growth, 2006, 294, 278–282 CrossRef CAS.
- G. Nazri and G. Pistoia, Technology, 2004, 453–455 Search PubMed.
- S. Pagés-Camagna, I. Reiche, C. Brouder, D. Cabaret, S. Rossano, B. Kanngießer and A. Erko, X-Ray Spectrom., 2006, 35, 141–145 CrossRef.
- W. Klysubun, Y. Thongkam, S. Pongkrapan, K. Won-in, T. Jiraroj and P. Dararutana, Anal. Bioanal. Chem., 2011, 399, 3033–3040 CrossRef CAS PubMed.
- Y. M. Lee, J. Y. Lee, H.-T. Shim, J. K. Lee and J.-K. Park, J. Electrochem. Soc., 2007, 154, A515–A519 CrossRef CAS.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- S. Grugeon, S. Laruelle, R. Herrera-Urbina, L. Dupont, P. Poizot and J. Tarascon, J. Electrochem. Soc., 2001, 148, A285–A292 CrossRef CAS.
- Z. Liu, Q. Yu, Y. Zhao, R. He, M. Xu, S. Feng, S. Li, L. Zhou and L. Mai, Chem. Soc. Rev., 2019, 48, 285–309 RSC.
- S. P. Ong, W. D. Richards, A. Jain, G. Hautier, M. Kocher, S. Cholia, D. Gunter, V. L. Chevrier, K. A. Persson and G. Ceder, Comput. Mater. Sci., 2013, 68, 314–319 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner and G. Ceder, APL Mater., 2013, 1, 011002 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00956g |
| This journal is © The Royal Society of Chemistry 2021 |