
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnexpected ortho C–H bond activation in coordinated 7,8-benzoquinoline: synthesis and characterisation of heteroleptic Ir(III)-7,8-benzoquinoline complexes†
Kahnu Charan Pradhan ,
Hemanta K. Kisan and
Satyanarayan Pal*
,
Hemanta K. Kisan and
Satyanarayan Pal*
P. G. Dept. of Chemistry, Utkal University, Bhubaneswar, India. E-mail: snpal@utkaluniversity.ac.in
First published on 30th March 2021
Abstract
Two iridium(III) complexes were isolated via the reaction of pyridine-2-aldoxime (Hpyrald) with 7,8-benzoquinoline (benzq)-derived iridium starting material, namely [(benzq)2Ir(μ-Cl)2Ir(benzq)2] (1). Among the two complexes, [IrIII(benzq)2(pyrald)] (2) and [IrIII(benzq-κN,κC10)(benzq-κC2)(Hpyrald)(Cl)] (3), the later displayed unusual ortho C–H bond activation in one of the coordinated 7,8-benzoquinoline rings. The complex (2) presented a usual structure as expected.
C–H bond activation has long been explored fruitfully in the formation of carbon–carbon1,2 and metal–carbon3,4 bonds during the synthesis of fascinating organic compounds and organometallic complexes. The C–H bond, though inert in nature, can be activated through the catalytic power of different metal-based catalysts.1,3 Among the several catalysts designed and synthesised so far, the platinum group metal-derived catalysts are well-known for their facile C–H bond activation in a vast range of organic molecules.3 A judicious choice of organic ligands in metal complex preparation and C–H bond activation therein could lead to the metal–carbon bond formation. The cyclometallating ligand, such as phenyl pyridine (ppy), is one such class of ligand, which binds the metal through pyridine-N and a benzene ring-C (κN, κC8) in the cyclometallated way (Fig. 1; complex (4)) involving the C–H bond activation by the coordinated metal ions.3 Iridium is well known for its cyclometallating properties, which causes C–H bond activation in the cyclometallating class of ligands. A vast range of phosphorescent Ir(III) complexes were prepared from cyclometallating ligands and were studied for their application in numerous fields, such as organic light emitting diodes (OLEDs),5,6 solar light harvesting devices,7,8 and anticancer agents.9,10
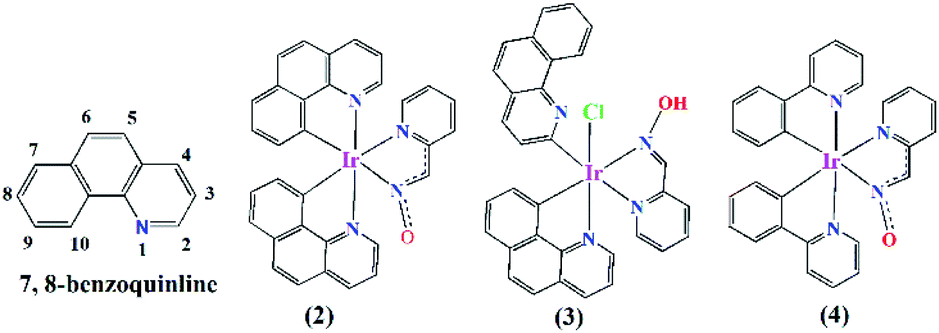 | ||
| Fig. 1 Coordination modes of 7,8-benzoquinoline and phenyl pyridine to Ir(III) in presence of pyridine-2-aldoxime. | ||
7,8-Benzquinoline (benzq) resembles phenyl pyridine in its structural and complexation behaviour (Fig. 1, complex (2)) to iridium and coordinates through N1 and C10. Nonoyama prepared the famous [(benzq)2Ir(μ-Cl)2Ir(benzq)2] (1) in 1974.11 The C–H bond activation of 7,8-benzoquinoline by iridium was observed at the C10 position and led to the formation of a dichloro bridged dimeric complex, where each iridium centre was coordinated to two 7,8-benzoquinolines in the cyclometallated form (κC10, κN). Since then numerous heteroleptic Ir(III)-7,8-benzoquinoline complexes were reported with very same coordination mode.12–18 The Ir-benzoquinoline complexation could be viewed as the formation of initial Ir–N(quinoline) bond, which selectively induces the C10–H bond activation and leads to Ir–C10 coordination. This results in a stable five-membered chelation by 7,8-benzoquinoline. Till date no other types of coordination modes including the ortho C–H activated Ir–C2 coordination in the Ir-7,8-benzoquinoline complex have been found. Moreover, ortho C–H bond activation as reported in 7,8-benzoquinoline in the Minisci type reaction needs designed synthesis to activate the ortho C–H bond19–26 and metalation with ortho-carbon reported only with Sc27 and Zr.28 Till date, literature lacks on any such ortho-carbon coordinated Ir(III)-7,8-benzoquinoline complex. Although no special design/reaction condition was adopted for the synthesis of complex (3), a coordinated 7,8-benzoquinoline (κC10, κN) ring in complex (2) displayed ortho C–H bond activation in the presence of ancillary ligand pyridine-2-aldoxime. The reaction yielded the expected complex with usual 7,8-benzoquinoline coordination, [Ir(benzq)2(pyrald)] (2) (Fig. 1), along with the ortho-carbon coordinated complex (3). Herein, we report the first iridium(III) complex with ortho-carbon coordination in a 7,8-benzoquinoline ring.
The reaction was designed to synthesize the complex [Ir(benzq-κN, κC10)2(pyrald)] (2) (pyrald = deprotonated pyridine 2-aldoxime) (Fig. 1) by employing pyridine-2-aldoxime with [(benzq)2Ir(μ-Cl)2Ir(benzq)2] (1) in the presence of triethylamine in ethanol under reflux conditions (see ESI†). Beside the complex (2), the reaction resulted in complex (3) with lesser yield than the former.
The formulation of complexes (2) and (3) were consistent with the molecular ion peaks found in the ESI mass spectra (Fig. S3 and S6†). The peaks at 670.86 (cald 670.13) of complex (2) and 706.83 (calcd 706.11) of complex (3) confirmed the presence of entire molecular frameworks.
The 1H NMR spectrum of complex (2) in DMSO-d6 (Fig. S4†) confirmed the presence of 21 aromatic protons and matched with the structural formula of (2). The 1H NMR spectrum of complex (3) in DMSO-d6 (Fig. S7†) clearly depicted all aromatic protons and supported the structure of complex (3). The oxime-OH proton however could not be located, which might be due to the involvement of the –N–OH group in strong intramolecular hydrogen bonding interactions29 with quinoline-N (Fig. S8 and Table S3†).
The formation of complex (3) was further confirmed via single crystal X-ray structure determination (Fig. 2). The slow evaporation of the dichloromethane hexane mixture of (3) afforded single crystals at room temperature for structural analysis. The selected bond lengths and bond angles associated with the metal centre and other crystallographic parameters (Tables S1 and S2†) are provided in the ESI.† The structure displayed a distorted octahedral arrangement of ligands around the iridium centre. The C11, C14, N4 and N3 atoms of coordinated ligands were arranged in a square plane with positioning of N1 and Cl1 in the axial positions. This arrangement of ligands was supported with equal bond lengths of corresponding Ir–C14 and Ir–C11 (2.023(5) and 2.031(5) Å) and Ir–N3 and Ir–N4 (2.124(4) & 2.118(4) Å) in the square plane. The Ir–N1 bond (2.043(3) Å) at the axial position was found to be shorter compare to the Ir–N3 and Ir–N4 bond lengths. This could be the result of strong trans effects exerted by coordinating C11 and C14 on Ir–N4 and Ir–N3 bond lengths30 respectively.
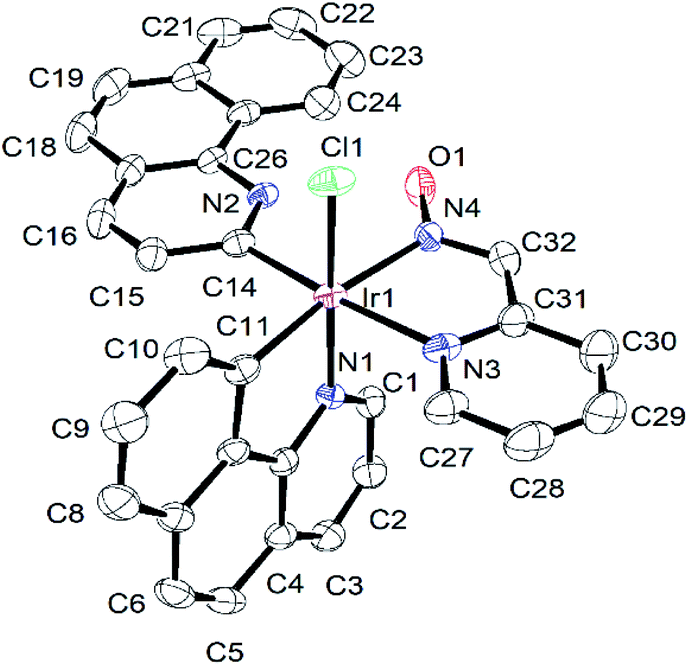 | ||
| Fig. 2 Ortep diagram of complex (3). Thermal ellipsoids are drawn at 40% probability level. Hydrogen atoms are omitted for clarity. | ||
In the axial positions, N1–Ir1–Cl1 was found to be lying almost linearly with an angle of 173.44(10)°. This structural arrangement of ligands was consistent with the reported Ir-pyridine-2-aldoxime complex, [Ir(ppy)2(pyrald)] (4),31 where the pyridine-2-aldoxime and two carbon atoms of phenyl pyridines (ppy) arranged on the square plane. The Ir–C, Ir–N and Ir–Cl [2.3683(12) Å] bond lengths in complex (2) were in good agreement with the values reported for several similar Ir(III) complexes.13,32
The structure of complex (3) displayed two benzoquinoline ligands in different coordination modes. One of them had the ortho C–H bond activation and coordinated in a monodentate way through C14 to the iridium centre, whereas the other benzoquinoline had acted as a bidentate ligand and chelated the Ir(III) centre through N1 and C11. The different coordination modes did not have noticeable effects on Ir–C11 and Ir–C14 bond lengths, which appeared to be indifferent with the values of 2.031(5) and 2.023(5) Å, respectively.
The monodentate benzoquinoline ring and the pyridine-2-aldoxime moiety were oriented in the same direction and appeared to be planar with each other with a dihedral angle of 2.829(114)°. This caused the two benzoquinoline rings to orient near the perpendicular plane with a dihedral angle of 84.752(74)° between them. The oxime-OH group exhibited strong intramolecular hydrogen bonding interactions33 with the bare N2 of the monodentate benzoquinoline ring with the O1⋯N2 distance measured at 2.609(5) Å and O1–H1A⋯N2 angle at 156.88° (Table S3†). Such interactions were expected as N2 with a free lone pair of electrons acted as a Lewis base and the hydrogen of oxime-OH is acidic in nature. The lone pairs of oxime-O1 are involved in the formation of another weak intramolecular hydrogen bond34 with C24–H24. The formation of the O1⋯H24–C24 hydrogen bond was found with O1⋯C24 distance at 3.211(7) Å and C24–H24⋯O1 angle of 159.61°. These two hydrogen bonding interactions might be the reason to orient the benzoquinoline ring along with the plane of coordinated pyridine-2-aldoxime.
The lone pairs of the oxime oxygen atom (O1) are also found to be involved in weak inter molecular hydrogen bonding interactions with C1–H1 and C2–H2 of a neighbouring molecule in the crystal lattice (Fig. S8†). The C1⋯O1 and C2⋯O1 distances were found to be 3.201(6) and 3.259 Å along with the C1–H1⋯O1 and C2–H2⋯O1 angles of 127° and 121.45°, respectively. Thus, the oxime oxygen atom (O1) was crowded with three weak O⋯H–C interactions and its two lone pairs got involved in the formation of trifurcated hydrogen bonds.34
The C1–H1⋯O1 and C2–H2⋯O1 hydrogen bonding interactions between two neighbouring molecules completed an inversion dimer via R22(12)/R22(14) rings (Fig. S8†). Each pair of dimeric units were further connected to each other through inverse C3–H3⋯Cl1 hydrogen bonds35 (Fig. S9†) expanding to a one-dimensional molecular assembly. The C3⋯Cl1 distance and C3–H3⋯Cl1 angle were found to be 3.677(5) Å and 165°, respectively. Further inspection revealed that chloride lone pairs were involved in the formation of another weak hydrogen bonding interaction with the C21–H21 of neighbouring 1D chain (Fig. S8 and S9†). The donor–acceptor (C21⋯Cl1) distance of 3.647(8) Å and the C21–H21⋯Cl1 angle of 155° were consistent with the C3–H3⋯Cl1 hydrogen bond.35 Thus, each chloride atom displayed bifurcated hydrogen bonds between the inverse dimeric unit and the ID molecular assembly (Fig. S10†). The one dimensional molecular chains in the crystal lattice were further stacked with C–H⋯π and π⋯π interactions to complete a two dimensional molecular network (Fig. S10†). The centroid to centroid (Cg–Cg) and C–H⋯π distances are tabulated in Table S4.†
Till date the available structural reports of Ir(III)-7,8-benzoquinoline complexes12–18 displayed similarity with complex (2), where the benzoquinolines chelated iridium centre through N1 and C10 in the cyclometallated form. The formation of the Ir–C10 bond resulted with C–H bond activation during the preparation of the starting material (1) from IrCl3. 3H2O and 7,8-benzoquinoline in 2-methoxy ethanol under reflux conditions.11 The subsequent reaction of starting material (1) with ancillary ligand, pyridine-2-aldoxime resulted in the opening up of one of the 7,8-benzoquinoline chelation and a new coordination of Ir–C(2) appeared as a result of ortho C–H bond activation at C2. This observation of the opening up of Ir(benzq-κN, κC10) chelate and formation of only Ir–C2 bond were highly unusual.
As mentioned hitherto Ir(III) −7,8-benzoquinoline complexes of same design, [Ir(benzq)2(bidentate ancillary ligand)], reported till date have not adopted such ortho C–H bond activation. A range of different bidentate ancillary ligands have been employed in the synthesis of arrays of Ir(III) complexes.12–18 We have employed pyridine-2-aldoxime as a bidentate ancillary ligand and expected the formation of a heteroleptic complex having the formula [Ir(benzq-κN, κC10)2(pyrald)] (2). We have already reported a phenyl pyridine-Ir(III) complex with pyridine-2-aldoxime having the formula [Ir(ppy)2(pyrald)] (4)31 (Fig. 1) (ppy = phenyl pyridine), where such ortho C–H bond activation in the phenyl pyridine (ppy) ring was not observed. It was found to be a bis-heteroleptic Ir(III) octahedral complex, where the two ppy ligands produced expected cyclometallation (κN, κC) to iridium along with the pyridine-N and oxime-N coordination of pyridine-2-aldoxime (Fig. 1). Compared to the other known complexes of Ir(III) −7,8 benzoquinoline, it appeared a definitive involvement of pyridine-2-aldoxime in causing ortho C–H bond activation as found in complex (3).
Alongside the role of pyridine-2-aldoxime, the reaction temperature factor was also taken into consideration and reactions were carried out in different temperatures. First the complexation reaction was carried out in the 2-methoxy ethanol medium at about 120 °C and isolation of complex (3) was linked to the high reaction temperature, which might have caused the modification of chelation of the 7,8-bezoquinoline ring in the starting material [(benzq)2Ir(μ-Cl)2Ir(benzq)2] (1) or complex (2) to induce ortho C–H bond activation.
Though the starting material (1) was obtained under the very same reaction conditions11 without causing any such ortho C–H bond activation in 7,8-benzoquinolines. Considering the above fact, the reactions were carried out comparatively at a low temperature. The synthesis of complex (2) was carried out in methanol and ethanol at the temperature range of 70–80 °C aiming to prevent the ortho C–H bond activation. The purification of the reaction mixture on the neutral aluminium oxide column always produced [Ir(benzq-κN, κC10)(benzq-κC2)(Hpyrald)(Cl)] (3) along with [Ir(benzq-κN, κC10)2(pyrald)] (2). It was also found that the latter always had higher yield than the former.
To further explore the possibility of conversion of complex (2) to (3), the former was heated in ethanol in the presence of LiCl and was found with the partial transformation of complex (2) to (3).
Thus, it appeared that the complexation reaction of pyridine-2-aldoxime with starting material (1) led to the formation of complex (2) first, which subsequently had undergone ortho C–H bond activation and formed (3). A plausible reaction mechanism for the formation of (3) from complex (2) is depicted in Scheme 1. It is suggested that the C10 atom of the one coordinated benzoquinoline ring got protonated by ethanol/pyridine-2-aldoxime/[NEt3H]Cl, which led to the formation of a five-coordinated intermediate Ir(III) complex (2.1) (Scheme 1). [NEt3H]Cl was formed in situ as NEt3 was used as the base to deprotonate the oxime-OH group of pyridine-2-aldoxime and was combined with the chloride obtained from the dichloro-bridged starting material (1). The geometry of the intermediate (2.1) brought –C2–H (ortho –CH) of benzoquinoline and ![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–O− of pyridine-2-aldoxime to a close proximity. The interaction of deprotonated oxime-O with C2–H in the intermediate (2.1) caused the C–H bond activation and led to the formation of intermediate (2.2). On further progress, chloride from [NEt3H]Cl coordinated (2.2) to produce complex (3).
N–O− of pyridine-2-aldoxime to a close proximity. The interaction of deprotonated oxime-O with C2–H in the intermediate (2.1) caused the C–H bond activation and led to the formation of intermediate (2.2). On further progress, chloride from [NEt3H]Cl coordinated (2.2) to produce complex (3).
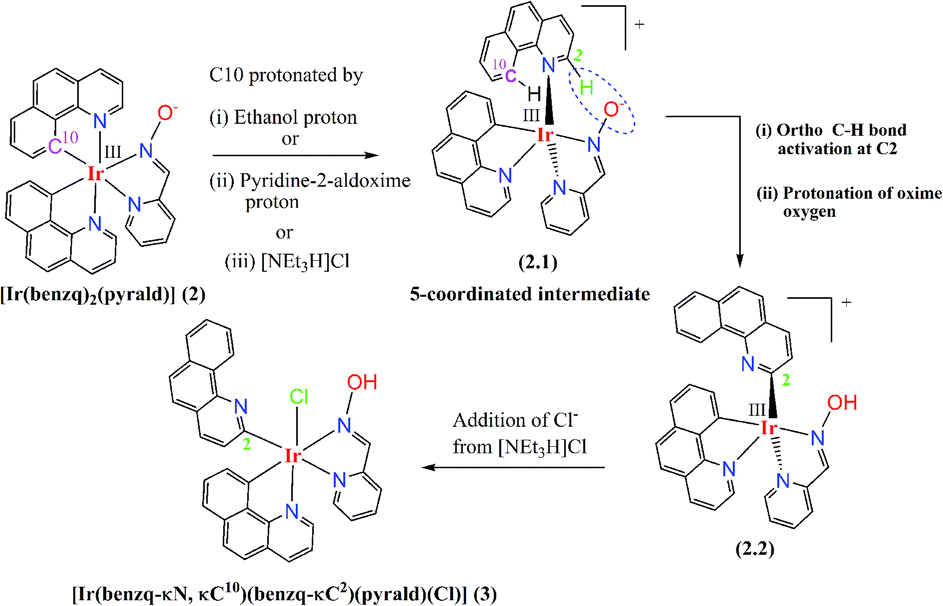 | ||
| Scheme 1 Proposed reaction mechanism for ortho C–H bond activation process. | ||
Recent report on visible light induced ortho C–H bond activation in 7,8-benzoquinoline36 was also considered. The reaction of complex preparation thus carried out in dark without the alteration of other reaction conditions. It was found with an unaffected reaction, which yielded both the complexes (2) and (3) as before.
The UV-Vis spectra of complex (2) and (3) are depicted in Fig. S12† and the data are tabulated in Table S6.† Both the complexes displayed several bands and shoulders in the range of 258–500 nm with a similar pattern of spectra.
The highest energy band at 258 nm of complex (2) and bands at 282 nm for both the complexes were assigned to spin-allowed intra-ligand (1IL) π–π* transitions. Except these bands, all other absorptions appeared as shoulders in the absorption spectra. The band positions at 355 and 384 nm of complex (3) were attributed to the mixture of MLCT (metal to ligand charge transfer) and LLCT (ligand to ligand charge transfer) transitions. This was supported from the theoretically predicted electronic transitions from HOMO−3 and HOMO−1 to the LUMO+1 level at 355 nm and 384 nm, respectively (Table S5†). Both the HOMO−1 and HOMO−3 constituted with orbitals of iridium, chloride and carbons of two 7,8-benzoquinoline rings (Fig. S11†). LUMO+1 is mainly constituted with the orbitals of the bidentate benzoquinoline ring (Fig. S11†). Therefore, the shoulders at 355 nm and 384 nm originated from 1MLCT, 1LLCT and Cl(dπ) to L(π*) transitions in complex (3). A similar assignment of the electronic transitions (except Cl(dπ) to L(π*)) for the observed band at 382 nm of complex (2) was quite feasible. The observed shoulder at 425 nm of complex (3) did not have a good agreement with the predicted theoretical bands at 407 nm and 443 nm. The difference of observed and calculated bands positions appeared at ±18 nm, which is in the range of typical accuracy of the TDDFT calculation of MLCT bands for transition metal complexes. The calculated bands at 407 nm and 443 nm predicted the transitions of HOMO−2 and HOMO−1 to LUMO, respectively. Unlike HOMO−1, HOMO−2 is largely constituted by d-orbitals of iridium and chloride atoms, whereas the LUMO orbital is found to be mainly constituted by the π* molecular orbitals of the pyridine-2-aldomixe moiety (L′) with the minimal contribution of iridium d-orbitals. Thus, the observed 425 nm band constituted with bigger contribution of 1ML′CT (L′ = pyridine-2-aldoxime, L= 7,8-benzoquinoline) transition compared to that of inter ligand transitions (LL′CT), Cl(dπ) to L′(π*) transition along with a part of d–d transition originated from iridium. The band at 428 nm of complex (2) could be considered for the similar electronic transitions except the chloride centre transition, as described for complex (3). Further the HOMO to the LUMO transition band in complex (3) was calculated to be at 452 nm, which largely differed with the observed lowest energy band at 500 nm. This band with low extinction coefficient was assigned to spin forbidden transitions (3MLCT, 3ML′CT, 3ILCT, 3LLCT, 3IL′CT and 3LL′CT) facilitated by the strong spin orbit coupling of the iridium centre.37 The complex (2) also presented a weak absorption at 500 nm and assigned to the transitions of similar origin as described above.
The photoluminescence spectra of these complexes were recorded in the degassed dichloromethane solution. The excitation of the 425 nm band of complex (3) resulted in the emission with two peaks positioned at 594 nm and 641 nm (Fig. S13†). A similar pattern of spectrum was observed for complex (2) with peak positions at 539 nm and 591 nm. Both complexes were found to be very weakly luminescent in the solution medium. The quantum yields were found to be 0.097 and 0.0077 for complex (2) and (3), respectively. This could be due to the nature of electronic transitions involved in the emissive decay in these complexes. As stated above, the band at 425 nm was due to the combined transitions of 1ML′CT, and 1LL′CT and metal centred d–d transitions. On excitation, the excited electron could find several radiative and nonradiative decay pathways due to the presence of several suitable low-lying energy levels arising out of these transitions.38 The dominant emissive decay from 3IL′CT and 3ILCT compared to that from 3MLCT and 3ML′CT levels could have led to the weak phosphorescence emission.37,39 The presence of dual peaks in the photoluminescence spectrum was also supportive of more3 π–π* character in the emissive decay process.39 The above explanation also suited to the observed weak luminescence behaviour of complex (2).
The cyclic voltammetry of the complexes (2), (3) and pyridine-2-aldoxime were investigated in an acetonitrile solution containing 0.1 M TBAP. Complexes (2) and (3) displayed two irreversible oxidation responses each (Fig. S14†). The first oxidation responses at 0.94 V of complex (2) and 1.27 V for complex (3) were assigned to the ligand centred oxidation. These oxidation potentials appeared in the vicinity of oxidation potential (1.06 V) of pure pyridine-2-aldoxime (Table S6†) under similar conditions. The peaks at 1.35 V of complex (2) and 1.59 V of complex (3) could be assigned to the metal centred IrIII–IrIV redox couple. The assignment is consistent with the reported values of similar Ir(III) complexes.30 The reduction peaks at −1.41 V and −1.32 V were assigned to the reduction of the coordinated pyridine-2-aldoxime moiety. These values were quite close to the reduction potential of free pyridine-2-aldoxime value at −1.14 V. The DFT calculations of complex (3) also predicted the large distribution of LUMO over pyridine-2-aldoxime making it susceptible to reduction and which was reflected from the observed low reduction potential.
Conclusions
In summary, we have first noticed the ortho C–H bond activation in a coordinated 7,8-benzoquinoline in an Ir(III) complex. The activation process was facilitated by an ancillary ligand, namely pyridine-2-aldoxime. The phenomenon was confirmed via the crystal structure analysis of the new complex and other related analytical data. The activation process was even taken place well below 100 °C. This interesting observation has instigated for further investigation to garner more insight into the role of pyridine-2-aldoxime in causing the C–H bond activation. A detailed theoretical study is currently underway to reveal the mechanism of ortho C–H bond activation in complex (2).Author contributions
(1) Satyanarayan Pal: Conceptualisation and writing original draft. (2) Kahnu Charan Pradhan: Investigation and data analysis. (3) Hemanta K. Kisan: Theoretical investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The work was supported by Science and Technology Department, Govt. of Odisha (Grant no. 27562800512017/201288/ST/Bhubaneswar, dt. 24.02.2018), DST-FIST program of Govt. India (SR/FST/CSI/-275/2016 (C) Dt. 01/02/2018) and Council of Scientific and Industrial Research (CSIR), New Delhi (Grant no. 01/(2860)/16/EMR-II, Dt. 12.05.2016). We thank the National Single Crystal Diffractometer Facility, School of Chemistry, University of Hyderabad for X-Ray crystallographic studies. We express our sincere thanks to Prof. Samudranil Pal, of University of Hyderabad for his constant support.Notes and references
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- J. Wencel-Delord, T. Dröge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740–4761 RSC.
- R. C. Evans, P. Douglas and C. J. Winscom, Coord. Chem. Rev., 2006, 250, 2093–2126 CrossRef CAS.
- J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401–2425 CrossRef CAS.
- Y. Im, S. Y. Byun, J. H. Kim, D. R. Lee, C. S. Oh, K. S. Yook and J. Y. Lee, Adv. Funct. Mater., 2017, 27, 1603007 CrossRef.
- T.-Y. Li, J. Wu, Z.-G. Wu, Y.-X. Zheng, J.-L. Zuo and Y. Pan, Coord. Chem. Rev., 2018, 374, 55–92 CrossRef CAS.
- E. Baranoff and P. Kumar, in Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons, Ltd, Chichester, UK, 2017, pp. 655–681 Search PubMed.
- R. Bevernaegie, S. A. M. Wehlin, E. J. Piechota, M. Abraham, C. Philouze, G. J. Meyer, B. Elias and L. Troian-Gautier, J. Am. Chem. Soc., 2020, 142, 2732–2737 CrossRef CAS PubMed.
- P.-Y. Ho, C.-L. Ho and W.-Y. Wong, Coord. Chem. Rev., 2020, 413, 213267 CrossRef CAS.
- C.-N. Ko, G. Li, C.-H. Leung and D.-L. Ma, Coord. Chem. Rev., 2019, 381, 79–103 CrossRef CAS.
- M. Nonoyama, Bull. Chem. Soc. Jpn., 1974, 47, 767–768 CrossRef CAS.
- S. Salinas, M. A. Soto-Arriaza and B. Loeb, Polyhedron, 2011, 30, 2863–2869 CrossRef CAS.
- B. Liu, M. A. Jabed, J. Guo, W. Xu, S. L. Brown, A. Ugrinov, E. K. Hobbie, S. Kilina, A. Qin and W. Sun, Inorg. Chem., 2019, 58, 14377–14388 CrossRef CAS PubMed.
- F. Zhang, C. Si, X. Dong, D. Wei, X. Yang, K. Guo, B. Wei, Z. Li, C. Zhang, S. Li, B. Zhai and G. Cao, J. Mater. Chem. C, 2017, 5, 9146–9156 RSC.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704–1711 CrossRef CAS PubMed.
- K. K.-W. Lo, K. Y. Zhang and S. P.-Y. Li, Pure Appl. Chem., 2011, 83, 823–840 CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, H.-E. Lee, C. Adachi, P. E. Burrows, S. R. Forrest and M. E. Thompson, J. Am. Chem. Soc., 2001, 123, 4304–4312 CrossRef CAS PubMed.
- J. Su, X. Sun, G. Gahungu, X. Qu, Y. Liu and Z. Wu, Synth. Met., 2012, 162, 1392–1399 CrossRef CAS.
- S. P. Pitre, M. Muuronen, D. A. Fishman and L. E. Overman, ACS Catal., 2019, 9, 3413–3418 CrossRef CAS.
- Z. Wang, Q. Liu, X. Ji, G.-J. Deng and H. Huang, ACS Catal., 2020, 10, 154–159 CrossRef CAS.
- Z. Wang, X. Ji, T. Han, G.-J. Deng and H. Huang, Adv. Synth. Catal., 2019, 361, 5643–5647 CrossRef CAS.
- D. Zheng and A. Studer, Angew. Chem., Int. Ed., 2019, 58, 15803–15807 CrossRef CAS PubMed.
- Y. Cheng, S. Yu, Y. He, G. An, G. Li and Z. Yang, Chem. Sci., 2021, 12, 3216–3225 RSC.
- S. Wu, Z. Wang, D. Ma, C. Chen and B. Zhu, Org. Chem. Front., 2020, 7, 1158–1163 RSC.
- S. V. Kumar, S. Banerjee and T. Punniyamurthy, Org. Chem. Front., 2020, 7, 1527–1569 RSC.
- A. Sagadevan, A. Charitou, F. Wang, M. Ivanova, M. Vuagnat and M. F. Greaney, Chem. Sci., 2020, 11, 4439–4443 RSC.
- R. F. Jordan and A. S. Guram, Organometallics, 1990, 9, 2116–2123 CrossRef CAS.
- C. T. Carver, B. N. Williams, K. R. Ogilby and P. L. Diaconescu, Organometallics, 2010, 29, 835–846 CrossRef CAS.
- S. Grzesiek and E. D. Becker, in Encyclopedia of Magnetic Resonance, ed. R. K. Harris, John Wiley & Sons, Ltd, Chichester, UK, 2011 Search PubMed.
- K. C. Pradhan, S. Barik, B. C. Singh, P. Mohapatra, H. K. Kisan and S. Pal, J. Mol. Struct., 2020, 1211, 128058 CrossRef CAS.
- S. Pal and B. C. Singh, Acta Crystallogr., 2013, E69, m159 Search PubMed.
- L. F. Gildea, A. S. Batsanov and J. A. G. Williams, Dalton Trans., 2013, 42, 10388 RSC.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, 1997 Search PubMed.
- G. R. Desirau and T. Steiner, The weak hydrogen bond in structural chemistry and biology, Oxford University Press, 2001 Search PubMed.
- V. Balamurugan, M. S. Hundal and R. Mukherjee, Chem.–Eur. J., 2004, 10, 1683–1690 CrossRef CAS PubMed.
- H. Tian, H. Yang, C. Tian, G. An and G. Li, Org. Lett., 2020, 22, 7709–7715 CrossRef CAS PubMed.
- A. Tsuboyama, H. Iwawaki, M. Furugori, T. Mukaide, J. Kamatani, S. Igawa, T. Moriyama, S. Miura, T. Takiguchi, S. Okada, M. Hoshino and K. Ueno, J. Am. Chem. Soc., 2003, 125, 12971–12979 CrossRef CAS PubMed.
- Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 761–784 RSC.
- M. S. Lowry, W. R. Hudson, R. A. Pascal and S. Bernhard, J. Am. Chem. Soc., 2004, 126, 14129–14135 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2050870. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00860a |
| This journal is © The Royal Society of Chemistry 2021 |