
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel one-pot synthesis of flavones†
Meng-Yang Chang *ab,
Min-Chen Tsaiab and
Chun-Yi Linab
*ab,
Min-Chen Tsaiab and
Chun-Yi Linab
aDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 80708, Taiwan. E-mail: mychang@kmu.edu.tw
bDepartment of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung 807, Taiwan
First published on 22nd March 2021
Abstract
In this paper, a one-pot facile route for the BiCl3/RuCl3-mediated synthesis of functionalized flavones is described, including: (i) intermolecular ortho-acylation of substituted phenols with cinnamoyl chlorides, and (ii) intramolecular cyclodehydrogenation of the resulting o-hydroxychalcones. The reaction conditions are discussed herein.
Introduction
For the flavonoid family, flavone (also called 2-arylchromone) with numerous biological activities is attracting many synthetic researchers to develop a number of versatile methodologies.1 A recent review article1a has revealed the synthetic history of functionalized flavones from the traditional well-known named reactions (e.g., Baker–Venkataraman,2 Karl von-Auwers,3 Algar–Flynn–Oyamada,4 Allan–Robinson,5 Kostanecki,6 Mentzer7 and Wittig8) to modern novel transition-metal promoted protocols (e.g., Suzuki–Miyaura9 and Sonogashira10). By the use of other reaction conditions such as microwave irradiation,11 ionic liquids improvement12 and photolytic annulation,13 the unique methodologies of flavones and their derivatives have been established. For the synthetic chemistry of diversified flavones, the two-step process for (1) ortho-acylation of substituted phenols and (2) cyclodehydrogenation of the resulting o-hydroxychalcones is a general and direct route to provide access to the core structure, as shown in Scheme 1.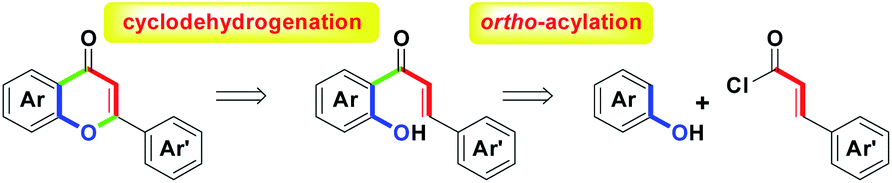 | ||
| Scheme 1 Synthetic route of flavones. | ||
By the involvement of transition metals, various ortho-acylations of phenols have been well-developed, including CuCl2,14a FeCl3,14b TiCl4,14c mercury lamp/photolysis,14d MsOH/microwave.14e For the following cyclodehydrogenation step, the uses of InCl3/SiO2,15a FeCl3·6H2O/MeOH,15b and CuI/ionic liquids15c have been studied. In addition, transition metal-free oxidants-mediated reaction systems, for example, I2/DMSO,16a DDQ/dioxane,16b NaIO4/DMSO,16c H2O2/NaOH,16d SeO2/dioxane16e and Br2/NaOH16f have been investigated in the cyclodehydrogenation step.
Among these reported routes with the synthetic sequence of intermolecular ortho-acylation followed by intramolecular cyclodehydrogenation, we found that all attempts adopted a two-step stepwise process as the major focused design in the flavone family formation. Despite the above elegant synthetic routes, to date, there are no reports on the one-pot synthesis of substituted flavones on the basis of the formal (3 + 3) annulation. Herein, we present metal chlorides-mediated one-pot synthesis of flavones 4 (Scheme 2) via the combination of BiCl3 andRuCl3-mediated intermolecular ortho-acylation of substituted phenols 1 with cinnamoyl chlorides 2 (one carbon–carbon bond formation, green), and intramolecular cyclodehydrogenation of the corresponding o-hydroxychalcones 3 (one carbon–oxygen bond formation, green). These starting materials, 1 and 2, were obtained from commercial sources and used without further purification.
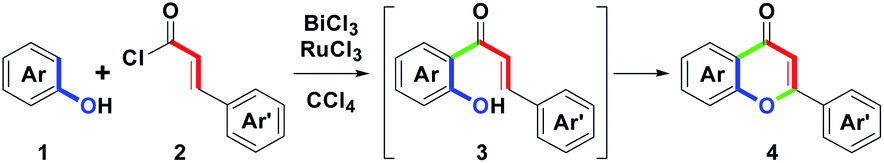 | ||
| Scheme 2 Our synthetic route towards flavones. | ||
Results and discussion
The initial study commenced with the treatment of the model substrate 1a (Ar = Ph, 1.0 mmol) with 2a (Ar′ = Ph, 1.0 mmol) and AlCl3 (2.0 equiv.) in CCl4 (20 mL) at reflux (77 °C) for 10 h. Only 3a was produced at a 54% yield. The results are shown in Table 1, entry 1. Entries 2–7 show that six trivalent transition-metal chlorides (2.0 equiv., InCl3, FeCl3, BiCl3, CeCl3, AuCl3 and RuCl3) were studied in CCl4 (20 mL) at reflux for 10 h. However, only InCl3 and FeCl3 obtained sole 3a in 67% and 70% yields, respectively. BiCl3 provided 65% yield of 3a along with trace amount of 4a. In particular, no reactions were observed for CeCl3 and AuCl3, while RuCl3 produced a complex mixture. According to the experimental results, we understood that InCl3 or FeCl3-mediated ortho-acylation of 1a proceeded well, but they could not promote the conversion from 3a to 4a (entries 2 and 3). In addition to forming 3a, trace amount of 4a (3%) could be obtained in the presence of BiCl3 (entry 4). For CeCl3 and AuCl3, the reactivity of Lewis acid was weak to a degree that no desired reaction was initiated (entries 5 and 6). Compared with other MCl3, RuCl3 with strong oxidation ability could force the ortho-acylation of 1a to complexation (entry 7). Next, we refocused the synthetic aim to study the equivalents of BiCl3; however, after increasing the stoichiometric from 2.0 to 3.0 equiv., the conversion efficiency from 3a to 4a was similar to that of 2.0 equiv. (5%, entry 8).
|
||||
|---|---|---|---|---|
| Entry | MCl3 (equiv.) | Solvent | Time (h) | 3a/4ab % |
| a The reactions were run on a 1.0 mmol scale with phenol 1a, cinnamoyl chloride 2a (1.0 equiv.), metal chlorides (MCl3, equiv.), solvent (20 mL), time (h), reflux.b Isolated yields.c No detection.d No reaction.e Unknown and unidentified complex mixture was isolated. | ||||
| 1 | AlCl3 (2.0) | CCl4 | 10 | 54/—c |
| 2 | InCl3 (2.0) | CCl4 | 10 | 67/—c |
| 3 | FeCl3 (2.0) | CCl4 | 10 | 70/—c |
| 4 | BiCl3 (2.0) | CCl4 | 10 | 65/3 |
| 5 | CeCl3 (2.0) | CCl4 | 10 | —d |
| 6 | AuCl3 (2.0) | CCl4 | 10 | —d |
| 7 | RuCl3 (2.0) | CCl4 | 10 | —e |
| 8 | BiCl3 (3.0) | CCl4 | 10 | 68/5 |
| 9 | BiCl3 (1.0) | CCl4 | 10 | 70/—c |
| 10 | BiCl3 (1.0), RuCl3 (1.0) | CCl4 | 10 | 15/68 |
| 11 | BiCl3 (1.0), RuCl3 (1.0) | MeNO2 | 10 | —e |
| 12 | BiCl3 (1.0), RuCl3 (1.0) | (CH2Cl)2 | 10 | 35/52 |
| 13 | BiCl3 (1.0), RuCl3 (1.0) | CH2Cl2 | 10 | 78/10 |
| 14 | BiCl3 (1.0), RuCl3 (1.0) | DMF | 10 | —e |
| 15 | BiCl3 (1.0), RuCl3 (1.0) | CCl4 | 15 | 3/81 |
| 16 | BiCl3 (1.0), RuCl3 (1.0) | CCl4 | 20 | —d/75 |
| 17 | BiCl3 (1.0), CuCl2 (1.0) | CCl4 | 15 | 60/8 |
| 18 | BiCl3 (1.0), FeCl3 (1.0) | CCl4 | 15 | 72/3 |
| 19 | BiCl3 (1.0), AuCl3 (1.0) | CCl4 | 15 | 70/—c |
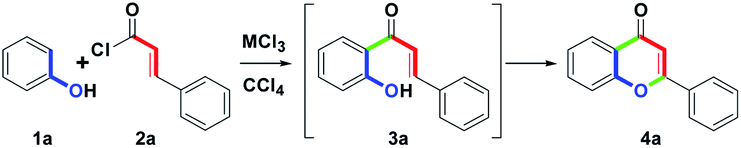
In decreasing the equivalence of BiCl3 from 2.0 to 1.0, no isolation of 4a was observed (entry 9). On the basis of the above phenomenon, we found that excess amounts of BiCl3 could not completely drive the conversion of 3a to 4a. For this reason, another promoter was required to enhance the reaction condition. Hence, we turned the synthetic focus to study the combination of BiCl3 and RuCl3-mediated synthesis of flavones. In entry 10, we found that the combination of BiCl3 (1 equiv.) and RuCl3 (1 equiv.) provided 4a as the major product (68%) along with a 15% of 3a. With these results in hand, we envisioned that RuCl3 could trigger the cyclodehydrogenation step to accomplish the synthesis of 4a. Three solvents having different boiling points, such as MeNO2, (CH2Cl)2, CH2Cl2, and DMF were tested next. Using MeNO2 (entry 11), complex unknown products were detected due to the high boiling temperature (101 °C). Entry 12 shows that (CH2Cl)2 produced a low conversion ratio (2/3) for 3a and 4a. For the low boiling point solvent, CH2Cl2 showed that only 10% of the amounts of 4a were isolated, and 3a was obtained as the major component (78%, entry 13). Among the three chloro-containing solvents, the temperature of boiling CH2Cl2 (40 °C) was low; as a result, the cyclodehydrogenation step could not be induced easier. Changing the solvent to DMF (entry 14), however, only unknown and unidentified complex mixture was isolated. Compared with CH2Cl2, CCl4 and (CH2Cl)2 with higher boiling points (77 °C and 84 °C) could initiate the occurrence of the cyclodehydrogenation step, besides DMF (153 °C). According to the results, CCl4 was the preferred solvent to obtain 4a. To achieve the exhaustive conversion, the reaction time was examined next. Elongating the times to 15 h and 20 h respectively, showed that the transformation from 3a to 4a was complete, and the afforded yields of 4a increased to 81% or 75%, respectively (entries 15 and 16). Even though the reaction time was elongated to 20 h, the yield of 4a could not be enhanced. Remarkably, adjusting the temperature from reflux to room temperature (25 °C), no reactions were observed by the combination of BiCl3 and RuCl3. To increase the yield of 4a, three combinations were examined next. In entry 17, the combination of BiCl3 and CuCl2 provided a 60% yield of 3a along with trace amount of 4a (8%). After changing CuCl2 to FeCl3, 72% yield of 3a and 3% yield of 4a were obtained for the combination of BiCl3 and CuCl2 (entry 18). The two results were similar to entries 4 and 8. Under the combination of BiCl3 and AuCl3 condition (entry 19), only 3a was isolated in a 70% yield, and no desired 4a was detected. Based on the results, we found that CuCl2, FeCl3 and AuCl3 could not trigger the cyclodehydrogenation step easily. From the above screening reaction conditions, we envisioned that the combination of BiCl3 (1.0 equiv.) and RuCl3 (1.0 equiv.) could perform better for the formation of 4a in refluxing CCl4 for 15 h (entry 15). All the conditions were routinely carried out under an atmosphere of air (open-vessel conditions). The heating mantle was used to provide a stable heat source.
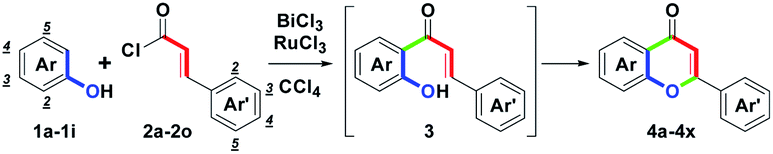
To study the scope and limitations of this one-pot route, substituted phenols 1a–1k and cinnamoyl chlorides 2a–2o were examined further. With optimal conditions established (Table 1, entry 14), we found that the one-pot two-step route could allow a direct synthesis of diversified flavones 4a–4y in moderate to good yields (60–82%), as shown in Table 2, entries 1–26. For the electronic character of different Ar substituents on 1a–1k and Ar′ substituents on 2a–2o, these various substituents included: (1) electron-donating mono-, di- or trioxygenated aryl groups, (2) electron-neutral phenyl, biphenyl or naphthyl groups, (3) electron-withdrawing nitroaryl groups, (4) haloaryl groups and (5) heterocyclic furyl and thienyl groups were highly appropriate. However, when the Ar′ substituent was the 2-furyl group, the by-product 3q was obtained in a 10% yield. Furthermore, with the use of RuCl3, the conversion from 3q to 4q was successful. Therefore, efficient formation of 4a–4y showed that these substituents (Ar and Ar′) did not affect the distribution of the provided yields, besides 4z (Ar = 4-NO2C6H4). The structures of 4a–4y could be determined by 1H NMR analysis. The structure of 4t was determined by single-crystal X-ray analysis.17
|
|||
|---|---|---|---|
| Entry | 1, Ar = | 2, Ar′ = | 4b, (%) |
| a All reactions were run on a 1.0 mmol scale with phenols 1a–1k, cinnamoyl chlorides (2a–2o, 1.0 equiv.), BiCl3 (315 mg, 1.0 equiv.), CCl4 (20 mL), 10 h, reflux (77 °C); then RuCl3·3H2O (261 mg, 1.0 equiv.) was added into the reaction mixture, 5 h, reflux (monitored by TLC).b Isolated yields.c 3q (10%) was obtained.d No detection. | |||
| 1 | 1a, Ph | 2a, Ph | 4a, 81 |
| 2 | 1a, Ph | 2b, 4-MeOC6H4 | 4b, 82 |
| 3 | 1a, Ph | 2c, 3-MeOC6H4 | 4c, 80 |
| 4 | 1a, Ph | 2d, 3,4,5-(MeO)3C6H2 | 4d, 78 |
| 5 | 1a, Ph | 2e, 2,3,4-(MeO)3C6H2 | 4e, 72 |
| 6 | 1a, Ph | 2f, 3,4-CH2O2C6H3 | 4f, 73 |
| 7 | 1a, Ph | 2g, 4-PhC6H4 | 4g, 80 |
| 8 | 1a, Ph | 2h, 3,4-(MeO)2C6H3 | 4h, 72 |
| 9 | 1a, Ph | 2i, 3,4-Cl2C6H3 | 4i, 78 |
| 10 | 1a, Ph | 2a, Ph | 4j, 80 |
| 11 | 1b, 4-MeOC6H4 | 2a, Ph | 4k, 76 |
| 12 | 1c, 3-MeOC6H4 | 2a, Ph | 4l, 68 |
| 13 | 1d, 3-MeO-5-HOC6H3 | 2a, Ph | 4m, 64 |
| 14 | 1e, 3,5-(MeO)2C6H3 | 2j, 4-NO2C6H4 | 4n, 76 |
| 15 | 1a, Ph | 2k, 2-naphthyl | 4o, 70 |
| 16 | 1a, Ph | 2l, 2-FC6H4 | 4p, 67 |
| 17 | 1a, Ph | 2m, 2-furyl | 4q, 60c |
| 18 | 1a, Ph | 2n, 2-thienyl | 4r, 62 |
| 19 | 1a, Ph | 2o, 1-naphthyl | 4s, 75 |
| 20 | 1f, 4-FC6H4 | 2h, 3,4-(MeO)2C6H3 | 4t, 73 |
| 21 | 1g, 4-ClC6H4 | 2h, 3,4-(MeO)2C6H3 | 4u, 72 |
| 22 | 1h, 4-BrC6H4 | 2h, 3,4-(MeO)2C6H3 | 4v, 66 |
| 23 | 1i, 4-MeC6H4 | 2h, 3,4-(MeO)2C6H3 | 4w, 74 |
| 24 | 1c, 3-MeOC6H4 | 2h, 3,4-(MeO)2C6H3 | 4x, 64 |
| 25 | 1j, 3-MeO-2-HOC6H3 | 2a, Ph | 4y, 60 |
| 26 | 1k, 2-NO2C6H4 | 2a, Ph | 4z, —d |
On the basis of the experimental results, a plausible mechanism for the formation of 4 is illustrated in Scheme 3. Initially, BiCl3-mediated complexation of 2 forms A by one bismuth–chloro (Bi–Cl) bond formation. Then, by the removal of ϴBiCl4, B with a styryl acylium center could be generated. Following the Friedel–Crafts ortho-acylation process, B reacts with 1 to lead 3 via one carbon–carbon (C–C) bond formation (green mark).18a Dubac et al. have reported similar reactions. In addition to BiCl3, other bismuth salts-mediated Friedel–Crafts reactions have been well-documented.18b-18d Then, by the involvement of RuCl3, C with a ruthenium(III)-chelated complex was produced. Furthermore, the intramolecular oxa-Michael addition provided D via one carbon-oxygen (C–O) bond formation (green mark). After the proton exchange, E could be formed along with the releasing HCl. Finally, dehydrogenation of E obtained 4 by the removal of RuCl and HCl.19 Under the cyclodehydrogenation step, Laurenczy et al. reported the redox conversion between Ru(III) and Ru(I).19a
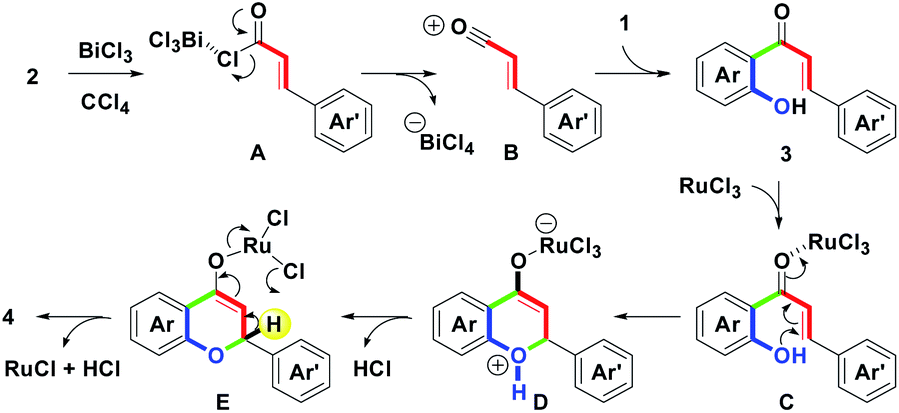 | ||
| Scheme 3 Plausible mechanism. | ||
On the other hand, as an extension of the one-pot two-step synthetic route, changing the combination from BiCl3/RuCl3 to BiCl3/ZnCl2 was examined. However, the flavanone skeleton 5a was isolated at a 90% yield, and no flavone 4a was detected. A possible reason could be that ZnCl2 lacks sufficient oxidative ability to completely promote the cyclodehydrogenation under the redox condition such that it only served a Lewis acid role in promoting the formation of 5a via an intramolecular annulation of 3a. Herein, we also developed the BiCl3/ZnCl2-mediated one-pot two-step, novel route for the synthesis of flavanone skeleton (Scheme 4).
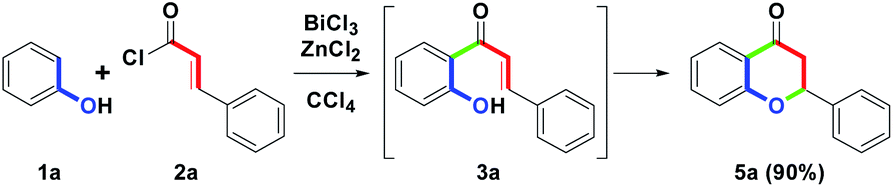 | ||
| Scheme 4 Synthesis of 5a. | ||
With the results in mind, cinnamoyl chlorides 2a–2o were adjusted next to crotonoyl chloride (2p) or acryloyl chloride (2q) under one-pot condition (Scheme 5). As shown in eqn (1), the BiCl3/RuCl3 mediated reaction of 1a with 2p provided 6a with the chromen-4-one skeleton in only a 31% yield. Compared with 4a (81%), the isolated yield of 6a (31%) was low. The possible reason could be that with the hydrogen on E (yellow, Scheme 3), the low acidity was not easy to abstract by the RuCl3 for the dehydrogenation step to be triggered efficiently. On the basis of the results, we demonstrated that benzylic hydrogen was acidic and easily eliminated. Furthermore, by the removal of the β-methyl group on 2, BiCl3/RuCl3 mediated reaction of 1a with 2q was shown in eqn (2). In particular, only 7a was generated in a 67% yield. However, an intramolecular dehydrogenation step was not initiated so that the predicted 7b was not obtained. The resulting phenomenon meant that cyclodehydrogenation only occurred in cinnamoyl substituents (with benzylic hydrogen). Although substrate 2 was limited to the cinnamoyl substituents, it still provided a novel one-pot synthesis of the flavone skeleton.
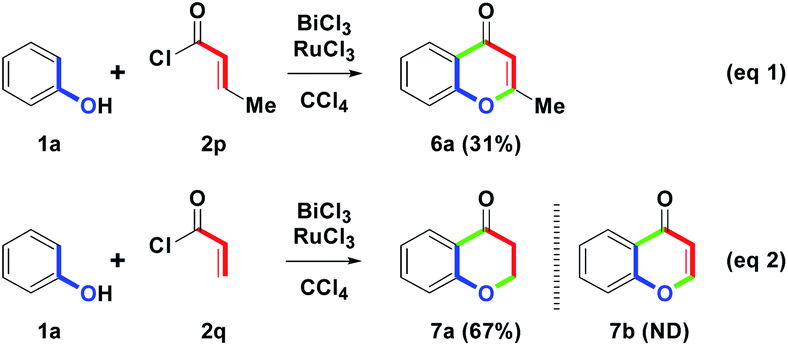 | ||
| Scheme 5 Synthesis of 6a and 7a. | ||
Conclusion
In summary, we have developed a concise route for the effective synthesis of functionalized flavones via BiCl3/RuCl3 mediating the one-pot, direct intermolecular ortho-acylation of substituted phenols with cinnamoyl chlorides followed by intramolecular cyclodehydrogenation of the resulting o-hydroxychalcones under refluxing CCl4 reaction conditions. The process provides a cascade pathway of one carbon–oxygen and one carbon–carbon bond formation. Related plausible mechanisms have been proposed. Further studies regarding the efficient synthetic routes towards flavones will be conducted and published in due course.Experimental
General
All reagents and solvents were obtained from commercial sources and used without further purification. Reactions were routinely carried out under an atmosphere of air with magnetic stirring. Products in organic solvents were dried with anhydrous magnesium sulfate before concentration in vacuo. Melting points were determined with a SMP3 melting apparatus. 1H and 13C NMR spectra were recorded on a Varian INOVA-400 spectrometer operating at 400 and at 100 MHz, respectively. Chemical shifts (δ) are reported in parts per million (ppm) and the coupling constants (J) are given in Hertz. High resolution mass spectra (HRMS) were measured with a mass spectrometer Finnigan/Thermo Quest MAT 95XL. X-ray crystal structures were obtained with an Enraf-Nonius FR-590 diffractometer (CAD4, Kappa CCD).A representative synthetic procedure of compounds 4a–4y and 3q is as follows
BiCl3 (315 mg, 1.0 mmol) was added to a solution of phenols 1a–1k (1.0 mmol) in CCl4 (20 mL) at 25 °C. The reaction mixture was stirred at 25 °C for 10 min. Cinnamoyl chlorides 2a–2o (1.0 mmol) was added to the reaction mixture at 25 °C. The reaction mixture was stirred at reflux for 10 h (monitored by TLC). Then, RuCl3·3H2O (261 mg, 1.0 mmol) was added to the reaction mixture (containing o-hydroxychalcones 3) at reflux. The reaction mixture was stirred at reflux for 5 h (monitored by TLC). The reaction mixture was cooled to 25 °C and the solvent was concentrated. The residue was diluted with water (10 mL) and the mixture was extracted with CH2Cl2 (3 × 30 mL). The combined organic layers were washed with brine (2 × 20 mL), dried (MgSO4), filtered and evaporated to afford crude product under reduced pressure. Purification on silica gel (hexanes/EtOAc = 20/1–4/1) afforded 4a–4y and 3q.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank the Ministry of Science and Technology of the Republic of China (Taiwan) for its financial support (MOST 109-2113-M-037-014-MY3).Notes and references
- Recent leading review articles with synthesis, see: (a) R. Kshatriya, V. P. Jejurkar and S. Saha, Tetrahedron, 2018, 74, 811–833 CrossRef CAS; (b) C. M. M. Snatos and A. M. S. Silva, Eur. J. Org. Chem., 2017, 2017, 3115–3133 CrossRef; (c) For recent review articles with the biological activities, see: M. Singh, M. Kaur and O. Silakari, Eur. J. Med. Chem., 2014, 84, 206–239 CrossRef CAS PubMed; (d) J. Reis, A. Gaspar, N. Milhazes and F. Borges, J. Med. Chem., 2017, 60, 7941–7957 CrossRef CAS PubMed.
- (a) W. Baker, J. Chem. Soc., 1933, 1381–1389 RSC; (b) D. C. Bhalla, H. S. Mahal and K. Venkataraman, J. Chem. Soc., 1934, 1933–1935 Search PubMed.
- K. V. Auwers and K. Muller, Ber. Dtsch. Chem. Ges., 1908, 41, 4233–4241 CrossRef.
- (a) A. J. Flynn, Proc. R. Ir. Acad., Sect. B, 1934, 42, 1–4 Search PubMed; (b) T. J. Oyamada, Bull. Chem. Soc. Jpn., 1935, 10, 182–186 CrossRef CAS.
- J. Allan and R. Robinson, J. Chem. Soc., Trans., 1924, 125, 2192–2195 RSC.
- (a) S. V. Kostanecki and T. Emilevickz, Eur. J. Inorg. Chem., 1898, 31, 696–705 Search PubMed; (b) S. V. Kostanecki and J. Thamobor, Eur. J. Inorg. Chem., 1895, 28, 2302–2309 Search PubMed.
- (a) C. Mentzer, W. Molho and P. Vercier, Compt. Rend., 1951, 232, 1488–1490 CAS; (b) C. Mentzer and P. Vercier, Compt. Rents., 1952, 232, 1674–1678 Search PubMed.
- (a) P. Kumar and M. S. Bodas, Org. Lett., 2000, 2, 3821–3823 CrossRef CAS PubMed; (b) G. Bose, E. Mondal, A. T. Khan and M. J. Bordoloi, Tetrahedron Lett., 2001, 42, 8907–8909 CrossRef CAS; (c) F. Lassagne and F. Pochat, Tetrahedron Lett., 2003, 44, 9283–9285 CrossRef CAS.
- M. A. Selepe and F. R. van Heerden, Molecules, 2013, 18, 4739–4765 CrossRef CAS PubMed.
- (a) E. Awuah and A. Capretta, Org. Lett., 2009, 11, 3210–3213 CrossRef CAS PubMed; (b) H. Miao and Z. Yang, Org. Lett., 2000, 2, 1765–1768 CrossRef CAS PubMed.
- J. A. Seijas, M. P. Vázquez-Tato and R. Carballido-Reboredo, J. Org. Chem., 2005, 70, 2855–2858 CrossRef CAS PubMed.
- Z. Du, H. Ng, K. Zhang, H. Zeng and J. Wang, Org. Biomol. Chem., 2011, 9, 6930–6935 RSC.
- J. Das and S. Ghosh, Tetrahedron Lett., 2011, 52, 7189–7194 CrossRef CAS.
- (a) Transition metal-mediated ortho-acylation of phenols, for Cu(II), see: J. Hu, E. A. Adogla, Y. Ju, D. Fan and Q. Wang, Chem. Commun., 2012, 48, 11256–11258 RSC; (b) For Fe(III), see: H. Naeimi and L. Moradi, Russ. J. Org. Chem., 2007, 43, 1757–1759 CrossRef CAS; (c) For Ti(IV), see: A. Bensari and N. T. Zaveri, Synthesis, 2003, 267–271 CAS; (d) For Hg lamp, see: F. Galindo, M. C. Jimenez, M. A. Miranda and R. Tormos, J. Photochem. Photobiol., A, 1997, 97, 151–153 CrossRef; (e) For microwave, see: H. Naeimi, A. Raeisi and M. Moradian, Arabian J. Chem., 2017, 10, S2723–S2728 CrossRef CAS.
- (a) Transition metal-mediated cyclodehydrogenation methods, for InCl3/SiO2, see: N. Ahmed, H. Ali and J. E. van Lier, Tetrahedron Lett., 2005, 46, 253–256 CrossRef CASand cited references therein (b) For FeCl3-6H2O/MeOH, see: K. H. Kumar and P. T. Perumal, Tetrahedron, 2007, 63, 9531–9535 CrossRef CAS; (c) For CuI/ionic liquids, see: Z. Du, H. Ng, K. Zhang, H. Zeng and J. Wang, Org. Biomol. Chem., 2011, 9, 6930–6933 RSC.
- (a) Metal-free-mediated cyclodehydrogenation methods, for I2/DMSO, see: M. D. L. de la Torre, G. L. Marcorin, G. Pirri, A. C. Tome, A. M. S. Silva and J. A. S. Cavaleira, Tetrahedron Lett., 2002, 43, 1689–1691 CrossRef CAS; (b) For DDQ/dioxane, see: K. Imafuku, M. Honda and J. F. W. McOmie, Synthesis, 1987, 199–201 CrossRef CAS; (c) For NaIO4/DMSO, see: N. Hans and S. K. Grover, Synth. Commun., 1993, 23, 1021–1023 CrossRef CAS; (d) For H2O2/NaOH, see: S. Gobbi, A. Rampa, A. Bisi, F. Belluti, L. Piazzi, P. Valent, A. Caputo, A. Zampiron and M. Carrara, J. Med. Chem., 2003, 46, 3662–3669 CrossRef CAS PubMed; (e) For SeO2/dioxane, see: M. E. Zwaagstra, H. Timmerman, A. C. van de Stolpe, F. J. J. de Kanter, M. Tamura, Y. Wada and M.-Q. Zhang, J. Med. Chem., 1998, 41, 1428–1438 CrossRef CAS; (f) For Br2/NaOH, see: J. R. Pfister, W. E. Wymann, M. E. Schuler and A. P. Roszkowski, J. Med. Chem., 1980, 23, 335–338 CrossRef CAS PubMed.
- CCDC 1957347 (4t) contains the supplementary crystallographic data for this paper.†.
- (a) BiCl3-mediated Friedel-Crafts acylations, see: S. Repichet, C. Le Roux, N. Roques and J. Dubac, Tetrahedron Lett., 2003, 44, 2037–2040 CrossRef CAS; (b) For recent reviews on bismuth salts-mediated reactions, see: P. Ondet, G. Lemiere and E. Dunach, Eur. J. Org. Chem., 2017, 2017, 761–780 CrossRef CAS; (c) B. Abnerjee, ChemistrySelect, 2017, 2, 6744–6757 CrossRef; (d) A. Gagnon, J. Dansereau and A. Le Roch, Synthesis, 2017, 49, 1707–1745 CrossRef CAS.
- (a) RuCl3-mediated dehydrogenations, see: V. Henricks, I. Yuranov, N. Autissier and G. Laurenczy, Catalysts, 2017, 7, 348–355 CrossRef; (b) Y. Kim, S. Ahn, J. Y. Hwang, D.-H. Ko and K.-Y. Kwon, Catalysts, 2017, 7, 7–17 CrossRef . For review on ruthenium salts-mediated reactions, see:; (c) M. Pagliao, S. Campestrini and R. Ciriminna, Chem. Soc. Rev., 2005, 34, 837–845 RSC.
- For 4a and 4s, see: M. Golshani, M. Khoobi, N. Jalalimanesh, F. Jafarpour and A. Ariafard, Chem. Commun., 2017, 53, 10676–10679 RSC.
- For 4b and 4n, see: R. Charugandla, R. S. Vangala, S. Chidara and R. B. Korupolu, Tetrahedron Lett., 2018, 59, 3283–3287 CrossRef CAS.
- For 4c, 4h, 4j, 4k, 4m, see: J. Lee, J. Yu, S. H. Son, J. Heo, T. Kim, J.-Y. An, K.-S. Inn and N.-J. Kim, Org. Biomol. Chem., 2016, 14, 777–784 RSC.
- For 4d, see: P. S. Kulkarni, D. D. Kondhare, R. Varala and P. Zubaidha, J. Serb. Chem. Soc., 2013, 78, 909–916 CrossRef CAS.
- For 4f, see: G. Priyadarshani, S. Amrutkar, A. Nayak, U. C. Banerjee, C. N. Kundu and S. K. Guchhait, Eur. J. Med. Chem., 2016, 122, 43–54 CrossRef CAS PubMed.
- For 4g, see: T. Rodrigues, A. S. Ressurreicao, F. P. da Cruz, I. S. Albuquerque, J. Gut, M. P. Carrasco, D. Goncalves, R. C. Guedes, D. J. V. A. dos Santos, M. M. M. Philip, R. Moreira, M. Prudencio and F. Lopes, Eur. J. Med. Chem., 2013, 69, 872–880 CrossRef CAS PubMed.
- For 4i, see: V. K. Rai, F. Verma, G. P. Sahu, M. Singh and A. Rai, Eur. J. Org. Chem., 2018, 48, 537–544 CrossRef.
- For 4l, see: S.-H. Wang, C.-H. Chen, C.-Y. Lo, J.-Z. Feng, H.-J. Lin, P.-Y. Chang, L.-L. Yang, L.-G. Chen, Y.-W. Liu, C.-D. Kuo and J.-Y. Wu, MedChemComm, 2015, 6, 1864–1873 RSC.
- For 4o, see: M. Pérez, D. Ruiz, J. Autino, A. Sathicq and G. Romanelli, Compt. Rendus Chem., 2016, 19, 551–555 CrossRef.
- For 4p, see: J. J. Ares, P. E. Outt, S. V. Kakodkar, R. C. Buss and J. C. Geiger, J. Org. Chem., 1993, 58, 7903–7905 CrossRef CAS.
- For 4q and 4r, see: D. Banerjee, U. Kayal and G. Maiti, Tetrahedron Lett., 2016, 57, 1667–1671 CrossRef CAS.
- For 4u, see: J. Zhao, Y. Zhao and H. Fu, Angew. Chem., Int. Ed., 2011, 50, 3769–3773 CrossRef CAS PubMed.
- For 4v, see: T.-L. Shih, C.-E. Chou, W.-Y. Liao and C.-A. Hsiao, Tetrahedron, 2014, 70, 3657–3664 CrossRef CAS.
- For 4w, see: B. R. Nawghare, S. V. Gaikwad, A. Raheem and P. D. Lokhande, J. Chil. Chem. Soc., 2014, 59, 2284–2286 CrossRef CAS.
- For 4x, see: M. Yoshida, Y. Fujino and T. Doi, Org. Lett., 2011, 13, 4526–4529 CrossRef CAS PubMed.
- For 5a, see: M. Shaikh, K. K. Atyam, M. Sahu and K. V. S. Ranganath, Chem. Commun., 2017, 53, 6029–6032 RSC.
- For 6a, see: C. Proençaa, H. M. T. Albuquerqueb, D. Ribeiroa, M. Freitasa, C. M. M. Santosb, A. M. S. Silvab and E. Fernandes, Eur. J. Med. Chem., 2016, 122, 381–392 CrossRef PubMed.
- For 7a, see: W. Li and X.-F. Wu, Eur. J. Org. Chem., 2015, 2015, 331–335 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Scanned photocopies of NMR spectral data for all compounds and X-ray analysis data of 4t were supported. CCDC 1957347. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00534k |
| This journal is © The Royal Society of Chemistry 2021 |