
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluorescent calix[4]arene with naphthalene units at the upper rim exhibits long fluorescence emission lifetime without fluorescence quenching†
Masaki
Takahashi
 *a,
Naoya
Tsuji
a,
Kohei
Yazaki
a,
Yoshihisa
Sei
b and
Makoto
Obata
a
*a,
Naoya
Tsuji
a,
Kohei
Yazaki
a,
Yoshihisa
Sei
b and
Makoto
Obata
a
aInterdisciplinary Graduate School of Medicine and Engineering, University of Yamanashi, 4-4-37 Takeda, Kofu, 400-8510, Japan. E-mail: tmasaki@yamanashi.ac.jp
bLaboratory for Chemistry and Life Science, Institute of Innovative Research, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan
First published on 22nd March 2021
Abstract
We synthesised a new compound with four naphthyl groups in the upper rims of calix[4]arene (1). Compared to the monomer unit, compound 1 has redshifted absorption and fluorescence, together with high fluorescence quantum yield and long fluorescence lifetime, which is extremely rare because long fluorescence lifetime emission tends to reduce the quantum yield. Single-crystal X-ray analysis and quantum calculations in the S1 state revealed π–π through-space interactions between naphthalene rings.
In 1954, Förster and Kasper first reported dimerised aromatic compounds in the excited state,1 which were termed ‘excimers’ by Stevens and Hutton.2 Excimers have attracted much attention in various fields such as organic solar cells,3–5 organic electronics,6 chemical sensors,7 and biotechnology8–12 because of their unique photophysical properties. In particular, with the development of time-resolved fluorescence imaging and stimulated emission depletion microscopy (STED) microscopes, there is a growing demand for fluorescent dyes with both high brightness and long fluorescence lifetime.9–12 Excimers with long emission lifetime are promising candidates for next-generation imaging probes.13
Many progresses have been made towards understanding the relationship between the molecular structure of organic dyes and their fluorescence intensity. However, there is little knowledge on the relationship between the fluorescence lifetime and molecular structure. Also, only a few fluorescent probes have achieved both high intensity and long lifetime.13–15 In recent years, it was reported that bright and long-lived fluorescence can be obtained from the excimer state13,16–19 that was conventionally thought to be the cause of quenching due to the low-energy excimer trap states with forbidden radiative transition and activated non-radiative process.20–24 However, most of these reports were in the solid state, where the molecular movement is suppressed and the luminescence occurs in a single crystal.16–19 In contrast, there are few reports of dyes with long fluorescent lifetime in solution systems with free molecular movement. The reported substances also suffer from low synthesis yields in the ring-forming reaction and difficulty in introducing functional groups (such as hydrophilic substituents).13 In general, the yield of the ring formation step is extremely low in the synthesis of cyclophanes.13,25–27 Therefore, there remains the need for new molecules that can be easily synthesised and chemically modified.
In this study, we adopted the calixarene skeleton as the macrocyclic structure. Calixarenes have been used in supramolecular chemistry,28 analytical chemistry,29 biochemistry,30 material science,31 and catalysts32 because of their easy molecular modification. Nevertheless, almost all studies introducing fluorescence sites do so at the lower rim of calixarene, while few reports considered incorporating fluorophores at the upper rim. Further, no researchers have investigated the fluorescence lifetime, and there was also no reported computational investigation of the excited state.33–35
Specifically, we synthesised a fluorescent molecule in which naphthyl group was introduced into the upper rim of calix[4]arene. The new molecule showed both a long fluorescence wavelength and a high quantum yield. After measuring the conformation of tetranaphthylcalix[4]arene in a single crystal, structural optimisation of the ground state and excited state (S1) was performed by time-dependent density functional theory (TD-DFT) at the DFT-D3-CAM-B3LYP/6-31G(d) level. The calculation takes into account effects such as dispersion force.36–39 Molecular orbital calculation confirmed that the π orbitals of the naphthalene rings of tetranaphthylcalixarene have a binding interactions in LUMO of the S1 state.
The synthesis of tetranaphthylcalix[4]arene (1) was carried out by deprotecting the tert-Bu groups of tetra-tert-butyl(tetrahydroxy)calix[4]arene, followed by introduction of substituents by Williamson ether synthesis of phenolic hydroxyl groups, and iodation by silver tetrafluoroacetate and iodine. The iodination was followed by Suzuki–Miyaura cross-coupling reaction with naphthylboronic acid pinacol ether (ESI†).
The target substance was characterised by 1H NMR, 13C NMR, HRMS of ESI-TOF-MS, and single-crystal X-ray diffraction (Fig. S7–S9 and Table S3, ESI†).
We also synthesised the phenylnaphthalene derivative 2 (Fig. 1), which is the unit molecule of 1. To compare its photophysical properties with that of 1, first we measured the absorption spectra. At 1 × 10−4 mol L−1 in chloroform solution, 1 and 2 have their maximum absorption wavelengths at 297 and 294 nm, and the molar absorption coefficients were 3.3 × 105 and 1.0 × 105 mol−1 L cm−1, respectively (Fig. 2). Their fluorescence spectra were measured in chloroform solution at 1 × 10−4 mol L−1 (Fig. 3). 1 has a broader fluorescence peak with maximum intensity at 389 nm, which is redshifted by 25 nm from that of 2. Furthermore, the fluorescence spectrum of the powder after grinding in a mortar was also measured and it was found that the fluorescence wavelength was increased by 22 nm (Fig. S14, ESI†). We also measured the absorption and fluorescence spectra at a lower concentration of 1 × 10−5 mol L−1 (Fig. S15, ESI†), and there was almost no change in the wavelength or shape of the peak. Therefore, the spectral changes in tetranaphthylcalix[4]arene from the unit model molecule are due to intramolecular rather than intermolecular interactions. Furthermore, the temperature dependence of fluorescence was investigated by measuring fluorescence by changing the temperature from 20 °C to 80 °C (Fig. S16†). As the temperature rose from 20 °C to 80 °C, a blue shift of the fluorescence wavelength of about 10 nm was observed, and the half-value width of the peak narrowed. These results indicate that as the temperature rises, the intramolecular interaction in the excited state weakens and the light emission becomes closer to that of 2.
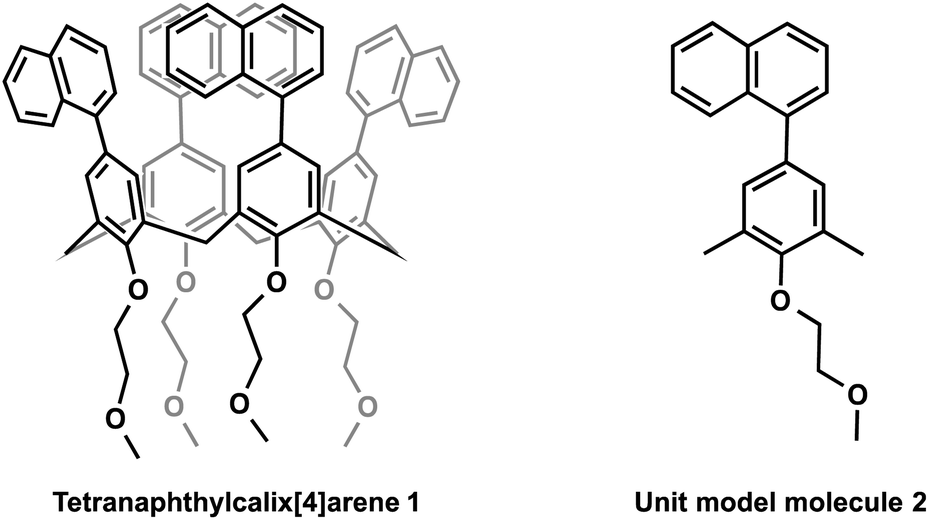 | ||
| Fig. 1 Molecular structures of tetranaphthylcalix[4]arene 1 and the unit model molecule 2. | ||
 | ||
| Fig. 2 Absorption spectra of tetranaphthylcalix[4]arene 1 and the unit model molecule 2. Both spectra were measured in chloroform at 1 × 10−4 mol L−1, and normalised to the maximum absorption in the longer wavelength region. | ||
 | ||
| Fig. 3 Fluorescence spectra of 1 and 2. Both spectra were measured in chloroform solution at 1 × 10−4 mol L−1, and normalised by the maximum intensity. 1: λex = 310.5 nm, 2: λex = 326.5 nm. | ||
Single-crystal X-ray crystal structure analysis of 1 revealed that two of the four naphthyl groups facing each other had an intramolecular stacking structure, with a distance of 3.54 Å between them (Fig. 4 and Table S3, ESI†).
 | ||
| Fig. 4 (a) Side and (b) top views of the structure of 1 determined by single-crystal X-ray crystallographic analysis. The structures were drawn by ORTEP program with the thermal ellipsoids set at 50% probability. | ||
The macrocyclic structure improved the fluorescence quantum yield from 0.38 in 2 to 0.46 in 1 (Table 1). The unit model molecule showed single-exponential fluorescence decay with a fluorescence lifetime of 2.0 ns. In contrast, 1 displayed non-single exponential fluorescence decay. When the data were fitted using a two-component exponential equation (Fig. S17† and Table 1), the fitted weight ratio and fluorescence lifetime were τ1 = 2.4 ns, A1 = 0.95, τ2 = 48.0 ns, and A2 = 0.05. The area-weighted fluorescence lifetime 〈τ〉 was 26 ns. These lifetimes are very long compared to current commercial dyes with long fluorescence lifetimes for STED microscopes and time-gate imaging: Alexa Fluor 488 (4.1 ns),11,40 azadioxatriangulenium (ADOTA+) (25 ns),14,41 and SeTau425 NHS (26.2 ns).15 To investigate this characteristic property, the fluorescence emission rate constant kf and the nonradiative decay rate constant knr were determined by the following equation:
 | (1) |
| Cmpd | Φ f a | τ 1 b [ns] | τ 2 b [ns] | 〈τ〉c [ns] | k f [ns−1] | k nr [ns−1] |
|---|---|---|---|---|---|---|
| a Absolute fluorescence quantum yields. b The area-weighted ratios (A1 and A2) are shown in parentheses. c The area-weighted mean fluorescence lifetime was calculated according to the function in ESI. | ||||||
| 1 | 0.46 | 2.4 (95%) | 48.0 (5%) | 26 | 0.018 | 0.021 |
| 2 | 0.38 | 2.0 | — | — | 0.19 | 0.31 |
 | ||
| Fig. 5 Top views of (a) the molecular structure of 1 in S1 excited state and with (b) superimposed LUMO. The S1 geometry was optimised by TD-DFT calculation. | ||
In summary, we synthesised tetranaphthylcalix[4]arene (1). Such a structure with naphthalene substituents on the upper rim has never been reported before. A unit model molecule of 1 was also synthesised. For the optical properties, it was found that 1 has a higher fluorescence quantum yield and a fluorescence lifetime at least 11 times longer than that of the unit model molecule. These differences were interpreted based on single-crystal X-ray structure analysis and molecular orbital calculations, which revealed binding interactions between the naphthalene rings in the S1 state. These results will provide new insights into the molecular design of dyes with high fluorescence quantum yields and long fluorescence lifetimes.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. K. Yamamoto, Dr T. Kambe, and Dr K. Takada of Tokyo Institute of Technology for PL quantum yield measurements (Hamamatsu Photonics C9920-02) and ESI-TOF-MS (Bruker micrOTOF II) support. This study was financially supported in part by the Japan Society for the Promotion of Science KAKENHI (Grants-in-Aid for Scientific Research, No. JP20K15243), the Izumi Science and Technology Foundation, and the Cooperative Research Program of the “Network Joint Research Center for Materials and Devices”.References
- T. Förster and K. Kasper, Z. Physiol. Chem., 1954, 1, 275–277 CrossRef
.
- B. Stevens and E. Hutton, Nature, 1960, 186, 1045–1046 CrossRef CAS
- B. J. Walker, A. J. Musser, D. Beljonne and R. H. Friend, Nat. Chem., 2013, 5, 1019 CrossRef CAS PubMed
- S. W. Eaton, L. E. Shoer, S. D. Karlen, S. M. Dyar, E. A. Margulies, B. S. Veldkamp, C. Ramanan, D. A. Hartzler, S. Savikhin and T. J. Marks, J. Am. Chem. Soc., 2013, 135, 14701–14712 CrossRef CAS PubMed
- C. M. Mauck, P. E. Hartnett, E. A. Margulies, L. Ma, C. E. Miller, G. C. Schatz, T. J. Marks and M. R. Wasielewski, J. Am. Chem. Soc., 2016, 138, 11749–11761 CrossRef CAS PubMed
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CrossRef CAS PubMed
- S. Karuppannan and J. C. Chambron, Chem.–Asian J., 2011, 6, 964–984 CrossRef CAS PubMed
- M. E. Østergaard and P. J. Hrdlicka, Chem. Soc. Rev., 2011, 40, 5771–5788 RSC
- M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed
- Y. Kodama, PLoS One, 2016, 11, e0152484 CrossRef PubMed
- G. Vicidomini, G. Moneron, K. Y. Han, V. Westphal, H. Ta, M. Reuss, J. Engelhardt, C. Eggeling and S. W. Hell, Nat. Methods, 2011, 8, 571 CrossRef CAS PubMed
- Y. Xie, M. C. Arno, J. T. Husband, M. Torrent-Sucarrat and R. K. O'Reilly, Nat. Commun., 2020, 11, 2460 CrossRef CAS PubMed
- H. Osaki, C. M. Chou, M. Taki, K. Welke, D. Yokogawa, S. Irle, Y. Sato, T. Higashiyama, S. Saito and A. Fukazawa, Angew. Chem., Int. Ed., 2016, 55, 7131–7135 CrossRef CAS PubMed
- R. M. Rich, D. L. Stankowska, B. P. Maliwal, T. J. Sørensen, B. W. Laursen, R. R. Krishnamoorthy, Z. Gryczynski, J. Borejdo, I. Gryczynski and R. Fudala, Anal. Bioanal. Chem., 2013, 405, 2065–2075 CrossRef CAS PubMed
- E. Garcia-Fernandez, M. C. Gonzalez-Garcia, S. Pernagallo, M. J. Ruedas-Rama, M. A. Fara, F. J. López-Delgado, J. W. Dear, H. Ilyine, C. Ress and J. J. Díaz-Mochón, Chem. Commun., 2019, 55, 14958–14961 RSC
- S. Sekiguchi, K. Kondo, Y. Sei, M. Akita and M. Yoshizawa, Angew. Chem., Int. Ed., 2016, 55, 6906–6910 CrossRef CAS PubMed
- H. Liu, L. Yao, B. Li, X. Chen, Y. Gao, S. Zhang, W. Li, P. Lu, B. Yang and Y. Ma, Chem. Commun., 2016, 52, 7356–7359 RSC
- Y. Liu, X. Tao, F. Wang, J. Shi, J. Sun, W. Yu, Y. Ren, D. Zou and M. Jiang, J. Phys. Chem. C, 2007, 111, 6544–6549 CrossRef CAS
- Y. Liu, X. Tao, F. Wang, X. Dang, D. Zou, Y. Ren and M. Jiang, J. Phys. Chem. C, 2008, 112, 3975–3981 CrossRef CAS
- J. Cornil, D. Dos Santos, X. Crispin, R. Silbey and J. Brédas, J. Am. Chem. Soc., 1998, 120, 1289–1299 CrossRef CAS
- G. C. Bazan, W. J. Oldham, R. J. Lachicotte, S. Tretiak, V. Chernyak and S. Mukamel, J. Am. Chem. Soc., 1998, 120, 9188–9204 CrossRef CAS
- R. J. Lindquist, K. M. Lefler, K. E. Brown, S. M. Dyar, E. A. Margulies, R. M. Young and M. R. Wasielewski, J. Am. Chem. Soc., 2014, 136, 14912–14923 CrossRef CAS PubMed
- S. P. Jagtap, S. Mukhopadhyay, V. Coropceanu, G. L. Brizius, J.-L. Brédas and D. M. Collard, J. Am. Chem. Soc., 2012, 134, 7176–7185 CrossRef CAS PubMed
- E. A. Chandross and C. J. Dempster, J. Am. Chem. Soc., 1970, 92, 3586–3593 CrossRef CAS
- Y. Yu, Y. Li, S. Chen, T. Liu, Z. Qin, H. Liu and Y. Li, Eur. J. Org. Chem., 2012, 4287–4292 CrossRef CAS
- H. Langhals and R. Ismael, Eur. J. Org. Chem., 1998, 1915–1917 CrossRef CAS
- P. Spenst and F. Würthner, Angew. Chem., 2015, 127, 10303–10306 CrossRef
- S. Shinkai, Tetrahedron, 1993, 49, 8933–8968 CrossRef CAS
- R. Ludwig, Fresenius. J. Anal. Chem., 2000, 367, 103–128 CrossRef CAS PubMed
- S. B. Nimse and T. Kim, Chem. Soc. Rev., 2013, 42, 366–386 RSC
- K. L. Chan, J. P. F. Lim, X. Yang, A. Dodabalapur, G. E. Jabbour and A. Sellinger, Chem. Commun., 2012, 48, 5106–5108 RSC
- D. M. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS PubMed
- J. S. Kim and D. T. Quang, Chem. Rev., 2007, 107, 3780–3799 CrossRef CAS PubMed
- E. Botana, E. Da Silva, J. Benet-Buchholz, P. Ballester and J. de Mendoza, Angew. Chem., Int. Ed., 2007, 46, 198–201 CrossRef CAS PubMed
- P. K. Lo and M. S. Wong, Sensors, 2008, 8, 5313–5335 CrossRef CAS PubMed
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- E. Wegel, A. Göhler, B. C. Lagerholm, A. Wainman, S. Uphoff, R. Kaufmann and I. M. Dobbie, Sci. Rep., 2016, 6, 1–13 CrossRef PubMed
- T. J. Sørensen, E. Thyrhaug, M. Szabelski, R. Luchowski, I. Gryczynski, Z. Gryczynski and B. W. Laursen, Methods Appl. Fluoresc., 2013, 1, 025001 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2058720. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra01743h |
| This journal is © The Royal Society of Chemistry 2021 |