DOI:
10.1039/D1RA00396H
(Paper)
RSC Adv., 2021,
11, 10497-10511
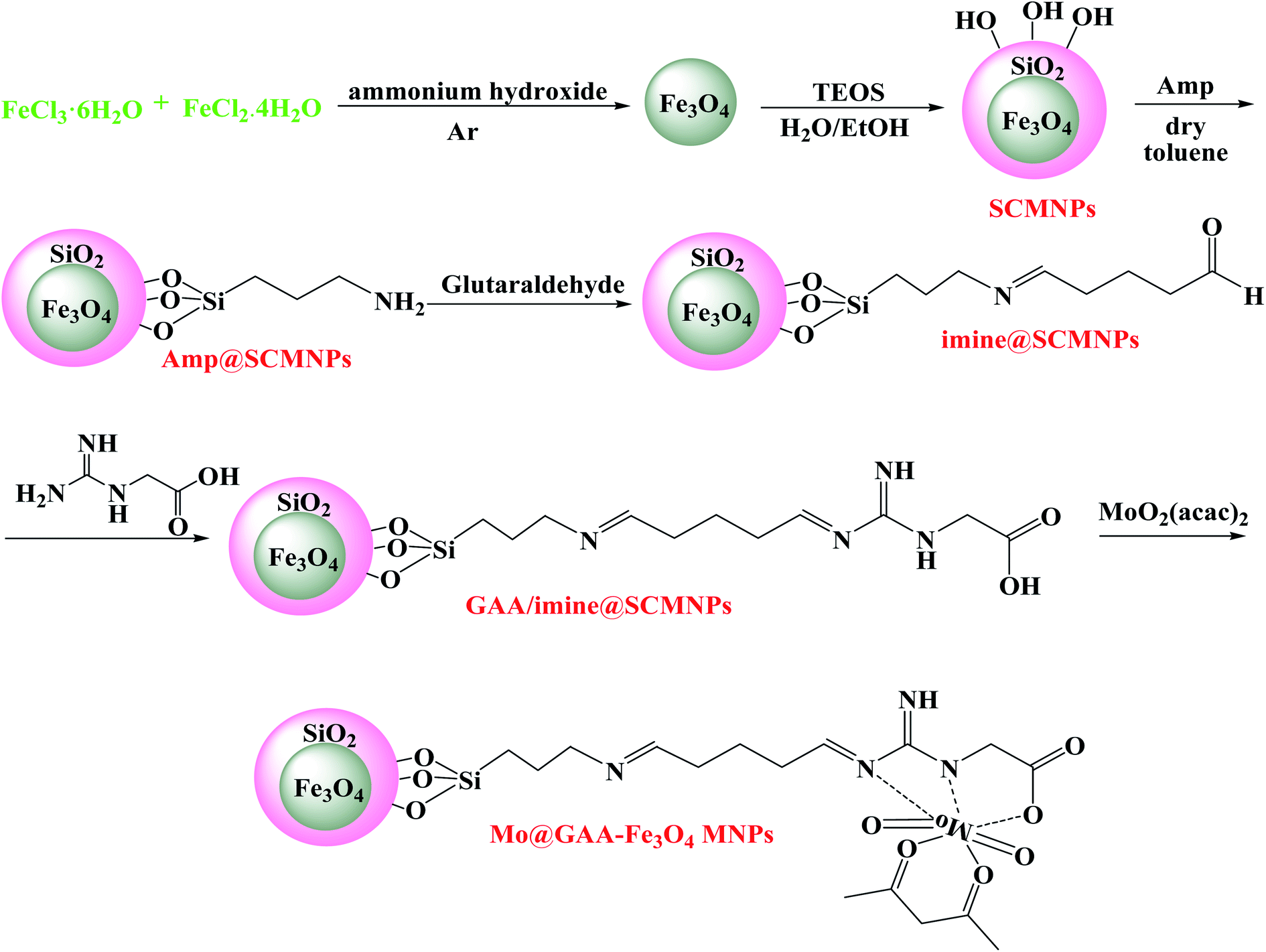
Mo@GAA-Fe3O4 MNPs: a highly efficient and environmentally friendly heterogeneous magnetic nanocatalyst for the synthesis of polyhydroquinoline derivatives
Received
16th January 2021
, Accepted 3rd March 2021
First published on 11th March 2021
Abstract
Polyhydroquinolines were efficiently obtained from a sequential four-component reaction between dimedone or 1,3-cyclohexandione, ethyl acetoacetate, or methyl acetoacetate as a β-ketoester, aldehydes, and ammonium acetate, under the catalysis of Mo@GAA-Fe3O4 MNPs as a green, effective, recyclable, and environmentally friendly nanocatalyst. Due to its magnetic nature the prepared catalyst can be easily separated from the reaction mixture by an external magnet and reused several times without significant changes in catalytic activity and reaction efficiency. The catalyst was characterized using energy dispersive X-ray spectroscopy (EDX), X-ray diffraction (XRD), thermogravimetric analysis (TGA), Fourier transform infrared spectroscopy (FTIR), vibrating sample magnetometry (VSM), scanning electron microscopy (SEM), and transmission electron microscopy (TEM).
1. Introduction
In recent decades, the preparation of environmentally friendly catalysts with recoverability and reusability are the main challenges amongst researchers in organic chemistry. Chemical reactions can be accelerated in the presence of homogeneous or heterogeneous catalysts, each of which has its advantages and disadvantages. Homogeneous catalysts, due to the greater interaction with the substrates compared to their heterogeneous counterparts, can generate a uniform layer with the starting materials in the organic solvent of the reaction which increases their activity and selectivity during the corresponding reaction.1–6 However, the main problem when using these catalysts is related to the difficulty in separating and reusing them, which can be largely solved by using heterogeneous types. Heterogeneous catalysts, despite their lower efficiency, can exhibit suitable recyclability by centrifugation or filtration and can be reused several times without a significant decrease in their catalytic behavior. Functionalization of the heterogeneous supports via a strong connection with homogeneous catalysts increase their efficiency while maintaining their simplicity in recovery and separation.7
Nanocatalysts exhibit high catalytic activity, which is provided by their large surface area, and this significantly increases the contact between reactants and catalyst.8,9 Nowadays, remarkable attention has been paid to the use of magnetic nanoparticles (MNPs), as a bridge between homogeneous and heterogeneous catalysts, by scientific and industrial researchers because of their potential importance in modern chemical research.10 Some excellent physical and chemical features of MNPs such as separation by an external magnet, superparamagnetism, high surface area, and strong adsorption ability11 have expanded their utilization in the removal of heavy metal ions in wastewater,12–14 catalysis,15 and drug delivery.16 However, some of the MNPs features such as a tendency to oxidize and accumulate during reactions, high initial chemical activity, and high surface area to volume ratio will reduce their catalytic activity and magnetic nature during the reaction period.17,18 Thus, to overcome this drawback and increase their chemical stability for the special application, functionalization and modification of their surface with organic or inorganic supports are necessary.
Molybdenum as a d-block transition metal has various oxidation states from –I to +VI and it has a significant role in the life evolution. Despite the little amount of molybdenum on the earth's crust, it could be known as an oxotransferases for the active places and cofactors of several enzymes that catalyzes the oxygen and electron transfer reactions on the layers of nitrogen, carbon, and sulfur.19,20 Mo(VI) is one of the main oxidation states that obtains a formal double bond between the metal and the oxo ligand via the donating σ and π bonds. There is a wide range of reports on the fabricated complexes using dioxomolybdenum(VI) obtained from varying the type and denticity of the remaining anionic ligands.21–24 Among those, MoO2(acac)2 is one of the most famous and major dioxomolybdenum(VI) complexes used as a catalyst for the organic transformations including epoxidation of alkenes and the oxidation of sulfides.25,26 Also, the coordination of molybdenum complexes by the organic groups (including oxygen, nitrogen, and sulfur) stabilized on the surfaces of magnetic nanoparticles has been used in many catalytic systems.27–30
Multicomponent reactions (MCRs), as a versatile synthetic strategy, combine at least three or more reactants31–34 via a single synthetic operation to generate several covalent bonds into structurally complex organic molecules containing most atoms of the available starting materials.35 Currently, MCRs are taken into consideration in the sustainable synthetic strategies, via which we can easily generate high compatibility with green chemistry due to their unique advantages, such as concomitant step economy, mild conditions, atom economy, and high convergence. Over the past decades and due to their various important applications in combinatorial chemistry,36 agrochemistry,37 medicinal chemistry,38–40 polymer chemistry,31 and natural product synthesis,41 MCRs have received considerable attention among organic chemists.
1,4-Dihydropyridine (1,4-DHP) core as an important class of nitrogen-containing heterocycles found in the nature have attracted considerable synthetic efforts over the past decade because of their diversity of biological functions, including antitubercular,42 neuroprotectant,43 antimicrobial,44 insecticidal,45 antiviral,46 antihypertension,47 anticancer,48 and antioxidant49,50 activities. Amongst them, polyhydroquinoline derivatives are very interesting heterocyclic molecules due to their pharmacological properties such as anti-inflammatory, antimalarial, antibacterial, anti-asthmatic, and tyrosine kinase inhibiting agents.51–53 Many procedures have been developed for the preparation of polyhydroquinolines using catalysts such as ionic liquids,54 microwaves,55 refluxing at high temperature,56 metal triflates,57 ceric ammonium nitrate (CAN),58 trimethylsilyl chloride (TMSCl),59 HY-zeolite,60 FeF3,61 silica perchloric acid (HClO4–SiO2),62 trifluoroethanol,63 montmorillonite K-10,64 iodine,65 autoclave,66 NiFe2O4 MNPs,67 heteropoly acid,68 L-proline,69 PTSA-SDS,70 NiCuMgFe2O4 MNPs,71 polymers,72 Sc(OTf)3,73 {Fe3O4@SiO2@(CH2)3Im}C(NO2)3,74 Yb(OTf)3,57 Fe3O4-adenine-Ni,75 BINOL-phosphoric acid derivatives,76 and Cu-SPATB/Fe3O4.77 Although most of these procedures offer distinct merits, some of these procedures suffer from one or more limitations, such as generating a large amount of waste, low yields of the desired product, poor recovery of the catalyst, long reaction times, and hard reaction conditions. Therefore, to avoid these limitation based on the green chemistry protocols, the discovery of simple, efficient, versatile, and environmentally friendly processes for the synthesis of polyhydroquinolines is still favored.
2. Experimental
All of the chemical substances used in this work were purchased from Merck, Aldrich, and Fluka Chemical Companies and used without further purification. Melting points of the products were determined with an Electrothermal 9100 apparatus. Energy-dispersive X-ray spectroscopy (EDX) was carried out on a FE-SEM (MIRA III, Detector of SAMX, France). Powder X-ray diffraction (XRD) was performed using Cu-Kα radiation (λ = 1.54 Å) on a Philips-PW1730 in the 2θ range of 10°–80°. Thermogravimetric analyses (TGA) were performed using a Linseis SATPT 100 thermoanalyzer at a heating rate of 10 °C min−1 under nitrogen atmosphere over a temperature range of 25–700 °C. The Fourier-transform infrared (FT-IR) spectra were recorded on a Perkin Elmer PXI spectrum, using pellets of the materials diluted with KBr in the range of 400–4000 cm−1. The magnetic features of the catalyst were determined using a vibrating sample magnetometer (VSM; MDK Co. Kashan, Iran) in the magnetic field range of −15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Oe to 15000 Oe at room temperature. Scanning electron microscopy (SEM) images were recorded using an SEM-LEO 1430VP analyzer. Transmission electron microscopy (TEM) was utilized on a Zeiss-EM 900 instrument.
000 Oe to 15000 Oe at room temperature. Scanning electron microscopy (SEM) images were recorded using an SEM-LEO 1430VP analyzer. Transmission electron microscopy (TEM) was utilized on a Zeiss-EM 900 instrument.
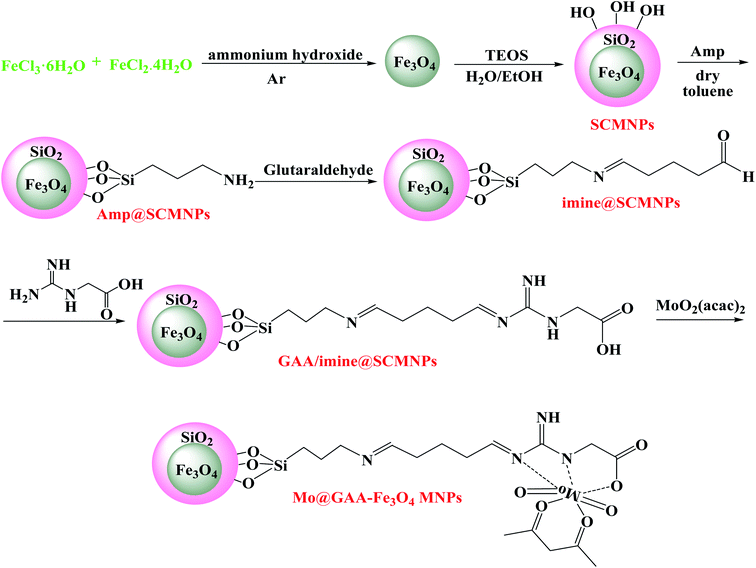
2.1. Synthesis of Fe3O4 nanoparticles (MNPs)
In the first step, a solution of FeCl2·4H2O (0.86 g) and FeCl3·6H2O (2.36 g) were dissolved in deionized water (40 mL). The mixture was heated to 90 °C, and mechanical stirring was done for 30 min under an argon atmosphere. Then, 10 mL of ammonia solution (25%) was added dropwise to the resulting mixture and stirred for another 20 min under argon flow. The precipitates were washed with distilled water and collected using a magnet. The product was dried under vacuum conditions.
2.2. Synthesis of Fe3O4@SiO2 (SCMNPs)
In a typical method, 1.0 g of obtained Fe3O4 nanoparticles was dispersed in a mixture of 60 mL ethanol, 20 mL deionized water, and 2 mL ammonia solution (25%) by sonication for 10 min. Then, 0.45 mL of tetraethylorthosilicate (TEOS) was added into the reaction system, sonicated for another 10 min, and stirred at ambient temperature for 14 h. The resultant precipitate was magnetically collected by a permanent magnetic field, washed three times with a mixture of ethanol and water (1:1), and dried in a vacuum oven.
2.3. Functionalization of SCMNPs by 3-aminopropyltriethoxysilane (Amp@SCMNPs)
1 g of core–shell Fe3O4@SiO2 nanoparticles was suspended in 20 mL of dry toluene by ultrasonication. After treatment for 20 min, 2 mL of 3-aminopropyltriethoxysilane (Amp) was added to the solution and refluxed under an argon atmosphere. After 24 h, the Amp@SCMNPs were collected using a permanent magnet, washed with ethanol and distilled water several times, and then dried under vacuum.
2.4. Functionalization of Amp@SCMNPs by glutaraldehyde (imine@SCMNPs)
2 g of prepared Amp@SCMNPs nanoparticles was added to 100 mL ethanol and dispersed for 30 min under ultrasonic irradiation. Then, 4 mmol of glutaraldehyde was added into the reaction solution and refluxed for 24 h. The prepared solid product (imine@SCMNPs) was then magnetically isolated and washed several times with ethanol to remove unreacted glutaraldehyde; finally, it was dried under vacuum.
2.5. Synthesis of GAA/imine@SCMNPs
1 g of the obtained imine@SCMNPs was dispersed in 25 mL DMF by the ultrasonic bath for 30 min. Then, 0.85 mmol of guanidineacetic acid (GAA) was added to the reaction solution. After the stirrer process for 8 h, the resulting substance was separated by a permanent magnet, rinsed with ethanol several times, and dried under vacuum.
2.6. Synthesis of Mo@GAA-Fe3O4 MNPs
MoO2(acac)2 was prepared using the literature method.78 2 g of GAA/imine@SCMNPs were added into 150 mL ethanol and a solution of MoO2(acac)2 (4 mmol) in 70 mL ethanol was added to this reaction solution and refluxed for 12 h. After magnetic separation, the resulting solid product was washed with dichloromethane to remove the unreacted MoO2(acac)2 and was dried under vacuum (Scheme 1).
 |
| | Scheme 1 The Mo@GAA-Fe3O4 MNPs synthesis. | |
2.7. General procedure for the preparation of polyhydroquinolines
Firstly, Mo@GAA-Fe3O4 MNPs (10 mg) was poured into the reaction mixture of dimedone or 1,3-cyclohexandione (1 mmol), ethyl acetoacetate, or methyl acetoacetate (1 mmol), aldehydes (1 mmol), and ammonium acetate (1.2 mmol), and the reaction mixture was heated for a specific time. Upon completion of the reaction, the catalyst was easily extracted using an external magnetic field; the resulting product was collected by filtration, rinsed, and recrystallized with ethanol to give pure polyhydroquinolines.
3. Results and discussion
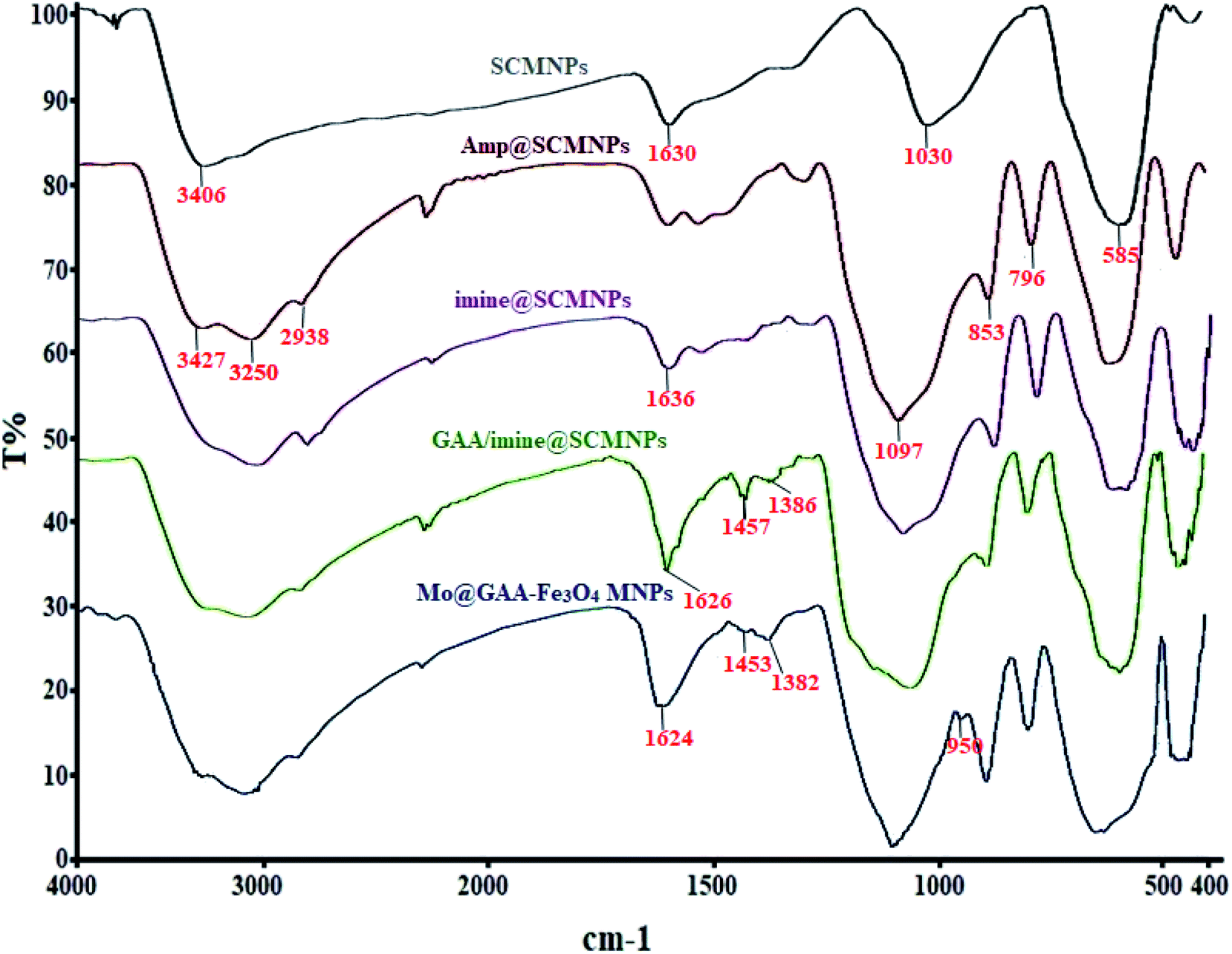
3.1. FTIR analysis Mo@GAA-Fe3O4 MNPs
FTIR spectroscopy is one of the main techniques for the qualitative determination of the molecular structures in different species and the identification of existing functional groups in the structure. Fig. 1 shows FTIR spectra for SCMNPs, Amp@SCMNPs, imine@SCMNPs, GAA/imine@SCMNPs, and Mo@GAA-Fe3O4 MNPs. In the case of SCMNPs, the band at 585 cm−1, from the Fe–O bond, verifies that the Fe3O4 lattice is formed. The presence of a broad peak at 3406 cm−1 is due to the O–H stretching vibrations, which are attached to the Fe3O4 surface. The above-mentioned structure has the symmetric stretching vibrations of Si–O–Si at around 1030 cm−1 and twisting bonds of Si–O–H and H–O–H at 1630 cm−1, indicating that the Fe3O4 nanoparticles (MNPs) contain silica layers. In the FT-IR spectrum of the Amp@SCMNPs can be observed the peaks for C–H (2938 cm−1), and NH2 (3250, and 3427 cm−1) stretching vibrations, which confirmed the successful covalent attachment of 3-aminopropyltriethoxysilane (Amp) to the SCMNPs surface. Also, vibrations at 796 and 853 cm−1 were probably attributed to the asymmetric stretching and in plane bending of Si–O–Si group, respectively. In about the imine@SCMNPs, the peak at 1636 cm−1 is assigned to the stretching vibrations of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond, revealing the functionalization of the Amp@SCMNPs with glutaraldehyde groups. The bands at 1457 and 1626 cm−1 of the FTIR spectrum of GAA/imine@SCMNPs can be assigned to the C–N and COO stretching vibrations. Also, a broad band about 3000–3400 cm−1 can be ascribed to the acidic OH stretching vibrations. The peak observed at 950 cm−1 corresponds to the MoO2 group, which indicates the formation of the Mo@GAA-Fe3O4 MNPs. Moreover, the FTIR spectra of the catalyst show a frequency shift for some bonds, indicating the coordination of the metal with the desired bonds.
N bond, revealing the functionalization of the Amp@SCMNPs with glutaraldehyde groups. The bands at 1457 and 1626 cm−1 of the FTIR spectrum of GAA/imine@SCMNPs can be assigned to the C–N and COO stretching vibrations. Also, a broad band about 3000–3400 cm−1 can be ascribed to the acidic OH stretching vibrations. The peak observed at 950 cm−1 corresponds to the MoO2 group, which indicates the formation of the Mo@GAA-Fe3O4 MNPs. Moreover, the FTIR spectra of the catalyst show a frequency shift for some bonds, indicating the coordination of the metal with the desired bonds.
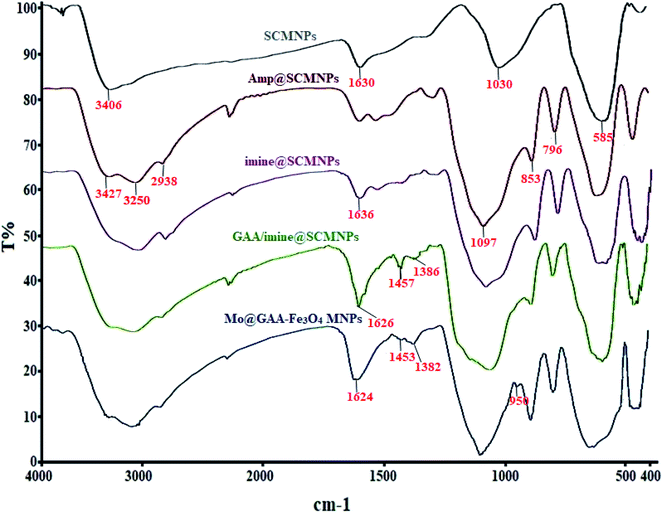 |
| | Fig. 1 FTIR spectra of SCMNPs, Amp@SCMNPs, imine@SCMNPs, GAA/imine@SCMNPs, and Mo@GAA-Fe3O4 MNPs. | |
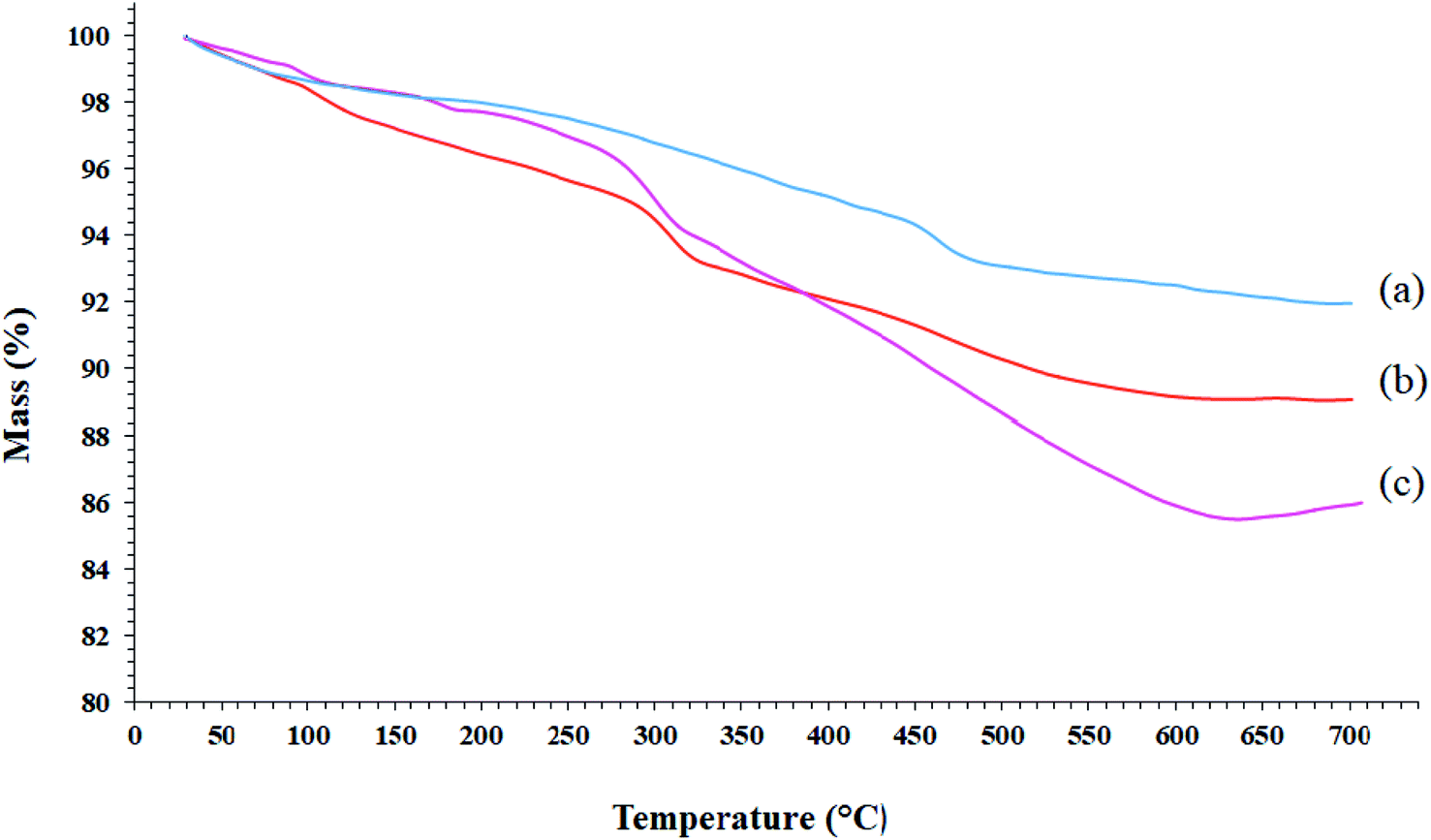
3.2. TGA analysis of Mo@GAA-Fe3O4 MNPs
TGA analysis can be used to determine the loading amount of the organic groups on the synthetic substrates. This measures the mass variations as a function of the temperature and time under a controlled atmosphere and indicates the thermal stability of the sample on a plot. The TGA analysis curves of SCMNPs, GAA/imine@SCMNPs, and Mo@GAA-Fe3O4 MNPs are shown in Fig. 2. All samples underwent a small weight loss below 200 °C due to water thermodesorption from the surface (drying). Another weight loss up to 700 °C was found in TGA curve of SCMNPs, which can be related to the release of hydroxyl ions from the nanoparticles and volatilization. In the TGA curves of GAA/imine@SCMNPs and Mo@GAA-Fe3O4 MNPs, another weight loss can be seen up to 650 °C, which can be attributed to the decomposition of the functionalized organic groups.
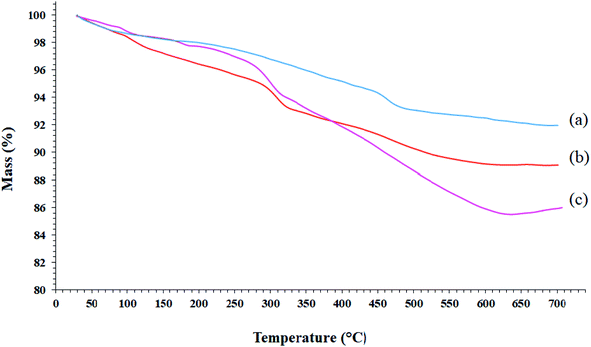 |
| | Fig. 2 TGA curves of (a) SCMNPs, (b) GAA/imine@SCMNPs, and (c) Mo@GAA-Fe3O4 MNPs. | |
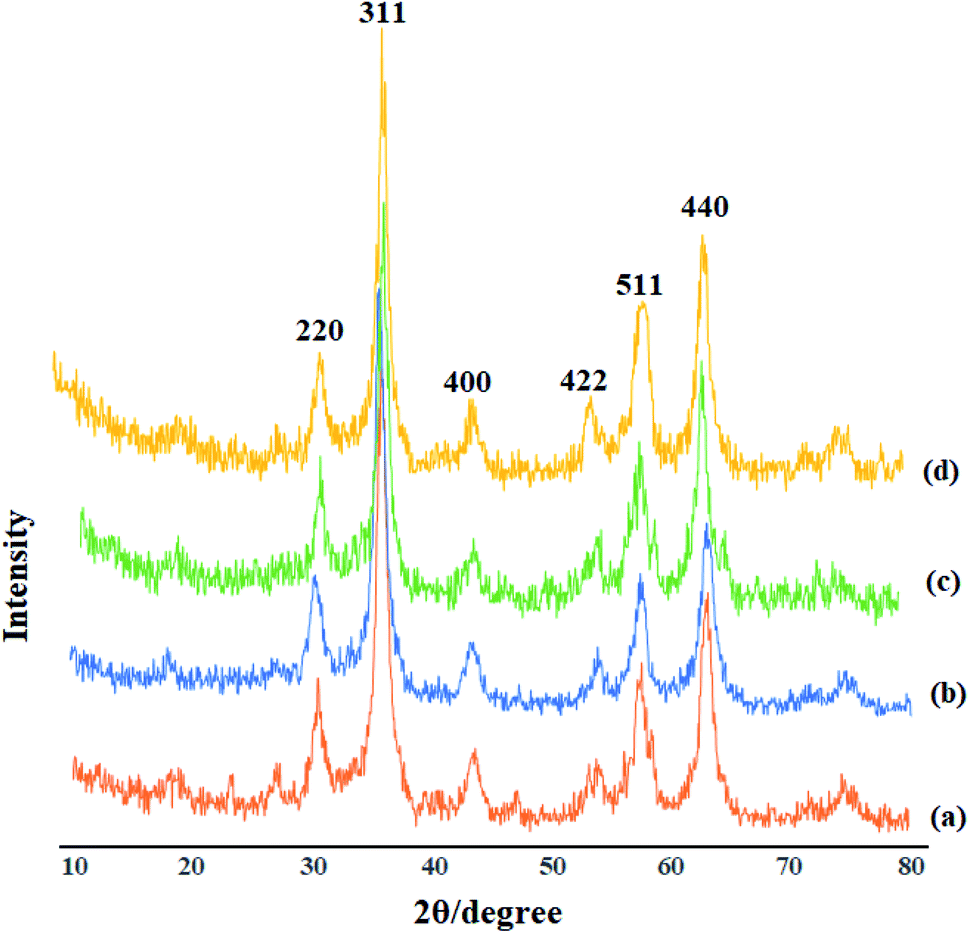
3.3. XRD analysis of Mo@GAA-Fe3O4 MNPs
Identification of the crystalline materials using X-ray is one of the most common and important analysis techniques. As the X-ray region is located between gamma and ultraviolet, useful data could be obtained from this spectral region including the crystalline structure, the material type, and the nanoparticle size. The XRD patterns for Fe3O4 (a), GAA/imine@SCMNPs (b), Mo@GAA-Fe3O4 MNPs (c) and recovered Mo@GAA-Fe3O4 MNPs (d) are depicted in Fig. 3. The X-ray diffraction patterns of Fe3O4 magnetic nanoparticles display six diffraction peaks at 2θ = 30.5°, 35.7°, 43.8°, 53.6°, 57.5°, and 63.1° (indexed as (220), (311), (400), (422), (511), and (440) reflection, associated with the crystalline structure of the spinel magnetic core). In addition, GAA/imine@SCMNPs and Mo@GAA-Fe3O4 MNPs have also similar diffraction peaks, indicating an unchanged crystalline structure of the above-mentioned products. It is necessary to mention that the XRD pattern of recovered Mo@GAA-Fe3O4 MNPs after the first recovery and reuse do not show any change in crystalline structure.
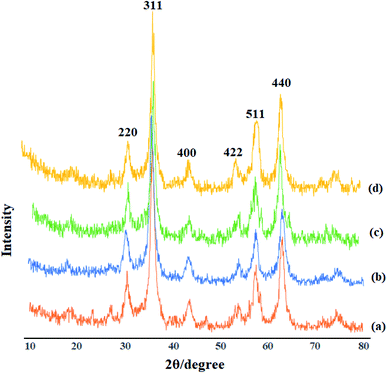 |
| | Fig. 3 XRD patterns of (a) Fe3O4, (b) GAA/imine@SCMNPs, (c) Mo@GAA-Fe3O4 MNPs, and recovered Mo@GAA-Fe3O4 MNPs (d). | |
3.4. VSM analysis of Mo@GAA-Fe3O4 MNPs
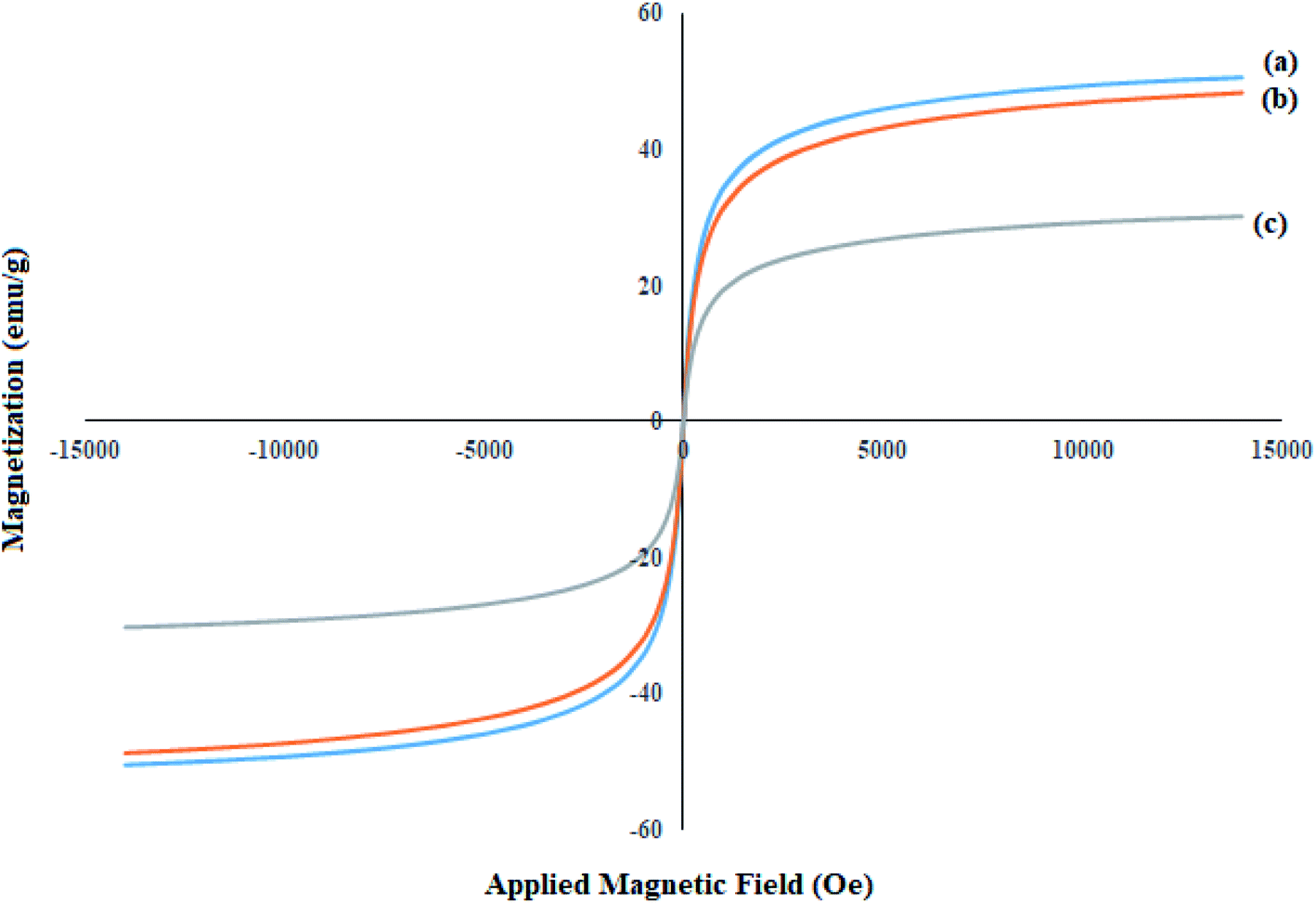
Magnetic properties and the behavior of the achieved nanoparticles are investigated by the VSM analysis. An external magnetic field is applied to evaluate the magnetization ability of the magnetic nanoparticles. The VSM analysis of the synthesized (a) Fe3O4, (b) SCMNPs, and (c) Mo@GAA-Fe3O4 MNPs were shown in Fig. 4. The saturation magnetization values (Ms) of magnetic materials revealed significant differences, which are 50.63 and 47.16 emu g−1 for bare Fe3O4 and SCMNPs, respectively, while for catalyst, the difference is 28.99 emu g−1. As can be seen, the value of Ms for the catalyst decreased, which can be attributed to the functionalization of the Fe3O4 core by silica layers, organic molecules, and metal groups.
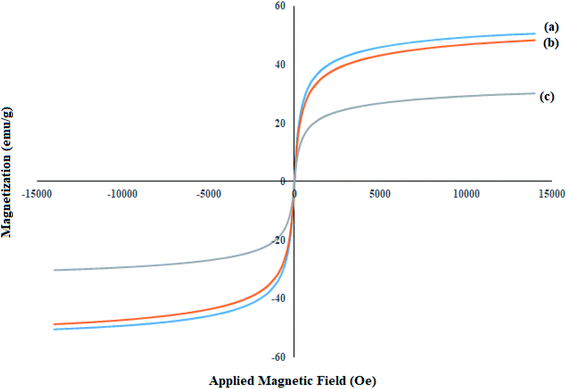 |
| | Fig. 4 VSM patterns of (a) Fe3O4, (b) SCMNPs, and (c) Mo@GAA-Fe3O4 MNPs. | |
3.5. SEM analysis of Mo@GAA-Fe3O4 MNPs
The obtained images from the SEM technique could be used to study the morphology, uniformity, and physical properties of nanoparticle surfaces. High magnifications are obtained by this device to precisely investigate the material details. As shown in Fig. 5, Fe3O4 and SCMNPs are nearly spherical with a smooth surface and their mean particle sizes are 19 and 26 nm, respectively. The obtained results clearly showed that the SCMNPs particles have grown compared to the Fe3O4. According to the represented images for GAA/imine@SCMNPs and Mo@GAA-Fe3O4 MNPs, the average size of the above-mentioned nanoparticles are in the range of 30–37 nm. The modification by organic and metallic groups can result in the growth and accumulation of the nanoparticles.
 |
| | Fig. 5 SEM image of (a) Fe3O4, (b) SCMNPs, (c) GAA/imine@SCMNPs, and (d) Mo@GAA-Fe3O4 MNPs. | |
3.6. TEM analysis of Mo@GAA-Fe3O4 MNPs
In TEM analysis, a focused electron beam is used for obtaining the images. In this technique, some information from the inner structure could be obtained by transmitting a high-energy electron beam through a thin sample. Fig. 6 shows the TEM image of Mo@GAA-Fe3O4 MNPs. TEM images indicate that the diameter of the obtained catalyst is about 28–35 nm and it has a nearly spherical morphology with a narrow particle size distribution.
 |
| | Fig. 6 TEM image of Mo@GAA-Fe3O4 MNPs. | |
3.7. EDX and elemental mapping analysis of Mo@GAA-Fe3O4 MNPs
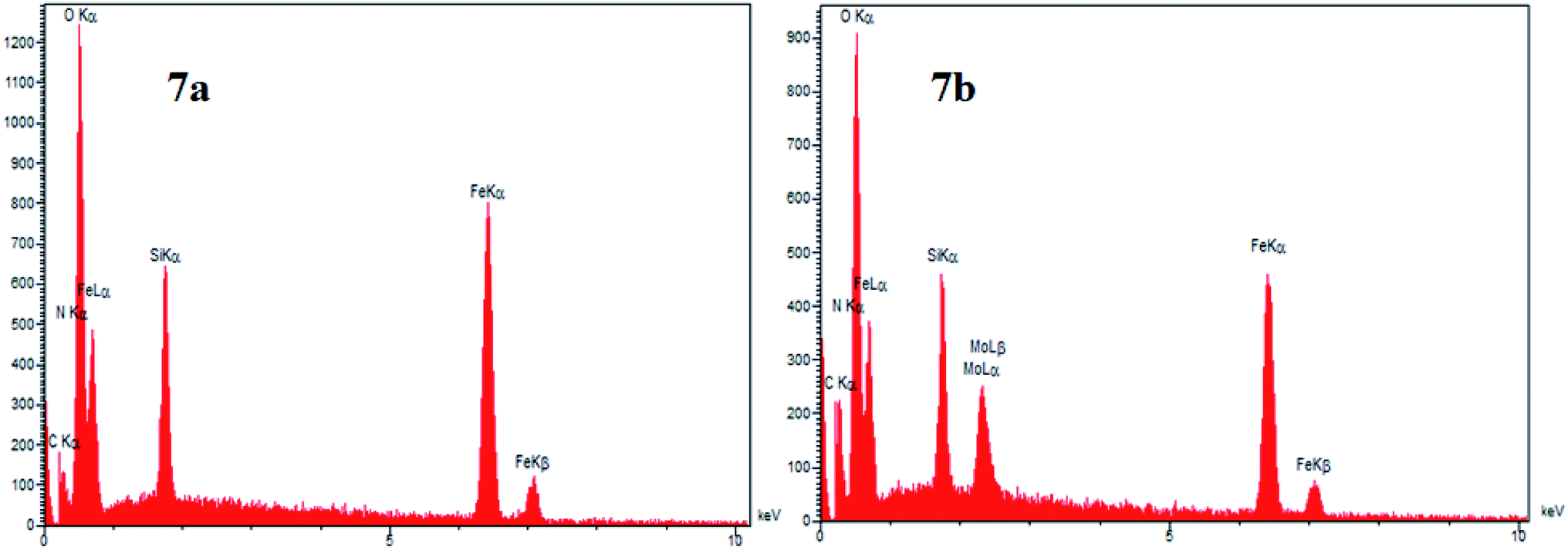
One of the main techniques to identify the elemental composition of the sample or a part of it is the EDX analysis. The EDX spectra of the GAA/imine@SCMNPs and Mo@GAA-Fe3O4 MNPs is shown in Fig. 7a and b, respectively. In the case of GAA/imine@SCMNPs, the presence of carbon, nitrogen, oxygen, iron, and silicon was confirmed (Fig. 7a). Also in Fig. 7b, the presence of the molybdenum element indicates coordination of Mo with nitrogen and oxygen electron pairs and thus the successful synthesis of the Mo@GAA-Fe3O4 MNPs. In addition, Fig. 8 shows the elemental map of the Mo@GAA-Fe3O4 MNPs nanocatalyst, which exhibits the presence of C, N, O, Fe, Si, and Mo elements.
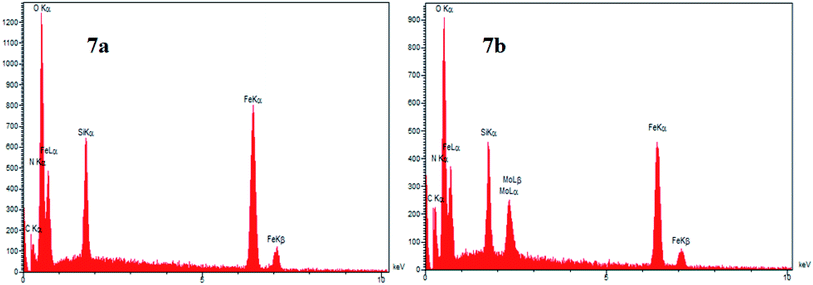 |
| | Fig. 7 EDX spectra of (a) GAA/imine@SCMNPs and (b) and Mo@GAA-Fe3O4 MNPs. | |
 |
| | Fig. 8 SEM image of Mo@GAA-Fe3O4 MNPs nanocatalyst and corresponding quantitative EDX element mapping of C, N, O, Fe, Si, and Mo. | |
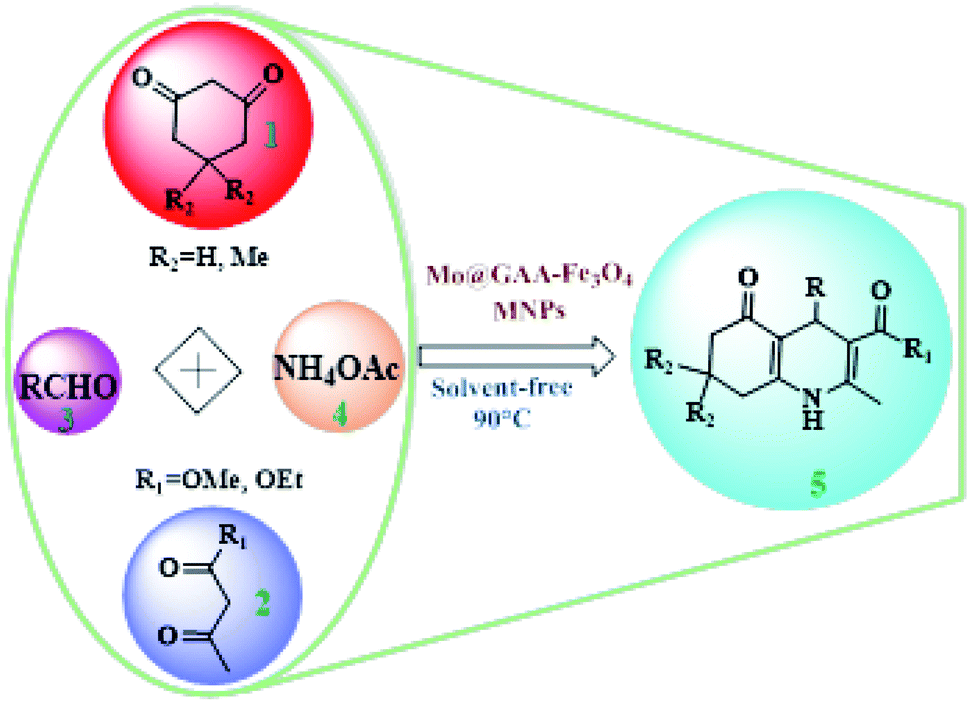
To synthesize new catalysts for certain organic processes,79–84 the synthesis of potentially active polyhydroquinoline derivatives (5) was investigated with various substituents from the reaction between dimedone or 1,3-cyclohexandione (1), ethyl acetoacetate, or methyl acetoacetate (2), aldehydes (3), and ammonium acetate (4) under solvent-free conditions using Mo@GAA-Fe3O4 MNPs as a novel, eco-friendly, reusable, and promising nanocatalyst (Scheme 2).
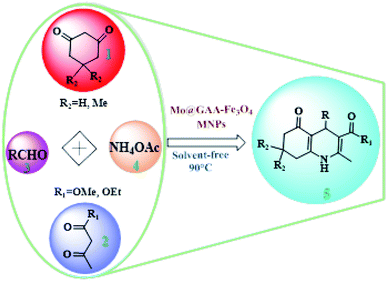 |
| | Scheme 2 Mo@GAA-Fe3O4 MNPs-catalyzed synthesis of polyhydroquinolines. | |
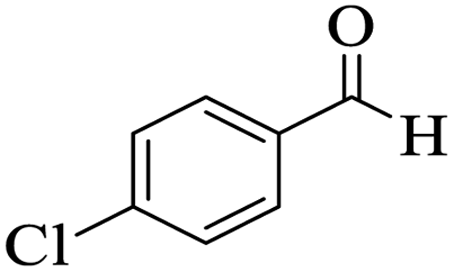
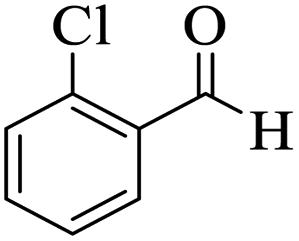
The catalytic activity of the Mo@GAA-Fe3O4 MNPs was surveyed in the preparation of polyhydroquinoline derivatives. Firstly, the condensation reaction of dimedone, ethyl acetoacetate, 4-chlorobenzaldehyde, and ammonium acetate in the presence of Mo@GAA-Fe3O4 MNPs was chosen as a model reaction. To optimize the reaction conditions, the synthesis of polyhydroquinolines we studied under various reaction conditions, including solvent, temperature, and the amount of catalyst. To optimize the reaction solvent, H2O, EtOH, THF, CH2Cl2, CH3CN, toluene, cyclohexane, and solvent-free conditions were tested (Entries 1–8). The results are summarized in Table 1, showing that carrying out the reaction in the absence of solvent led to the formation of the desired product 5p in the highest yield (Entry 8). However, a good yield of the product was obtained in EtOH after 50 min from the start of the reaction (Entry 2). To study the role of catalyst amount, the model reaction was performed in the presence of 5, 10, 15, and 20 mg of Mo@GAA-Fe3O4 MNPs (Entries 8 and 10–12). These observations showed that carrying out the reaction in the absence of the catalyst gave any yield for the desired product (Entry 9). The reaction in presence of 5 mg of the catalyst provided a good yield of the product (Entry 10), while 10, 15, and 20 mg gave excellent-to-high yields of the corresponding product (Entries 8 and 11–12). It was found that using 10 mg of the Mo@GAA-Fe3O4 MNPs is appropriate to carry out the reaction under solvent-free conditions with a 96% yield. Afterward, the influence of temperature on the model reaction was investigated. The reaction was carried out under different temperatures (25, 40, 60, 80, 90, and 100 °C) (Entries 8 and 13–17), and the best result was achieved at 90 °C under solvent-free conditions (Entry 8). Finally, when the model reaction was done in the existence of 10 mg of imine@SCMNPs, GAA/imine@SCMNPs under the optimized conditions, the product yield were 86 and 88%, respectively (Entries 18–19). Comparing the product efficiencies for the inputs of 8 and 18–19, it could be fully understood that the coordination of the compound Mo@GAA-Fe3O4 MNPs by free electron pair of nitrogen with the molybdenum complex increased the catalytic activity.
Table 1 Optimization one-pot four-component condensation of dimedone, ethyl acetoacetate, 4-chlorobenzaldehyde, and ammonium acetate, under different conditionsa
| Entry |
Solvent |
Catalyst (mg) |
Temp. (°C) |
Time (min) |
Yieldb (%) |
| Reaction conditions: dimedone (1 mmol), ethyl acetoacetate (1 mmol), 4-chlorobenzaldehyde (1 mmol), and ammonium acetate (1.2 mmol) and required amount of the catalyst. Isolated yield. |
| 1 |
H2O |
10 |
Reflux |
100 |
45 |
| 2 |
EtOH |
10 |
Reflux |
50 |
83 |
| 3 |
THF |
10 |
Reflux |
160 |
18 |
| 4 |
CH2Cl2 |
10 |
Reflux |
360 |
25 |
| 5 |
CH3CN |
10 |
Reflux |
220 |
35 |
| 6 |
Toluene |
10 |
Reflux |
360 |
39 |
| 7 |
Cyclohexane |
10 |
Reflux |
200 |
40 |
| 8 |
— |
10 |
90 |
12 |
96 |
| 9 |
— |
— |
90 |
100 |
— |
| 10 |
— |
5 |
90 |
35 |
81 |
| 11 |
— |
15 |
90 |
12 |
95 |
| 12 |
— |
20 |
90 |
12 |
92 |
| 13 |
— |
10 |
100 |
12 |
95 |
| 14 |
— |
10 |
80 |
20 |
88 |
| 15 |
— |
10 |
60 |
40 |
76 |
| 16 |
— |
10 |
40 |
90 |
65 |
| 17 |
— |
10 |
25 |
120 |
21 |
| 18 |
— |
10 (imine@SCMNPs) |
90 |
25 |
86 |
| 19 |
— |
10 (GAA/imine@SCMNPs) |
90 |
25 |
88 |
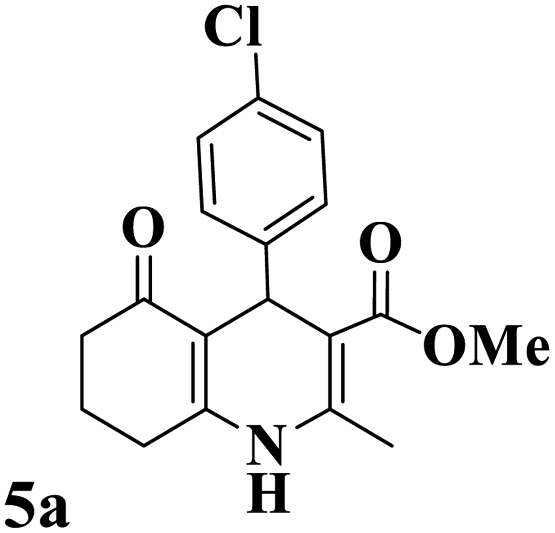
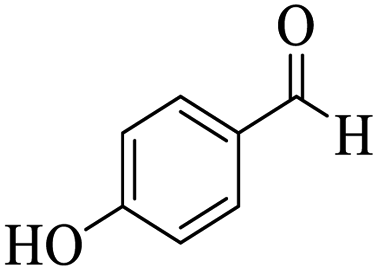
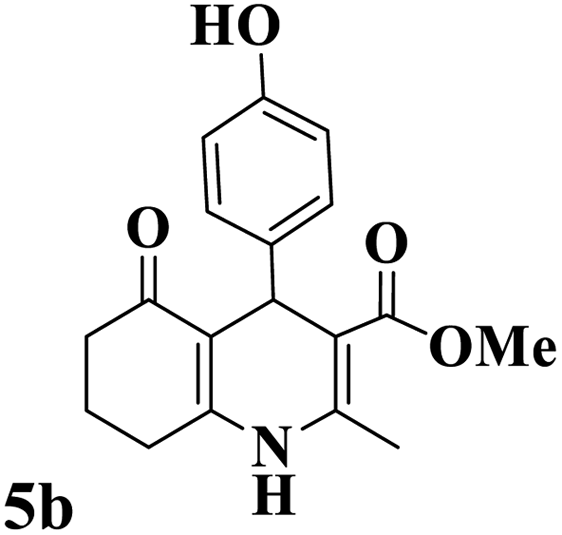
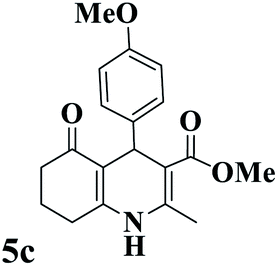
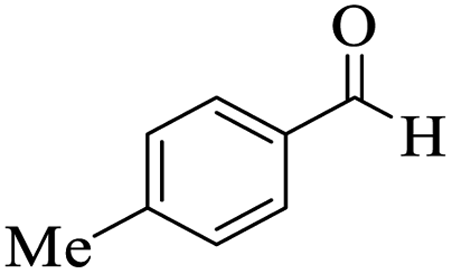
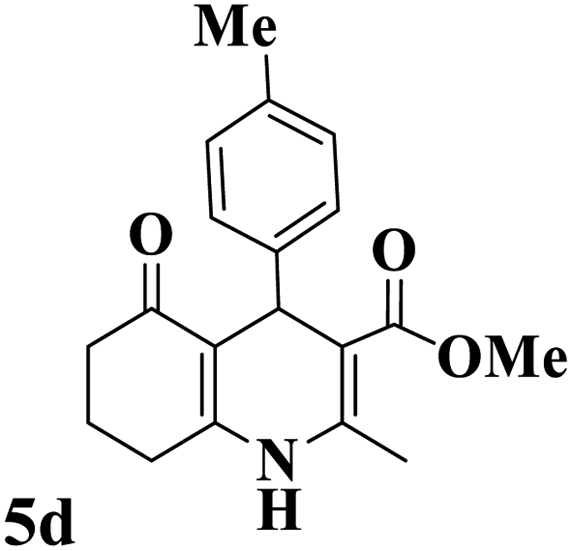
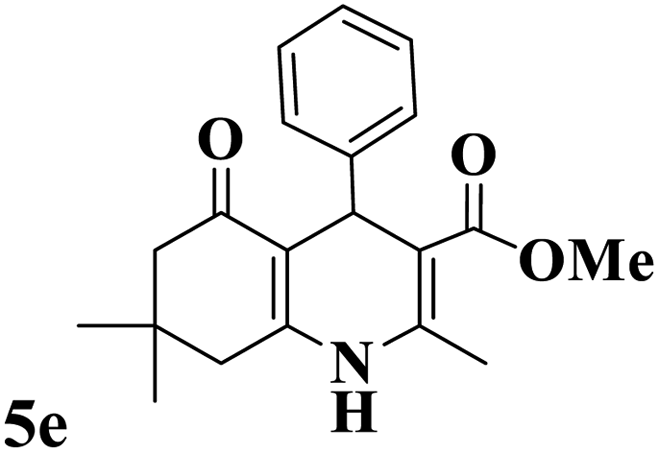
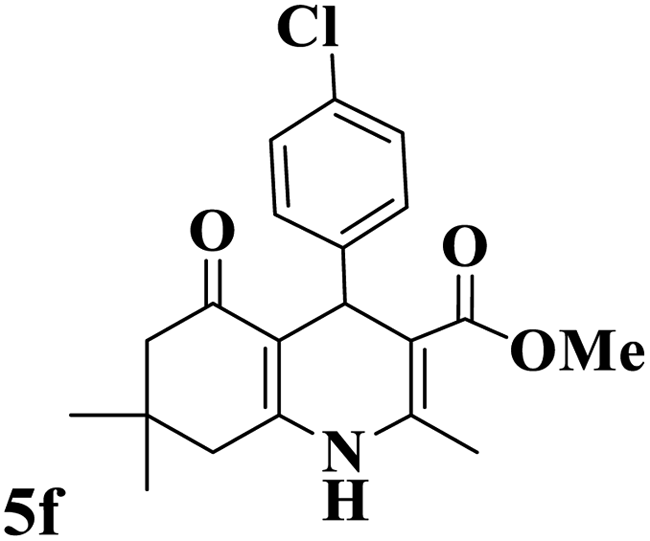
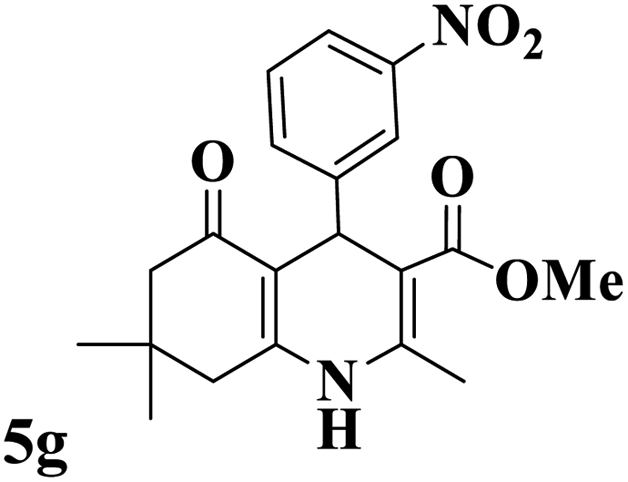
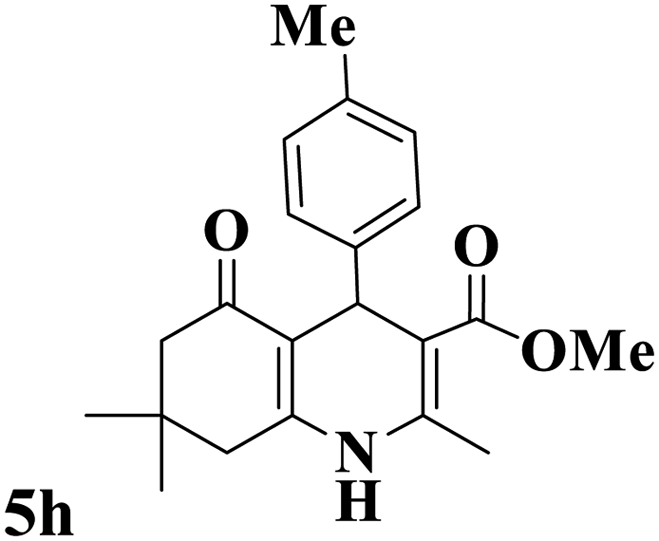
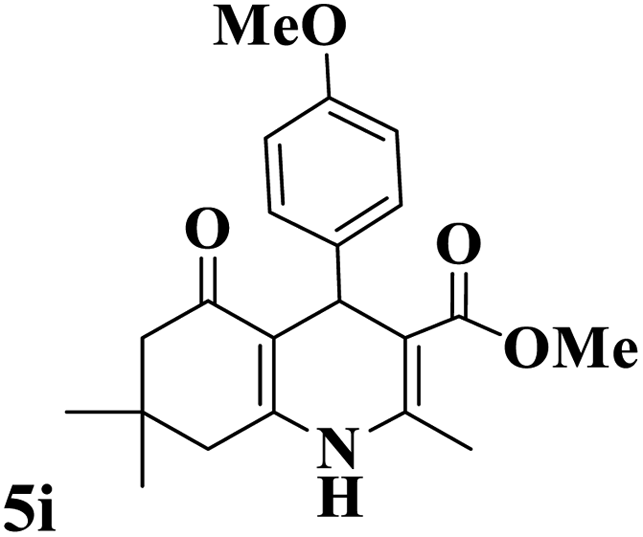
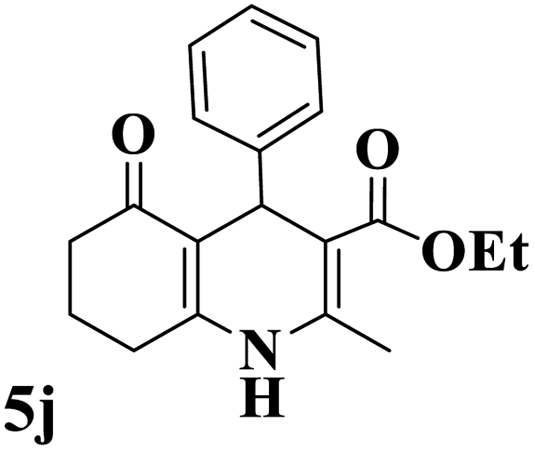
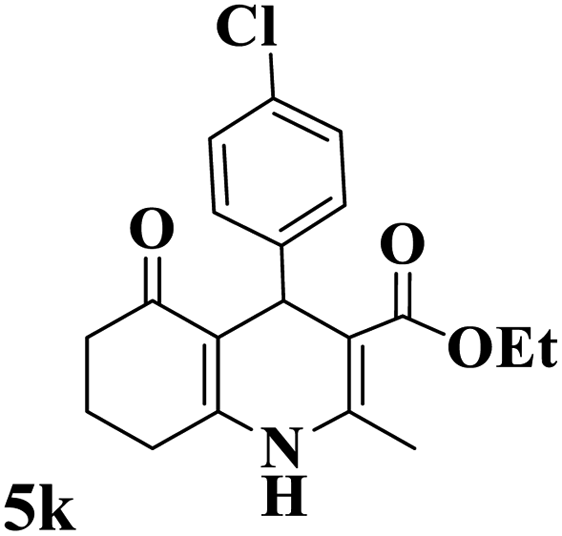
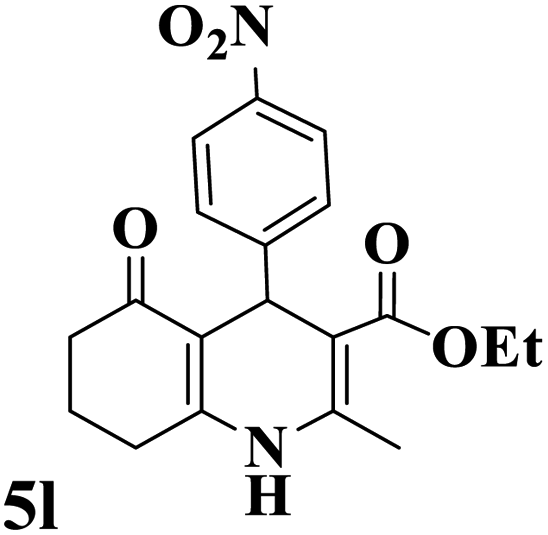
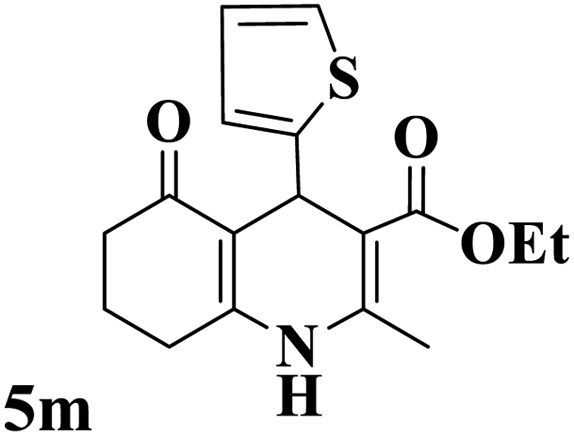
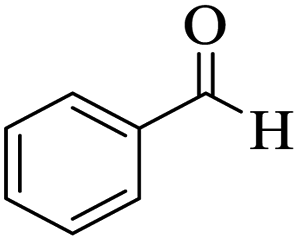
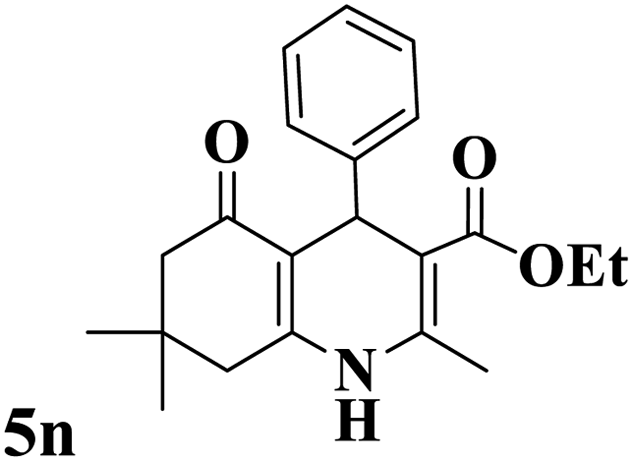
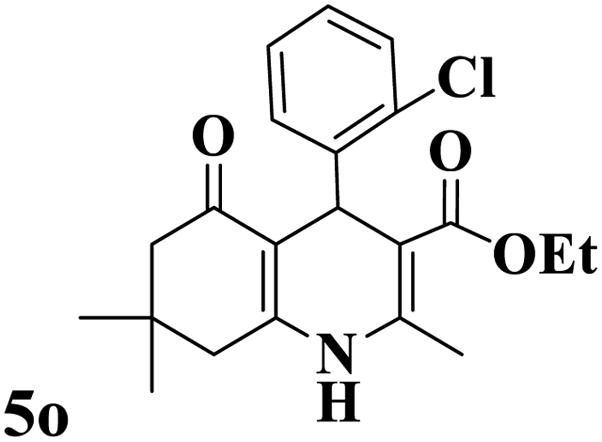
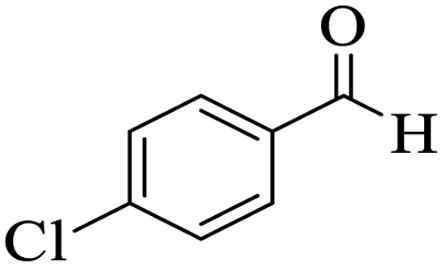
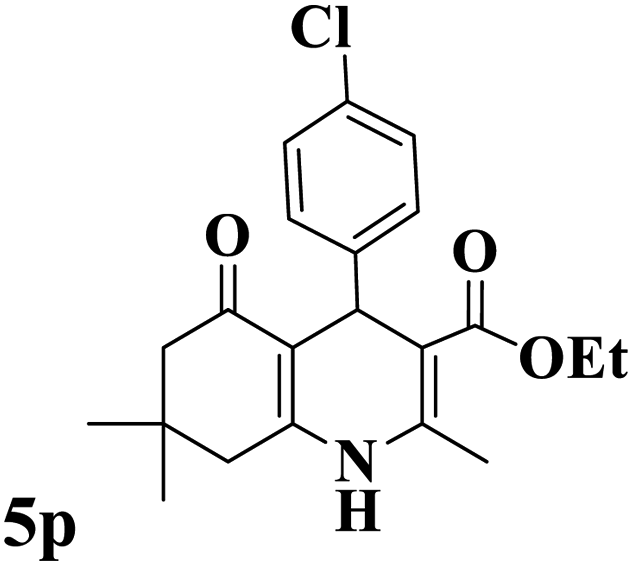
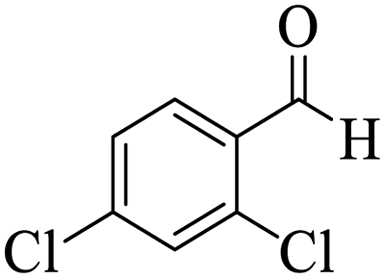
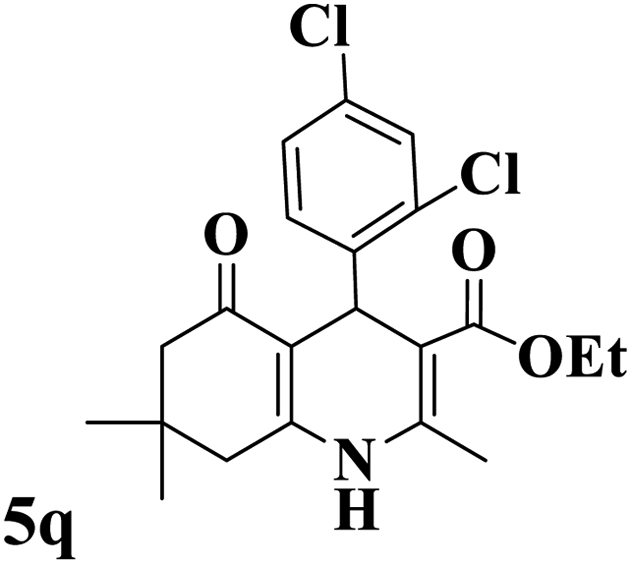
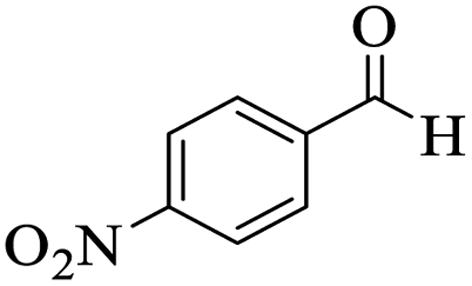
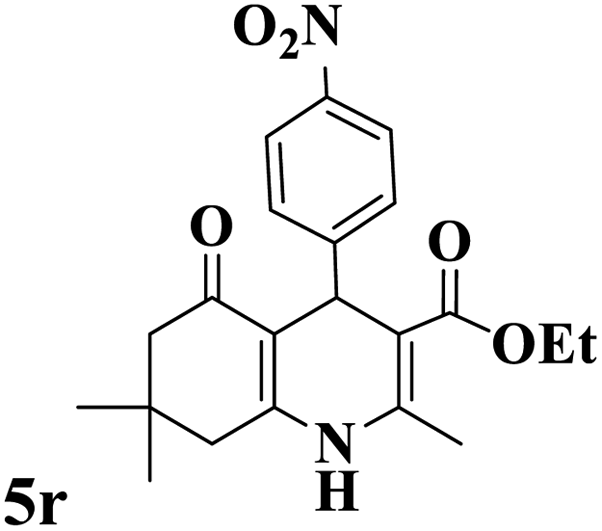
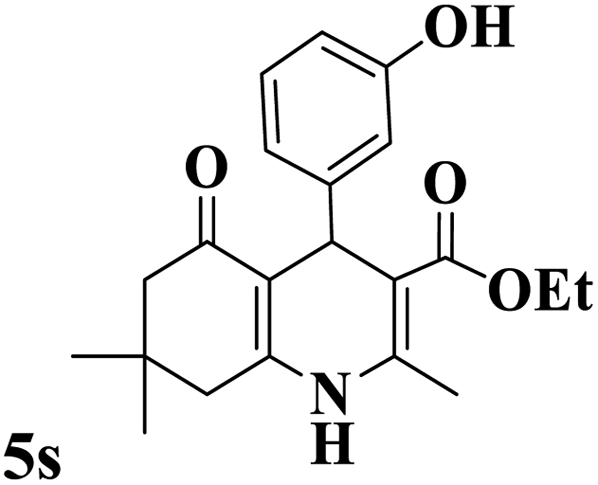
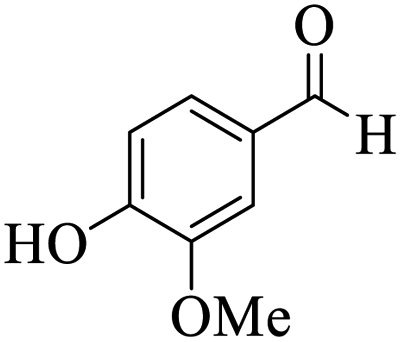
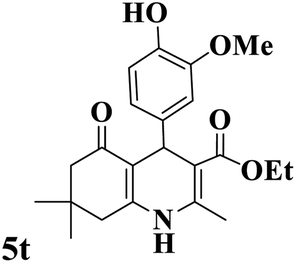
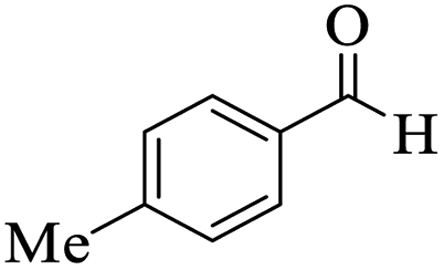
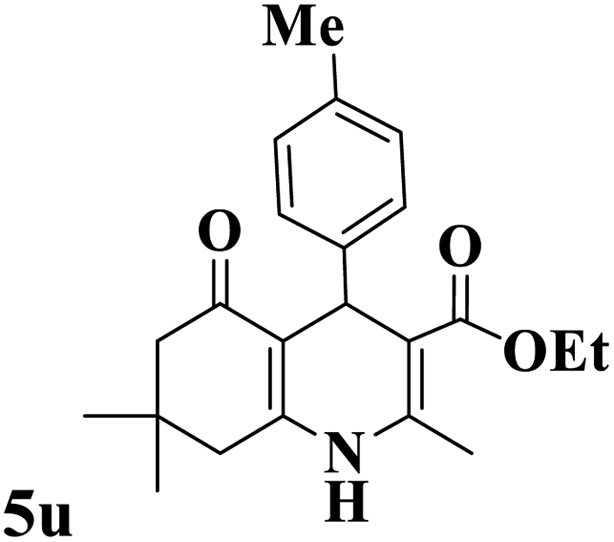
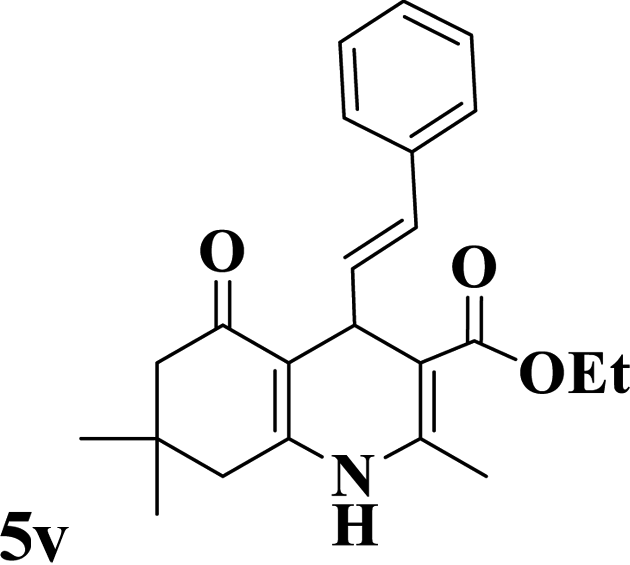
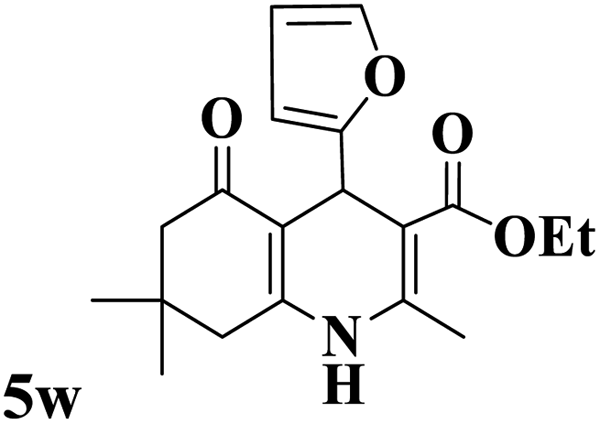
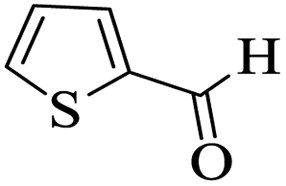
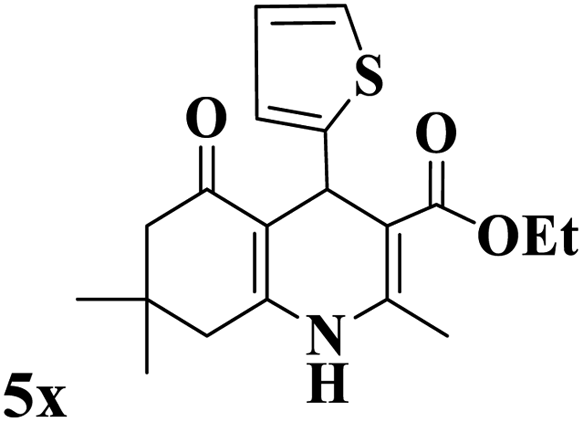
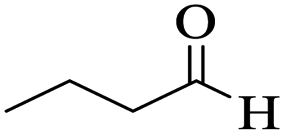
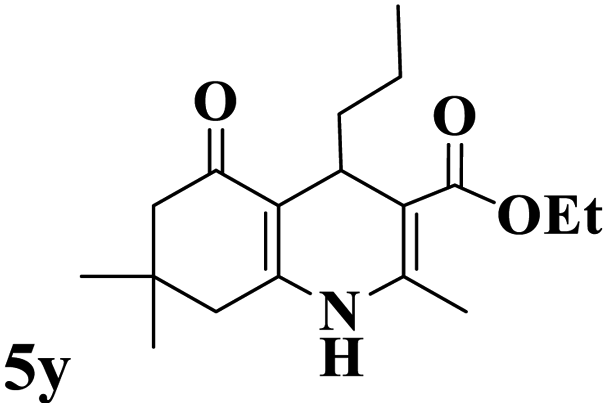
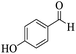
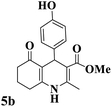
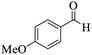
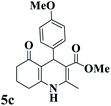
Utilizing the optimal reaction conditions, various polyhydroquinolines were prepared by the four-component reaction of dimedone, or 1,3-cyclohexandione, ethyl acetoacetate or methyl acetoacetate, a wide range of aldehydes, and ammonium acetate (Table 2). Aromatic aldehydes with electron-donating or electron-withdrawing groups tolerate smooth transformation to the corresponding products without the formation of by-products at better yields and in short reaction times. The reaction was also carried out with aliphatic aldehyde instead of aromatic aldehyde but did not afford the title products in considerable amounts (5y).
Table 2 Preparation of polyhydroquinoline derivatives using desired catalysta
| Entry |
RCHO (3) |
Product |
Time (min) |
Yieldb (%) |
M.P. (Obsd) (°C) |
M.P. (Lit) (°C) |
| Reaction conditions: dimedone or 1,3-cyclohexandione (1 mmol), ethyl acetoacetate or methyl acetoacetate (1 mmol), aldehydes (1 mmol), ammonium acetate (1.2 mmol) and catalyst (10 mg) at 90 °C. Isolated yields. |
| 1 |
 |
 |
13 |
93 |
222–224 |
221–223 (ref. 85) |
| 2 |
 |
 |
15 |
93 |
253–256 |
256–257 (ref. 85) |
| 3 |
 |
 |
20 |
92 |
208–211 |
208–211 (ref. 86) |
| 4 |
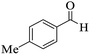 |
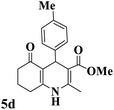 |
17 |
90 |
236–238 |
238–240 (ref. 87) |
| 5 |
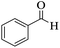 |
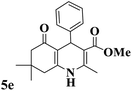 |
13 |
93 |
257–259 |
257–259 (ref. 88) |
| 6 |
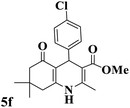
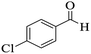 |
 |
15 |
93 |
219–221 |
220–222 (ref. 89) |
| 7 |
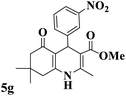
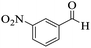 |
 |
20 |
90 |
223–225 |
222–223 (ref. 90) |
| 8 |
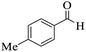 |
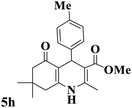 |
20 |
93 |
269–272 |
270–274 (ref. 91) |
| 9 |
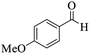 |
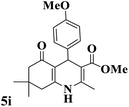 |
15 |
95 |
256–258 |
256–259 (ref. 88) |
| 10 |
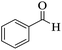 |
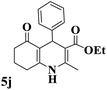 |
14 |
94 |
239–241 |
240–241 (ref. 91) |
| 11 |
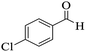 |
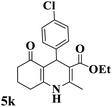 |
15 |
94 |
234–236 |
234–235 (ref. 91) |
| 12 |
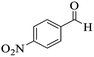 |
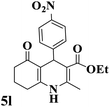 |
18 |
91 |
202–205 |
204–205 (ref. 91) |
| 13 |
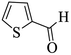 |
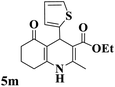 |
20 |
88 |
231–235 |
233–234 (ref. 91) |
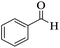
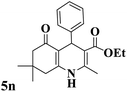
| 14 |
 |
 |
13 |
95 |
201–203 |
202–204 (ref. 57) |
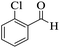
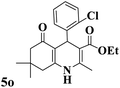
| 15 |
 |
 |
15 |
90 |
207–208 |
206–208 (ref. 92) |
| 16 |
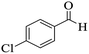 |
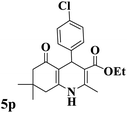 |
12 |
96 |
243–245 |
244–246 (ref. 93) |
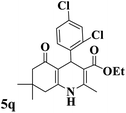
| 17 |
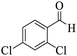 |
 |
14 |
92 |
240–242 |
241–243 (ref. 57) |
| 18 |
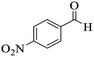 |
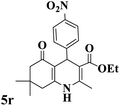 |
14 |
89 |
245–247 |
244–246 (ref. 92) |
| 19 |
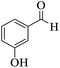 |
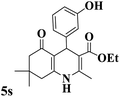 |
15 |
91 |
218–221 |
218–220 (ref. 92) |
| 20 |
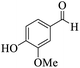 |
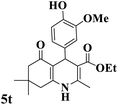 |
20 |
92 |
210–213 |
211–212 (ref. 72) |
| 21 |
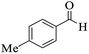 |
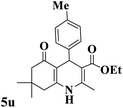 |
18 |
89 |
259–262 |
260–261 (ref. 57) |
| 22 |
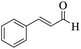 |
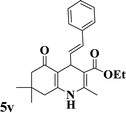 |
18 |
94 |
205–207 |
204–206 (ref. 57) |
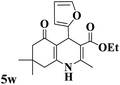
| 23 |
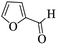 |
 |
20 |
90 |
247–249 |
246–248 (ref. 57) |
| 24 |
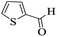 |
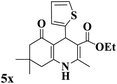 |
20 |
88 |
237–240 |
238–240 (ref. 94) |
| 25 |
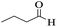 |
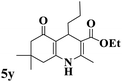 |
45 |
72 |
148–150 |
147–148 (ref. 57) |
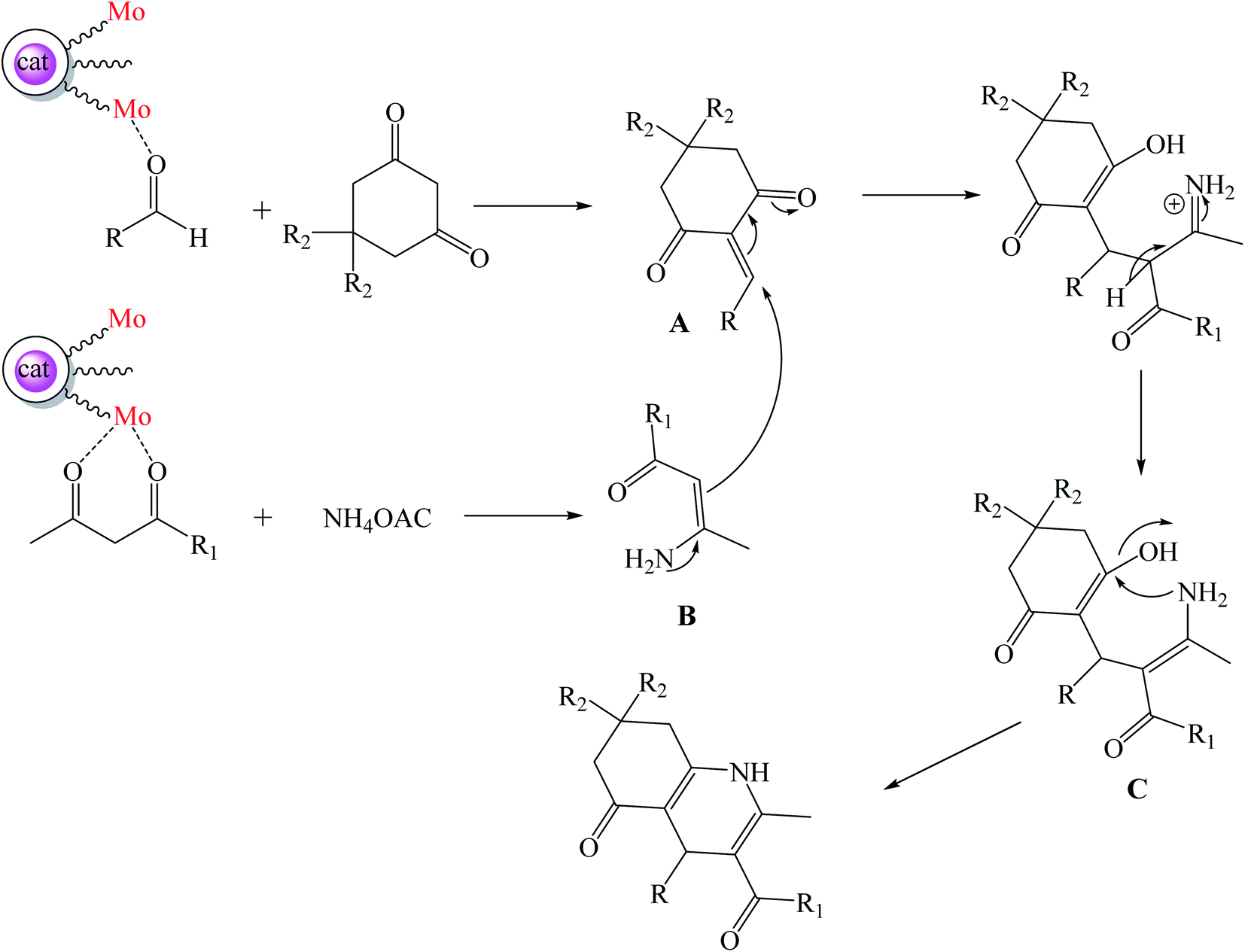
The plausible mechanism for the synthesis of polyhydroquinoline derivatives using Mo@GAA-Fe3O4 MNPs has been shown in Scheme 3. The metallic parts of the catalyst play a noticeable role in this organic transformation. Initially, Mo groups of the catalyst activate the CO functional groups of aldehyde and facilitate the condensation with dimedone or 1,3-cyclohexandione and afford intermediate A. Then, Mo groups of the catalyst also activate CO functional groups of ethyl acetoacetate or methyl acetoacetate and increase the activity of this group. The condensation of ethyl acetoacetate or methyl acetoacetate and ammonium acetate resulted in intermediate B. The Michael addition of intermediate A with intermediate B resulted in intermediate C. The achieved compound is unstable and is converted to the resulting product in subsequent dehydration and cyclization.
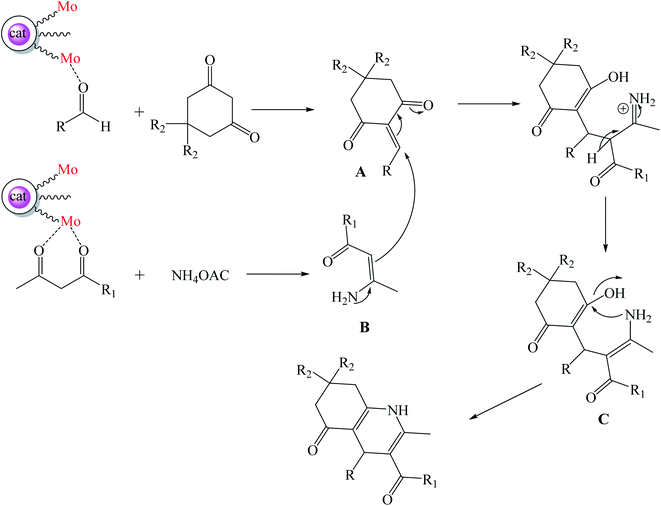 |
| | Scheme 3 Proposed mechanism for the synthesis of polyhydroquinoline derivatives. | |
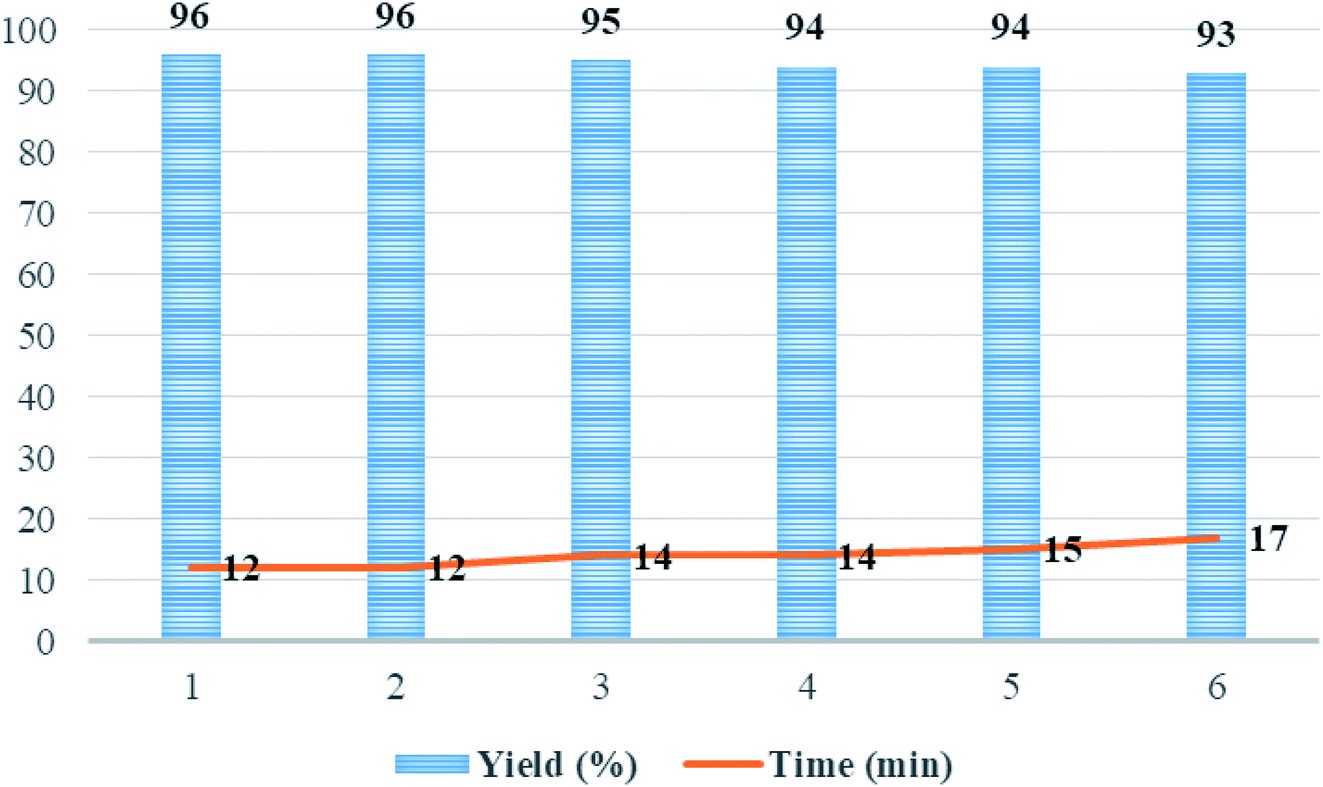
After completion of the reaction as indicated by thin layer chromatography (TLC), the catalyst precipitate was easily filtered off from the product using an external magnetic field, washed with water/ethanol (1:1) to eliminate the residual product, dried under the vacuum oven, and reused under the same experimental conditions. The recycled catalyst was reused with a negligible reduction in catalytic activity and the product yield for six runs (Fig. 9). The yields of the product 5p for each of the six runs were 96, 96, 95, 94, 94, and 93%, respectively.
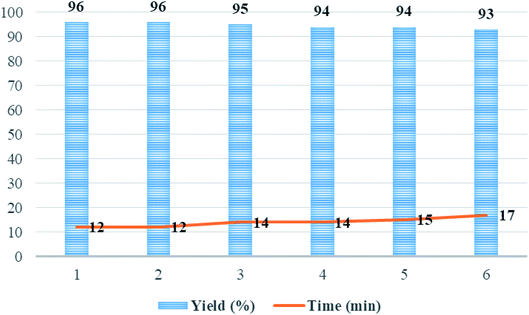 |
| | Fig. 9 The recycling of Mo@GAA-Fe3O4 MNPs as catalysts under solvent-free conditions in the synthesis of 5p. | |
To show the merits of the Mo@GAA-Fe3O4 MNPs in comparison with previously reported catalysts in the literature for the synthesis of polyhydroquinoline derivatives, some of the results are tabulated in Table 3. As indicated in Table 3, the low catalyst loading of Mo@GAA-Fe3O4 MNPs can develop a suitable methodology in terms of the reaction times and yields, green chemistry, and compatibility with the environment.
Table 3 Comparison of the results of the production of products 5n
| Entry |
Catalyst and conditions |
Catalyst loading |
Time (h) |
Yield (%) |
Ref. |
| 1 |
Yb(OTf)3/EtOH/r.t. |
5 mol% |
5 |
90 |
57 |
| 2 |
(NH4)6[MnIVMo9O32]/EtOH/r.t. |
2 mol% |
1.5 |
90 |
95 |
| 3 |
MgMnO3/solvent-free/100 °C |
8 mg |
33 min |
88 |
96 |
| 4 |
FePO4/EtOH/reflux |
2 mol% |
1 |
96 |
97 |
| 5 |
La2O3/TFE/r.t. |
10 mol% |
1 |
90 |
98 |
| 6 |
L-Proline/EtOH/reflux |
10 mol% |
5 |
92 |
99 |
| 7 |
Sc(OTf)3/EtOH/r.t. |
25 mg |
4 |
93 |
73 |
| 8 |
Cu(II)-DCC-CMK-3/solvent-free/80 °C |
30 mg |
30 min |
94 |
100 |
| 9 |
Al2(SO4)3/EtOH/reflux |
10 mol% |
4 |
92 |
101 |
| 10 |
Aluminized polyborate/solvent-free/100 °C |
75 mg |
20 min |
92 |
102 |
| 11 |
Mo@GAA-Fe3O4 MNPs/solvent-free/90 °C |
10 mg |
13 min |
95 |
This work |
4. Conclusion
In summary, the Mo@GAA-Fe3O4 MNPs were synthesized as an effective, recyclable, and environmentally friendly heterogeneous magnetic nanocatalyst that catalyzed the synthesis of biologically and pharmacologically interesting functionalized polyhydroquinoline derivatives under solvent-free conditions. The catalyst can be magnetically recovered from the reaction media by an external magnet and reused several times without any significant changes in the reaction efficiency. Moreover, high yields of products, short reaction times, lower loading of the catalyst, clean procedure, easy work-up, and heterogeneous reaction conditions are several advantages of this straightforward and efficient strategy.
Conflicts of interest
There are no conflicts to declare.
References
- G. A. Somorjai and J. Y. Park, Angew. Chem., Int. Ed., 2008, 47, 9212 CrossRef CAS.
- Y. Liu, J. Zhang, X. Zhang, B. Li, X. Wang, H. Cao, D. Wei, Z. Zhou and A. K. Cheetham, J. Mater. Chem. A, 2016, 4, 4280 RSC.
- G. Schmid, Nanoparticles: From Theory to Application, Wiley-VCH, Weinheim, 1st edn, 2004 Search PubMed.
- S. Duan, G. Han, Y. Su, X. Zhang, Y. Liu, X. Wu and B. Li, Langmuir, 2016, 32, 6272 CrossRef CAS PubMed.
- A. H. Lu, E. L. Salabas and F. Schuth, Angew. Chem., Int. Ed., 2007, 46, 1222 CrossRef CAS PubMed.
- C. Xing, Y. Liu, Y. Su, Y. Chen, S. Hao, X. Wu, X. Wang, H. Cao and B. Li, ACS Appl. Mater. Interfaces, 2016, 8, 15430 CrossRef CAS PubMed.
- K. Niknam and D. Saberi, Tetrahedron Lett., 2009, 50, 5210 CrossRef CAS.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef CAS PubMed.
- Y. Piao, Y. Jang, M. Shokouhimehr, I. S. Lee and T. Hyeon, Small, 2007, 3, 255 CrossRef CAS PubMed.
- M. Kidwai, A. Jain and S. Bhardwaj, Mol. Diversity, 2012, 16, 121 CrossRef CAS.
- X. F. Shen, Q. Wang, W. L. Chen and Y. H. Pang, Appl. Surf. Sci., 2014, 317, 1028 CrossRef CAS.
- D. P. Li, Y. R. Zhang, X. Zhao and B. X. Zhao, Chem. Eng. J., 2013, 232, 425 CrossRef CAS.
- Y. Y. Xu, M. Zhou, H. J. Geng, J. J. Hao, Q. Q. Ou, S. D. Qi, H. L. Chen and X. G. Chen, Appl. Surf. Sci., 2012, 258, 3897 CrossRef CAS.
- Y. R. Zhang, S. Q. Wang, S. L. Shen and B. X. Zhao, Chem. Eng. J., 2013, 233, 258 CrossRef CAS.
- T. Zeng, W. W. Chen, C. M. Cirtiu, A. Moores, G. Song and C. J. Li, Green Chem., 2010, 12, 570 RSC.
- M. Arruebo, R. Fernandez-Pacheco, M. R. Ibarra and J. Santamaria, J. Pharm. Sci., 2008, 97, 2948 CrossRef PubMed.
- R. Y. Hong, B. Feng, G. Liu, S. Wang and H. Z. Li, J. Alloys Compd., 2009, 476, 612 CrossRef CAS.
- L. Vayssières, C. Chanéac, E. Tronc and J. P. Jolivet, J. Colloid Interface Sci., 1998, 205, 205 CrossRef PubMed.
- S. Metz and W. Thiel, Coord. Chem. Rev., 2011, 255, 1085 CrossRef CAS.
- R. Hille, Dalton Trans., 2013, 42, 3029 RSC.
- R. Sanz and M. R. Pedrosa, Curr. Org. Synth., 2009, 6, 239 CrossRef CAS.
- K. Jeyakumar and D. K. Chand, J. Chem. Sci., 2009, 121, 111 CrossRef CAS.
- R. Sanz and M. R. Pedrosa, Adv. Org. Synth., 2012, 4, 183 Search PubMed.
- D. K. Chand and R. D. Chakravarthy, Molybdenum Chloride Oxide, in Encyclopedia of Reagents for Organic Synthesis, Wiley, 2012 Search PubMed.
- I. Sheikhshoaie, A. Rezaeifard, N. Monadi and S. Kaafi, Polyhedron, 2009, 28, 733 CrossRef CAS.
- M. Bagherzadeh, M. M. Haghdoost and A. Ghanbarpour, Inorg. Chim. Acta, 2014, 411, 61 CrossRef CAS.
- M. Masteri-Farahani and N. Tayyebi, J. Mol. Catal. A: Chem., 2011, 348, 83 CrossRef CAS.
- M. Masteri-Farahani and Z. Kashef, J. Magn. Magn. Mater., 2012, 324, 1431 CrossRef CAS.
- N. Divsalar, N. Monadi and M. Tajbakhsh, J. Nanostruct., 2016, 6, 312 CAS.
- N. Sadri, M. Moghadam and A. Abbasi, J. Mater. Sci.: Mater. Electron., 2018, 29, 11991 CrossRef CAS.
- R. Kakuchi, Angew. Chem., Int. Ed., 2014, 53, 46 CrossRef CAS PubMed.
- A. Domling and I. Ugi, Angew. Chem., Int. Ed., 1993, 32, 563 CrossRef.
- O. Kreye, T. Toth and M. A. R. Meier, J. Am. Chem. Soc., 2011, 133, 1790 CrossRef CAS PubMed.
- I. Bae, H. Han and S. Chang, J. Am. Chem. Soc., 2005, 127, 2038 CrossRef CAS PubMed.
- J. Zhu and H. Bienaymé, Multicomponent Reactions, Wiley-VCH, Weinheim, 2005 Search PubMed.
- W. H. Moos, C. R. Hurt and G. A. Morales, Mol. Diversity, 2009, 13, 241 CrossRef CAS PubMed.
- C. Lamberth, A. Jeanguenat, F. Cederbaum, A. De Mesmaeker, M. Zeller, H. J. Kempf and R. Zeun, Bioorg. Med. Chem., 2008, 16, 1531 CrossRef CAS PubMed.
- I. V. Magedov and A. Kornienko, Chem. Heterocycl. Compd., 2012, 48, 33 CrossRef CAS PubMed.
- I. Akritopoulou-Zanze, Curr. Opin. Chem. Biol., 2008, 12, 324 CrossRef CAS PubMed.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083 CrossRef PubMed.
- B. B. Touré and D. G. Hall, Chem. Rev., 2009, 109, 4439 CrossRef PubMed.
- A. Trivedi, D. Dodiya, B. Dholariya, V. Kataria, V. Bhuva and V. Shah, Chem. Biol. Drug Des., 2011, 78, 881 CrossRef CAS PubMed.
- V. Klusa, Drugs Future, 1995, 20, 135 CrossRef.
- Y. L. N. Murthy, A. Rajack, M. Taraka-Ramji, J. Jeson-Babu, C. Praveen and K. Aruna-Lakshmi, Bioorg. Med. Chem. Lett., 2012, 22, 6016 CrossRef CAS PubMed.
- C. Sun, Y. Chen, T. Liu, Y. Wu, T. Fang, J. Wang and J. Xing, Chin. J. Chem., 2012, 30, 1415 CrossRef CAS.
- A. Hilgeroth, Mini-Rev. Med. Chem., 2002, 2, 235 CrossRef CAS PubMed.
- D. J. Triggle, D. D. Langs and R. A. Janis, Med. Res. Rev., 1989, 9, 123 CrossRef CAS PubMed.
- M. Kawase, A. Shah, H. Gaveriya, N. Motohashi, H. Sakagami, A. Varga and J. Molnar, Bioorg. Med. Chem., 2002, 10, 1051 CrossRef CAS PubMed.
- R. P. Mason, I. T. Mak, M. W. Trumbore and P. E. Mason, Am. J. Cardiol., 1999, 84, 16 CrossRef.
- O. Aruoma, C. Smith, R. Cecchini, P. Evans and B. Halliwell, Biochem. Pharmacol., 1991, 42, 735 CrossRef CAS PubMed.
- Y. L. Chen, K. C. Fang, J. Y. Sheu, S. L. Hsu and C. C. Tzeng, J. Med. Chem., 2000, 44, 2374 CrossRef PubMed.
- G. Roma, M. D. Braccio, G. Grossi and M. Chia, Eur. J. Med. Chem., 2000, 35, 1021 CrossRef CAS.
- M. P. Maguire, K. R. Sheets, K. McVety, A. P. Spada and A. Zilberstein, J. Med. Chem., 1994, 37, 2129 CrossRef CAS PubMed.
-
(a) S. J. Ji, Z. Q. Jiang, J. Lu and T. P. Loh, Synlett, 2004, 5, 831 CrossRef;
(b) R. Sridhar and P. T. Perumal, Tetrahedron, 2005, 61, 2465 CrossRef CAS;
(c) B. X. Du, Y. L. Li, X. S. Wang, M. M. Zhang, D. Q. Shi and S. J. Tu, Chin. J. Org. Chem., 2006, 26, 698 CAS.
-
(a) L. Ohberg and J. Westman, Synlett, 2001, 8, 1296 CrossRef;
(b) A. Agarwal and P. M. S. Chauhan, Tetrahedron Lett., 2005, 46, 1345 CrossRef CAS;
(c) S. Tu, J. Zhang, X. Zhu, Y. Zhang, Q. Wang, J. Xu, B. Jiang, R. Jia, J. Zhang and F. Shi, J. Heterocycl. Chem., 2006, 43, 985 CrossRef CAS.
-
(a) A. P. J. Phillips, J. Am. Chem. Soc., 1949, 71, 4003 CrossRef CAS;
(b) G. Anderson and G. J. Berkelhammer, J. Am. Chem. Soc., 1958, 80, 992 CrossRef;
(c) H. Singh, D. S. S. Chimni and S. Kumar, Tetrahedron, 1995, 51, 12775 CrossRef CAS.
- L. M. Wang, J. Sheng, L. Zhang, J. W. Han, Z. Fan, H. Tian and C. T. Qian, Tetrahedron, 2005, 61, 1539 CrossRef CAS.
- S. Ko and C. F. Yao, Tetrahedron, 2006, 62, 7293 CrossRef CAS.
- G. Sabitha, G. S. K. Reddy, C. S. Reddy and J. S. Yadav, Tetrahedron Lett., 2003, 44, 4129 CrossRef CAS.
- B. Das, B. Ravikanth, R. Ramu and B. Vittal Rao, Chem. Pharm. Bull., 2006, 54, 1044 CrossRef CAS PubMed.
- R. Surasani, D. Kalita, A. V. Dhanunjaya Rao, K. Yarbagi and K. B. Chandrasekhar, J. Fluorine Chem., 2012, 135, 91 CrossRef CAS.
- M. Maheswara, V. Siddaiah, G. L. Damu and C. Venkata Rao, ARKIVOC, 2006, 2, 201 Search PubMed.
- A. Heydari, S. Khaksar, M. Tajbakhsh and H. R. Bijanzadeh, J. Fluorine Chem., 2009, 130, 609 CrossRef CAS.
- G. Song, B. Wang, X. Wu, Y. Kang and L. Yang, Synth. Commun., 2005, 35, 2875 CrossRef CAS.
- S. Ko, M. N. V. Sastry, C. Lin and C. F. Yao, Tetrahedron Lett., 2005, 46, 5771 CrossRef CAS.
- Y. Watanabe, K. Shiota, T. Hoshiko and S. Ozaki, Synthesis, 1983, 9, 761 CrossRef.
- H. Ahankar, A. Ramazani and S. W. Joo, Res. Chem. Intermed., 2016, 42, 2487 CrossRef CAS.
- M. M. Heravi, K. Bakhtiri, N. M. Javadi, F. F. Bamoharram, M. Saeedi and H. A. Oskooi, J. Mol. Catal. A: Chem., 2007, 264, 50 CrossRef CAS.
- A. Kumar and R. A. Maurya, Tetrahedron, 2007, 63, 1946 CrossRef CAS.
- A. Kumar and R. A. Maurya, Synlett, 2008, 6, 883 CrossRef.
- S. T. Fardood, A. Ramazani and S. Moradi, J. Sol-Gel Sci. Technol., 2017, 82, 432 CrossRef CAS.
- J. G. Brietenbucher and G. Figliozzi, Tetrahedron Lett., 2000, 41, 4311 CrossRef.
- J. L. Donelson, A. Gibbs and S. K. De, J. Mol. Catal. A: Chem., 2006, 256, 309 CrossRef CAS.
- M. Yarie, M. A. Zolfigol, Y. Bayat, A. Asgari, D. A. Alonso and A. Khoshnood, RSC Adv., 2016, 6, 82842 RSC.
- T. Tamoradi, M. Ghadermazi and A. Ghorbani-Choghamarani, Appl. Organomet. Chem., 2018, 32, e3974 CrossRef.
- C. G. Evans and J. E. Gestwicki, Org. Lett., 2009, 11, 2957 CrossRef CAS PubMed.
- A. Ghorbani-Choghamarani, B. Tahmasbi, P. Moradi and N. Havasi, Appl. Organomet. Chem., 2016, 30, 619 CrossRef CAS.
- G. J. Chen, J. W. McDonald and W. E. Newton, Inorg. Chem., 1976, 15, 2612 CrossRef CAS.
- E. Abbaspour-Gilandeh, S. C. Azimi and A. Mohammadi-Barkchai, RSC Adv., 2014, 4, 54854 RSC.
- E. Abbaspour-Gilandeh, M. Aghaei-Hashjin, A. Yahyazadeh and H. Salemi, RSC Adv., 2016, 6, 55444 RSC.
- E. Abbaspour-Gilandeh, A. Yahyazadeh and M. Aghaei-Hashjin, RSC Adv., 2018, 8, 40243 RSC.
- A. Khorshidi, K. Tabatabaeian, H. Azizi, M. Aghaei-Hashjin and E. Abbaspour-Gilandeh, RSC Adv., 2017, 7, 17732 RSC.
- A. Yahyazadeh, E. Abbaspour-Gilandeh and M. Aghaei-Hashjin, Catal. Lett., 2018, 148, 1254 CrossRef CAS.
- K. Rad-Moghadam, S. C. Azimi and E. Abbaspour-Gilandeh, Tetrahedron Lett., 2013, 54, 4633 CrossRef CAS.
- A. Maleki, M. Kamalzare and M. Aghaei, J. Nanostruct. Chem., 2015, 5, 95 CrossRef CAS.
- H. N. Peng, D. G. Zheng and X. M. Peng, Asian J. Chem., 2011, 23, 1833 CAS.
- X. Y. Qin, T. S. Jin, Z. X. Zhou and T. S. H. Li, Asian J. Chem., 2010, 22, 1179 CAS.
- A. R. Moosavi-Zare, M. A. Zolfigol, M. Zarei, A. Zare and J. Afsar, Appl. Catal., A, 2015, 505, 224 CrossRef CAS.
- M. Nasr-Esfahani, D. Elhamifar, T. Amadeh and B. Karimi, RSC Adv., 2015, 5, 13087 RSC.
- A. Zare, F. Abi, A. R. Moosavi-Zare, M. H. Beyzavi and M. A. Zolfigol, J. Mol. Liq., 2013, 178, 113 CrossRef CAS.
- M. Nasr-Esfahani, S. J. Hoseini, M. Montazerozohori, R. Mehrabi and H. Nasrabadi, J. Mol. Catal. A: Chem., 2014, 382, 99 CrossRef CAS.
- S. B. Sapkal, K. F. Shelke, B. B. Shingate and M. Shingare, Tetrahedron Lett., 2009, 50, 1754 CrossRef CAS.
- C. S. Reddy and M. Raghu, Chin. Chem. Lett., 2008, 19, 775 CrossRef CAS.
- B. Maleki, R. Tayebee, M. Kermanian and S. Ashrafi, J. Mex. Chem. Soc., 2013, 57, 290 CAS.
- R. S. Amit and S. G. Gavisiddappa, Open Catal. J., 2009, 2, 61 CrossRef.
- F. Soleimani, M. Salehi and A. Gholizadeh, Iran. J. Polym. Sci. Technol., 2020, 44, 1011 CrossRef.
- F. K. Behbahani and M. Homafar, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2012, 42, 291 CrossRef CAS.
- S. U. Tekale, V. P. Pagore, S. S. Kauthale and R. P. Pawar, Chin. Chem. Lett., 2014, 25, 1149 CrossRef CAS.
- N. N. Karade, V. H. Budhewar, S. V. Shinde and W. N. Jadhav, Lett. Org. Chem., 2007, 4, 16 CrossRef CAS.
- R. Ghafouri-Nejad and M. Hajjami, React. Kinet., Mech. Catal., 2020, 129, 371 CrossRef CAS.
- P. Kulkarni and J. Chil, Chem. Soc., 2014, 59, 2319 CAS.
- D. Aute, A. Kshirsagar, B. Uphade and A. Gadhave, Res. Chem. Intermed., 2020, 46, 3491 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b
*b