
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFTIR product study of the Cl-initiated oxidation products of CFC replacements: (E/Z)-1,2,3,3,3-pentafluoropropene and hexafluoroisobutylene
Cynthia B. Rivelaa,
Rodrigo G. Gibiliscob,
Carmen M. Tovarb,
Ian Barnes†
b,
Peter Wiesenb,
María B. Blanco a and
Mariano A. Teruel*a
a and
Mariano A. Teruel*a
aInstituto de Investigaciones en Fisicoquímica de Córdoba (INFIQC), CONICET, Dpto. de Fisicoquímica, Facultad de Ciencias Químicas, Universidad Nacional de Córdoba, Ciudad Universitaria, 5000 Córdoba, Argentina. E-mail: mteruel@fcq.unc.edu.ar
bPhysikalische Chemie/FBC, Bergische Universitaet Wuppertal, Wuppertal, Germany
First published on 1st April 2021
Abstract
A product study of the reactions of (E/Z)-1,2,3,3,3-pentafluoropropene ((E/Z)-CF3CF![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHF) and hexafluoroisobutylene ((CF3)2CCH2) initiated by Cl atoms were developed at 298 ± 2 K and atmospheric pressure. The experiments were carried out in a 1080 L quartz-glass environmental chamber coupled via in situ FTIR spectroscopy to monitor the reactants and products. The main products observed and their yields were as follows: CF3C(O)F (106 ± 9)% with HC(O)F (100 ± 8)% as a co-product for (E/Z)-CF3CFCHF, and CF3C(O)CF3 (94 ± 5)% with HC(O)Cl (90 ± 7)% as a co-product for (CF3)2CCH2. Atmospheric implications of the end-product degradation are assessed in terms of their impact on ecosystems to help environmental policymakers consider HFOs as acceptable replacements.
CHF) and hexafluoroisobutylene ((CF3)2CCH2) initiated by Cl atoms were developed at 298 ± 2 K and atmospheric pressure. The experiments were carried out in a 1080 L quartz-glass environmental chamber coupled via in situ FTIR spectroscopy to monitor the reactants and products. The main products observed and their yields were as follows: CF3C(O)F (106 ± 9)% with HC(O)F (100 ± 8)% as a co-product for (E/Z)-CF3CFCHF, and CF3C(O)CF3 (94 ± 5)% with HC(O)Cl (90 ± 7)% as a co-product for (CF3)2CCH2. Atmospheric implications of the end-product degradation are assessed in terms of their impact on ecosystems to help environmental policymakers consider HFOs as acceptable replacements.
1 Introduction
The adverse effects of chlorofluorocarbons (CFCs) on the stratospheric ozone layer are known and have been extensively studied.1,2 Several viable CFC replacements have been proposed in the last decade, such as hydrofluorocarbons (HFCs), hydrofluoroethers (HFEs), and hydrofluoroalcohols (HFAs). Moreover, recently, hydrofluoroolefins (HFOs) have been proposed as the fourth-generation replacements for CFCs.3 Hydrofluoroolefins (HFOs) have been proposed as replacements mainly of refrigerants.4,5 In addition, HFOs have numerous other industrial applications as foaming agents, aerosol propellants, mobile air conditioning systems, precision solvent cleaners, MDIs (metered dose inhalers), medical propellants, coatings, and as basic components in the production of fluoropolymers, etc.4,6,7 HFOs are known to be highly reactive due to the presence of a C–C double bond, resulting in short atmospheric lifetimes and therefore zero ozone depletion potentials (ODP) and minimized global warming potentials (GWP).Several experiments determined that the reactions of fluorinated alkenes in the troposphere are mainly controlled by OH radicals,8,9 while oxidation via Cl atoms can also be an important homogeneous loss process at dawn in coastal or marine regions and has also been observed in the central and urban areas.10,11
This present work aims to perform a detailed study on the atmospheric degradation mechanism of the reaction of E/Z-isomeric mixture of 1,2,3,3,3-pentafluoropropene (PFP) and hexafluoroisobutylene (HFIB) initiated by Cl atoms.
Only one product study related to OH and Cl-initiated oxidation of individual (Z) and (E) isomers of 1,2,3,3,3-pentafluoropropene has been reported in the literature using a Pyrex reactor and the FTIR technique as a detection system.12 The authors have reported for both Cl and OH radical-initiated oxidations of CF3CFCHF, and the presence of CF3C(O)F and HC(O)F in 100% indistinguishable molar yields for both (Z) and (E) isomers.
Only a product identification study of the reactions of hexafluoroisobutylene with OH radicals and Cl atoms was previously reported.13 This study was performed at 296 ± 1 K and atmospheric pressure using a Pyrex reactor and FTIR spectroscopy to monitor the reactants and products. HC(O)H and CF3C(O)CF3 were identified as final oxidation products in both OH- and Cl-initiated oxidation reactions, while HC(O)Cl, HCl, and CO were observed in the Cl-initiated oxidation reaction. In addition, a theoretical study of the reaction of hexafluoroisobutylene (HFIB) with OH radicals has been performed, where two different reaction channels of HFIB with OH radicals were postulated: addition to the ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CC
CC![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) bond and one H-abstraction reaction channel. These authors reported three possible reaction pathways for this reaction: two addition channels associated with the addition of OH radicals to the CC bond and the abstraction of the H atom from the –CH2 group. They further observed that while the addition channels dominate the kinetics of this reaction, the contribution of the H abstraction channel cannot be completely neglected. This observation was particularly shown at the high temperature range of 250–1000 K used this study.9
bond and one H-abstraction reaction channel. These authors reported three possible reaction pathways for this reaction: two addition channels associated with the addition of OH radicals to the CC bond and the abstraction of the H atom from the –CH2 group. They further observed that while the addition channels dominate the kinetics of this reaction, the contribution of the H abstraction channel cannot be completely neglected. This observation was particularly shown at the high temperature range of 250–1000 K used this study.9
To the best of our knowledge, previous product quantification studies of the reactions of Cl atoms with the isomeric mixture (E/Z) of 1,2,3,3,3-pentafluoropropene, or for hexafluoroisobutylene, are not available in literature. Consequently, the current work is the first determination of the product distribution for the title reactions.
Before the widespread use of these species, it is essential to investigate the potential environmental effects of its degradation products. For this purpose, the reaction products of the Cl atom-initiated gas phase degradation of the E/Z-isomeric mixture of 1,2,3,3,3-pentafluoropropene (PFP) and hexafluoroisobutylene (HFIB) have been studied:
|
(E/Z)-CF3CFCHF + Cl → products
| (1) |
|
(CF3)2CCH2 + Cl → products
| (2) |
2 Experimental
The product studies were performed in a quartz chamber of 1080 L capacity in synthetic air at 298 ± 2 K and 760 Torr coupled with in situ FTIR spectroscopy. The reactor consists of two quartz glass container cylinders, each 3 m long and 45 cm internal diameter, joined in the middle with both open ends closed by aluminum flanges. The photolysis system consists of 32 super actinic black lamps (Philips TL05 40 W: 320–480 nm, λmax = 360 nm) and 32 low-pressure mercury vapor germicidal lamps (Philips TUV 40 W; λmax = 254 nm). The quartz chamber reactor has been used previously at the Wuppertal laboratory, and more relevant details are described by Barnes et al.14,15Chlorine atoms were generated by the photolysis of molecular chlorine (Cl2) using the black lamps:
| Cl2 + hν (λ = ∼360 nm) → 2Cl˙ | (3) |
The initial mixing ratios used in the experiments for HFOs in ppm (1 ppm = 2.46 × 1013 molecule cm3 at 298 K and 760 Torr total pressure) were 1.5 ppm for 1,2,3,3,3-pentafluoropropene, (E/Z) mixture and 1.5 ppm of hexafluoroisobutylene. The initial concentration for the precursor of the oxidant was typically around 6 ppm of Cl2. The infrared absorption frequencies at 1407.8 and 1383.7 cm−1 were used to monitor the concentration–time behavior of 1,2,3,3,3-pentafluoropropene, (E/Z) mixture and hexafluorobutylene, respectively. Readily identifiable products were monitored at the following absorption frequencies (in cm−1): CF3C(O)F at 1897.7; HC(O)F at 1837.7; CF3C(O)CF3 at 974.7 and HC(O)Cl at 738.6.
The chemicals used in the experiments had the following purities as given by the manufacturer and were used as supplied: synthetic air (Air Liquide, 99.999%); 1,2,3,3,3-pentafluoropropene (Apollo Scientific, mixture (E/Z) isomers 97%, no manufacture details on isomer ratio), hexafluorobutylene (Apollo Scientific, purity not specified), and Cl2 (Messer Griesheim, ≥99.8%). Wall deposition, reaction with the precursor of Cl, or photolysis was found to be negligible for both HFOs studied.
3 Results and discussion
To investigate the oxidation mechanism of HFOs initiated by Cl atoms, a mixture of HFO, Cl2, and air was irradiated for the time period of 10–30 minutes, during which infrared spectra were recorded on an FTIR spectrometer. Typically, up to 128 interferograms were co-added per spectrum for approximately 40 s, and 15–20 spectra were collected. Prior to the initiation of the reaction by Cl atoms, 5 spectra were collected in dark to check the homogeneity and the unexpected dark decay of the compounds (e.g., wall losses or dark reactions).(E/Z)-isomeric mixture of 1,2,3,3,3-pentafluoropropene + Cl reaction
In Fig. 1, a schematic representation of the expected reaction mechanism of PFP with Cl atoms and the obtained products, trifluoroacetyl fluoride (CF3C(O)F) and formyl fluoride (HC(O)F), is presented. The proposed mechanism suggests that the Cl atom could be added to the carbon atom C1 or C2 of the double bond, followed by the subsequent reactions with O2 to form with further 1,2-chloroalkoxy radical production. This radical formed could decompose to give CF3C(O)F and HC(O)F.
with further 1,2-chloroalkoxy radical production. This radical formed could decompose to give CF3C(O)F and HC(O)F.
 | ||
| Fig. 1 Reaction mechanism of (E/Z)-1,2,3,3,3-pentafluoropropene with Cl atoms via the addition to the double bond. | ||
Fig. 2 trace A shows the infrared spectrum of a PFP/Cl2/air reaction mixture after irradiation and subtraction of residual PFP. The spectra of the identified products are presented in Fig. 2, where trace B shows a reference spectrum of trifluoroacetyl fluoride and trace C shows a formyl fluoride spectrum. Trace D shows the residual spectrum obtained after the subtraction of the spectra in traces A, B, and C. The presence of unidentified absorption bands was negligible in the residual product spectrum. Therefore, it is assumed that trifluoroacetyl fluoride and formyl fluoride are the main reaction products of PFP with Cl atoms. This result is in agreement with results obtained in previous studies on the atmospheric degradation of PFP.16,17 The concentration–time profiles of PFP and its identified products are shown in Fig. 3. This graph supports the result that trifluoroacetyl fluoride and formyl fluoride are both primary products.
 | ||
| Fig. 2 Trace A shows the infrared spectrum obtained from a PFP/Cl2/air reaction mixture after UV irradiation. Traces B and C show the reference spectra of trifluoroacetyl fluoride and formyl fluoride, respectively. Trace D shows the residual product spectrum obtained after the subtraction of features due to the reference spectra from the spectrum in trace A. | ||
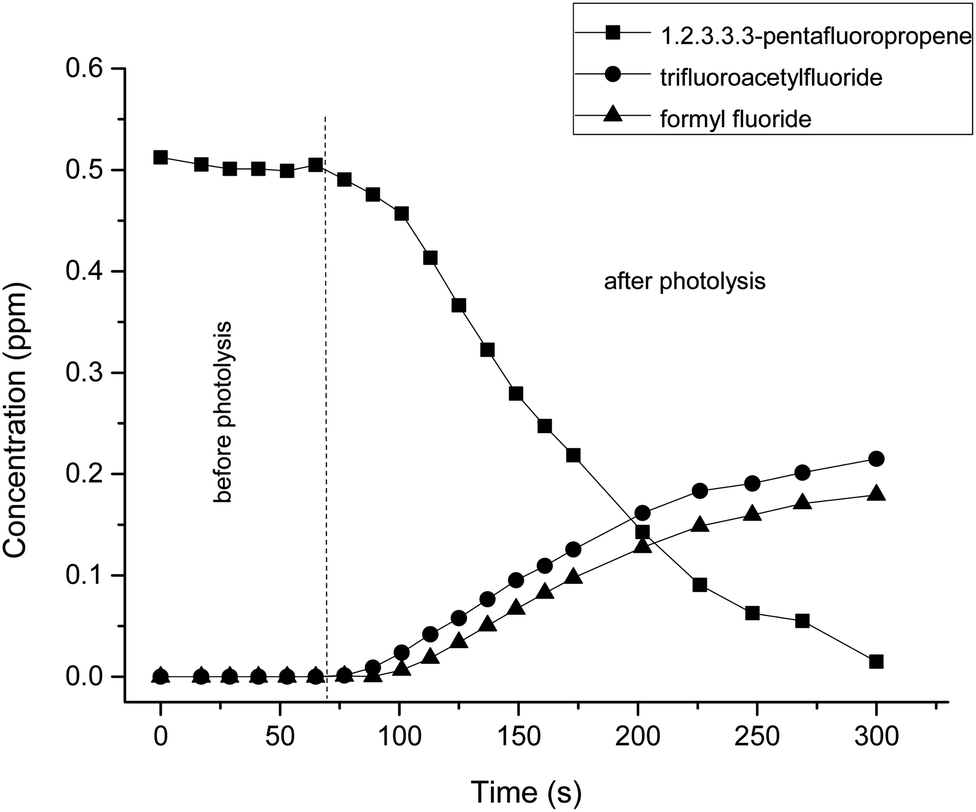 | ||
| Fig. 3 Concentration–time profiles of PFP and the reaction products, trifluoroacetyl fluoride and formyl fluoride, obtained from the UV photolysis of the PFP/Cl2/air reaction mixture. | ||
In Fig. 4, the concentrations of the products formed versus the consumed PFP in the reaction with Cl atoms show reasonable linearity. The average product yields obtained from the two experiments are CF3C(O)F (106 ± 9)% and HC(O)F (100 ± 8)% (Table 1). These results indicate that the products formed are the result of the decomposition of the CF3CF(Cl)COHF˙ and CF3COF˙CHF(Cl) radicals formed via the C1 and C2 bond scission.
 | ||
| Fig. 4 Plots of the concentrations of the reaction products, trifluoroacetyl fluoride and formyl fluoride, as a function of reacted PFP obtained from the UV photolysis of PFP/Cl2/air reaction mixtures. | ||
| HFOs | Product | Yield (%) |
|---|---|---|
| a The errors quoted are 2σ statistical errors from the linear regression analysis.b This work.c Hurley et al.18d Papadimitriou et al.16 | ||
| (E/Z)-CF3CFCHF |
CF3C(O)F | (106 ± 9)b |
| HC(O)F | (100 ± 8)b | |
| (E)-CF3CFCHF |
CH3C(O)F | (98 ± 4)c |
| (Z)-CF3CFCHF |
HC(O)F | (100 ± 5)c |
| (Z)-CF3CFCHF |
CH3C(O)F | (108 ± 8)d |
| HC(O)F | (112 ± 8)d | |
| (CF3)2CCH2 |
CF3C(O)CF3 | (94 ± 5)b |
| HC(O)Cl | (90 ± 7)b | |
The same results are expected for both (E) and (Z) isomers in the mixture of 1,2,3,3,3-pentafluoropropene.
Previous studies for the reactions of the (Z) and (E) isomers of CF3CFCHF initiated by OH radicals, Cl atoms, and O3 molecules by Hurley et al.18 using a 140 L Pyrex reactor interfaced to a Mattson Sirius 100 FTIR spectrometer reported yields of (98 ± 4)% for CF3C(O)F and (100 ± 5)% for HC(O)F for the Cl reactions with both CF3CFCHF isomers. These results are in agreement with those obtained in the present study. Furthermore, Papadimitriou et al.16 reported rate coefficients and product yields for the gas-phase reactions of Cl atoms and NO3 radicals with (Z)-CF3CFCHF using a temperature-regulated Pyrex reactor with a pulsed excimer laser photolysis source to produce Cl atoms and FTIR to monitor the reactant and product concentrations. The experiments were carried out in the presence of O2 over a range of temperatures (247–380) K and pressures ranging from (50–630) Torr. As a result, the authors observed that the reaction was independent of temperature and pressure. The product yields obtained were (108 ± 8)% for CF3C(O)F and (112 ± 8)% for HC(O)F. The present study is in excellent agreement with all previous determinations for the reactions of similar HFOs from the literature.
Hexafluoroisobutylene + Cl reaction
In the same way as the reaction described above, the reaction of Cl atoms with HFIB follows the addition of the Cl atom to the C1–C2 double bond to form chloroalkyl radicals, which react rapidly under experimental conditions with O2 to form the peroxy radicals, and after further reduction, chloroalkoxy radicals are formed. The proposed reaction mechanism is displayed in Fig. 5. | ||
| Fig. 5 Reaction mechanism of hexafluoroisobutylene with Cl atoms via the addition to the double bond. | ||
Fig. 6 trace A shows the infrared spectrum of an HFIB/Cl2/air mixture recorded after UV irradiation and subtraction of residual absorptions of HFIB. The presence of the CO vibration band centered at 2140 cm−1 can also be observed. Fig. 6 trace B shows a reference spectrum of hydrochloric acid (HCl), trace C is the reference formyl chloride (HC(O)Cl) spectrum, trace D is the hexafluoroacetone reference spectrum, and trace E is the residual product spectrum after the subtraction of all four identified products from trace A. In the residual spectrum, the unidentified absorption bands were negligible. No other reaction products were expected, and again, these results are consistent with the previous study of the reaction of HFIB initiated by Cl atoms rather than atmosphere.13
 | ||
| Fig. 6 Trace A shows the infrared product spectrum obtained from the UV photolysis of the HFIB/Cl2/air reaction mixture. Traces B, C, and D show the reference spectra of hydrochloric acid, hexafluoroacetone, and formyl chloride, respectively. Trace E shows the residual product spectrum obtained after the subtraction of features due to the reference spectra from the spectrum in trace A. | ||
The CO and HCl infrared absorptions observed could be due to the rapid thermal decomposition of HC(O)Cl via a reaction in the walls. This has been previously observed by Libuda et al.19 and Papadimitriou et al.13 This decomposition can also be observed in the concentration–time profiles shown in Fig. 7. In Fig. 8, the concentrations of carbonyls formed versus the consumed HFIB in the reaction with Cl atoms show reasonable linearity. The average product yields obtained from the two experiments are CF3C(O)CF3 (94 ± 5)% and HC(O)Cl (90 ± 7)% (Table 1).
 | ||
| Fig. 7 Concentration–time profiles of hexafluoroisobutylene and the reaction products, hexafluoroacetone and formyl chloride, obtained from the UV photolysis of the HFIB/Cl2/air reaction mixture. | ||
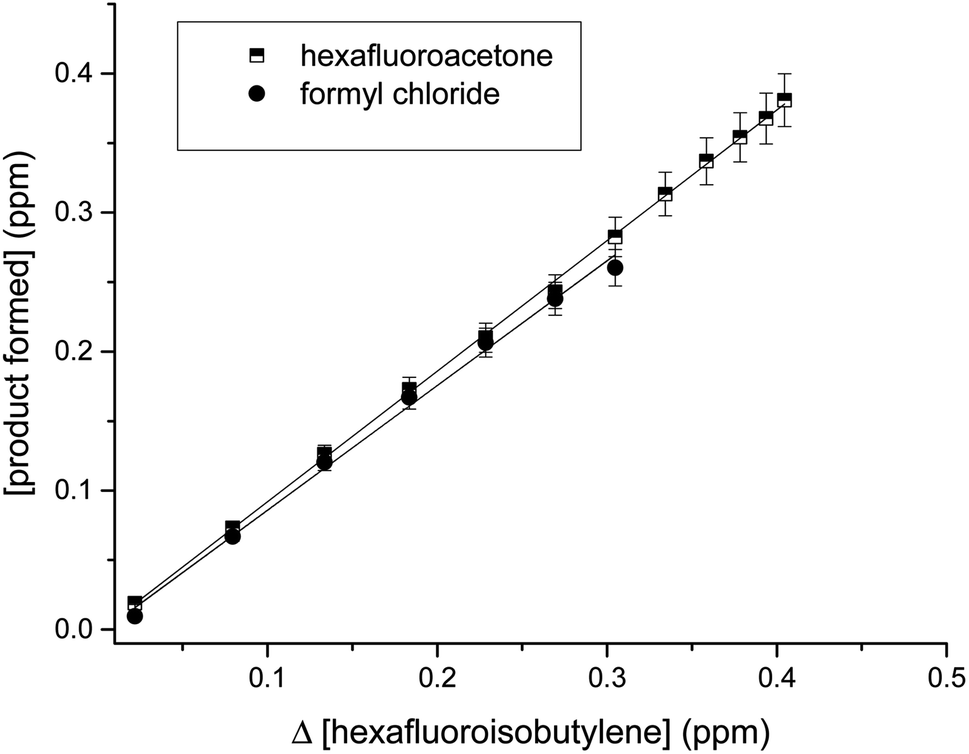 | ||
| Fig. 8 Plots of the concentrations of the reaction products, formyl chloride and hexafluoroacetone, as a function of the reacted hexafluoroisobutylene obtained from the UV photolysis of HFIB/Cl2/air reaction mixtures. | ||
In a previous product identification study performed by Papadimitriou et al., the experiments were carried out under atmospheric conditions in two different collapsible Teflon FEP reaction chambers of 7300 L and 140 L, respectively. Chlorine atoms were generated by the photolysis of molecular chlorine using fluorescent lamps, which provided radiation sources centered at 365 nm. The reagents and oxidation products were detected using a multiple reflection target-type mirror system (path length 143 m), which is mounted inside the reactor and connected to an FT-IR spectrometer (Nicolet Magna 5700).13 CF3C(O)CF3, HC(O)Cl, HCl, and CO were identified as HFIB degradation products in the reaction with Cl atoms, although no product yields were reported. The experiments were carried out at room temperature and atmospheric pressure using two atmospheric simulation chambers and a static photochemical reactor. The identified products are in accordance with the identified and quantified products reported in this study. To the best of our knowledge, no other study has been conducted on the atmospheric degradation mechanism of HFIB with Cl atoms. Consequently, this is the first product quantification study of the reaction of HFIB with Cl atoms.
The end-products found in both HFO reactions with Cl atoms suggest that all reactions proceed mainly via an electrophilic addition to the double bond. Subsequently, in the absence of NOx, the alkyl radicals formed react with O2 to form peroxy radicals. These peroxy radicals will undergo self- and cross-peroxy reactions, which will result in the formation of 1,2-chloroalkoxy radicals, as suggested by Calvert et al. for the atmospheric oxidation of alkenes.20 This behavior is consistent with a previous study that reported the degradation reactions of similar HFOs with the atmospheric oxidants.21–27
Atmospheric implications
Atmospheric lifetimes of 1,2,3,3,3-pentafluoropropene (PFP) and hexafluoroisobutylene (HFIB) with OH radicals and Cl atoms have been previously reported by our research group,28,29 and they were also measured by Papadimitriou et al. and Tokuhashi et al.13,30 Considering these results, it was possible to observe that the reaction with OH radicals is the main atmospheric sink of these species (few days). Besides, other sinks could also be relevant, such as reactions with Cl (days) mainly in areas with significant concentrations of Cl.10,11,31 It is also known that ozonolysis reactions generally represent a minor or insignificant atmospheric loss for halo-olefins.32 The atmospheric lifetimes of both compounds were calculated in previous studies, and the values obtained were τOH = 4 and τCl = 51 days for PFP and τOH = 18 and τCl = 66 days for HFIB by Tovar et al.28 Moreover, the values obtained by Rivela et al.29 were τOH = 2 and τCl = 18 days for PFP, and τOH = 9 and τCl = 33 days for HFIB. Therefore, it is expected that HFOs will react close to their emission sources and have a local impact due to their short lifetimes. OH-initiated degradation will be the main atmospheric sink of these species, although in industrial areas or marine troposphere where Cl atoms could reach a peak, the Cl-initiated oxidation could be a significant sink of HFO studied.Degradation products found in the studied reactions (1) and (2): HC(O)Cl and HC(O)F have relatively small reaction rate coefficients towards OH radicals (3.2 × 10−13 and <5 × 10−15 cm−3 per molecule per s).19,33 Thus, it is reasonable to assume that this sink is of minor importance for their atmospheric removal. Both compounds are expected to hydrolyze when in contact with cloud–rain–seawater on a time scale of ∼10–15 days. HC(O)F hydrolyzes to give HC(O)OH and HF,34 while in the case of HC(O)Cl, it could be hydrolyzed and removed by wet deposition in ∼5–15 days. The hydrolysis of HC(O)Cl also gives HC(O)OH, which is a ubiquitous component of the environment.35
CH3C(O)F (acetyl fluoride) quantum photodissociation yields are relatively low, giving an averaged atmospheric lifetime in the order of 41 years. Therefore, it is possible to assume that photolysis does not represent a significant atmospheric removal process. The small rate coefficient of the reaction with OH radicals of kCH3C(O)F = (0.74 ± 0.05) × 10−14 cm3 per molecule per s gives an estimated atmospheric lifetime of 4.3 years. These results also suggest a negligible contribution to the atmospheric removal of acetyl fluoride.36,37 Tropospheric removal of acetyl fluoride is expected to occur primarily by absorption in the aqueous phase with an estimated lifetime of 30 days.38 Acetyl fluoride hydrolyzes in cloud droplets to produce CH3C(O)OH and HF. Although it is known that CH3C(O)OH is ubiquitous, abundant in the troposphere, and one of the major contributors to cloud water acidity in remote regions,39 while HF is not considered a main air-pollutant in the EU, and they are among the substances normally considered as potential contributors to acidification.40 On the other hand, a recent study of Lindley et al. concerning the emissions of hydrofluorocarbons (HFCs), hydrofluoroolefins (HFOs), and hydrochlorofluoroolefins (HCFOs) concluded that by 2030 these compounds would make a negligible contribution to acidification.41
CF3(O)CF3 will most likely lead to the formation of trifluoroacetic acid (TFA), and although it cannot alter the global atmospheric amount, it is important to identify and quantify its sources.42,43 Probably, this fluorinated VOC will be removed via heterogeneous wet and dry deposition processes. On the other hand, its photolysis generates CF3 and CO. CF3 radicals further will be oxidized to produce CF2O and subsequently be hydrolyzed to HF and CO2.44
These compounds have high vapor pressures, and most of them are gases at 25 °C. Consequently, these materials will not accumulate in water and will partition rapidly into air. Furthermore, it is known that the reactions of HFOs in the atmosphere occur primarily with OH radicals where they are removed from the atmosphere in the order of days.8,9 This phenomenon occurs in marine and coastal regions and in urban areas as well, where the oxidation through Cl atoms could be a significant loss.45–47
For the aforementioned, HFOs are not expected to enter aquatic environments, including aquifers, which would prevent their pollution.48
The short-chain HFOs studied in this work have short atmospheric lifetimes, in the order of days, when reacting with the main atmospheric oxidants (OH/Cl). Their ability to contribute to the ozone photochemical potentials (POCP) is negligible as well as the ozone depletion potential (ODP), the global warming potentials (GWP), and the contribution to rain acidification. As for the primary oxidation products, they are benign, and they would have an unimportant impact on ecosystems.49,50
This study aims to provide more information about the degradation mechanism of the HFOs mentioned above and the possible environmental impact of their final degradation products. This is necessary since HFOs have been proposed for several industrial uses, such as eco-friendly refrigerants, aerosol propellants, and the production of fluoropolymers.4,6,7
Further experiments to study the product distribution of PFP and HFIB in the presence of NOx would be desirable to compare degradation mechanisms and performance of the reaction pathways with the chemistry of polluted areas and with large amounts of NOx.
4 Conclusions
For the first time, in this study, the degradation products of the reactions of Cl atoms with hydrofluoroolefins (HFOs), isomeric mixture (E/Z)-1,2,3,3,3-pentafluoropropene and hexafluoroisobutylene, in atmospheric conditions have been studied. The end-products observed and their yields were: CF3C(O)F (106 ± 9)% with HC(O)F (100 ± 8)% as a co-product for (E/Z)-1,2,3,3,3-pentafluoropropene, and CF3C(O)CF3 (94 ± 5)% with HC(O)Cl (90 ± 7)% as a co-product for hexafluoroisobutylene. The proposed mechanism for both compounds suggest that the Cl atom could be added to the C1 or C2 carbon atom of the double bond, followed by subsequent reactions with O2 to give with additional production of 1,2-chloroalkoxy radicals. The radicals formed could be decomposed to give the corresponding products. However, for the HFIB + Cl reaction, the formation of CO + HC(O)Cl gives values of approximately 100%, indicating that chlorine atoms attacked the C1 of the CC bond preferentially.
with additional production of 1,2-chloroalkoxy radicals. The radicals formed could be decomposed to give the corresponding products. However, for the HFIB + Cl reaction, the formation of CO + HC(O)Cl gives values of approximately 100%, indicating that chlorine atoms attacked the C1 of the CC bond preferentially.
It is known that short-chain halo-olefins have a short atmospheric life in the order of days when they react with OH radicals or Cl atoms. Therefore, their ability to contribute towards photochemical ozone potentials, global warming potentials, and acidification contribution from rain are negligible. With respect to the final oxidation products, as discussed above, short-chain halo-olefins would not have a significant impact on ecosystems.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
C. B. R. wishes to acknowledge CONICET for a doctoral fellowship and support. R. G. G. and M. B. B. wish to acknowledge the Alexander von Humboldt Foundation (AvH). The authors wish to acknowledge Deutsche Forschungsgemeinschaft (DFG), DAAD-PROALAR (Germany), the EU project EUROCHAMP2, SECYT (Argentina), and CONICET (Argentina) for financial support of this research.References
- M. J. Molina and F. S. Rowland, Predicted present stratospheric abundances of chlorine species from photodissociation of carbon tetrachloride, Geophys. Res. Lett., 1974, 1(7), 309–312 CrossRef CAS.
- S. A. Montzka, S. Reimannander, A. Engel, K. Kruger, W. T. Sturges, D. R. Blake, M. Dorf Dorf, P. Fraser, L. Froidevaux, K. Jucks, K. Kreher, M. Kurylo III, A. Mellouki, J. Miller, O. Nielsen, V. Orkin, R. Prinn, R. Rhew, M. Santee, M. Stohl and D. Verdonik, Ozone-Depleting Substances (ODSs) and Related Chemicals, in Scientific Assessment of Ozone Depletion: 2010, Global Ozone Research and Monitoring Project-Report No. 52, World Meteorological Organization, Geneva, Switzerland, 2011, ch. 1, p. 516 Search PubMed.
- J. S. Brown, Introduction to hydrofluoro-olefin alternatives for high global warming potential hydrofluorocarbon refrigerants, HVACR Res., 2013, 19(6), 693–704 CAS.
- J. S. Brown, HFOs: new, low global warming potential refrigerants, ASHRAE J., 2009, 51(8), 22 Search PubMed.
- Y. Fang, S. Croquer, S. Poncet, Z. Aidoun and Y. Bartosiewicz, Drop-in replacement in a R134 ejector refrigeration cycle by HFO refrigerants, Int. J. Refrig., 2017, 77, 87–98 CrossRef CAS.
- P. J. Atkins, Dry powder inhalers: an overview, Respir. Care, 2005, 50(10), 1304–1312 Search PubMed.
- G. G. Belen'Kii, V. A. Petrov, S. A. D. Postovoi, P. R. Resnick and Y. V. Zeifman, US Pat., 6703533, U.S. Patent and Trademark Office, Washington, DC, 2004 Search PubMed.
- E. Jiménez, S. González, M. Cazaunau, H. Chen, B. Ballesteros, V. Daële, J. Albaladejo and A. Mellouki, Atmospheric Degradation Initiated by OH Radicals of the Potential Foam Expansion Agent, CF3(CF2)2CH=CH2 (HFC-1447fz): Kinetics and Formation of Gaseous Products and Secondary Organic Aerosols, Environ. Sci. Technol., 2016, 50(3), 1234–1242 CrossRef PubMed.
- B. Baidya and A. K. Chandra, Theoretical investigation on atmospheric chemistry of (CF3)2C=CH2: reaction with OH radical, Chem. Phys. Lett., 2020, 749, 137409 CrossRef CAS.
- J. Thornton, J. Kercher, T. Riedel, L. Wagner, J. Cozic, J. Holloway, W. Dubé, G. Wolfe, P. Quinn, A. Middlebrook, B. Alexander and S. Brown, A large atomic chlorine source inferred from mid-continental reactive nitrogen chemistry, Nature, 2010, 464(7286), 271 CrossRef CAS PubMed.
- C. B. Faxon and D. T. Allen, Chlorine chemistry in urban atmospheres: a review, Environ. Chem., 2013, 10(3), 221–233 CrossRef CAS.
- T. Nakayama, K. Takahashi, Y. Matsumi, A. Toft, M. P. Sulbaek Andersen, O. J. Nielsen, R. L. Waterland, R. C. Buck, M. D. Hurley and T. J. Wallington, Atmospheric Chemistry of CF3CH=CH2 and C4F9CH=CH2: Products of the Gas-Phase Reactions with Cl Atoms and OH Radicals, J. Phys. Chem. A, 2007, 111(5), 909–915 CrossRef CAS PubMed.
- V. C. Papadimitriou, C. S. Spitieri, P. Papagiannakopoulos, M. Cazaunau, M. Lendar, V. Daële and A. Mellouki, Atmospheric chemistry of (CF3)2C=CH2: OH radicals, Cl atoms and O3 rate coefficients, oxidation end-products and IR spectra, Phys. Chem. Chem. Phys., 2015, 17(38), 25607–25620 RSC.
- I. Barnes, K. H. Becker and T. Zhu, Near UV absorption spectra and photolysis products of difunctional organic nitrates: possible importance as NOx reservoirs, J. Atmos. Chem., 1993, 17(4), 353–373 CrossRef CAS.
- I. Barnes, K. H. Becker and N. Mihalopoulos, An FTIR product study of the photooxidation of dimethyl disulfide, J. Atmos. Chem., 1994, 18(3), 267–289 CrossRef CAS.
- V. C. Papadimitriou, Y. G. Lazarou, R. K. Talukdar and J. B. Burkholder, Atmospheric Chemistry of CF3CF=CH2 and (Z)-CF3CF=CHF: Cl and NO3 Rate Coefficients, Cl Reaction Product Yields, and Thermochemical Calculations, J. Phys. Chem. A, 2011, 115(2), 167–181 CrossRef CAS PubMed.
- C. B. Rivela, C. M. Tovar, R. Gibilisco, M. A. Teruel, I. Barnes, P. Wiesen and M. B. Blanco, Product distribution and mechanism of the OH – initiated tropospheric degradation of three CFC replacement candidates: CH3CF=CH2, (CF3)2C=CH2 and (E/Z)-CF3CF=CHF, RSC Adv., 2019, 9(10), 5592–5598 RSC.
- M. D. Hurley, J. C. Ball and T. J. Wallington, Atmospheric Chemistry of the Z and E Isomers of CF3CF=CHF; Kinetics, Mechanisms, and Products of Gas-Phase Reactions with Cl Atoms, OH Radicals, and O3, J. Phys. Chem. A, 2007, 111(39), 9789–9795 CrossRef CAS PubMed.
- H. G. Libuda, F. Zabel, E. H. Fink and K. H. Becker, Formyl chloride: UV absorption cross sections and rate constants for the reactions with chlorine atom and hydroxyl radical, J. Phys. Chem., 1990, 94(15), 5860–5865 CrossRef CAS.
- J. G. Calvert, R. G. Derwent, J. J. Orlando, T. J. Wallington and G. S. Tyndall, The Mechanisms of Atmospheric Oxidation of the Alkenes, Mechanisms of atmospheric oxidation of the alkanes, OUP, USA, 2008 Search PubMed.
- M. Antiñolo, I. Bravo, E. Jiménez, B. Ballesteros and J. Albaladejo, Atmospheric Chemistry of E- and Z-CF3CH=CHF (HFO-1234ze): OH Reaction Kinetics as a Function of Temperature and UV and IR Absorption Cross Sections, J. Phys. Chem. A, 2017, 121(43), 8322–8331 CrossRef PubMed.
- F. F. Østerstrøm, S. T. Andersen, T. I. Sølling, O. J. Nielsen and M. P. Sulbaek Andersen, Atmospheric chemistry of Z- and E-CF3CH=CHCF3, Phys. Chem. Chem. Phys., 2017, 19(1), 735–750 RSC.
- P. K. Rao and S. P. Gejji, Molecular insights for the HFO-1345fz + X (X = Cl, O3 or NO3•) reaction and fate of alkoxy radicals initiated by Cl: DFT investigations, J. Fluorine Chem., 2017, 204, 65–75 CrossRef CAS.
- F. Qing, Q. Guo, L. Chen, H. Quan and J. Mizukado, Atmospheric chemistry of E-CF3CH=CHCF3: reaction kinetics of OH radicals and products of OH-initiated oxidation, Chem. Phys. Lett., 2018, 706, 93–98 CrossRef CAS.
- F. Jabeen, A. Kumar and B. Rajakumar, Kinetics, thermochemistry and atmospheric implications for the reaction of OH radicals with CH3CF=CF2 (HFO-1243yc), Chem. Phys. Lett., 2020, 758, 137933 CrossRef CAS.
- N. K. Gour, K. Borthakur, S. Paul and R. Chandra Deka, Tropospheric degradation of 2-fluoropropene (CH3CF=CH2) initiated by hydroxyl radical: reaction mechanisms, kinetics and atmospheric implications from DFT study, Chemosphere, 2020, 238, 124556 CrossRef CAS PubMed.
- N. K. Gour, R. C. Deka and S. Paul, Atmospheric oxidation of 2-fluoropropene (CH3CFCH2) with Cl atom and aerial degradation of its product radicals by computational study, New J. Chem., 2020, 44(8), 3434–3444 RSC.
- C. M. Tovar, M. B. Blanco, I. Barnes, P. Wiesen and M. A. Teruel, Gas-phase reactivity study of a series of hydrofluoroolefins (HFOs) toward OH radicals and Cl atoms at atmospheric pressure and 298 K, Atmos. Environ., 2014, 88, 107–114 CrossRef CAS.
- C. B. Rivela, C. M. Tovar, M. A. Teruel, I. Barnes, P. Wiesen and M. B. Blanco, CFCs replacements: reactivity and atmospheric lifetimes of a series of hydrofluoroolefins towards OH radicals and Cl atoms, Chem. Phys. Lett., 2019, 714, 190–196 CrossRef CAS.
- K. Tokuhashi, K. Takizawa and S. Kondo, Rate constants for the reactions of OH radicals with CF3CX=CY2 (X=H, F, CF3, Y = H, F, Cl), Environ. Sci. Pollut. Res., 2018, 25(15), 15204–15215 CrossRef CAS PubMed.
- C. Faxon, J. Bean and L. Ruiz, Inland Concentrations of Cl2 and ClNO2 in Southeast Texas Suggest Chlorine Chemistry Significantly Contributes to Atmospheric Reactivity, Atmosphere, 2015, 6(10), 1487–1506 CrossRef CAS.
- J. B. Burkholder, R. A. Cox and A. R. Ravishankara, Atmospheric Degradation of Ozone Depleting Substances, Their Substitutes, and Related Species, Chem. Rev., 2015, 115(10), 3704–3759 CrossRef CAS PubMed.
- T. J. Wallington and M. D. Hurley, Atmospheric Chemistry of HC(O)F: Reaction with OH Radicals, Environ. Sci. Technol., 1993, 27(7), 1448–1452 CrossRef CAS.
- T. J. Wallington, W. F. Schneider, D. R. Worsnop, O. J. Nielsen, J. Sehested, W. J. Debruyn and J. A. Shorter, The Environmental Impact of CFC Replacements—HFCs and HCFCs, Environ. Sci. Technol., 1994, 28(7), 320–326 Search PubMed.
- T. J. Wallington, M. P. Sulbaek Andersen and O. J. Nielsen, Atmospheric chemistry of halogenated organic compounds, in Advances in Atmospheric Chemistry, World Scientific Publishing Co. Pte. Ltd., 2016, vol. 1, pp. 305–402 Search PubMed.
- O. V. Rattigan, O. Wild, R. L. Jones and R. A. Cox, Temperature-dependent absorption cross-sections of CF3COCl, CF3COF, CH3COF, CCl3CHO and CF3COOH, J. Photochem. Photobiol., A, 1993, 73(1), 1–9 CrossRef CAS.
- X. Song, G. L. Zügner, M. Farkas, A. Illés, D. Sarzyński, T. Rozgonyi, B. Wang and S. Dóbé, Experimental and Theoretical Study on the OH-Reaction Kinetics and Photochemistry of Acetyl Fluoride (CH3C(O)F), an Atmospheric Degradation Intermediate of HFC-161 (C2H5F), J. Phys. Chem. A, 2015, 119(28), 7753–7765 CrossRef CAS PubMed.
- W. J. De Bruyn, J. A. Shorter, P. Davidovits, D. R. Worsnop, M. S. Zahniser and C. E. Kolb, Uptake of Haloacetyl and Carbonyl Halides by Water Surfaces, Environ. Sci. Technol., 1995, 29(5), 1179–1185 CrossRef CAS PubMed.
- F. Paulot, D. Wunch, J. D. Crounse, G. Toon, D. B. Millet, P. F. DeCarlo, C. Vigouroux, N. M. Deutscher, G. Gonzalez Abad, J. Notholt, T. Warneke, J. Hannigan, C. Warneke, J. A. De Gouw, E. Dunlea, M. De Maziere, D. W. Griffith, P. Bernath, J. L. Jimenez and P. O. Wennberg, Importance of secondary sources in the atmospheric budgets of formic and acetic acids, Atmos. Chem. Phys., 2011, 11, 1989–2013 CrossRef CAS PubMed.
- H. K. Stranddorf, L. Hoffmann and A. Schmidt, LCA technical report: impact categories, normalization and weighting in LCA, Updat Sel EDIP97-data, 2005 Search PubMed.
- A. Lindley, A. McCulloch and T. Vink, Contribution of Hydrofluorocarbons (HFCs) and Hydrofluoro-Olefins (HFOs) Atmospheric Breakdown Products to Acidification (“Acid Rain”) in the EU at Present and in the Future, Open J. Air Pollut., 2019, 8(4), 81–95 CrossRef CAS.
- J. C. Boutonnet, P. Bingham, D. Calamari, C. Rooij, J. Franklin, T. Kawano, J.-M. Libre, A. McCul-loch, G. Malinverno, J. Odom, G. Rusch, K. Smythe, I. Sobolev, R. Roy Thompson and J. Tiedje, Environmental Risk Assessment of Trifluoroacetic Acid, Hum. Ecol. Risk Assess., 1999, 5(1), 59–124 CrossRef CAS.
- V. C. Papadimitriou, R. K. Talukdar, R. W. Portmann, A. R. Ravishankara and J. B. Burkholder, CF3CF=CH2 and (Z)-CF3CF=CHF: temperature dependent OH rate coefficients and global warming potentials, Phys. Chem. Chem. Phys., 2008, 10(6), 808–820 RSC.
- T. J. Wallington, W. F. Schneider, D. R. Worsnop, O. J. Nielsen, J. Sehested, W. J. Debruyn and J. A. Shorter, The Environmental Impact of CFC Replacements HFCs and HCFCs, Environ. Sci. Technol., 1994, 28(7), 320A–326A CAS.
- C. W. Spicer, E. G. Chapman, B. J. Finlayson-Pitts, R. A. Plastridge, J. M. Hubbe, J. D. Fast and C. M. Berkowitz, Unexpectedly high concentrations of molecular chlorine in coastal air, Nature, 1998, 394(6691), 353–356 CrossRef CAS.
- P. L. Tanaka, D. D. Riemer, S. Chang, G. Yarwood, E. McDonald-Buller, E. Apel, J. Orlando, P. Silva, J. Jimenez, C. Canagaratna, J. Neece, C. Buddie Mullins and D. Allen, Direct evidence for chlorine-enhanced urban ozone formation in Houston, Texas, Atmos. Environ., 2003, 37(9–10), 1393–1400 CrossRef CAS.
- M. J. Lawler, R. Sander, L. J. Carpenter, J. D. Lee, R. Von Glasow, R. Sommariva and E. S. Saltzman, HOCl and Cl2 observations in marine air, Atmos. Chem. Phys., 2011, 11, 7617–7628 CrossRef CAS.
- T. J. Wallington and J. E. Anderson, Comment on “Environmental Fate of the Next Generation Refrigerant 2,3,3,3-Tetrafluoropropene (HFO-1234yf)”, Environ. Sci. Technol., 2015, 49(13), 8263–8264 CrossRef CAS PubMed.
- T. J. Wallington, M. P. Sulbaek Andersen and O. J. Nielsen, Atmospheric chemistry of short-chain haloolefins: photochemical ozone creation potentials (POCPs), global warming potentials (GWPs), and ozone depletion potentials (ODPs), Chemosphere, 2015, 129, 135–141 CrossRef CAS PubMed.
- G. M. Rusch, The development of environmentally acceptable fluorocarbons, Crit. Rev. Toxicol., 2018, 48(8), 615–665 CrossRef CAS PubMed.
Footnote |
| † Deceased 1 January 2018. |
| This journal is © The Royal Society of Chemistry 2021 |