
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of novel artemisone–piperazine–tetronamide hybrids†
Meng-Xue Wei ,
Jia-Ying Yu,
Xin-Xin Liu,
Xue-Qiang Li*,
Jin-Hui Yang,
Meng-Wei Zhang,
Pei-Wen Yang,
Si-Si Zhang and
Yu He
,
Jia-Ying Yu,
Xin-Xin Liu,
Xue-Qiang Li*,
Jin-Hui Yang,
Meng-Wei Zhang,
Pei-Wen Yang,
Si-Si Zhang and
Yu He
State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering, Ningxia Engineering Research Center for Natural Medicine, National Demonstration Center for Experimental Chemistry Education, College of Chemistry and Chemical Engineering, Ningxia University, 489 Helanshan West Road, Yinchuan 750021, China. E-mail: lixq@nxu.edu.cn
First published on 21st May 2021
Abstract
For the first time, six novel artemisone–piperazine–tetronamide hybrids (12a–f) were efficiently synthesised from dihydroartemisinin (DHA) and investigated for their in vitro cytotoxicity against some human cancer cells and benign cells. All the targets showed good cytotoxic activity in vitro. Hybrid 12a exhibited much better inhibitory activity against human liver cancer cell line SMMC-7721 (IC50 = 0.03 ± 0.04 μM for 24 h) than the parent DHA (IC50 > 0.7 μM), and two references, vincristine (VCR; IC50 = 0.27 ± 0.03 μM) & cytosine arabinoside (ARA; IC50 = 0.63 ± 0.04 μM). Furthermore, hybrid 12a had low toxicity against human benign liver cell line LO2 (IC50 = 0.70 ± 0.02 μM for 24 h) compared with VCR, ARA, and DHA in vitro. Moreover, the inhibitory activity of hybrid 12a was obviously enhanced when human liver cancer cell line MHCC97H absorbed Fe2+ in vitro.
1. Introduction
Artemisinin (ARS, Fig. 1) obtained from Artemisia annua is used to treat chills and fever.1 In the last century, it was found that ARS has anticancer activity,2 and a large volume of studies have highlighted the potential of ARS as a novel therapeutic agent for cancer.3–7 Cancer is often life-threatening and hinders health and social progress.8 Artemisinin and its derivatives are highly selective and cytotoxic to tumor cells.9 ARS combined with cisplatin, can induce apoptosis of ovarian cancer cells.10 Dihydroartemisinin (DHA, 1) exhibited high anticancer activity in lewis lung carcinoma (LLC) cell lines in vitro.11 In 2006, Haynes et al. reported the discovery, synthesis and preclinical evaluation of artemisone (3) as an effective oral antimalarial drug.12 In 2011, the same group reported 3 combined with oxaliplatin can enhance anticancer drug activity in breast cancer cells MCF-7, colon cancer cells HCT116, and SW480.13 The mechanism of action of artemisinin-type drugs is still under research. Our group also reported a series of dihydroartemisinin ramifications, some of which showed good cytotoxic activity against cancer cells.14 Tetronamides are an important class of β-heterosubstituted butenolides and has extensive biological activities.15 In previous research, our group has synthesised some tetronamides16 and some of them have good anticancer activity. The combination of artemisinin-type drugs with anticancer drugs can enhance the activity of the anticancer drugs in vitro or in vivo.9,10,17,18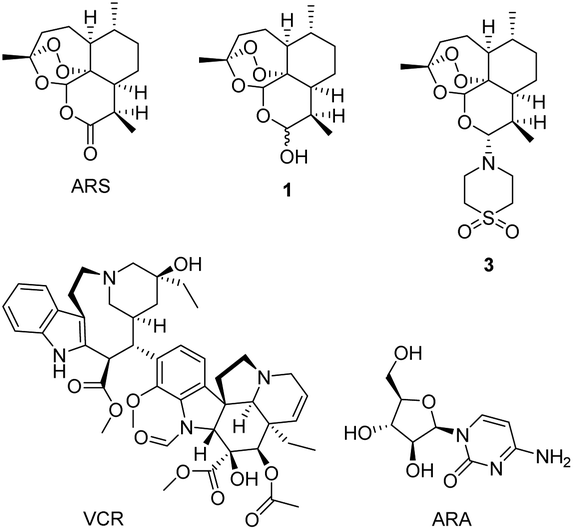 | ||
| Fig. 1 Structures of artemisinin (ARS), dihydroartemisinin (DHA, 1), artemisone (3), vincristine (VCR), and cytosine arabinoside (ARA). | ||
Given the therapeutic properties of tetronamides and the lack of artemisone–piperazine–tetronamide hybrids reported in the literature, we designed and synthesised a series of hybrid tetronamide derivatives possessing an artemisone–piperazine moiety (12a–f, Scheme 4) to evaluate their in vitro cytotoxicity and compare with VCR and ARA. Then we also research the effects of iron ions (Fe2+) on cytotoxic activity.19–22
2. Results and discussion
2.1. Chemistry
Artemiside 2 was synthesised through substitution reaction using dry dimethyl sulfoxide (DMSO) as a catalyst (shown in Scheme 1). First, 1 and oxaloyl chloride formed chloroartemisinin. Then the amine of thiomorpholine attacked chloroartemisinin to produce 2.23 2 was converted to 3 by mild oxidation of hydrogen peroxide and Na2WO4, and this reaction rate was affected by temperature; the higher the temperature, the faster the speed, but the more by-products were produced. Finally, we performed the reaction at room temperature (rt) with a 91% yield.24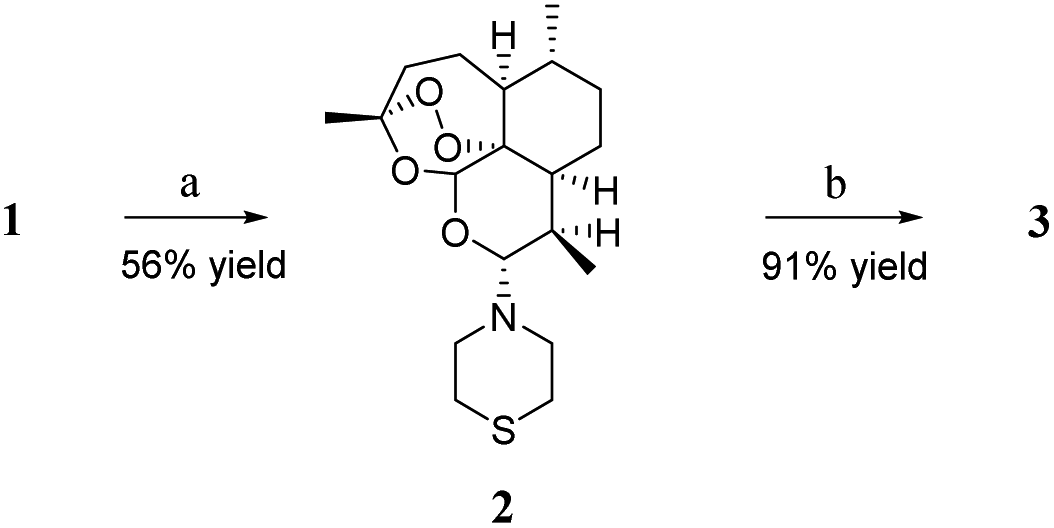 | ||
Scheme 1 Synthesis of artemisone 3. Reagents and conditions: (a) dry DMSO, oxalyl chloride, dry xylene, rt, 7–10 min, then drop in dry DCM solution for thiomorpholine, rt; (b) Na2WO4, H2O2, THF: water = 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, rt. DMSO = dimethyl sulfoxide; DCM = dichloromethane; THF = tetrahydrofuran. :1, rt. DMSO = dimethyl sulfoxide; DCM = dichloromethane; THF = tetrahydrofuran. | ||
As shown in Scheme 2, one hydroxyl group of 1,6-hexanediol 4 was protected with tert-butyldimethylsilyl chloride to produce compound 5, and then compound 6 was obtained through Appel reaction. The synthesis of 7α and 7β needed a strong base to promote the reaction at −78 °C, and the yield was low. Unfortunately, we did not find another way to get a better yield. However, 7α and 7β could be separated through chromatography on silica gel column; 7α made up about 71 percent of the total products 7α and 7β. We chose 7α as the major material of the next reactions. 7α was converted to compound 8 through a two-step series reaction. First, compound 7α was deprotected with tetrabutylammonium fluoride (TBAF), and then iodine substituted the hydroxyl through Appel reaction. Compound 8, piperazine and diisopropylethylamine (DIPEA) were dissolved in methanol and tetrahydrofuran (v/v = 1:20), then heated to 61 °C, and stirred for about 4 hours to obtain artemisone-piperazine 9 with 94% yield.
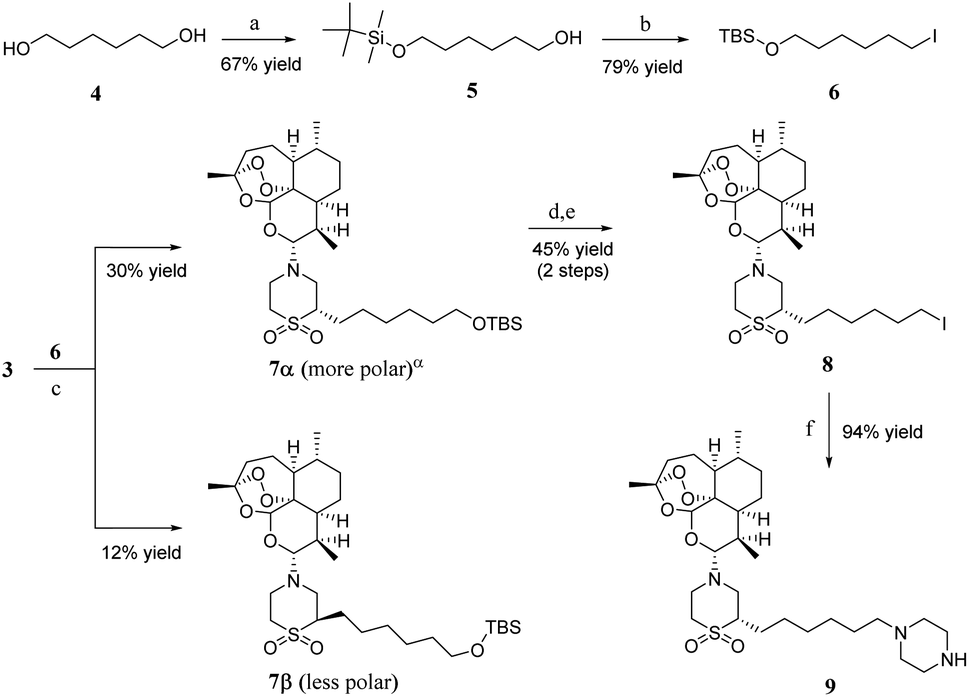 | ||
| Scheme 2 Synthesis of artemisone–piperazine 9. Reagents and conditions: (a) tert-butyldimethylsilyl chloride, imidazole, dry DMF, rt; (b) triphenylphosphine, imidazole, dry THF, rt, then add iodine, keep in dark place; (c) LDA, dry THF, −78 °C to rt; (d) TBAF, THF, rt; (e) triphenylphosphine, imidazole, dry THF, rt, then add iodine, keep in dark place; (f) piperazine, DIPEA, dry THF: dry MeOH = 20:1, rt to reflux. DMF = N,N-dimethylformamide; LDA = lithium diisopropylamide; TBAF = tetrabutylammonium fluoride; DIPEA = diisopropylethylamine. α The stereochemistry of 7α was assigned on the basis of X-ray analysis of a closely related artemisone derivative prepared by the same method. These findings will be reported in a future publication. | ||
As shown in Scheme 3, we synthesised the halogenated furan ethers 11a–f with mucochloric acid or mucobromic acid 10 and alcohol as raw materials, and concentrated sulphuric acid as a catalyst.25
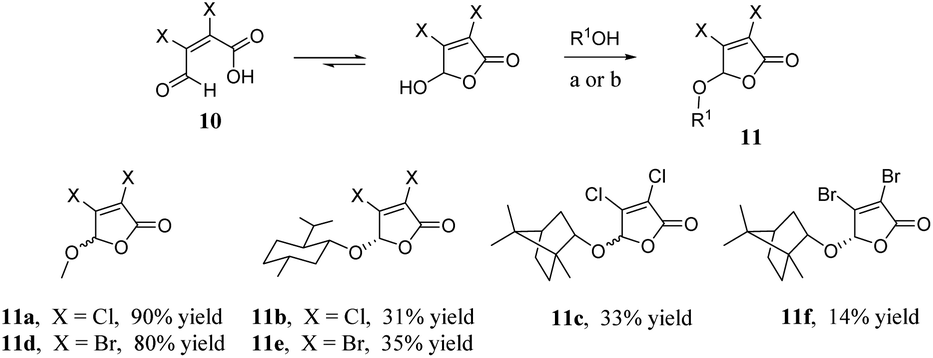 | ||
| Scheme 3 Synthesis of furan ethers 11a–f. Reagents and conditions: (a) MeOH, 1,2-dichloroethane, H2SO4, rt then reflux; (b) menthol or borneol, cyclohexane, H2SO4, rt then reflux. | ||
As shown in Scheme 4, a mixture of artemisone–piperazine 9 and DIPEA in DCM was added to a stirred solution of furan ethers 11 and stirred for 4 hours at room temperature. Then, it was purified by chromatography on a silica gel column to give hybrids 12a–f in good yields.
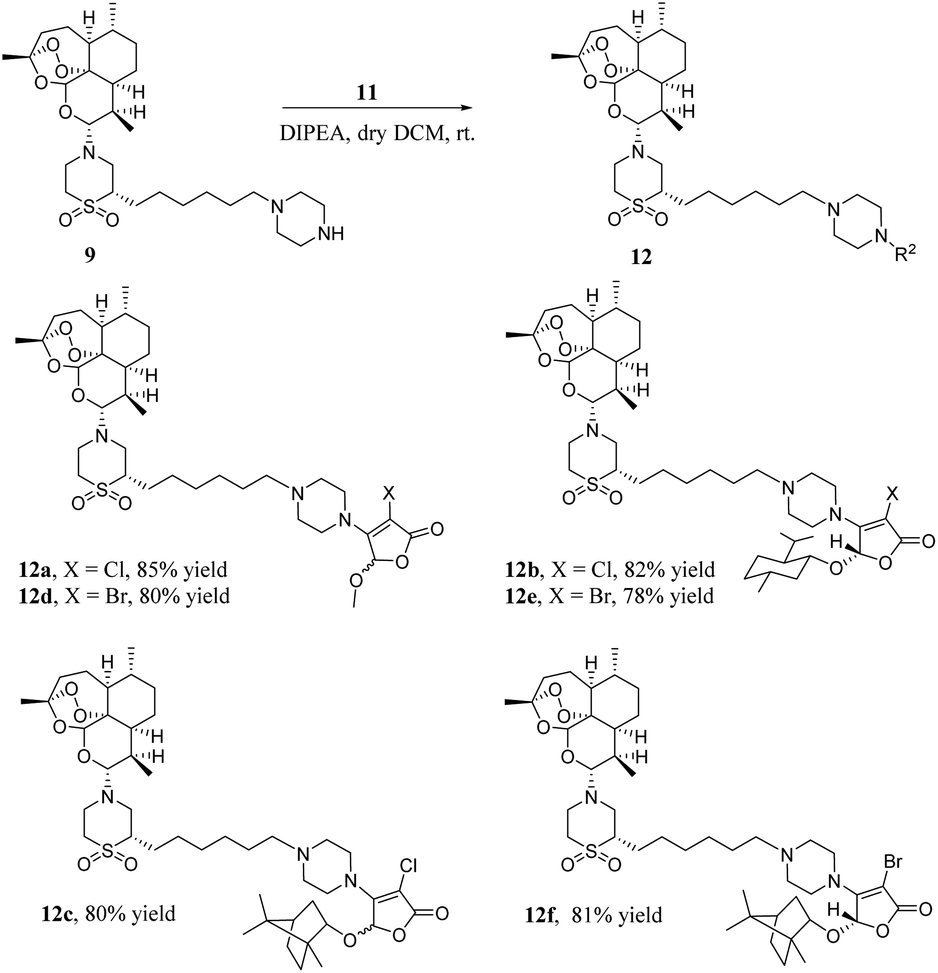 | ||
| Scheme 4 Synthesis of hybrids 12a–f. | ||
2.2. Biological activity
The cytotoxic activities of all the target hybrids 12a–f were tested. The results are given in Table 1. It is clear that 12a (IC50 = 0.03 ± 0.04 μM, 24 h) shows better cytotoxic activity against human liver cancer cell lines SMMC-7721 than the anticancer drugs VCR (IC50 = 0.27 ± 0.03 μM, 24 h), ARA (IC50 = 0.63 ± 0.04 μM, 24 h) and DHA (IC50 > 0.7 μM, 24 h) in vitro, and it is gratifying that 12a (IC50 = 0.70 ± 0.02 μM, 24 h) had lower cytotoxicity against human benign liver cells LO2 compared with VCR (IC50 = 0.30 ± 0.02 μM, 24 h), ARA (IC50 = 0.43 ± 0.04 μM, 24 h) and DHA (IC50 = 0.22 ± 0.04 μM, 24 h) in vitro. As a whole, hybrid 12a showed far more cytotoxic against human liver cancer cells SMMC-7721 than benign cells LO2 compared with VCR, ARA and DHA in vitro.| Compounds | IC50 (μM)/24 h | |
|---|---|---|
| SMMC-7721 | LO2 | |
| a SMMC-7721 = human liver cancer cell lines; LO2 = human benign liver cell lines; VCR = vincristine; ARA = cytosine arabinoside; μM = μmol mL−1; IC50 values represent the compound concentration (μM) required to inhibit tumor cell proliferation by 50% and were calculated using concentrations from triplicate measurements. | ||
| 12a | 0.03 ± 0.04 | 0.70 ± 0.02 |
| 12b | 0.32 ± 0.03 | 0.36 ± 0.03 |
| 12c | 0.07 ± 0.04 | 0.07 ± 0.03 |
| 12d | 0.34 ± 0.02 | 0.37 ± 0.04 |
| 12e | 0.21 ± 0.02 | 0.33 ± 0.03 |
| 12f | 0.22 ± 0.03 | 0.30 ± 0.03 |
| VCR | 0.27 ± 0.03 | 0.30 ± 0.02 |
| ARA | 0.63 ± 0.04 | 0.43 ± 0.04 |
| DHA | >0.7 | 0.22 ± 0.04 |
As can be seen from Fig. 2, hybrid 12a showed the best inhibitory activity against SMMC-7721 cell lines, and its inhibitory rate increased with increasing concentration. Moreover, hybrid 12a was less toxic against LO2 cell lines compared with DHA, ARA, and VCR. As shown in Fig. 3A, the annexin V-FITC (AV) and propidium iodide (PI) staining were used and performed on flow cytometry to explore the apoptotic cells mediated by 12a. MHCC97H cells were treated with 12a at different concentrations of 0.01, 1, and 25 μg mL−1, respectively. As can be seen from Fig. 3B, as the concentration of 12a increased, the percentage of apoptosis increased significantly from 7.67% to 59.76%. Hybrid 12a could induce the apoptosis of MHCC97H cell lines in an appropriate concentration-dependent manner. The results indicated that the antiproliferative effect of 12a might be related to the induction of apoptosis.
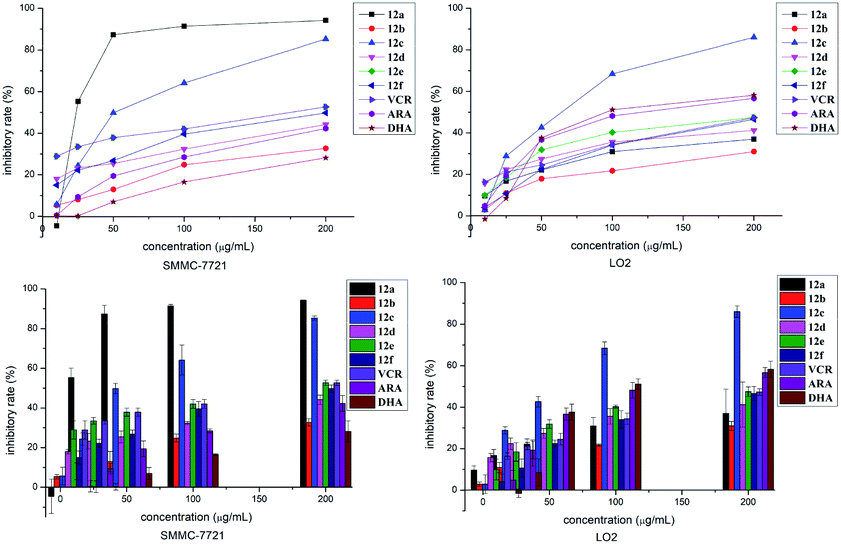 | ||
| Fig. 2 Inhibitory effect of 12a–f, VCR, ARA and DHA with various concentrations on SMMC-7721 and LO2 cells at 24 h. | ||
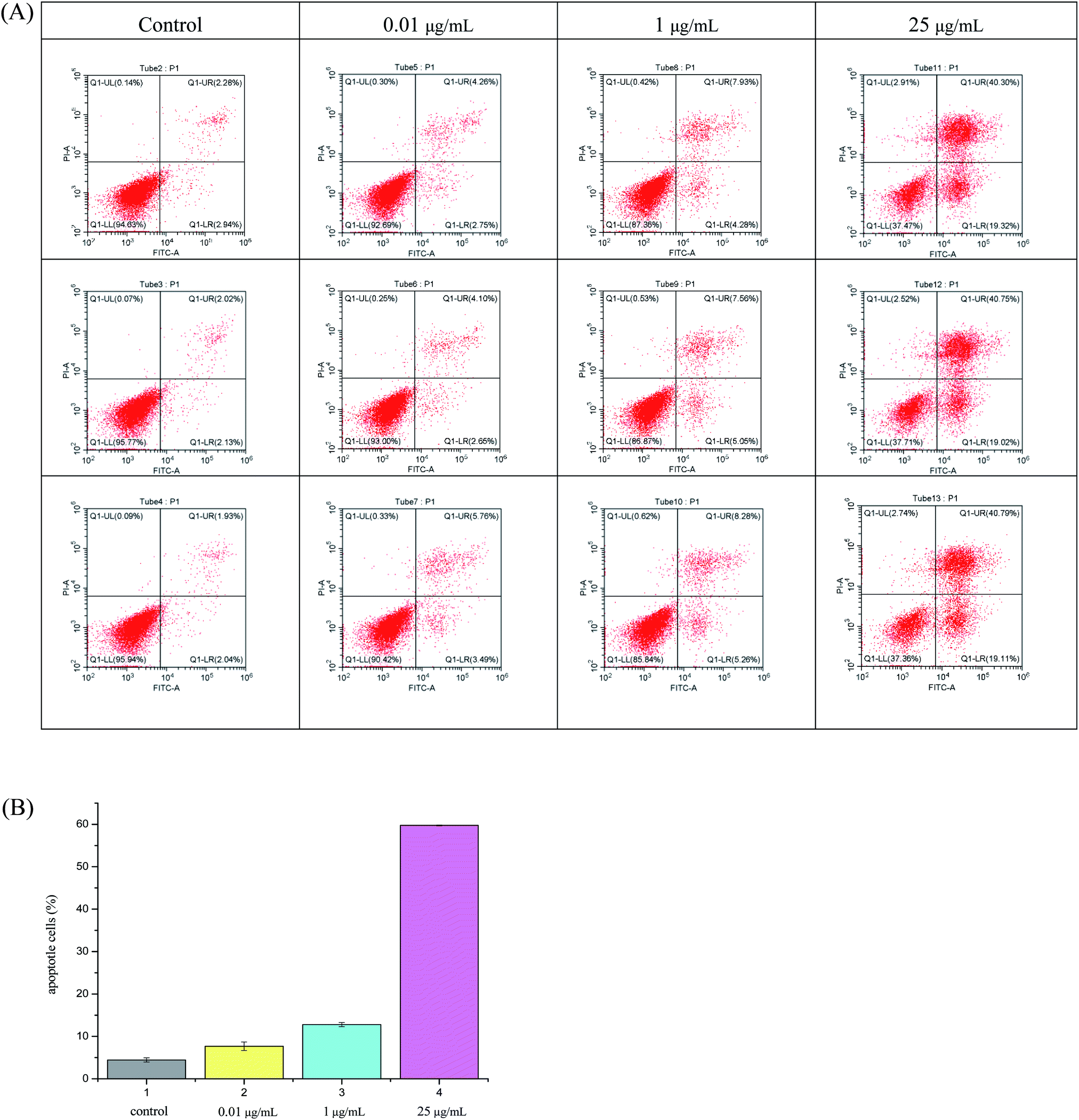 | ||
| Fig. 3 (A) Flow cytometric analysis of MHCC97H cell lines using different concentrations of hybrid 12a (0.01, 1 and 25 μg mL−1) for 48 h and each concentration was detected three times in parallel. The percentage of cells positive for AV and/or PI was reported in the quadrants including cells in the lower left quadrant: live cells; lower right quadrant: early apoptotic cells; upper right quadrant: late apoptotic cells; upper left quadrant: necrotic cells. (B) Apoptotic assay by flow cytometry. MHCC97H cell was treated with 12a at 0.01, 1, and 25 μg mL−1 for 48 h. Then AV and PI staining were used and performed on flow cytometry. | ||
In addition, we chose 12a as a sample to test the cytotoxic activity against other cancer cells and benign cells. The results are shown in Table 2. It's clear that hybrid 12a had a good cytotoxic activity against human liver cancer cells MHCC97H (IC50 = 0.01 ± 0.02 μM), colon cancer cells HCT116 (IC50 = 0.01 ± 0.04 μM), breast cancer cells MCF-7 (IC50 = 0.01 ± 0.04 μM), and prostate cancer cells DU145 (IC50 = 0.02 ± 0.04 μM) for 48 h in vitro. The cytotoxicity against liver cancer cells SMMC-7721 of hybrid 12a had no discernible change as time prolonged to 48 h (IC50 = 0.03 ± 0.03 μM; Table 2) compared with that for 24 h (IC50 = 0.03 ± 0.04 μM; Table 1). It is reported that some cancer cells can import Fe2+ through abnormal iron transporters in cancer cell membranes, while benign cells seldom have abnormal iron transporters.19,20 We choose 12a as a sample to test the cytotoxic activity against human liver cancer cells MHCC97H and SMMC-7721 under an environment with Fe2+ for 48 h in vitro. Instead of using a medium of the mixture of Fe2+ and 12a, Fe2+ was added when culturing cells, which is a good way to simulate the process of cancer cells absorbing Fe2+. Hybrid 12a showed better cytotoxic activity against human liver cancer cells with Fe2+ than without Fe2+. We believe that hybrid 12 may have better antitumor activity in vivo. The morphological changes of human liver cancer cells induced by hybrid 12a are shown in Fig. 4. It is clear that hybrid 12a can effectively inhibit tumor cells' proliferation in vitro.
| Cells | IC50 (μM)/48 h |
|---|---|
| a Cancer cells were cultured with 100 μmol L−1 FeSO4. MHCC97H = human liver cancer cell lines; HCT116 = human colon cancer cell lines; CCD18CO = human benign colon cell lines; MCF-7 = human breast cancer cell lines; MCF-10A = human benign breast cell lines; DU145 = human prostate cancer cell lines; RWPE-1 = human benign prostate cell lines. | |
| MHCC97H | 0.01 ± 0.02 |
| MHCC97H | 0.003 ± 0.002a |
| SMMC-7721 | 0.03 ± 0.03 |
| SMMC-7721 | 0.02 ± 0.03a |
| HCT116 | 0.01 ± 0.04 |
| CCD18CO | 0.06 ± 0.04 |
| MCF-7 | 0.01 ± 0.04 |
| MCF-10A | 0.04 ± 0.04 |
| DU145 | 0.02 ± 0.04 |
| RWPE-1 | 0.10 ± 0.03 |
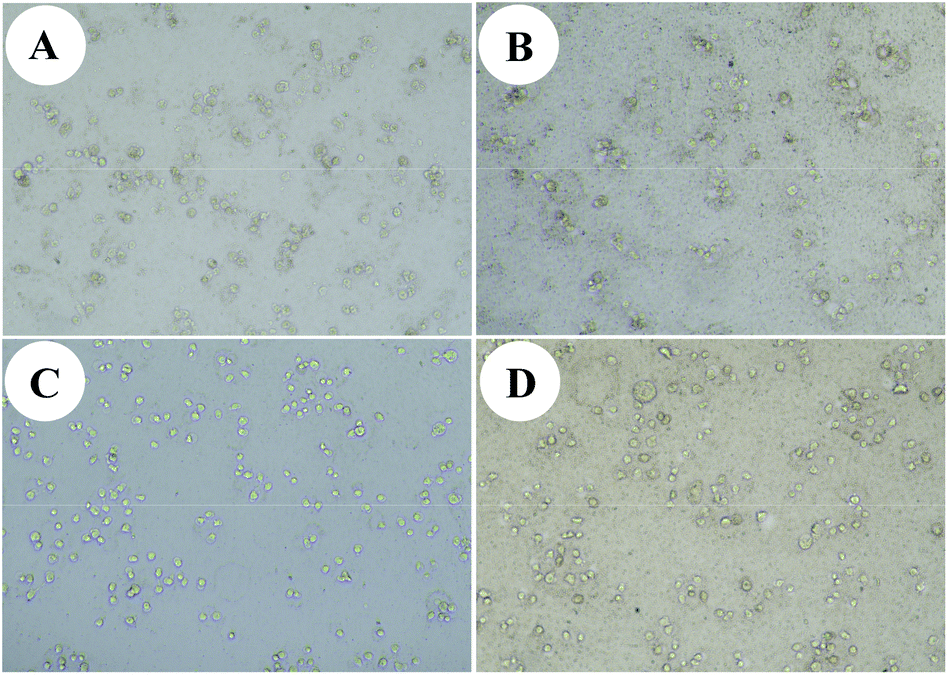 | ||
| Fig. 4 Morphological changes of human liver cancer cells induced by hybrid 12a at a concentration of 10 μg mL−1 after 48 h in (A) MHCC97H cells; (B) MHCC97H cells with Fe2+; (C) SMMC-7721 cells; (D) SMMC-7721 cells with Fe2+. | ||
3. Conclusions
For the first time, a series of artemisone–piperazine–tetronamide hybrids were synthesised and their cytotoxic activities were represented in this study. Artemisone was synthesised through substitution and oxidation using DHA as the raw material. Halogenated furan ethers were synthesised from mucochloric acid or mucobromic acid, and some chiral furan ethers were obtained through recrystallisation in petroleum ether, water and ethanol. Then artemisone and furan ethers were combined using piperazine as a linker to get the target hybrids through 5 steps reaction. Epimers of 7 can be separated by chromatography on a silica gel column. All target hybrids showed good cytotoxic activity in vitro. 12a was the most active hybrid, with better antiproliferative action against SMMC-7721 cell lines (IC50 = 0.03 ± 0.04 μM) and lower toxicity against LO2 cell lines (IC50 = 0.70 ± 0.02 μM) compared with VCR, ARA, and DHA for 24 h in vitro. The cytotoxicity of hybrid 12a against human liver cancer cells SMMC-7721 and MHCC97H was enhanced when cancer cells were cultured with Fe2+ in vitro. The results could provide evidence for the antitumor mechanism of artemisinin-type drugs with Fe2+.4. Experimental
4.1. General
Thin-layer chromatography (TLC) was carried out on silica GF254 plates (Qingdao Haiyang Chemical Co., Ltd, China). IR spectra were recorded on a FTIR-8400S spectrometer as KBr discs. 1H NMR and 13C NMR spectra were obtained with a Bruker Avance III 400 MHz spectrometer in chloroform-d (CDCl3) and tetramethylsilane (TMS) was used as an internal standard. High resolution mass spectrums (HRMS) of targets were recorded on a Thermo Fisher LTQ Orbitrap XL. All chemicals were used as received without further purification unless otherwise stated.4.2. Synthesis
:1). Artemiside 2 was formed in 56% yield as a white solid. 1H NMR (400 MHz, CDCl3) δ 5.28 (s, 1H), 4.18 (d, J = 10.2 Hz, 1H), 3.45–3.26 (m, 1H), 3.07–2.95 (m, 1H), 2.89 (t, J = 4.9 Hz, 3H), 2.55 (ddd, J = 10.9, 9.4, 5.8 Hz, 1H), 2.49–2.35 (m, 2H), 2.35–2.26 (m, 2H), 2.06–1.95 (m, 2H), 1.87 (ddt, J = 10.0, 6.4, 3.4 Hz, 1H), 1.71 (dq, J = 10.2, 3.3 Hz, 1H), 1.56 (dt, J = 13.7, 4.3 Hz, 1H), 1.46 (q, J = 7.4, 4.9 Hz, 3H), 1.31 (dq, J = 10.8, 6.4, 5.2 Hz, 1H), 1.24 (dt, J = 11.0, 5.5 Hz, 1H), 1.02 (td, J = 13.6, 13.2, 3.9 Hz, 1H), 0.95 (d, J = 6.2 Hz, 3H), 0.79 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 105.3, 93.7, 91.3, 80.5, 52.8, 51.8, 47.2, 44.6, 37.2, 35.9, 34.2, 29.6, 25.2, 24.3, 21.8, 20.2, 13.9.:1) were added to a flask (100 mL), stirred at room temperature for 5 min, then H2O2 (1.5 mL, 33%) added in 0.5 h and stirred for another 2 h at room temperature. After the compound 2 had disappeared, the mixture was diluted with ethyl acetate (100 mL), the organic phase was separated and washed with brine (3 × 30 mL), dried over anhydrous Na2SO4 and the solvent removed under diminished pressure. The residue was purified by chromatography on a silica gel column, eluting with petroleum ether–ethyl acetate 3:1. Compound 3 was formed in 91% yield as a white solid. mp 145.7–146.1 °C; [α]20D +34 (c 0.5, CH2Cl2); 1H NMR (400 MHz, CDCl3) δ 5.27 (s, 1H), 4.21 (d, J = 10.3 Hz, 1H), 3.49 (dt, J = 14.6, 5.3 Hz, 2H), 3.37 (dt, J = 13.8, 5.3 Hz, 2H), 3.20 (s, 4H), 2.58 (dqd, J = 14.4, 7.1, 4.4 Hz, 1H), 2.34 (ddd, J = 14.6, 13.3, 4.0 Hz, 1H), 2.00 (ddd, J = 14.6, 5.0, 2.9 Hz, 1H), 1.87 (ddq, J = 13.6, 6.8, 3.2 Hz, 1H), 1.76–1.65 (m, 2H), 1.57 (dt, J = 13.8, 4.3 Hz, 1H), 1.47 (dd, J = 5.1, 2.4 Hz, 1H), 1.45–1.41 (m, 1H), 1.36 (s, 3H), 1.33 (d, J = 3.7 Hz, 1H), 1.24 (dt, J = 11.1, 5.5 Hz, 1H), 1.06–0.97 (m, 1H), 0.94 (d, J = 6.1 Hz, 3H), 0.79 (d, J = 7.1 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 103.9, 92.4, 91.6, 80.3, 51.7, 50.5, 45.8, 37.3, 36.3, 34.2, 28.3, 26.0, 24.7, 21.6, 20.3, 13.5.:1 petroleum ether–ethyl acetate. Compound 5 was formed in 67% yield as a colourless liquid. 1H NMR (CDCl3) δ 3.67–3.57 (m, 4H), 1.63–1.47 (m, 4H), 1.36 (p, J = 3.4 Hz, 4H), 0.89 (s, 9H), 0.04 (s, 6H); 13C NMR (CDCl3) δ 63.6, 62.9, 33.0, 33.1, 26.3, 26.0, 18.7, −4.9.:1. Compound 6 was formed in 79% yield as a colourless liquid. 1H NMR (400 MHz, CDCl3): δ 3.593 (t, J = 6.4 Hz, 2H), 3.175 (t, J = 7 Hz, 2H), 1.773–1.856 (m, 2H), 1.476–1.544 (m, 2H), 1.307–1.436 (m, 4H), 0.883 (s, 9H), 0.038 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 63.0, 33.5, 32.6, 30.3, 26.0, 24.8, 18.4, 7.1, −5.2.:1. Compound 7α was formed in 30% yield as a faint yellow liquid. 1H NMR (400 MHz, CDCl3) δ 5.28 (s, 1H), 4.18 (d, J = 10.3 Hz, 1H), 3.58 (t, J = 6.6 Hz, 2H), 3.48–3.26 (m, 5H), 3.02 (t, J = 12.5 Hz, 1H), 2.96–2.85 (m, 1H), 2.55 (tt, J = 11.5, 7.0 Hz, 1H), 2.39–2.28 (m, 1H), 2.04–1.95 (m, 2H), 1.87 (ddt, J = 13.6, 6.7, 3.6 Hz, 1H), 1.71 (dq, J = 10.2, 3.4 Hz, 2H), 1.67–1.60 (m, 1H), 1.56 (dt, J = 13.9, 4.4 Hz, 1H), 1.51 (s, 1H), 1.36–1.28 (m, 3H), 1.28–1.22 (m, 1H), 1.03 (ddd, J = 15.5, 10.8, 4.2 Hz, 1H), 0.95 (t, J = 5.1 Hz, 4H), 0.88 (s, 12H), 0.85–0.81 (m, 1H), 0.79 (d, J = 7.1 Hz, 3H), 0.04 (s, 7H); 13C NMR (100 MHz, CDCl3) δ 104.0, 91.7, 90.8, 63.0, 60.4, 51.5, 51.3, 45.4, 37.2, 36.0, 34.0, 32.5, 29.1, 28.8, 26.5, 25.8, 25.7, 25.3, 24.6, 23.1, 21.4, 20.1, 18.2, 13.3, −5.4.:1. Compound 8 was formed in 45% yield as a faint yellow liquid for two steps. 1H NMR (400 MHz, CDCl3) δ 5.28 (s, 1H), 4.19 (d, J = 10.3 Hz, 1H), 3.56–3.26 (m, 6H), 3.17 (t, J = 7.0 Hz, 2H), 3.00 (dd, J = 14.4, 10.9 Hz, 1H), 2.94–2.84 (m, 1H), 2.54 (ddt, J = 13.1, 7.1, 3.6 Hz, 1H), 2.34 (ddd, J = 14.7, 13.3, 4.0 Hz, 1H), 2.00 (ddt, J = 14.3, 5.2, 3.3 Hz, 2H), 1.92–1.77 (m, 3H), 1.71 (ddq, J = 13.8, 6.5, 3.5 Hz, 2H), 1.57 (dt, J = 13.8, 4.4 Hz, 1H), 1.48 (dt, J = 7.5, 5.0 Hz, 1H), 1.45–1.39 (m, 1H), 1.37 (s, 8H), 1.31 (dt, J = 15.1, 5.0 Hz, 1H), 1.23 (dt, J = 11.2, 5.6 Hz, 1H), 1.02 (ddd, J = 15.9, 11.5, 4.2 Hz, 1H), 0.95 (d, J = 6.2 Hz, 3H), 0.91 (t, J = 7.3 Hz, 1H), 0.79 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 104.2, 91.9, 91.0, 80.1, 60.4, 51.7, 51.4, 45.6, 37.4, 36.1, 34.1, 33.3, 30.1, 29.0, 28.3, 26.5, 25.9, 24.7, 23.2, 21.6, 20.2, 13.5, 7.1.:1. Compound 9 was formed in 94% yield as a faint yellow gum. 1H NMR (400 MHz, CDCl3) δ 5.29 (d, J = 7.4 Hz, 1H), 4.18 (d, J = 10.2 Hz, 1H), 3.46–3.25 (m, 5H), 3.08–2.95 (m, 1H), 2.89 (t, J = 4.9 Hz, 4H), 2.55 (ddd, J = 10.9, 9.4, 5.8 Hz, 2H), 2.37 (d, J = 4.0 Hz, 4H), 2.35–2.25 (m, 3H), 2.06–1.94 (m, 3H), 1.92–1.81 (m, 2H), 1.71 (dq, J = 10.2, 3.3 Hz, 3H), 1.56 (dt, J = 13.7, 4.3 Hz, 4H), 1.48 (d, J = 9.5 Hz, 6H), 1.30 (dd, J = 15.6, 10.8 Hz, 6H), 1.26–1.18 (m, 1H), 1.12 (d, J = 6.5 Hz, 1H), 1.06–0.98 (m, 1H), 0.95 (d, J = 6.2 Hz, 3H), 0.79 (d, J = 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 104.2, 91.9, 91.0, 80.1, 59.3, 54.5, 53.2, 51.7, 51.4, 50.8, 46.0, 45.6, 37.4, 36.1, 34.2, 29.4, 29.0, 27.2, 26.6, 26.4, 25.9, 24.8, 23.2, 21.6, 20.2, 13.5.:1 to 4:1 to give hybrids 12a–f.
4.2.9.1. 3-Chloro-4-(4-(6-((2S)-1,1-dioxido-4-((3R,5aS,6R,8aS,9R,10R,12aR)-3,6,9-trimethyldecahydro-3H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thiomorpholin-2-yl)hexyl)piperazin-1-yl)-5-methoxyfuran-2(5H)-one (12a). Hybrid 12a was formed in 85% yield (99.4% HPLC purity) as a yellow gum. [α]20D +19 (c 1.0, EtOH); 1H NMR (400 MHz, CDCl3) δ 5.69 (s, 1H), 5.29 (s, 1H), 4.20 (d, J = 10.2 Hz, 1H), 3.73 (s, 4H), 3.56–3.24 (m, 7H), 3.01 (t, J = 12.6 Hz, 1H), 2.96–2.80 (m, 1H), 2.65–2.46 (m, 5H), 2.34 (ddd, J = 18.2, 11.5, 4.5 Hz, 3H), 2.01 (dt, J = 14.3, 4.1 Hz, 2H), 1.88 (dq, J = 10.2, 3.3 Hz, 1H), 1.73 (ddt, J = 13.5, 6.3, 3.3 Hz, 2H), 1.62–1.19 (m, 18H), 1.10–0.99 (m, 1H), 0.96 (d, J = 6.1 Hz, 3H), 0.80 (d, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 171.3, 167.9, 153.6, 104.3, 97.5, 92.1, 91.2, 80.3, 60.6, 58.3, 58.3, 54.9, 54.8, 52.8, 51.9, 51.6, 45.8, 37.6, 36.3, 34.3, 29.8, 29.5, 29.2, 26.9, 26.7, 26.1, 24.9, 23.5, 21.7, 20.4, 14.3, 13.6; IR (KBr) ν/cm−1: 2934, 2869, 1744, 1638, 1438, 1301, 1259, 1041, 848, 746; HRMS m/z: calcd for C34H55ClN3O9S 716.3342, found 716.3337 [M + H]+.
4.2.9.2. (5S)-3-Chloro-4-(4-(6-((2S)-1,1-dioxido-4-((3R,5aS,6R,8aS,9R,10R,12aR)-3,6,9-trimethyldecahydro-3H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thiomorpholin-2-yl)hexyl)piperazin-1-yl)-5-(((1S,2S,5R)-2-isopropyl-5-methylcyclohexyl)oxy)furan-2(5H)-one (12b). Hybrid 12b was formed in 82% yield (98.6% HPLC purity) as a yellow solid. mp 75.6–76.1 °C; [α]20D −2 (c 1.0, EtOH); 1H NMR (400 MHz, CDCl3) δ 6.73 (s, 0.2H), 5.78 (s, 0.8H), 5.29 (s, 1H), 4.20 (d, J = 10.2 Hz, 1H), 3.82–3.59 (m, 4H), 3.59–3.27 (m, 5H), 3.01 (t, J = 12.5 Hz, 1H), 2.95–2.87 (m, 1H), 2.61–2.29 (m, 8H), 2.28–2.12 (m, 1H), 2.02 (q, J = 8.3, 6.5 Hz, 3H), 1.89 (ddd, J = 14.3, 7.0, 3.6 Hz, 2H), 1.71 (tdd, J = 17.9, 8.0, 3.7 Hz, 5H), 1.51 (tdd, J = 16.6, 11.5, 6.8 Hz, 5H), 1.43–1.22 (m, 13H), 1.13 (q, J = 12.2 Hz, 1H), 0.93 (dt, J = 15.2, 7.2 Hz, 11H), 0.78 (dq, J = 12.1, 6.8, 5.5 Hz, 7H); 13C NMR (101 MHz, CDCl3) δ 168.4, 155.0, 104.3, 97.3, 92.1, 91.2, 87.6, 81.0, 80.3, 60.7, 58.5, 52.9, 51.9, 51.6, 48.2, 47.7, 45.8, 42.6, 40.5, 37.6, 36.3, 34.3, 34.0, 31.8, 29.8, 29.5, 29.2, 27.1, 26.8, 26.1, 25.2, 24.9, 23.5, 22.8, 22.4, 21.7, 21.3, 20.4, 15.9, 13.6; IR (KBr) ν/cm−1: 2933, 2888, 2870, 1760, 1634, 1455, 1302, 1126, 974, 879, 746; HRMS m/z: calcd for C43H71ClN3O9S 840.4594, found 840.4596 [M + H]+.
4.2.9.3. 3-Chloro-4-(4-(6-((S)-1,1-dioxido-4-((3R,5aS,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-3H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thiomorpholin-2-yl)hexyl)piperazin-1-yl)-5-(((2S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)oxy)furan-2(5H)-one (12c). Hybrid 12c was formed in 80% yield (96.9% HPLC purity) as a yellow gum. [α]20D +10 (c 1.0, EtOH); 1H NMR (400 MHz, CDCl3) δ 6.73 (s, 0.7H), 5.72 (s, 0.2H), 5.29 (s, 1H), 4.19 (d, J = 10.3 Hz, 1H), 3.74 (s, 1H), 3.56–3.27 (m, 8H), 3.02 (t, J = 12.5 Hz, 1H), 2.90 (d, J = 11.1 Hz, 1H), 2.68–2.17 (m, 8H), 2.05–1.98 (m, 2H), 1.98–1.83 (m, 1H), 1.80–1.67 (m, 4H), 1.62–1.42 (m, 4H), 1.43–1.19 (m, 16H), 1.11–1.00 (m, 1H), 0.96 (d, J = 6.3 Hz, 5H), 0.92–0.84 (m, 9H), 0.80 (d, J = 7.1 Hz, 3H), 0.61 (t, J = 7.1 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 162.9, 161.8, 132.3, 129.1, 104.3, 92.1, 91.2, 83.5, 80.3, 60.7, 58.5, 53.0, 52.0, 51.6, 49.2, 48.1, 45.8, 44.9, 37.6, 36.4, 34.3, 32.1, 29.8, 29.4, 29.2, 28.5, 28.1, 27.2, 27.1, 26.8, 26.5, 26.1, 24.9, 24.4, 23.5, 22.8, 21.8, 20.4, 19.8, 19.0, 13.6; IR (KBr) ν/cm−1: 2932, 2887, 2867, 1758, 1628, 1449, 1377, 1302, 1127, 1040, 960, 847, 744; HRMS m/z: calcd for C43H69ClN3O9S 838.4438, found 838.4432 [M + H]+.
4.2.9.4. 3-Bromo-4-(4-(6-((2S)-1,1-dioxido-4-((3R,5aS,6R,8aS,9R,10R,12aR)-3,6,9-trimethyldecahydro-3H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thiomorpholin-2-yl)hexyl)piperazin-1-yl)-5-methoxyfuran-2(5H)-one (12d). Hybrid 12d was formed in 80% yield (98.8% HPLC purity) as a yellow solid. mp 78.2–79.2 °C; [α]20D +6 (c 1.0, EtOH); 1H NMR (400 MHz, CDCl3) δ 5.74 (s, 1H), 5.29 (s, 1H), 4.16 (dd, J = 29.7, 8.7 Hz, 1H), 3.90–3.79 (m, 1H), 3.77 (s, 4H), 3.75–3.63 (m, 1H), 3.48 (s, 1H), 3.47–3.36 (m, 3H), 3.33 (d, J = 14.4 Hz, 1H), 3.02 (t, J = 12.8 Hz, 1H), 2.95–2.87 (m, 1H), 2.64–2.54 (m, 1H), 2.58–2.51 (m, 4H), 2.44–2.35 (m, 2H), 2.32 (dd, J = 14.2, 4.3 Hz, 1H), 2.08–1.95 (m, 2H), 1.89 (ddd, J = 14.0, 6.8, 3.4 Hz, 1H), 1.73 (ddt, J = 13.5, 6.2, 3.4 Hz, 2H), 1.61–1.44 (m, 6H), 1.42–1.17 (m, 12H), 1.09–0.98 (m, 1H), 0.96 (d, J = 6.2 Hz, 3H), 0.80 (d, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 171.3, 168.7, 156.6, 104.3, 97.8, 92.1, 91.2, 80.3, 64.0, 60.7, 60.5, 58.4, 53.0, 51.9, 51.6, 47.6, 45.8, 37.6, 36.3, 34.3, 29.4, 29.2, 27.0, 26.8, 26.5, 26.1, 24.9, 23.5, 21.7, 20.4, 15.1, 13.6; IR (KBr) ν/cm−1: 2934, 2892, 2872, 1762, 1636, 1454, 1303, 1127, 974, 646; HRMS m/z: calcd for C34H55BrN3O9S 760.2837, found 760.2833 [M + H]+.
4.2.9.5. (5S)-3-Bromo-4-(4-(6-((2S)-1,1-dioxido-4-((3R,5aS,6R,8aS,9R,10R,12aR)-3,6,9-trimethyldecahydro-3H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thiomorpholin-2-yl)hexyl)piperazin-1-yl)-5-(((1S,2S,5R)-2-isopropyl-5-methylcyclohexyl)oxy)furan-2(5H)-one (12e). Hybrid 12e was formed in 78% yield as a yellow gum. [α]20D −3 (c 1.0, EtOH); 1H NMR (400 MHz, CDCl3) δ 5.79 (s, 1H), 5.28 (s, 1H), 4.19 (d, J = 10.2 Hz, 1H), 3.86–3.50 (m, 4H), 3.47–3.28 (m, 4H), 3.01 (t, J = 12.6 Hz, 1H), 2.90 (d, J = 11.5 Hz, 1H), 2.53 (ddt, J = 21.4, 6.8, 3.9 Hz, 4H), 2.43–2.28 (m, 4H), 2.25–2.13 (m, 1H), 2.04–1.84 (m, 3H), 1.75–1.63 (m, 4H), 1.59–1.43 (m, 6H), 1.35 (d, J = 12.6 Hz, 10H), 1.28–1.05 (m, 6H), 1.03–0.85 (m, 12H), 0.80–0.73 (m, 7H); 13C NMR (100 MHz, CDCl3) δ 168.7, 157.6, 104.2, 98.2, 92.0, 91.1, 80.7, 80.2, 73.8, 58.4, 52.8, 51.8, 51.5, 48.2, 47.8, 45.7, 42.5, 37.5, 36.2, 34.2, 34.0, 31.7, 31.5, 29.4, 29.1, 27.0, 26.7, 26.0, 25.1, 24.8, 23.4, 22.8, 22.3, 21.6, 21.2, 20.3, 19.4, 15.8, 13.5; IR (KBr) ν/cm−1: 2932, 1761, 1635, 1454, 1302, 1126, 934, 879; HRMS m/z: calcd for C43H71BrN3O9S 884.4089, found 884.4088 [M + H]+.
4.2.9.6. (5S)-3-Bromo-4-(4-(6-((S)-1,1-dioxido-4-((3R,5aS,6R,8aS,9R,10R,12R,12aR)-3,6,9-trimethyldecahydro-3H-3,12-epoxy[1,2]dioxepino[4,3-i]isochromen-10-yl)thiomorpholin-2-yl)hexyl)piperazin-1-yl)-5-(((2S)-1,7,7-trimethylbicyclo[2.2.1]heptan-2-yl)oxy)furan-2(5H)-one (12f). Hybrid 12f was formed in 81% yield (96.8% HPLC purity) as a yellow solid. mp 77.9–78.9 °C; [α]20D +11 (c 1.0, EtOH); 1H NMR (400 MHz, CDCl3) δ 6.95 (s, 0.3H), 5.73 (d, J = 10.1 Hz, 0.4H), 5.29 (s, 1H), 4.20 (d, J = 10.3 Hz, 1H), 3.79–3.73 (m, 2H), 3.67 (s, 1H), 3.54–3.24 (m, 6H), 3.02 (t, J = 12.7 Hz, 1H), 2.91 (d, J = 11.3 Hz, 1H), 2.63–2.25 (m, 8H), 2.03 (d, J = 6.9 Hz, 3H), 1.94–1.83 (m, 2H), 1.79–1.65 (m, 4H), 1.58 (dd, J = 13.1, 4.5 Hz, 1H), 1.53–1.46 (m, 7H), 1.36 (d, J = 11.3 Hz, 8H), 1.33–1.21 (m, 5H), 1.05 (ddt, J = 18.1, 13.4, 5.9 Hz, 2H), 0.96 (d, J = 6.3 Hz, 3H), 0.87 (dd, J = 11.0, 5.2 Hz, 8H), 0.81 (t, J = 10.0 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 163.3, 162.1, 136.2, 118.9, 104.3, 92.0, 91.1, 83.5, 80.2, 60.7, 58.5, 58.4, 53.0, 52.4, 51.9, 51.6, 49.5, 49.2, 48.0, 47.6, 45.7, 45.0, 44.9, 37.6, 36.3, 34.3, 29.4, 29.1, 28.1, 28.0, 27.1, 26.8, 26.0, 24.9, 23.4, 21.7, 20.4, 19.8, 18.9, 14.2, 13.6; IR (KBr) ν/cm−1: 2932, 2890, 2871, 1760, 1628, 1453, 1304, 1126, 879, 744; HRMS m/z: calcd for C43H69BrN3O9S 882.3932, found 882.3921 [M + H]+.
4.3. Pharmacology
| Cell inhibition rate = [A568 (negative control well) − A568 (dosing well)]/A568 (negative control well) × 100%. |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge financial support from the Natural Science Foundation of Ningxia (2021AAC05009, 2019AAC03018), the National Natural Science Foundation of China (21462032), the College Students' Innovative and Entrepreneurship Training Program of Ningxia University (G2020107490003), the Third Batch of Ningxia Youth Talents Supporting Program (TJGC2018100), the Discipline Project of Ningxia (NXYLXK2017A04), and the Institute Local Cooperation Project of Chinese Academy of Engineering (2019NXZD1). Thanks to Wuhan Bafeier Biotechnology Service Co., Ltd For testing the biological activity.Notes and references
- U. Eckstein-Ludwig, R. J. Webb, I. D. A. van Goethem, J. M. East, A. G. Lee, M. Kimura, P. M. O'Neill, P. G. Bray, S. A. Ward and S. Krishna, Nature, 2003, 424, 957–961 CrossRef CAS PubMed.
- Y. K. Wong, C. Xu, K. A. Kalesh, Y. He, Q. Lin, W. S. F. Wong, H.-M. Shen and J. Wang, Med. Res. Rev., 2017, 37, 1492–1517 CrossRef CAS PubMed.
- Z. Jiang, J. Chai, H. H. F. Chuang, S. Li, T. Wang, Y. Cheng, W. Chen and D. Zhou, Anti-Cancer Drugs, 2012, 23, 606–613 CrossRef CAS PubMed.
- H. Sun, X. Meng, J. Han, Z. Zhang, B. Wang, X. Bai and X. Zhang, Tumor Biol., 2013, 34, 3791–3800 CrossRef CAS PubMed.
- A. Bhaw-Luximon and D. Jhurry, Cancer Chemother. Pharmacol., 2017, 79, 451–466 CrossRef CAS PubMed.
- P. C. H. Li, E. Lam, W. P. Roos, M. Z. Zdzienicka, B. Kaina and T. Efferth, Cancer Res., 2008, 68, 4347–4351 CrossRef CAS PubMed.
- Q. Y. Pan, S. Z. Wang, J. J. Lu, L. H. Meng and Z. J. Yao, Sci. China. Chem., 2010, 53, 119–124 CrossRef CAS.
- A. Abu Ammar, R. Raveendran, D. Gibson, T. Nassar and S. Benita, J. Med. Chem., 2016, 59, 9035–9046 CrossRef CAS PubMed.
- T. Efferth, Biochem. Pharmacol., 2017, 139, 56–70 CrossRef CAS PubMed.
- X. Feng, L. Li, H. Jiang, K. Jiang, Y. Jin and J. Zheng, Biochem. Biophys. Res. Commun., 2014, 444, 376–381 CrossRef CAS PubMed.
- H.-J. Zhou, J.-L. Zhang, A. Li, Z. Wang and X.-E. Lou, Cancer Chemother. Pharmacol., 2010, 66, 21–29 CrossRef CAS PubMed.
- R. K. Haynes, B. Fugmann, J. Stetter, K. Rieckmann, H. Heilmann, H.-W. Chan, M.-K. Cheung, W.-L. Lam, H.-N. Wong, S. L. Croft, L. Vivas, L. Rattray, L. Stewart, W. Peters, B. L. Robinson, M. D. Edstein, B. Kotecka, D. E. Kyle, B. Beckermann, M. Gerisch, M. Radtke, G. Schmuck, W. Steinke, U. Wollborn, K. Schmeer and A. Römer, Angew. Chem., Int. Ed., 2006, 45, 2082–2088 CrossRef CAS PubMed.
- A. M. Gravett, W. M. Liu, S. Krishna, W.-C. Chan, R. K. Haynes, N. L. Wilson and A. G. Dalgleish, Cancer Chemother. Pharmacol., 2011, 67, 569–577 CrossRef CAS PubMed.
- J.-Y. Yu, X.-Q. Li and M.-X. Wei, Eur. J. Med. Chem., 2019, 169, 21–28 CrossRef CAS PubMed.
- M. Karak, L. C. A. Barbosa, J. A. M. Acosta, A. M. Sarotti and J. Boukouvalas, Org. Biomol. Chem., 2016, 14, 4897–4907 RSC.
- M.-X. Wei, J. Zhang, F.-L. Ma, M. Li, J.-Y. Yu, W. Luo and X.-Q. Li, Eur. J. Med. Chem., 2018, 155, 165–170 CrossRef CAS PubMed.
- H. N. Wong, A. Lewies, M. Haigh, J. M. Viljoen, J. F. Wentzel, R. K. Haynes and L. H. d. Plessis, Front. Pharmacol., 2020, 11, 558894 CrossRef CAS PubMed.
- T. Fröhlich, A. Kiss, J. Wölfling, E. Mernyák, Á. E. Kulmány, R. Minorics, I. Zupkó, M. Leidenberger, O. Friedrich, B. Kappes, F. Hahn, M. Marschall, G. Schneider and S. B. Tsogoeva, ACS Med. Chem. Lett., 2018, 9, 1128–1133 CrossRef PubMed.
- J. Chen, Z. Guo, H.-B. Wang, J.-J. Zhou, W.-J. Zhang and Q.-W. Chen, Biomaterials, 2014, 35, 6498–6507 CrossRef CAS PubMed.
- T. Efferth, A. Benakis, M. R. Romero, M. Tomicic, R. Rauh, D. Steinbach, R. Häfer, T. Stamminger, F. Oesch, B. Kaina and M. Marschall, Free Radical Biol. Med., 2004, 37, 998–1009 CrossRef CAS PubMed.
- X.-R. Deng, Z.-X. Liu, F. Liu, L. Pan, H.-P. Yu, J.-P. Jiang, J.-J. Zhang, L. Liu and J. Yu, J. Huazhong Univ. Sci. Technol., Med. Sci., 2013, 33, 862–865 CrossRef CAS PubMed.
- T. Efferth, World J. Tradit. Chin. Med., 2015, 1, 2–25 Search PubMed.
- W. C. Chan, D. H. W. Chan, K. W. Lee, W. S. Tin, H. N. Wong and R. K. Haynes, Tetrahedron, 2018, 74, 5156–5171 CrossRef CAS.
- J. Xu, M.-X. Wei, G.-M. Li and X.-Q. Li, Chem. J. Chin. Univ., 2015, 36, 919–926 CAS.
- M.-X. Wei, J.-Y. Yu, X.-X. Liu, X.-Q. Li, M.-W. Zhang, P.-W. Yang and J.-H. Yang, Eur. J. Med. Chem., 2021, 215, 113295 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Spectra data. See DOI: 10.1039/d1ra00750e |
| This journal is © The Royal Society of Chemistry 2021 |