
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing Li-ion capacity and rate capability in cation-defective vanadium ferrite aerogels via aluminum substitution†
Christopher N. Chervin a,
Ryan H. DeBlockb,
Joseph F. Parkera,
Bethany M. Hudakc,
Nathaniel L. Skeelea,
Jesse S. Kod,
Debra R. Rolisona and
Jeffrey W. Long*a
a,
Ryan H. DeBlockb,
Joseph F. Parkera,
Bethany M. Hudakc,
Nathaniel L. Skeelea,
Jesse S. Kod,
Debra R. Rolisona and
Jeffrey W. Long*a
aSurface Chemistry Branch, Code 6170, U.S. Naval Research Laboratory, Washington, DC 20375, USA. E-mail: jeffrey.long@nrl.navy.mil
bNRC Postdoctoral Associate at the Naval Research Laboratory, Washington, DC 20375, USA
cMaterials & Systems Branch, Code 6360, U.S. Naval Research Laboratory, Washington, DC 20375, USA
dFormer NRC Postdoctoral Associate at the Naval Research Laboratory, Washington, DC 20375, USA
First published on 19th April 2021
Abstract
Cation-defective iron oxides have proven to be effective Li-ion charge-storage hosts in nonaqueous electrolytes, particularly when expressed in disordered, nanoscale forms such as aerogels. Replacing a fraction of Fe sites in ferrites with high-valent cations such as V5+ introduces cation-vacancy defects that increase Li-ion capacity. Herein, we show that compositional substitution with electroinactive Al3+ further increases Li-ion capacity by 30% when incorporated within a disordered VFe2Ox aerogel, as verified by electrochemical tests in a two-terminal Li half-cell. We use electroanalytical techniques to show that both Al-VFe2Ox and VFe2Ox aerogels exhibit many of the hallmarks of pseudocapacitive materials, including fast charge–discharge and surface-controlled charge-storage kinetics. These disordered, substituted ferrites also provide the high specific capacity expected from battery-type electrode materials, up to 130 mA h g−1 for Al-VFe2Ox. Our findings are discussed in the context of related Li-insertion hosts that blur the distinctions between battery-like and capacitor-like behavior.
1 Introduction
Next-generation electrochemical energy storage requires active materials with increased capacity, high-power response, and cycling stability. One strategy to achieve these goals is to deliberately introduce cation defects (e.g., structural vacancies) into charge-storing metal oxides.1 This modification effectively increases specific capacity for Li-ion and Na-ion insertion in nonaqueous electrolytes.1–6 For example, we explored a series of disordered, cation-defective spinel-type iron oxides in which a fraction of the Fe sites are substituted with high-valent cations such as Mo6+ and V5+, whose presence induces formation of proton-stabilized charge-compensating cation vacancies.7,8 The positive impact on charge storage with these deliberately defective iron oxides, with increases in both capacity and thermodynamic potential, is further enhanced by expressing them in high surface-area aerogel forms,1,8 which innately amplify structural and surface defects.9Aluminum-substitution of cation sites offers another strategy to improve cycle stability and performance of positive electrode materials.10–14 In the well-known example of lithium nickel cobalt aluminum oxide (NCA), replacing a small number of cobalt sites with aluminum boosts the electrochemical and thermal stability by preventing unwanted phase change.12–14 In some examples, Al-substitution increases cation-insertion capacity and operating voltage.10,11 For Na3V2(PO4)3 (NVP), a common insertion material for sodium-ion batteries, higher levels of Al-substitution (up to 50% of the V3+) increase specific capacity by replacing redox-inactive V3+ sites with the lighter Al3+.11 Typically, NVP redox involves the V3+/4+ redox couple at a potential of ∼3.4 V vs. Na|Na+. When substituted with Al3+, however, the V4+/5+ redox couple is partially accessible at ∼3.95 V vs. Na|Na+ due to a decreased energy penalty for extraction of a second Na ion.11 Aluminum-free phosphates undergo significant lattice shrinkage and corresponding mechanical deterioration with extended cycling, whereas Al-substituted analogs remain robust under similar cycling conditions.10 To our knowledge, the effects of Al-substitution on the Li-insertion properties of ferrite spinels have yet to be explored, although Al-substituted lanthanum and copper-cobalt ferrites demonstrate increased specific capacitance in aqueous electrochemical capacitors.15,16
In this report, we show that vanadium ferrite (VFe2Ox) aerogels synthesized by an epoxide-driven sol–gel method readily undergo Al-substitution with a targeted 10% exchange of Al for V. This simple synthetic variation yields aerogels (designated 10Al-VFe2Ox) that exhibit ∼40% higher specific surface area (after thermal treatment at 300 °C), but otherwise similar structural characteristics to unsubstituted VFe2Ox aerogels. Aluminium compositional substitution into VFe2Ox further enhances specific capacity (by >30%) and high-rate performance for nonaqueous Li-ion storage when these materials are incorporated in powder-composite electrodes and electrochemically evaluated in two-terminal Li half-cells.
2 Experimental
2.1. Materials synthesis and processing
The VFe2Ox and 10Al-VFe2Ox aerogels were synthesized using a modification of the method described previously.8 Briefly, for 10Al-VFe2Ox, 1.195 mL (5.13 mmol) of vanadium isopropoxide pre-chilled in a refrigerator at ∼5 °C was added to 20 mL of ethanol that was chilled in an aqueous ice bath in a covered beaker. The vanadium solution was then placed in a dry ice-ethanol bath to which was added a solution of FeCl3·6H2O (3.0814 g; 11.4 mmol) and Al(NO3)3·9H2O (0.2138 g; 0.57 mmol), dissolved in 10 mL of ethanol pre-chilled in a dry ice-ethanol bath. An additional 10 mL of pre-chilled ethanol was used to rinse residual solution from the vial into the main reaction vessel. Propylene oxide (15 mL; 222 mmol), pre-chilled in a refrigerator, was added and the vessel was agitated for ∼5 min with occasional swirling and then moved to an aqueous ice bath and stirred for ∼30 min. The vessel was warmed to room temperature under ambient conditions without stirring. As the solution warmed, it changed colour from light brown to dark brown and formed a monolithic gel within ∼45 min. The vessel was covered and the gel was aged in its mother liquor overnight under ambient conditions and then rinsed with ethanol for two days, exchanging the ethanol three times each day. Following ethanol rinsing, the solvent in the gel's pores was exchanged with acetone over ∼3 days, exchanging three times per day.The wet gel was transferred to an autoclave and the acetone was exchanged with liquid CO2 at ∼10 °C, flushing the autoclave once every 30 min over ∼4 h. The autoclave was heated to ∼41 °C, while the internal pressure increased to ∼8.6 MPa, bringing the CO2 past its critical point (Tc = 31 °C; Pc = 7.4 MPa). The autoclave was vented slowly, yielding a dried aerogel. For VFe2Ox, a similar method was used with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry of V:Fe and no added Al(NO3)3·9H2O. The as-dried VFe2Ox and 10Al-VFe2Ox aerogels were heated under flowing O2 to 300 °C at 2 °C min−1 with a 4 h hold followed by a 2 °C min−1 cooling ramp.
:2 stoichiometry of V:Fe and no added Al(NO3)3·9H2O. The as-dried VFe2Ox and 10Al-VFe2Ox aerogels were heated under flowing O2 to 300 °C at 2 °C min−1 with a 4 h hold followed by a 2 °C min−1 cooling ramp.
2.2. Materials characterization
X-ray diffraction patterns of VFe2Ox and 10Al-VFe2Ox powders heated to 300 °C in flowing O2 were collected from 20–80° 2θ using a Rigaku Smartlab X-ray diffractometer using Cu Kα radiation (λ = 1.5406 Å). Attenuated total reflectance infrared spectra were collected on powdered samples using a Thermo Scientific Nicolet iS50 FTIR with an iS50 ATR diamond crystal. Simultaneous thermogravimetric analysis/differential scanning calorimetry (TGA/DSC) of the as-dried VFe2Ox and 10Al–VFe2Ox aerogels was collected at 10 °C min−1 under a 60/40 ratio of O2-to-Ar using a Netzsch Jupiter STA 449 F1 thermal analyzer. Surface area and pore volume of VFe2Ox and 10Al-VFe2Ox aerogels were measured with N2-sorption porosimetry using a Micromeritics ASAP 2020. Samples were degassed for 12 h at 80 °C under vacuum prior to analysis. Specific surface area was calculated using the Brunauer–Emmett–Teller (BET) method based on the linear portion of the adsorption isotherm. The average pore widths and pore volumes were calculated using the Barrett–Joyner–Halenda (BJH) method, fitting data across the entire range of the adsorption isotherm. Elemental analysis was determined using inductively coupled plasma atomic emission spectroscopy (ICP-AES) by Galbraith Laboratories, Inc. The ICP-AES results confirm that aluminum substitutes ∼10% of vanadium in 10Al-VFe2Ox and that the ratio of iron to vanadium and iron to vanadium + aluminum is ∼2:1 for VFe2Ox and 10Al-VFe2Ox, respectively.
Scanning electron micrographs of VFe2Ox and 10Al-VFe2Ox aerogel powders were collected at 10 keV on a Carl Zeiss Leo Supra MM scanning electron microscope. Samples were attached to aluminum SEM holders with conductive carbon tape. Scanning transmission electron microscopy (STEM) imaging and STEM-EDS were performed in a Nion UltraSTEM-X operated at 200 kV. The microscope is equipped with a Bruker XFlash windowless silicon drift detector, and EDS data were analysed using Bruker Esprit software. The VFe2Ox and 10Al-VFe2Ox aerogel samples were dispersed in distilled water and dropcast onto Cu TEM grids with lacey carbon films.
X-ray photoelectron spectroscopy (XPS) was used to examine the chemical state of Fe, V, Al, and O using a Thermo Scientific K-Alpha X-ray photoelectron spectrometer at a chamber pressure <5.0 × 10−8 torr and equipped with a monochromatic Al Kα source (1486.68 eV) using a 400 μm elliptical spot size. The VFe2Ox and 10Al-VFe2Ox aerogel powders were packed into wells of the powder module sample holder. For electrochemically cycled samples, powder-composite electrodes were scanned voltammetrically five times from 2.0–3.8 V vs. Li|Li+ and then held potentiostatically in either the charged or discharged state for 1 h. Electrodes from the cycled cells were removed inside an Ar glovebox, rinsed thoroughly with methanol, dried, and transferred into the XPS chamber with <1 min of exposure to ambient atmosphere. The spectra were obtained at an energy step-size of 0.15 eV by averaging scans for the Fe 2p, V 2p, Al 2p, O 1s, and C 1s regions. The spectra were analysed with Avantage® software version 5.99 using a Shirley-derived “Smart” background and peak-shifted relative to O 1s at 530.0 eV for V oxidation state analysis as described previously.17 The ratio of V5+:V4+ in VFe2Ox and 10Al-VFe2Ox was determined by deconvolving the V 2p3/2 envelope for each sample. Fitting parameters included: (i) allowing the ΔBE (V5+/V4+) to float between 0.69 and 1.75 eV; (ii) fixing the FWHM range for V5+ to remain between 0.5–2.0 eV and that of V4+ to remain between 2.2 eV–4.0 eV; and (iii) anchoring the Lorentzian–Gaussian percentage to be fixed at 42% for V5+ and 26% for V4+.17,18
2.3. Electrochemical characterization and testing
Powder-composite electrodes were fabricated by grinding either VFe2Ox or 10Al-VFe2Ox aerogel powders with Super P carbon black and polyvinylidene fluoride (PVDF) binder in a mortar and pestle in n-methyl-2-pyrrolidone (NMP) solvent. The mass ratio was 80:10:10 active material:carbon:binder. Electrodes were cast using a doctor blade to achieve an active material loading of 1–2 mg cm−2. Coin cells were constructed in an Ar-filled glovebox using a lithium metal counter/reference electrode, glass-fiber separator, and 1 M LiPF6 ethylene carbonate/dimethyl carbonate (EC/DMC) electrolyte. Galvanostatic experiments were performed on an Arbin BT2000 battery cycler and cyclic voltammetry experiments were conducted using a Gamry Reference 600 potentiostat.
3 Results and discussion
Aluminium substitution of VFe2Ox is readily achieved by modifying the epoxide-driven sol–gel chemistry previously reported for the synthesis of VFe2Ox aerogels.8 In this synthetic method, the epoxide extracts a proton from water molecules coordinated to precursor metal cations (H2O–M); the protonated epoxide undergoes a ring-opening reaction stabilized by nucleophilic attack from chloride ions.19,20 A slow rise in pH initiates hydrolysis and condensation reactions at HO–M centers resulting in a networked oxyhydroxide gel. In the modification reported here, vanadium alkoxide and aluminium nitrate are used in place of chloride analogs to avoid two obstacles: precipitation of vanadium oxide and insolubility of AlCl3·6H2O. Sufficient chloride is present in the solution from the FeCl3·6H2O precursor to stabilize the epoxide ring-opening step. As a second modification to the synthesis, we mix the precursors cold (−78 °C) to prevent rapid precipitation of VOxHy that would otherwise occur; homogeneous gelation ensues as the chilled solution warms to room temperature under ambient conditions. The dark-brown gels are solvent-exchanged with acetone and then dried in an autoclave with supercritical CO2 to yield the corresponding VFe2Ox and 10Al-VFe2Ox aerogels. Further thermal processing of these aerogels is required to remove residual organic byproducts.21,22Thermogravimetric and calorimetric heating profiles demonstrate similarity between VFe2Ox and 10Al-VFe2Ox. Heating under an oxygen atmosphere shows an exothermic weight loss from 80 to 300 °C for both oxides (Fig. 1a). The weight loss for VFe2Ox and 10Al-VFe2Ox is 24.5 and 23.5%, respectively, and is associated with dehydration of hydroxyl functionality and oxidation of residual organics.21,22 Based on these results, the VFe2Ox and Al-substituted VFe2Ox aerogels are calcined under flowing oxygen at 300 °C for 4 h to prevent crystallization, which diminishes charge-storage capacity.8
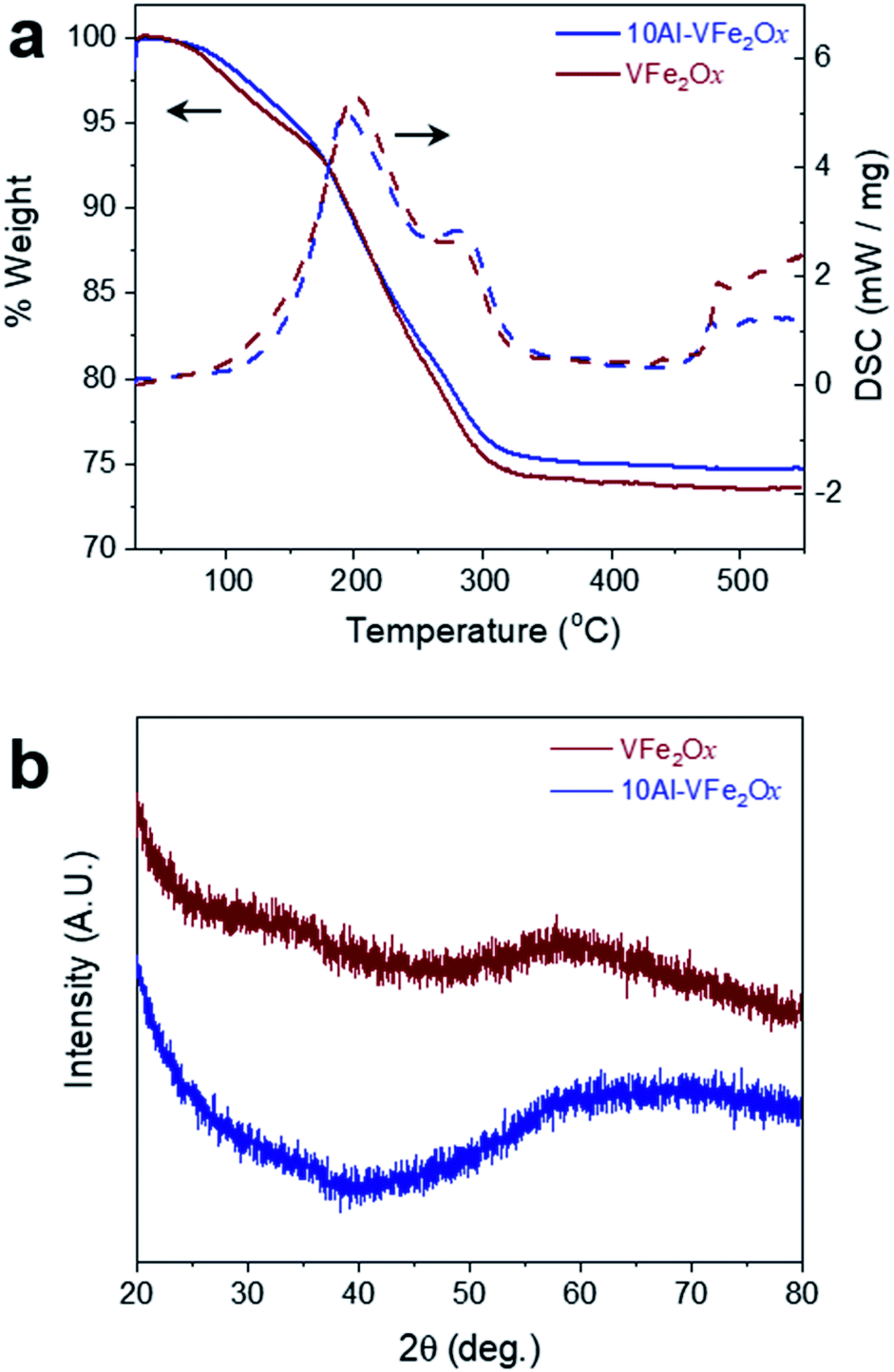 | ||
| Fig. 1 (a) Simultaneous thermogravimetric analysis/differential scanning calorimetry scans of as-prepared VFe2Ox and 10Al-VFe2Ox aerogels; (b) X-ray diffraction patterns of VFe2Ox and 10Al-VFe2Ox aerogels calcined at 300 °C in flowing O2. | ||
X-ray diffraction of the VFe2Ox and 10Al-VFe2Ox aerogels confirms that the oxides remain amorphous after the 300 °C treatment in flowing oxygen (Fig. 1(b)), a result consistent with our previously reported synthesis of VFe2Ox.8 Characterization by attenuated total-reflectance infrared spectroscopy (ATR-IR) shows that both oxides have comparable absorption bands in the mid-infrared region that represent the stretching vibrations of adsorbed water, structural hydroxide, and residual organics (Fig. S1†). The fingerprint region below 1000 cm−1 is also invariant for both samples, indicating that within the limits of ATR-IR analysis, the two compositions have similar bonding motifs.
When substituting 10 atom% Al into VFe2Ox, average pore size and specific pore volume are comparable (Table S1†), but the specific surface area increases by ∼43%: 327 vs. 229 m2 g−1 for 10Al-VFe2Ox and VFe2Ox. Similarly, enhancements in specific surface area have been noted with various aluminum-substituted cobalt ferrites.23–25 Scanning electron micrographs of calcined VFe2Ox and Al-VFe2Ox aerogels show that both oxides comprise highly porous and networked nanostructures (Fig. 2(a and b) and S2a, b†), but VFe2Ox has larger particles within its 3D solid network. High-angle annular dark field scanning transmission electron microscopy (HAADF-STEM) confirms the smaller feature size of 10Al-VFe2Ox relative to VFe2Ox, as well as the amorphous nature of both aerogels (Fig. S3a–d†). Energy dispersive spectroscopy (STEM-EDS) shows homogenous elemental distribution of Al, V, and Fe in 10Al-VFe2Ox without evidence of elemental clustering (Fig. 2). The larger secondary networked particle sizes in VFe2Ox indicates nucleation and growth kinetics is altered in the presence of Al(NO3)3·9H2O.
 | ||
| Fig. 2 Scanning electron micrographs of calcined aerogels (a) VFe2Ox and (b) 10Al-VFe2Ox; (c) HAADF-STEM micrograph of 10Al-VFe2Ox aerogel and the corresponding STEM-EDS spectra for (d) Al, (e) V, and (f) Fe. | ||
The oxidation states of V and Fe in the VFe2Ox and 10Al-VFe2Ox aerogels are assessed by XPS from the high-resolution spectra for the V 2p/O 1s (Fig. 3) and Fe 2p regions (Fig. S4†). Binding energies (BE) are referenced to the O 1s peak for oxide anion (O2−) at 530.0 eV. The ΔBEpeak between O 1s and V 2p3/2 provides a more accurate measure of the V oxidation state than relying on the BE of the V 2p3/2 peak alone.17 For VFe2Ox, ΔBEpeak is 12.7 eV, consistent with V5+. The ΔBEpeak for 10Al-VFe2Ox increases slightly to 13.1 eV, suggesting it is a mixed-valent oxide with predominantly V5+ character.17 Peak fitting the V 2p3/2 envelope reveals no distinguishable V4+ in VFe2Ox and a 65:35 atomic ratio of V5+:V4+ in 10Al-VFe2Ox (Fig. 3; see Experimental for fitting parameters).17 The position of the Fe 2p3/2 satellite in both samples (718–720 eV) suggests that the predominant iron oxidation state is Fe3+ (Fig. S4a†).26
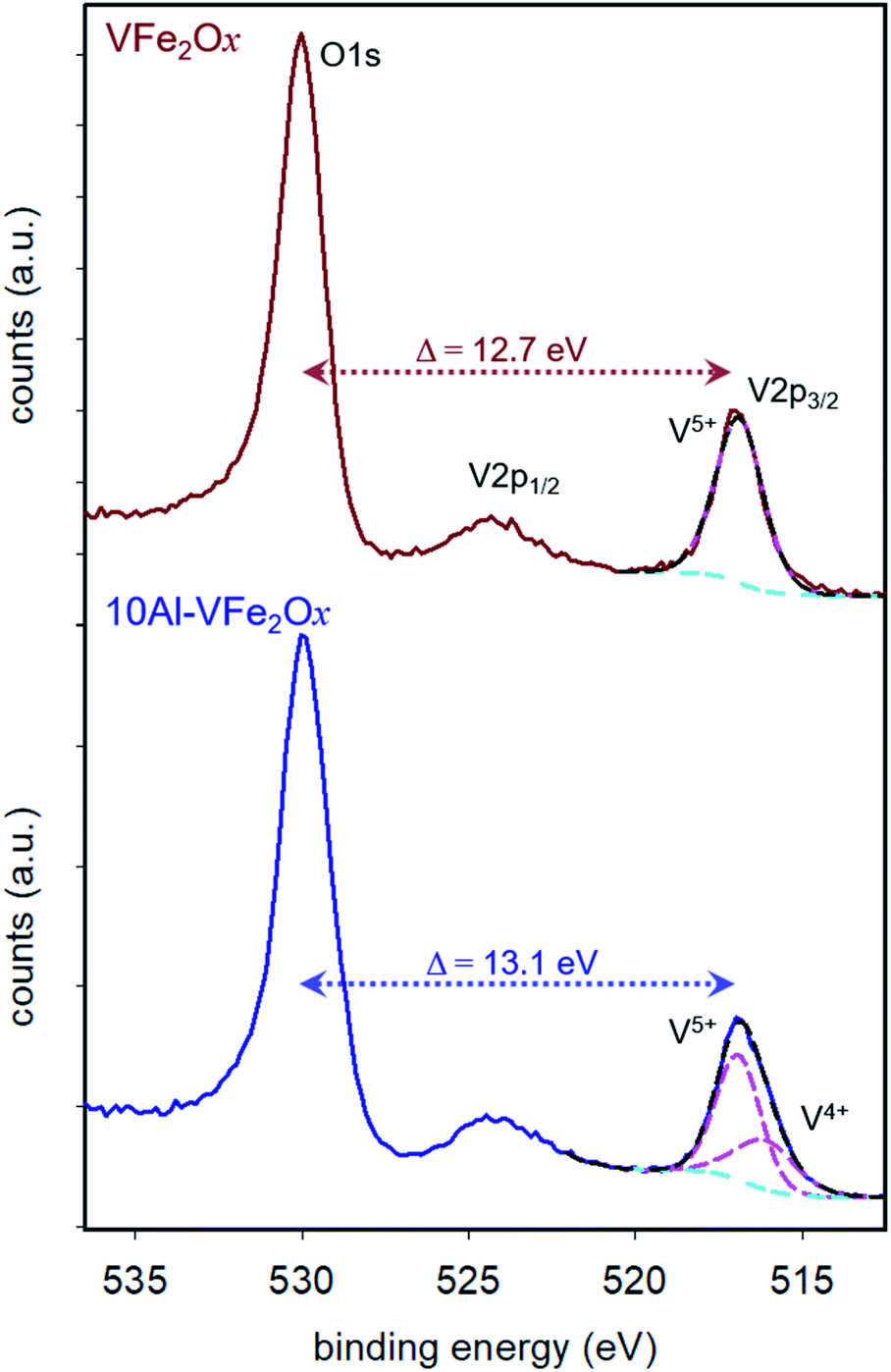 | ||
| Fig. 3 X-ray photoelectron spectra of the O1s and V2p region for 300 °C-calcined VFe2Ox and 10Al-VFe2Ox aerogels. | ||
For electrochemical evaluation, VFe2Ox and 10Al-VFe2Ox aerogels are processed into standard powder-composite electrodes (active material loading: 1–2 mg cm−2) and then assembled into coin cells with Li metal counter/reference electrodes and an electrolyte of 1 M LiPF6 ethylene carbonate/dimethyl carbonate (EC/DMC) electrolyte. Charge-storage characteristics are first assessed by voltammetrically scanning between 2.0–3.8 V (vs. Li|Li+), a range where these materials undergo Li-insertion, but not the conversion reactions that often occur at lower voltage for iron and vanadium oxides.27–29 In both cases, the voltammograms lack well-defined peaks with current broadly distributed across the voltage axis between 2.0 V and 3.4 V vs. Li|Li+ for all scan rates investigated (0.1–1 mV s−1), Fig. 4(a and b). The Al-substituted variant is distinguished from its VFe2Ox counterpart only in the magnitude of its current response, with peak currents ∼30% higher.
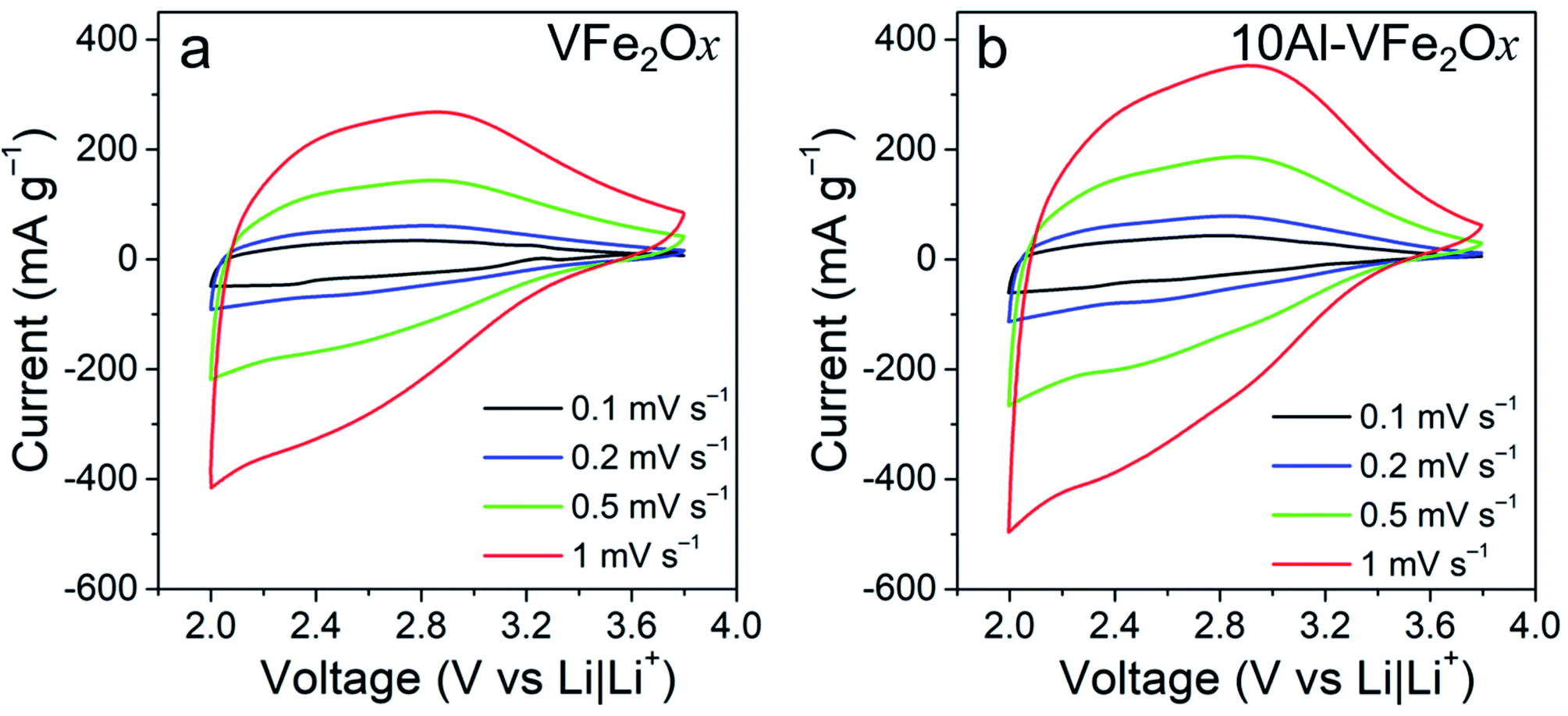 | ||
| Fig. 4 Cyclic voltammograms at various scan rates in 1 M LiPF6 EC/DMC for cells equipped with powder-composite cathodes containing either (a) VFe2Ox or (b) 10Al-VFe2Ox aerogels. | ||
Broad voltammetric features are one signature of materials that exhibit redox-based “pseudocapacitance”, although in this case deviating from the box-like voltammograms of ideal electrochemical capacitor systems,30–32 but reminiscent of other nominally pseudocapacitive materials such as conducting polymers.33 Another hallmark of pseudocapacitive materials is surface-controlled charge storage, which can be determined by applying power-law analysis to voltammetric data. In this analysis, the current for a redox peak (ipeak) is described by eqn (1) (ref. 34) as a function of sweep rate (ν) and two constants (a, b):
| ipeak=aνb | (1) |
For quantitative assessment of electrochemical performance, cells with VFe2Ox or 10Al-VFe2Ox aerogels were cycled under galvanostatic charge–discharge conditions. In both cases, the resulting cells exhibit sloping charge–discharge curves (Fig. 5(a, b) and S5†), qualitatively consistent with the broad voltammetry noted above. At the lowest current density examined (70 mA g−1), Al substitution increases specific capacity by >25%: 128 mA h g−1 for 10Al-VFe2Ox vs. 102 mA h g−1 for VFe2Ox. Compositional substitution of Al3+ into VFe2Ox also improves two other critical electrochemical properties: capacity retention with increasing current/rate demand and lower charge–discharge hysteresis with cycling.
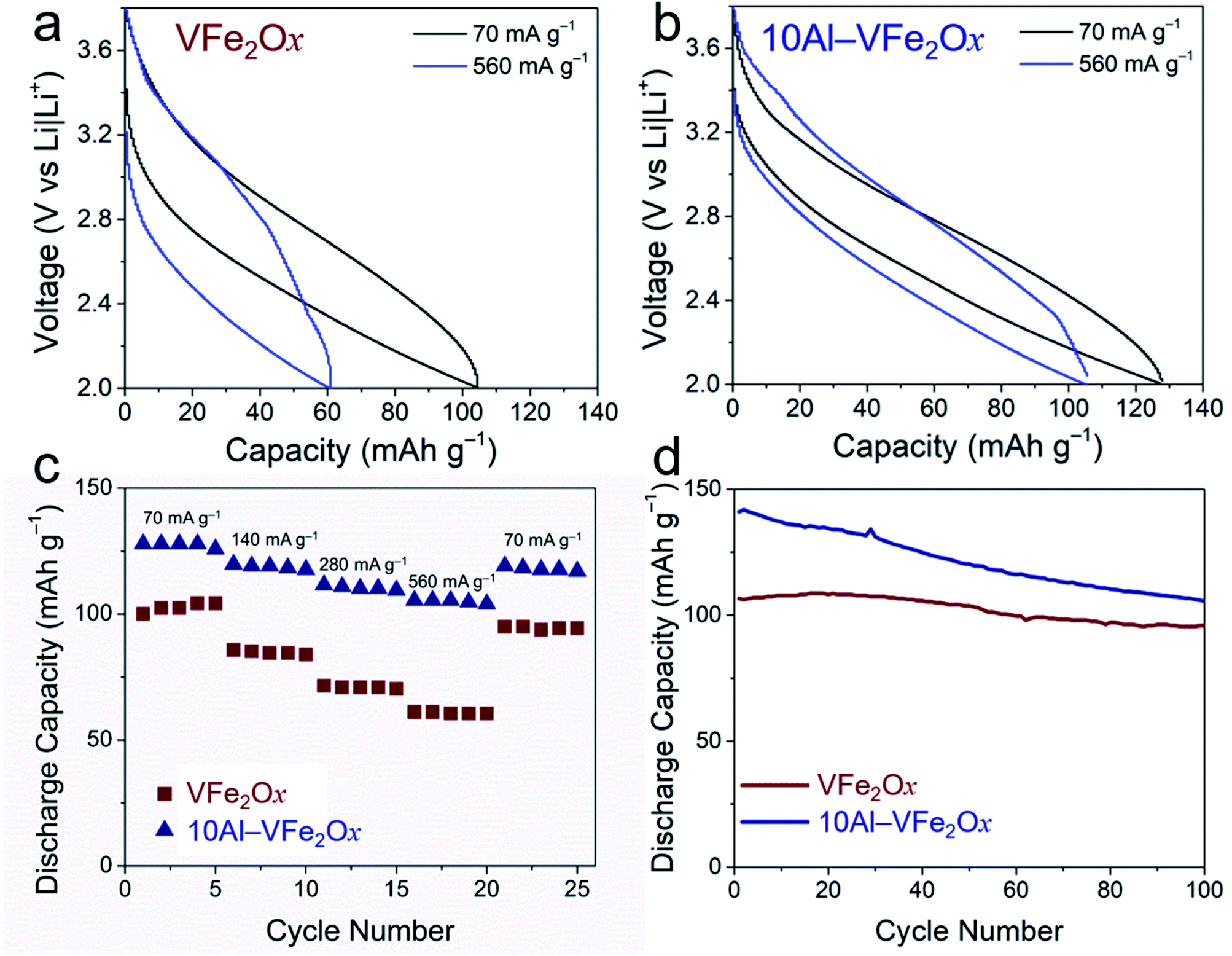 | ||
| Fig. 5 Galvanostatic charge–discharge curves (fifth cycle) of (a) VFe2Ox and (b) 10Al-VFe2Ox aerogels at 70 and 560 mA g−1 with current density normalized to the mass of active material in the powder-composite electrode; (c) rate retention and (d) long-term cycling at 70 mA g−1 for both materials. | ||
Increased capacity is delivered in the latter case despite replacing redox-active V4+/5+ for inactive Al3+ and nominally decreasing the number of charge-compensating cation vacancies in the substituted oxide. Assuming one-electron V-based redox and the stoichiometry obtained from the elemental analysis (Table S1†), the expected theoretical capacities are essentially identical for VFe2Ox and 10Al-VFe2Ox at 118 and 119 mA h g−1, respectively. While VFe2Ox does not reach theoretical capacity, 10Al-VFe2Ox displays excess capacity. This disparity in capacity becomes further pronounced with increasing current demand. At a challenging 560 mA g−1, corresponding to a charge–discharge time of ∼10 min, VFe2Ox capacity falls 40% to 61 mA h g−1, while 10Al-VFe2Ox maintains 105 mA h g−1. Strikingly, 10Al-VFe2Ox provides the same specific capacity in 11 min as that stored by unsubstituted VFe2Ox in 88 min. The 10Al-VFe2Ox delivers capacity comparable to that of other pseudocapacitve materials used in nonaqueous Li-ion electrolytes such as T-Nb2O5 (140 mA h g−1 at 1C) and MoS2 (167 mA h g−1 at 1C);31 however, 10Al-VFe2Ox provides capacity at a much higher potential enabling its use as a cathode.
We observe additional improvement in charge–discharge hysteresis, an important factor in determining round-trip energy efficiency in batteries. As clearly demonstrated by the galvanostatic curves in Fig. 5(a, b) and Table S2,† the hysteresis between charge and discharge profiles significantly diminishes for 10Al-VFe2Ox. The difference in hysteresis becomes more apparent at high current density: 1.1 V vs. 0.66 V for VFe2Ox and 10Al-VFe2Ox, respectively, at 560 mA g−1.
The performance enhancement we observe for 10Al-VFe2Ox has multiple plausible sources. The first explanation originates from the increased surface area of 10Al-VFe2Ox (330 m2 g−1) compared to VFe2Ox (220 m2 g−1). Based on defect-mediated and surface-controlled redox behaviour, an increase in surface area should lead to more active sites and shorter diffusional pathways. A second explanation could be the considerable presence of V4+ in 10Al-VFe2Ox (∼35% from XPS in Fig. 3), a vanadium mixed valency not found in VFe2Ox. Decades of solid-state electrochemistry validate the importance of mixed valency on improving charge transport.36 Various examples report increases in lithium diffusion, rate retention, and insertion capacity upon the introduction of V4+ into predominantly V5+ oxides (e.g., V2O5) due to the formation of oxygen vacancies and a significant increase in the electronic conductivity.37–40 Based on our EXAFS analysis of VFe2Ox aerogels, oxygen vacancies are not present.8 Also, substitution of Al3+ into VFe2Ox does not appear to create oxygen vacancies based on the mass stability observed when heating to 550 °C in oxygen (Fig. 1(a)).
The presence of Al3+ may also alter the local coordination and/or the electron density at vanadium and iron sites in ways beneficial for Li-ion-based redox.41 As a preliminary exploration of the charge-storage mechanisms, we performed ex situ XPS on VFe2Ox and 10Al-VFe2Ox powder-composite electrodes after electrochemical conditioning in both lithiated (2.0 V vs. Li|Li+) and delithiated (3.8 V vs. Li|Li+) states. The Fe 2p spectra (Fig. 6(a, b)) demonstrate clear satellite peaks for Fe3+ in delithiated samples whereas we observe an Fe2+ satellite peak for lithiated samples in both VFe2Ox and 10Al-VFe2Ox. However, due to the small difference in Fe 2p3/2 binding energy between Fe2+ and Fe3+ and their large FWHM, we cannot accurately deconvolve them. Qualitatively, VFe2Ox and 10Al-VFe2Ox exhibit similar Fe 2p envelopes in the lithiated and delithiated states.
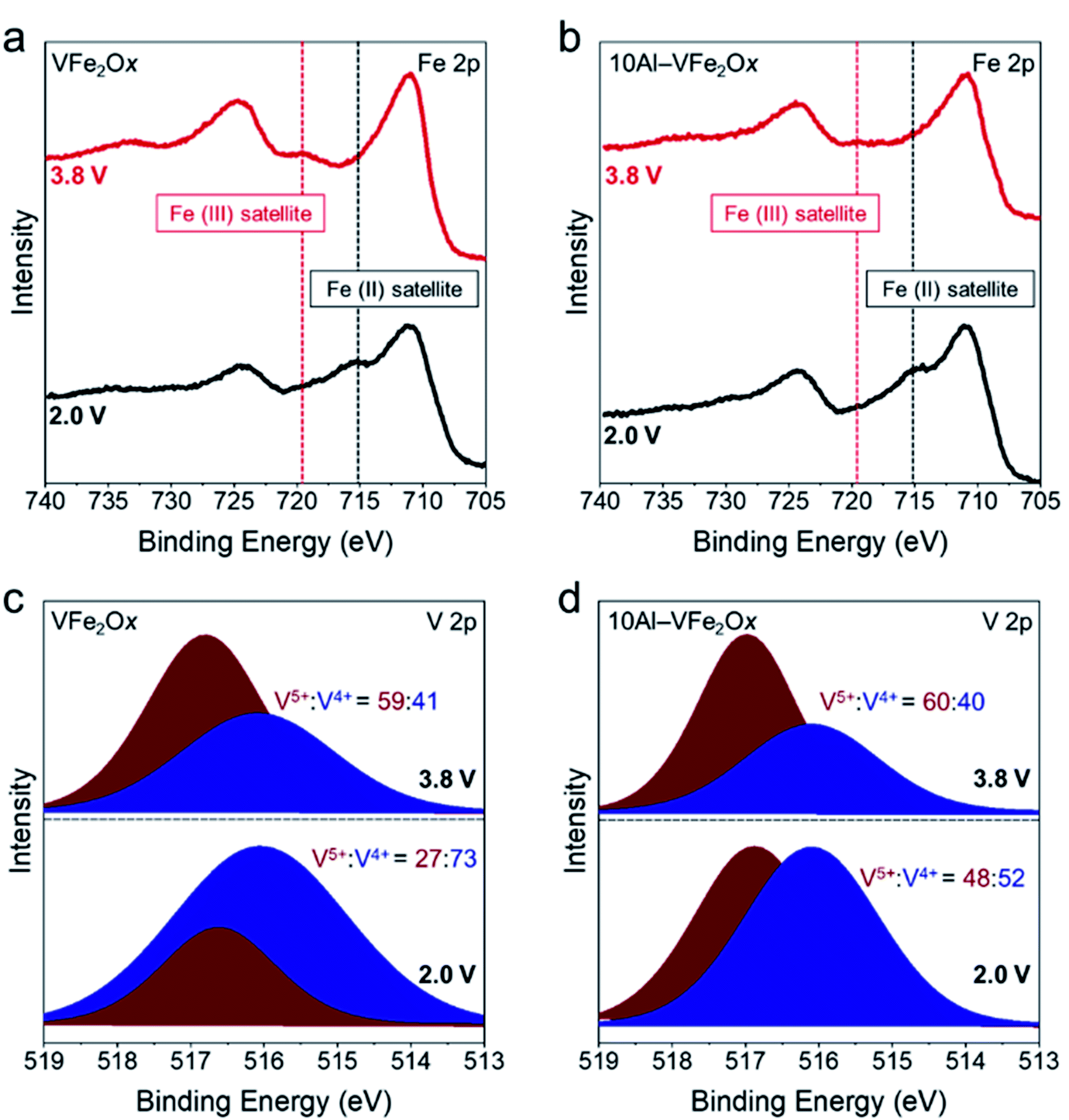 | ||
| Fig. 6 X-ray photoelectron spectra for VFe2Ox and 10Al-VFe2Ox for lithiated (2.0 V vs. Li|Li+) and delithiated (3.8 V vs. Li|Li+) electrodes. (a, b) Fe 2p and (c, d) V 2p regions. | ||
In contrast, the XPS response for the other redox-active element, vanadium (Fig. 6(c, d)), can be quantitatively deconvolved. The main discrepancy between VFe2Ox and 10Al-VFe2Ox lies in the V5+:V4+ ratio after lithiation. 10Al-VFe2Ox retains nearly twice the amount of V5+ after lithiation (48%) than VFe2Ox (27%) even though it stores 30% more charge. Together, these findings suggest that not only are both iron and vanadium redox-active in this system, but that aluminum substitution may alter the extent each transition metal contributes to charge. Proton-stabilized cation vacancies7,42 also likely play a role in the additional capacity seen upon Al3+ substitution. Both experimental43 and ab initio calculations44 conclude that the presence of Al3+ guest ions stabilize proton-compensated cation defects in iron oxyhydroxides, presumably introducing redox-active lattice –OH groups. Due to the amorphous and multi-metallic nature of VFe2Ox aerogels, precise determination of the chemical state and local atomic structure will require a thorough investigation in future work using such techniques as X-ray absorption spectroscopy and synchrotron X-ray scattering with pair-distribution function analysis.
4 Conclusions
We show that Al3+ substitution into disordered VFe2Ox aerogels enhances three critical electrochemical properties for Li-ion storage: total specific capacity, capacity retention with increasing current/rate demand, and lessened charge–discharge hysteresis. This performance improvement is achieved through a simple modification of a well-established sol–gel synthesis. Compositional substitution with 10% Al3+ into VFe2Ox generates ∼40% higher specific surface area and introduces mixed valency with ∼35% V4+ in place of V5+, while retaining similar morphology and bonding motifs characteristic of unsubstituted VFe2Ox. Increased specific surface area should enhance pseudocapacitive charge-storage while the presence of V4+ may increase electronic conductivity and battery-like charge-storage. Lessons learned from the choice of substituent coupled to local structural analyses will inform strategies to tune defective ferrites for stable, high-capacity electrochemical energy storage.5 Author contributions
The manuscript was written through contributions of all authors. C. N. Chervin, R. H. DeBlock, J. F. Parker, B. M. Hudak, N. L. Skeele, and J. S. Ko conducted materials synthesis, materials characterization, and electrochemistry. J. W. Long and D. R. Rolison provided intellectual contributions to data analysis and research design. J. S. K. was an NRL-National Research Council (NRC) Postdoctoral Associate (2016–2018); N. L. S. is a pathways student (2020–present); and R. H. D. was a Naval Underwater Research (NURP) fellow (2015–2020), NREIP student (2018–2019), and NRL-NRC Postdoctoral Associate (2020–present).Conflicts of interest
D. R. R. and J. W. L. are inventors on patents (issued to the U.S. Navy) that are related to increasing Li-ion capacity of battery active materials by deliberately forming cation-vacant metal oxides: US Patents no 7,618,609, 8,388,867, 8,790,612, and 9,466,831.Acknowledgements
This work was funded by the Office of Naval Research.References
- B. P. Hahn, J. W. Long and D. R. Rolison, Acc. Chem. Res., 2013, 46, 1181–1191 CrossRef CAS PubMed.
- J. Farcy, J. P. Pereiraramos, L. Hernan, J. Morales and J. L. Tirado, Electrochim. Acta, 1994, 39, 339–345 CrossRef CAS.
- K. E. Swider-Lyons, C. T. Love and D. R. Rolison, Solid State Ionics, 2002, 152, 99–104 CrossRef.
- G. W. Li, G. R. Blake and T. T. M. Palstra, Chem. Soc. Rev., 2017, 46, 1693–1706 RSC.
- P. Gao, Z. Chen, Y. X. Gong, R. Zhang, H. Liu, P. Tang, X. H. Chen, S. Passerini and J. L. Liu, Adv. Energy Mater., 2020, 10, 1903780 CrossRef CAS.
- Y. Q. Zhang, L. Tao, C. Xie, D. D. Wang, Y. Q. Zou, R. Chen, Y. Y. Wang, C. K. Jia and S. Y. Wang, Adv. Mater., 2020, 32, 1905923 CrossRef CAS PubMed.
- B. P. Hahn, J. W. Long, A. N. Mansour, K. A. Pettigrew, M. S. Osofsky and D. R. Rolison, Energy Environ. Sci., 2011, 4, 1495–1502 RSC.
- C. N. Chervin, J. S. Ko, B. W. Miller, L. Dudek, A. N. Mansour, M. D. Donakowski, T. Brintlinger, P. Gogotsi, S. Chattopadhyay, T. Shibata, J. F. Parker, B. P. Hahn, D. R. Rolison and J. W. Long, J. Mater. Chem. A, 2015, 3, 12059–12068 RSC.
- D. R. Rolison and B. Dunn, J. Mater. Chem., 2001, 11, 963–980 RSC.
- Y. J. Chen, Y. L. Xu, X. F. Sun and C. Wang, J. Power Sources, 2018, 375, 82–92 CrossRef CAS.
- F. Lalere, V. Seznec, M. Courty, R. David, J. N. Chotard and C. Masquelier, J. Mater. Chem. A, 2015, 3, 16198–16205 RSC.
- F. Zhou, X. M. Zhao, C. Goodbrake, J. W. Jiang and J. R. Dahn, J. Electrochem. Soc., 2009, 156, A796–A801 CrossRef CAS.
- P. Y. Hou, H. Z. Zhang, X. L. Deng, X. J. Xu and L. Q. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 29643–29653 CrossRef CAS PubMed.
- E. Jo, J.-H. Park, J. Park, J. Hwang, K. Y. Chung, K.-W. Nam, S. M. Kim and W. Chang, Nano Energy, 2020, 78, 105367 CrossRef CAS.
- B. Bhujun, M. T. T. Tan and A. S. Shanmugarn, Ceram. Int., 2016, 42, 6457–6466 CrossRef CAS.
- A. Rai, A. L. Sharma and A. K. Thakur, Solid State Ionics, 2014, 262, 230–233 CrossRef CAS.
- J. Mendialdua, R. Casanova and Y. Barbaux, J. Electron Spectrosc. Relat. Phenom., 1995, 71, 249–261 CrossRef CAS.
- G. Silversmit, D. Depla, H. Poelman, G. B. Marin and R. De Gryse, J. Electron Spectrosc. Relat. Phenom., 2004, 135, 167–175 CrossRef CAS.
- A. E. Gash, J. H. Satcher and R. L. Simpson, Chem. Mater., 2003, 15, 3268–3275 CrossRef CAS.
- A. E. Gash, J. H. Satcher and R. L. Simpson, J. Non-Cryst. Solids, 2004, 350, 145–151 CrossRef CAS.
- C. N. Chervin, B. J. Clapsaddle, H. W. Chiu, A. E. Gash, J. H. Satcher and S. M. Kauzlarich, Chem. Mater., 2005, 17, 3345–3351 CrossRef CAS.
- J. W. Long, C. N. Chervin, R. B. Balow, S. Jeon, J. B. Miller, M. E. Helms, J. C. Owrutsky, D. R. Rolison and K. P. Fears, Ind. Eng. Chem. Res., 2020, 59, 19584–19592 CrossRef CAS.
- K. P. Chae, W. H. Kwon and J. G. Lee, J. Magn. Magn. Mater., 2012, 324, 2701–2705 CrossRef CAS.
- S. P. Waghmare, D. M. Borikar and K. G. Rewatkar, Mater. Today: Proc., 2017, 4, 11866–11872 Search PubMed.
- N. Abbas, N. Rubab, N. Sadiq, S. Manzoor, M. I. Khan, J. F. Garcia, I. B. Aragao, M. Tariq, Z. Akhtar and G. Yasmin, Water, 2020, 12, 2285 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- S. H. Yu, S. H. Lee, D. J. Lee, Y. E. Sung and T. Hyeon, Small, 2016, 12, 2146–2172 CrossRef CAS PubMed.
- J. B. Lee, J. Moon, O. B. Chae, J. G. Lee, J. H. Ryu, M. Cho, K. Cho and S. M. Oh, Chem. Mater., 2016, 28, 5314–5320 CrossRef CAS.
- B. Long, M. S. Balogun, L. Luo, Y. Luo, W. T. Qiu, S. Q. Song, L. Zhang and Y. X. X. Tong, Small, 2017, 13, 1702081 CrossRef PubMed.
- T. Brousse, D. Belanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- C. Choi, D. S. Ashby, D. M. Butts, R. H. DeBlock, Q. L. Wei, J. Lau and B. Dunn, Nat. Rev. Mater., 2020, 5, 5–19 CrossRef.
- H. J. Huang and M. Niederberger, Nanoscale, 2019, 11, 19225–19240 RSC.
- A. M. Bryan, L. M. Santino, Y. Lu, S. Acharya and J. M. D'Arcy, Chem. Mater., 2016, 28, 5989–5998 CrossRef CAS.
- H. Lindstrom, S. Sodergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S. E. Lindquist, J. Phys. Chem. B, 1997, 101, 7717–7722 CrossRef.
- C. H. Lai, D. Ashby, M. Moz, Y. Gogotsi, L. Pilon and B. Dunn, Langmuir, 2017, 33, 9407–9415 CrossRef CAS PubMed.
- P. G. Pickup and R. W. Murray, J. Am. Chem. Soc., 1983, 105, 4510–4514 CrossRef CAS.
- H. Q. Song, C. F. Liu, C. K. Zhang and G. Z. Cao, Nano Energy, 2016, 22, 1–10 CrossRef CAS.
- S. Y. Li, X. F. Li, Y. W. Li, B. Yan, X. S. Song, L. L. Fan, H. Shan and D. J. Li, J. Alloys Compd., 2017, 722, 278–286 CrossRef CAS.
- F. Liu, Z. X. Chen, G. Z. Fang, Z. Q. Wang, Y. S. Cai, B. Y. Tang, J. Zhou and S. Q. Liang, Nano-Micro Lett., 2019, 11, 25 CrossRef CAS PubMed.
- Y. Song, T.-Y. Liu, B. Yao, T.-Y. Kou, D.-Y. Feng, X.-X. Liu and Y. Li, Small, 2017, 13, 1700067 CrossRef PubMed.
- L. H. B. Nguyen, A. Iadecola, S. Belin, J. Olchowka, C. Masquelier, D. Carlier and L. Croguennec, J. Phys. Chem. C, 2020, 124, 23511–23522 CrossRef CAS.
- K. E. Swider-Lyons, C. T. Love and D. R. Rolison, Solid State Ionics, 2002, 152, 99–104 CrossRef.
- H. D. Ruan, R. L. Frost, J. T. Kloprogge and L. Duong, Spectrochim. Acta, Part A, 2002, 58, 479–491 CrossRef CAS.
- N. Pinney and D. Morgan, Geochim. Cosmochim. Acta, 2013, 114, 94–111 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: AT-FTIR, SEM, TEM, XPS, electrochemical measurements. See DOI: 10.1039/D1RA00819F |
| This journal is © The Royal Society of Chemistry 2021 |