DOI:
10.1039/D1RA00175B
(Paper)
RSC Adv., 2021,
11, 11304-11317
Lithium calix[4]arenes: structural studies and use in the ring opening polymerization of cyclic esters†
Received
8th January 2021
, Accepted 8th March 2021
First published on 17th March 2021
Abstract
We have structurally characterized a number of lithiated calix[4]arenes, where the bridge in the calix[4]arene is thia (–S–, LSH4), sulfinyl (–SO–, LSOH4), sulfonyl (–SO2–, LSO2H4), dimethyleneoxa (–CH2OCH2–, LCOCH4) or methylene (–CH2–, LH4). In the case of L4SH4, interaction with LiOtBu led to the isolation of the complex [Li8(L4S)2(THF)4]·5THF (1·5THF), whilst similar interaction of L4SOH4 led to the isolation of [Li6(L4SOH)2(THF)2]·5(THF) (2·5THF). Interestingly, the mixed sulfinyl/sulfonyl complexes [Li8(calix[4]arene(SO)(SO2)(SO1.68)2)2(THF)6]·8(THF) (3·8THF) and [Li5Na(LSO/3SO2H)2(THF)5]·7.5(THF) (4·7.5(THF) have also been characterized. Interaction of LiOtBu with LSO2H4 and LCOCH4 afforded [Li5L4SO2(OH)(THF)4]·2THF (5·2THF) and [Li6(LCOC)2(HOtBu)2]·0.78THF·1.22hexane (6·0.78THF·1.22hexane), respectively. In the case of LH4, reaction with LiOtBu in THF afforded a monoclinic polymorph [LH2Li2(thf)(OH2)2]·3THF (7·3THF) of a known triclinic form of the complex, whilst reaction of the de-butylated analogue of LH4, namely de-BuLH4, afforded a polymeric chain structure {[Li5(de-BuL)(OH)(NCMe)3]·2MeCN}n (8·2MeCN). For comparative catalytic studies, the complex [Li6(LPr)2(H2O)2]·hexane (9 hexane), where LPr2H2 = 1,3-di-n-propyloxycalix[4]areneH2, was also prepared. The molecular crystal structures of 1–9 are reported, and their ability to act as catalysts for the ring opening (co-)/polymerization (ROP) of the cyclic esters ε-caprolactone, δ-valerolactone, and rac-lactide has been investigated. In most of the cases, complex 6 outperformed the other systems, allowing for higher conversions and/or greated polymer Mn.
Introduction
The search for alternative plastics to petroleum-based products continues at a pace. One avenue of exploration is to develop new, greener biodegradable polymers, but which retain the desirable features of traditional plastics.1 With this in mind, one promising route is to exploit the ring opening polymerization (ROP) of cyclic esters, a process which can be controlled by a metal-based catalyst.2 For the catalyst, it is important that the ancillary ligands present can be easily adjusted in terms of their steric and electronic properties. There has been considerable interest in the use of the phenolic macrocycles called calixarenes in a broad range of catalysis.3 Indeed, we have been interested in employing such species in the ROP of cyclic esters to afford biodegradable polymers.4 Both the upper- and lower-rim of a calixarene can readily be modified, and for the latter the introduction of ether groups allows the charge to be altered.5 Moreover, different bridging groups can be used to link the phenols, and the range of conformations available to calixarenes allows for further structural flexibility.6 In terms of the metal employed, it is important it is relatively cheap and non-toxic. Lithium is the 25th most abundant element, and lithium systems have shown promise as ROP catalysts.7 Over the last decade or so, a number of routes to lithiated calix[4]arenes and their thia-bridged analogues have been developed. The early work on calix[n]arenes involved the use of 7Li NMR spectroscopy,8 whilst for p-tert-butylcalix[4]arene (L4H4), early structural work involved the use of lithium amide,9 whilst Davidson et al. employed nBuLi in the presence of either wet or dried hexamethylphosphoramide (hmpa) and isolated Li4L4·LiOH·4hmpa or [Li4L4·2hmpa]2, respectively.10 Floriani et al. also employed nBuLi in the presence of naphthalene to prepare lithiated calix[4]arenes.11 Fromm et al. structurally characterized the complex [Li4(LH2)2(THF)4]·3THF, possessing a core containing two face-shared Li4O4 cubes from the reaction of L4H4 and LiOtBu; the partial hydrolysis product [Li2(L4H2)(H2O)(μ-H2O)(THF)]·3THF was also characterized.12 Hanna et al. also studied reactions of calix[4 and 8]arene as well as p-tert-butylcalix[4 and 8]arene with LiOtBu or LiOSiMe3 respectively, and isolated monoanions.13 In later studies, Hanna et al. extended their studies to the reaction of p-tert-butylcalix[5]arene with a number of lithiated reagents, namely LiOH, nBuLi and LiH.14 Moreover, a number of molybdocalix[4]arenes incorporating lithium, primarily as part of a bridging ligand have also been reported,15 as have mixed lithium–strontium complexes.16 We have previously structurally characterized the two complexes [L4H2(OMe)(OLi)]2·4MeCN and {L4H2(OLi)[OLi(NCMe)2]}2·8MeCN resulting from the use of nBuLi.17
For p-tert-butyltetrathiacalix[4]arene (LS4H4), Zeller and Radius employed nBuLi to access LS4Li4, which proved problematic to crystallize and only the decomposition product [LS4Li5(OH)(THF)4] was structurally characterized; the complex could be isolated directly using LiOH·H2O/LS4H4.18 We have reacted a lower rim 1,3-diacid calix[4]arene with either Li2CO3 or tBuLi and isolated helical nanotubes or infinite chains, respectively.19 The structure of a supramolecular lithium calix[4]arene complex has also been reported.20
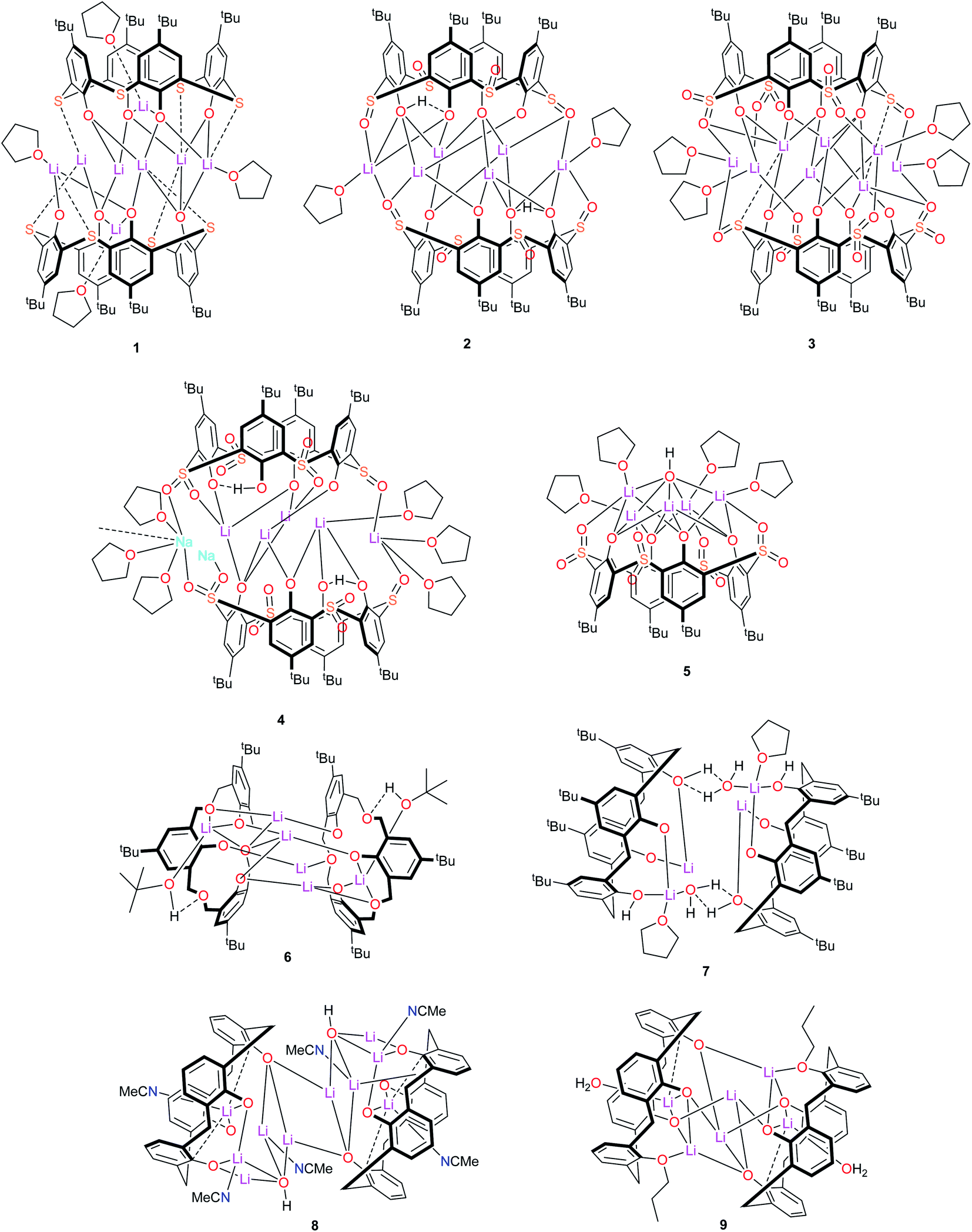
Despite this synthetic activity, applications of such lithiated calixarene species have not been forthcoming. With this in mind, we have embarked upon a programme to screen the potential for lithiated calixarenes for the ring opening polymerization (ROP) of cyclic esters. Herein, we present our findings on calix[4]arene systems for which the calixarene bridge has been varied, viz –S– (L4SH4), –SO– (L4SOH4), –SO2– (L4SO2H4), –CH2OCH2– (LCOCH4), –CH2– (L4H4), and in the case of the latter 1,3-n-propoxide groups have also been introduced at the lower-rim. Structural studies and the ROP of the cyclic esters ε-caprolactone, δ-valerolactone and rac-lactide are reported. The nine lithium-calixarenes prepared herein are shown in Chart 1. We note that a number of lithium-containing cages, rings, and ladders, supported primarily by phenolate-type ligation, have previously been employed for the ROP of cyclic esters.21 The use of calixarenes though, is somewhat limited.4,22
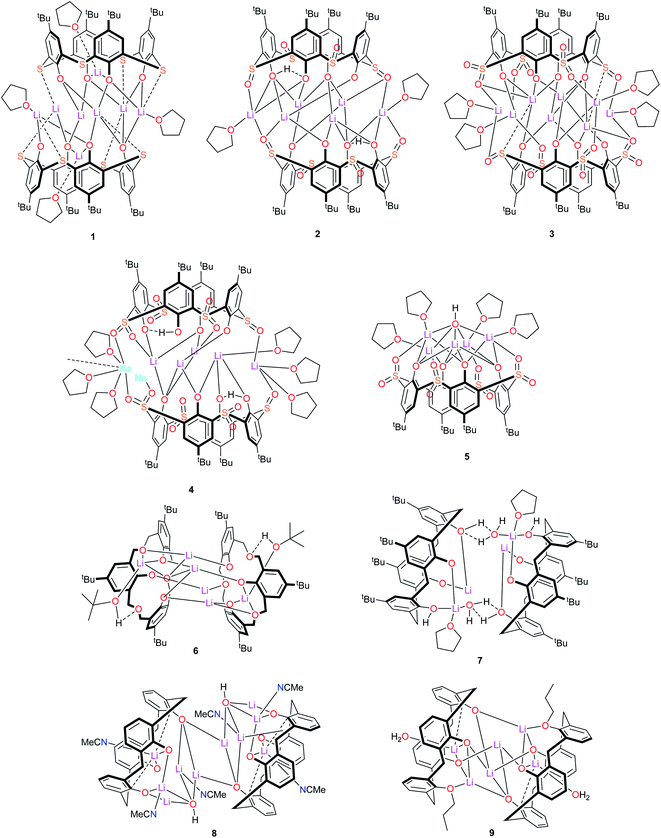 |
| | Chart 1 Lithium-calixarene complexes 1–9 prepared herein. | |
Results and discussion
Syntheses and solid-state structures
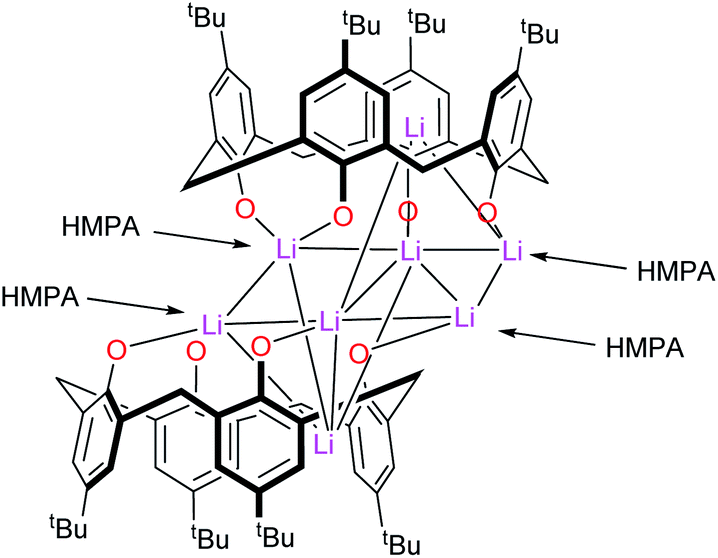
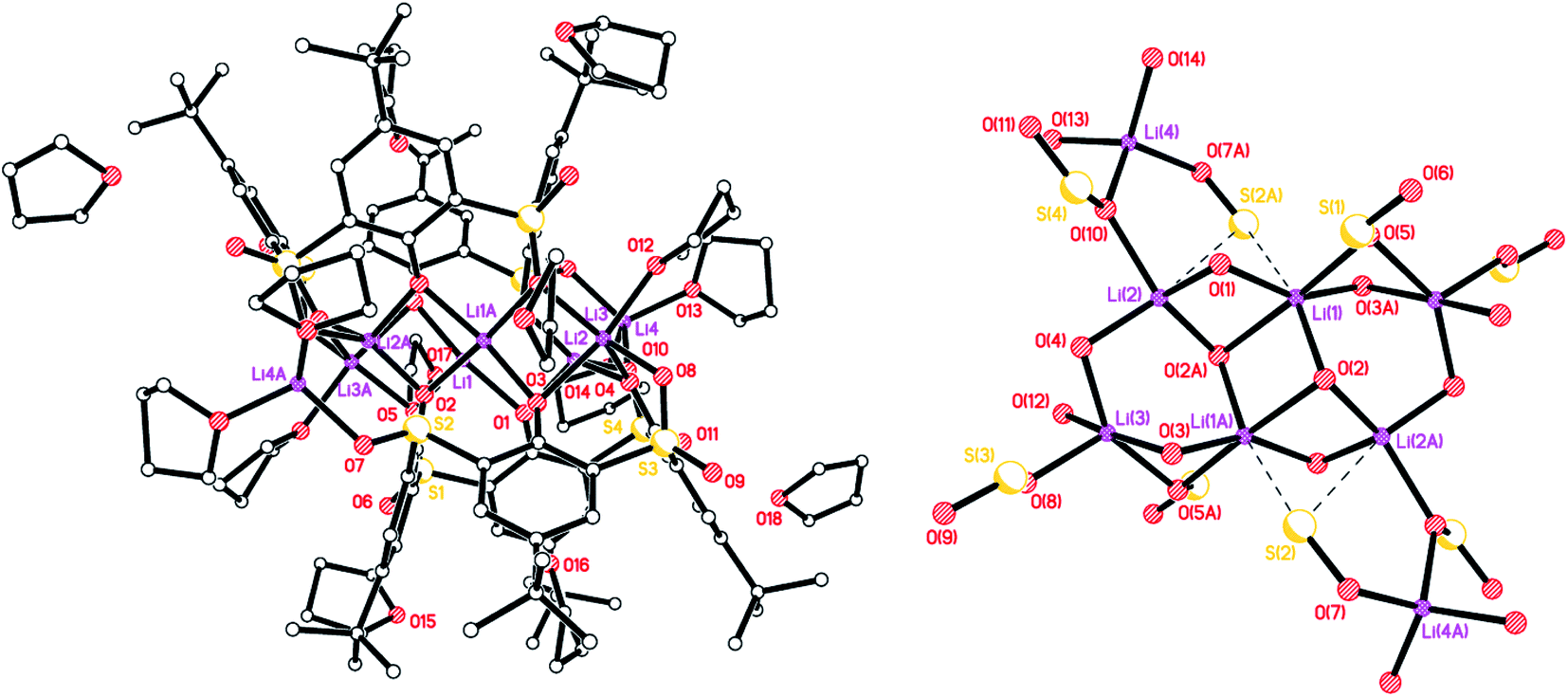
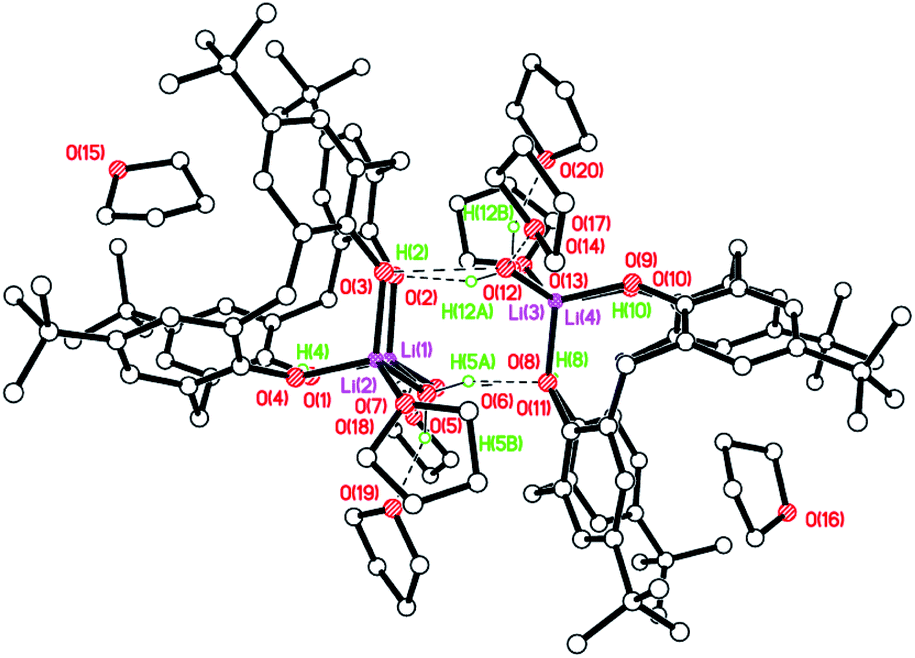
–Thia–. Of the number of synthetic routes to lithiated calixarenes outlined above,9–14 we chose as our entry point the use of LiOtBu. Reaction of LiOtBu (5 equiv.) with L4SH4 led to the isolation of the complex [Li8(L4S)2(THF)4]·5THF (1·5THF). Single crystals were grown, in ca. 65% isolated yield, from a saturated THF solution at ambient temperature and an X-ray structure determination revealed the structure shown in Fig. 1; for an alternative view of the core see Fig. S1, ESI.† Table 6 presents the crystal data for this and the other crystal structures reported herein. A closely related structural motif has been reported previously for [Li4L4·2hmpa]2 (Chart 2), whilst related sodium and potassium-containing motifs have also been reported.4,12,13
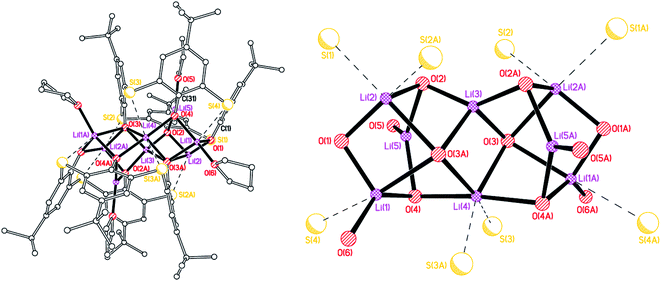 |
| | Fig. 1 Molecular structure (left) and core (right) of [Li8(L4S)2(THF)4]·5THF (1·5THF). H atoms, minor disorder components and non-coordinated THFs omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(1) 1.810(16), Li(1)–O(3A) 2.193(16), Li(1)–O(4) 1.947(18), Li(1)–O(6) 1.967(18), Li(3)–O(2) 1.882(8), Li(3)–O(3) 1.966(13), Li(4)–O(4) 2.046(6), Li(5)–O(2) 1.930(16), Li(5)–O(4) 1.951(17), Li(5)–O(5) 1.964(17); Li(1)–O(1)–Li(2) 96.8(8), Li(3)–O(3)–Li(4) 78.8(6), O(4)–Li(4)–O(4A) 174.3(11), O(2)–Li(5)–O(4) 119.9(8). | |
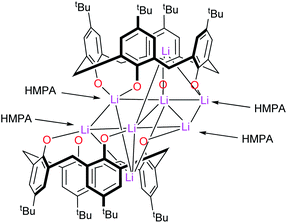 |
| | Chart 2 Structure of [Li4L4·2hmpa]2.10 | |
The molecule lies on a 2-fold axis which passes through Li(3)⋯Li(4). Thus, half of the molecule is unique as are 2½ THF molecules of crystallization. The ½ THF was modelled using Platon Squeeze.23 All four S atoms make contacts with one or more lithium centres, namely Li(1)⋯S(4) = 2.720(15), Li(2)⋯S(1) = 2.610(17), Li(2)⋯S(2A) = 2.644(15), Li(4)⋯S(3) = 2.734(10), Li(4)⋯S(3A) = 2.734(10) Å. Furthermore, Li(5) is involved in C(π) contacts, viz Li(5)⋯C(1) = 2.74(2) and Li(5)⋯C(31) = 2.670(18) Å. In the packing of 1, the calixarene complexes are well separated with THFs in the voids. The compound is not so soluble in toluene, which is problematic for the ROP studies vide infra.
Sulfinyl. Interaction of LiOtBu with L4SOH4 afforded small prisms of [Li6(L4SOH)2(THF)2]·5(THF) (2·5THF) in ca. 30% yield on standing (2 days) at ambient temperature. The molecular structure was determined using synchrotron radiation, and is shown in Fig. 2; the core is shown in Fig. S2, ESI.† Whilst the data was weak, the connectivity was clearly established. The molecule lies on a centre of symmetry, and so half is unique. Given there are six lithium ions present, and analysis of the bond lengths and relative positions of the phenolate oxygens, we conclude that O(3) is protonated; the position of O(2) also suggests it forms an H-bond with O(3). Two of the SO oxygens bind to lithium centres, namely Li(1) and Li(3), whilst Li(2) remains 4-coordinate. The phenolate oxygens O(2) and O(3) bridge three lithium centres, whilst O(1) and O(4) bridge only two Li centres. The SO groups have the O partially disordered at the alternative tetrahedral site on the S with minor component occupancies between ca. 0.1 and 0.2.
 |
| | Fig. 2 Molecular structure of [Li6(L4SOH)2(THF)2]·5(THF) (2·5THF). H atoms and minor disorder components omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(1) 1.947(12), Li(1)–O(2) 1.973(10), Li(1)–O(2A) 1.956(14), Li(1)–O(3A) 1.940(12), Li(1)–O(5) 2.038(12), Li(2)–O(1) 1.791(17), Li(2)–O(2A) 1.956(14), Li(2)–O(3) 2.037(15), Li(2)–O(4) 1.847(18), Li(3)–O(3) 2.228(12), Li(3)–O(5A) 2.072(12), Li(3)–O(7) 1.936(14), Li(3)–O(9) 1.996(12); O(1)–Li(1)–O(2) 94.9(5), O(1)–Li(2)–O(4) 119.5(8), O(3)–Li(3)–O(7) 96.6(5). | |
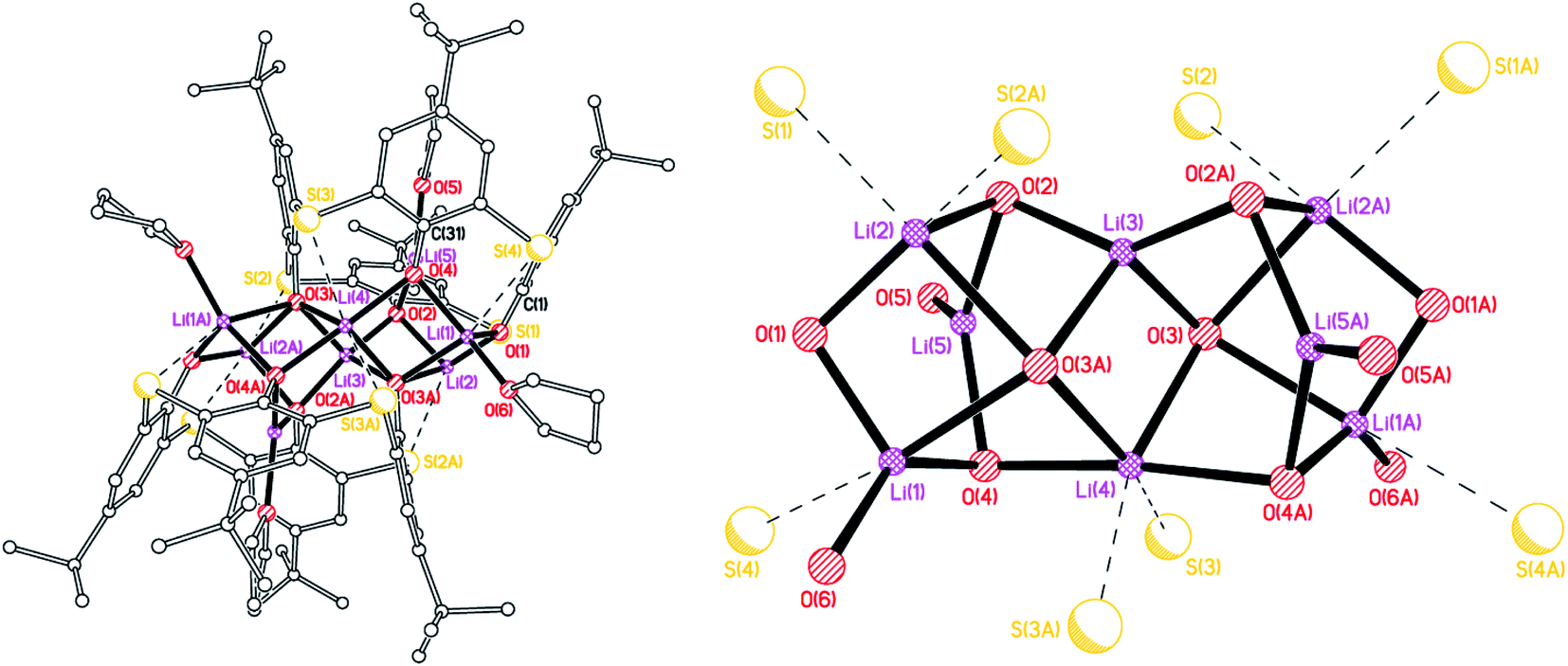
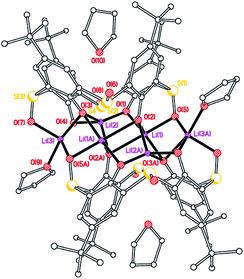
On one occasion, reaction of L4SOH4 with LiOtBu led to the isolation of a mixed sulfinyl/sulfonyl complex, namely [Li8(calix[4]arene(SO)(SO2)(SO1.68)2)2(THF)6]·8(THF) (3·8THF). The molecular structure is shown in Fig. 3 (for an alternative view, see Fig. S3, ESI†), with selected bond lengths and angles given in the caption. The molecule lies on a centre of symmetry, and so half of this formula is unique. Interestingly, only one of the bridging S centers on each calix {S(2)} has just one O group bound (i.e. a sulfinyl), whilst the other three have two oxygens (i.e. sulfonyls), though it should be noted two of these actually have ca. 1.68 oxygens rather than 2, indicating some slight variability in the degree of oxidation at sulfur in these calixarenes. There are weak Li(1)⋯S(2′) and Li(2)⋯S(2′) interactions of 2.929(5) and 3.099(5) Å, respectively, which are both longer than seen in 1·5THF. Each calixarene possesses a cone conformation. The molecules pack forming layers in the b/c plane (see Fig. S4, ESI†). The presence of the sulfonyl [–SO2–] bridges was thought to arise from adventitious oxidation during the reaction; mass spectra of the parent L4SOH4 did not exhibit higher peaks associated with the presence of –SO2– bridges.
 |
| | Fig. 3 Molecular structure (left) and core (right) of [Li8(LSO/3SO2)2(THF)6]·8(THF) 3·8(THF). H atoms, and minor disorder components omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(1) 1.957(5), Li(1)–O(2) 1.954(5), Li(1)–O(2A) 2.247(6), Li(1)–O(5) 2.145(6), Li(2)–O(1) 1.870(6), Li(2)–O(2A) 1.931(6), Li(2)–O(4) 1.852(6), Li(2)–O(10) 2.041(6); Li(1)–O(1)–Li(2) 89.4(2), Li(1)–O(2)–Li(2A) 110.7(2), Li(2)–O(10)–Li(4) 121.8(2). | |
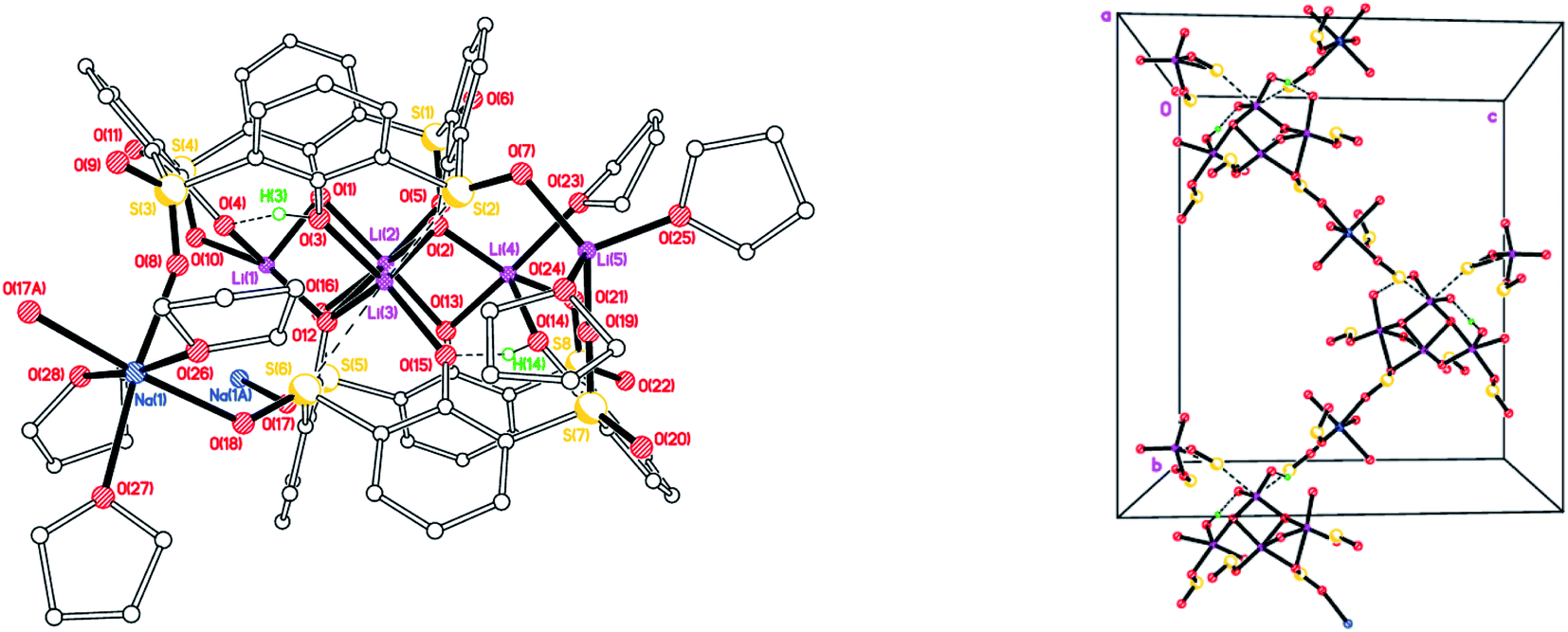
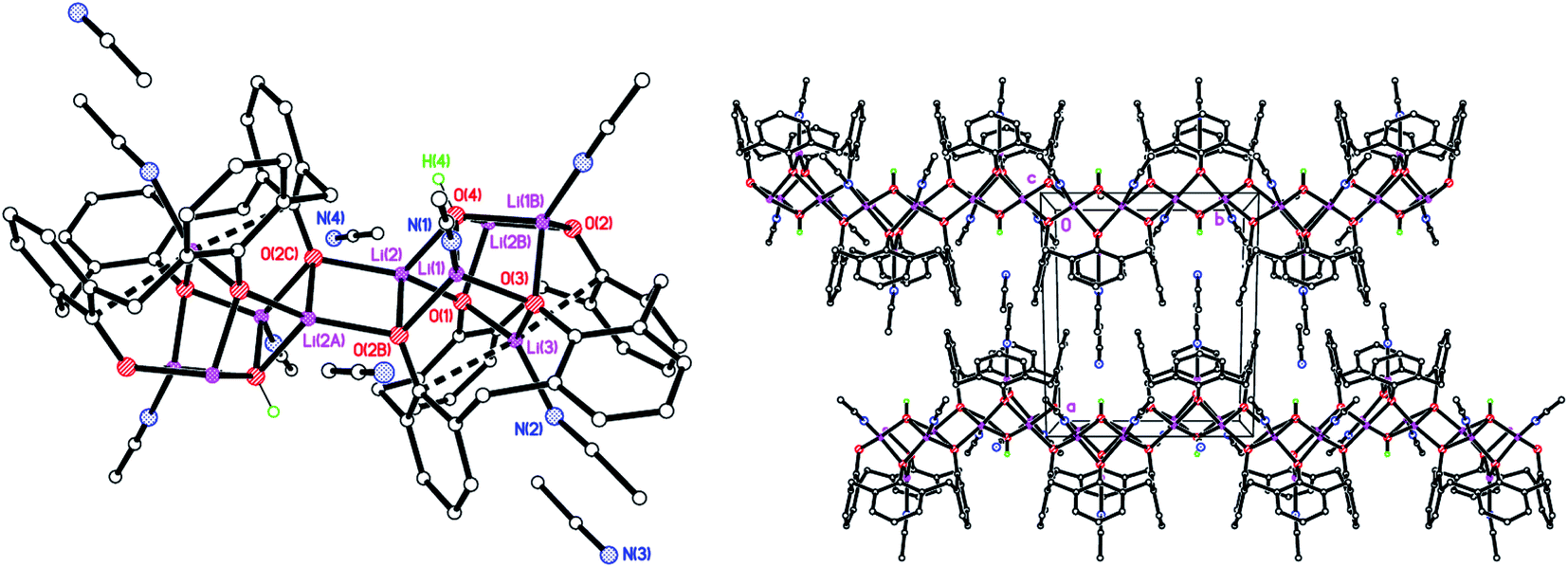
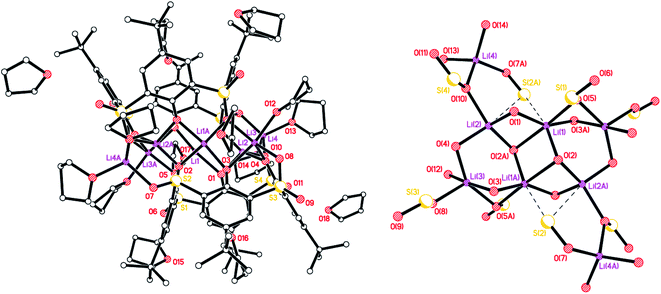
We also isolated a Na-bridged polymer [Li5Na(LSO/3SO2H)2(THF)5]·7.5(THF) (4·7.5(THF) from the reaction of L4SOH4 with LiOH. The molecular structure is shown in Fig. 4, with selected bond lengths and angles given in the caption. Both Li(1) and Li(4) are distorted squared-based pyramidal, whilst Li(2) is distorted octahedral and Li(5) distorted tetrahedral. Li(3), whilst forming 4 bonds to neighbouring oxygens, also forms two weaker interactions to S(2) and S(6). For all but one of the SO2 groups present, one of the oxygen atoms binds to Li or Na, whilst for S(5) one O bridges Li(1) and Li(2) and the other bonds to Na(1). In the case of the SO groups, the S binds weakly to Li(3) and the O binds to either Li(5) or Na(1). The slight variability in oxidation at S is manifested here with O(20) being 78.0(18)% occupied.
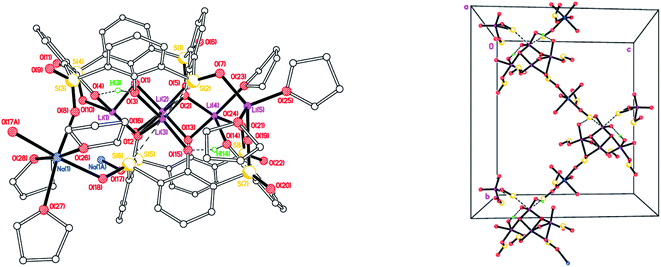 |
| | Fig. 4 Repeating unit (left) and 1D zig–zag polymer chains (right) in the structure of [Li5Na(LSO/3SO2H)2(THF)5]·7.5(THF) (4·7.5THF). H atoms not involved in H-bonding, minor disorder components and non-coordinated THFs omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(1) 1.876(8), Li(1)–O(4) 1.970(8), Li(1)–O(10) 1.988(8), Li(1)–O(12) 1.969(8), Li(1)–O(16) 2.352(8), Li(1)–S(4) 2.932(7), Li(2)–O(1) 1.974(8), Li(2)–O(2) 2.081(8), Li(2)–O(5) 2.135(9), Li(2)–O(12) 2.259(8), Li(2)–O(16) 2.147(8), Li(3)–O(2) 1.945(8), Li(3)–O(3) 1.996(7), Li(3)–O(12) 1.926(7), Li(3)–O(15) 2.016(7), Li(3)–S(2) 2.837(7), Li(3)–S(6) 2.968(7); Li(1)–O(1)–Li(2) 83.5(3), Li(1)–O(12)–Li(2) 74.3(3), Li(1)–O(12)–Li(3) 116.8(3), Li(2)–O(2)–Li(3) 87.2(3), Li(2)–O(12)–Li(3) 82.8(3). | |
Complex 4 forms 1D chains linked via octahedral Na+ ions (Fig. 4, right). We have previously noted the incorporation of Na from drying agents in metallocalixarene chemistry.24 In 4, to balance the overall charge, two phenols remain protonated, one on each calix[4]arene.
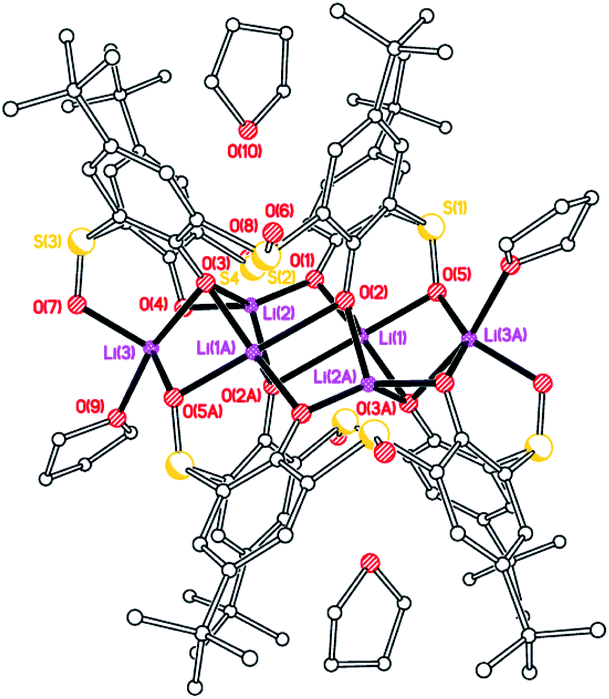
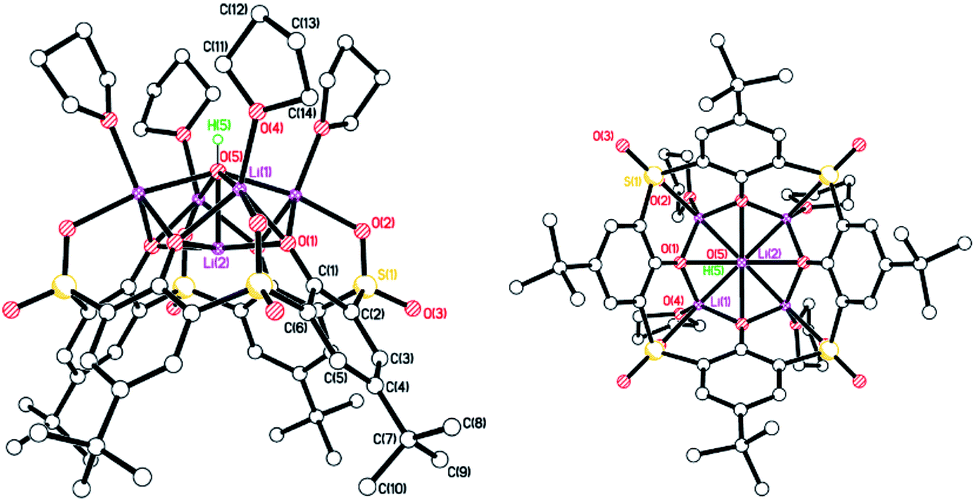
Sulfonyl. Extension of the LiOtBu methodology to the sulfonyl-(–SO2–) bridged calix[4]arene system, afforded single crystals suitable for X-ray diffraction from a saturated tetrahydrofuran solution at 0 °C in ca. 60% isolated yield. The molecular structure of [Li5L4SO2(OH)(THF)4]·2THF (5·2THF) is shown in Fig. 5, with selected bond lengths and angles given in the caption. The complex contains the Li5OH motif noted by Davidson et al.,10 and Zeller and Radius,18 and, like the latter, lies on a 4-fold axis and possesses four lithium-bound THF ligands. The difference in 5 is that an oxygen of each of the bridging sulfonyl groups is bound to each of the ‘outer’ lithium centres. The lithium atom Li(2) is 0.580 Å above the plane formed by the phenolate oxygens, whilst the Li(2)–O(5) bond length is 1.958(13) Å cf. 2.055(10) Å for the Zeller and Radius structure.18 Each of the lithium centres is 5-coordinate with the other four adopting what can best be described as trigonal bipyramidal geometries, whilst the inner lithium centre Li(2) adopts a square pyramidal geometry. Both the THFs of crystallization are substantially disordered on the 4-fold axis and were modelled as diffuse regions of electron density by using Platon Squeeze.23 One THF resides in the calixarene cavity, the other lies above the OH beyond the four coordinated THF molecules.
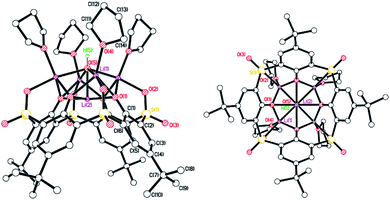 |
| | Fig. 5 Two perpendicular views of the molecular structure of [Li5L4SO2(OH)(THF)4]·2THF (5·2THF). H atoms, minor disorder components and non-cordinated THF omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(1) 2.002(6), Li(1)–O(4) 1.988(6), Li(1)–O(5) 2.138(6), Li(2)–O(1) 2.018(2), Li(2)–O(5) 1.958(13); Li(1)–O(5)–Li(2) 74.3(2), O(1)–Li(2)–O(5) 87.3(3). | |
Dimethyleneoxa. Reaction of LCOCH4 with LiOtBu led, following work-up, to the complex [Li6(LCOC)2(HOtBu)2]·0.78THF·1.22hexane (6·0.78THF·1.22hexane) in ca. 45% isolated yield. Two views of the molecular structure are shown in Fig. 6, with selected bond lengths and angles given in the caption. The core of the complex comprises a pseudo-cubane Li6O6 unit, which contains four, boat-shaped, six-membered faces of two Li3O3 units. A tBuOH molecule is bound to Li(2) and resides within the calixarene cavity and, being disordered, H-bonds to either O(2) (for the major component) or O(6) (minor). Pairs of molecules sit in a slipped face-to-face or cavity-to-cavity motif with tBuOH molecules pointing approximately towards each other.
 |
| | Fig. 6 Two views of the molecular structure of [Li6(LCOC)2(HOtBu)2]·THF·hexane (6 THF·hexane). H atoms not involved in H-bonding, minor disorder components and non-coordinated solvent of crystallisation omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(1) 1.861(3), Li(1)–O(2) 2.075(3), Li(1)–O(3) 1.922(3), Li(2)–O(3) 1.940(3), Li(2)–O(5) 1.937(4), Li(2)–O(7) 1.931(4), Li(3)–O(1) 1.859(3), Li(3)–O(3A) 1.915(3); O(1)–Li(1)–O(2) 94.87(13), O(1)–Li(1)–O(3) 127.32(17), O(3)–Li(2)–O(5) 112.94(15). | |
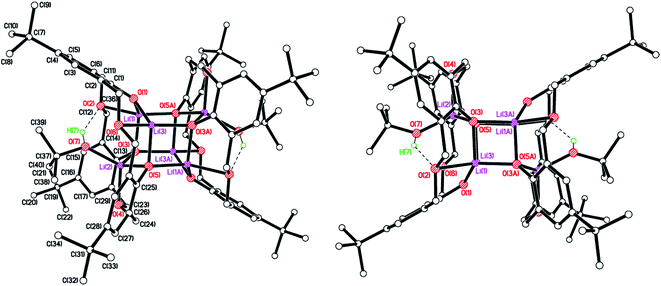
Methylene. In the case of LH4, reaction with LiOtBu (2.2 equiv.) in THF afforded [LH2Li2(THF)(OH2)2]·3THF (7·3THF), which is a monoclinic polymorph of a known, triclinic, structure reported by Fromm et al.12 The crystals were very small and very weakly diffracting, even at the synchrotron, which resulted in the poor R factor. The absolute structure could not be determined due to the combination of radiation wavelength and lack of an anomalous scatterer. The H atoms on the terminal water molecule could not be located in difference maps, but the proximity of this water molecule and two H-bond acceptors is perfectly logical. The H atoms on the bridging waters were placed geometrically, while those between calixarene oxygens are heavily restrained. In reality they are probably more asymmetric. Their presence is required for charge balance and supported by the better, known structure. There are two molecules of the Li2 complex and six THFs of crystallisation in the asymmetric unit. They form an H-bonded cluster involving both complexes and four THFs (see Fig. 7; a view of a single molecule of 7·3THF is shown in Fig. S5, ESI†), with an additional THF in each calixarene cavity. The same motif is seen in the published structure. Although the authors suggest that their structure does not have a THF in the cavity, closer inspection reveals it actually has. Also, for each complex, two THFs H-bond to water hydrogens.
 |
| | Fig. 7 Molecular structure of the H-bonded dimer of [LH2Li2(THF)(OH2)2]·3THF (7·3THF). H atoms not involved in H-bonding and minor disorder components omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(1) 1.86(3), Li(1)–O(2) 1.99(3), Li(1)–O(5) 1.97(3), Li(1)–O(6) 1.94(3), Li(1)⋯Li(2) 3.06(4), Li(2)–O(3) 1.95(3), Li(2)–O(4) 1.89(3), Li(2)–O(5) 2.00(3), Li(2)–O(7) 1.92(3), Li(3)⋯Li(4) 3.10(4), Li(1)–O(5)–Li(2) 100.7(12), O(1)–Li(1)–O(2) 104.2(14), O(3)–Li(2)–O(4) 105.3(14). | |
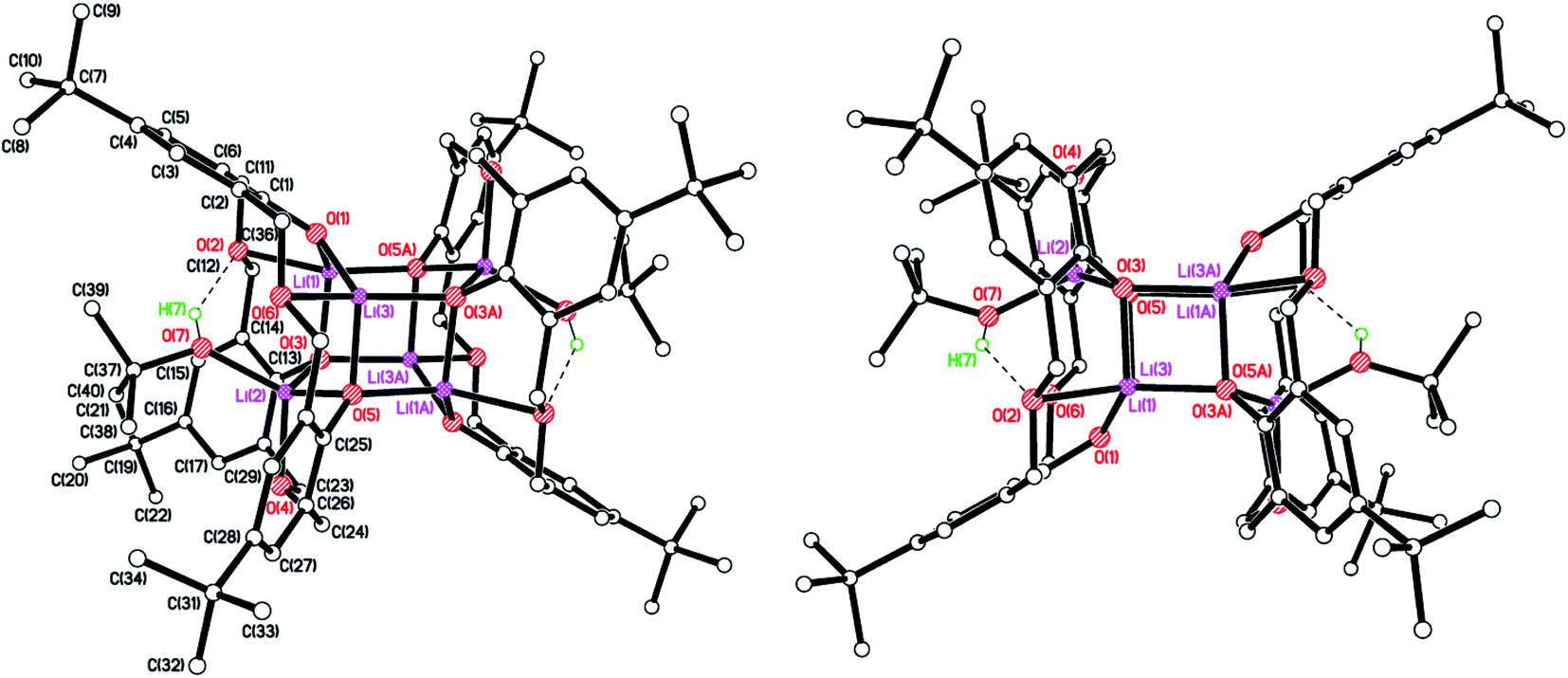
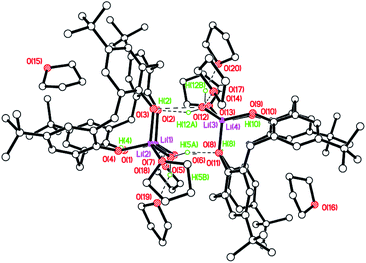
Similar reaction of the de-butylated analogue of LH4, namely de-BuLH4, afforded, following work-up (MeCN), the polymeric chain structure {[Li5(de-BuL)(OH)(NCMe)3]·2MeCN}n (8·2MeCN), and half of this is the asymmetric unit. The molecular structure is shown in Fig. 8 (left), with selected bond lengths and angles given in the caption. The structure is a 1D zig–zag chain polymer and a mirror plane bisects the calixarene. The structure was non-merohedrally twinned via a 180° rotation about real axis 0 0 1 with twin components 52.2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :47.8(4)%. Four lithium ions at the lower rim of the calixarene are each bridging a pair of oxygens. One lithium ion is in the cavity of the calixarene, bound to two oxygens and π-bonded to two ipso carbons and, as a result, a pinched-cone conformation for the calixarene is observed. The calixarenes are linked via centro-symmetric Li2O2 diamonds involving Li(2) and O(2). The Li(3)⋯C(6) distance is 2.734(6) Å. Li(1), which is not involved in the chain propagation, bears an NCMe ligand, as does Li(3) in the cavity. The complex packs with the 1D zig–zag chain polymer propagating in the b direction (Fig. 6, right).
:47.8(4)%. Four lithium ions at the lower rim of the calixarene are each bridging a pair of oxygens. One lithium ion is in the cavity of the calixarene, bound to two oxygens and π-bonded to two ipso carbons and, as a result, a pinched-cone conformation for the calixarene is observed. The calixarenes are linked via centro-symmetric Li2O2 diamonds involving Li(2) and O(2). The Li(3)⋯C(6) distance is 2.734(6) Å. Li(1), which is not involved in the chain propagation, bears an NCMe ligand, as does Li(3) in the cavity. The complex packs with the 1D zig–zag chain polymer propagating in the b direction (Fig. 6, right).
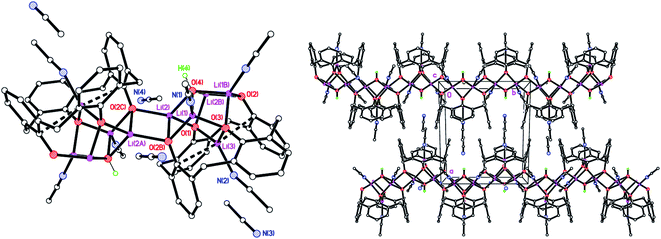 |
| | Fig. 8 (Left) Molecular structure of the repeating unit in the polymeric structure {[Li5(de-BuL)(OH)(NCMe)3]·2MeCN}n (8·2MeCN); (right) 1D zig–zag chain polymer propagating in the b direction. Most H atoms and minor disorder components omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(2B) 1.999(15), Li(1)–O(3) 1.910(16), Li(1)–O(4) 1.988(14), Li(1)–N(1) 2.071(16), Li(2)–O(1) 1.961(14), Li(2)–O(2B) 1.991(16), Li(2)–O(2) 2.058(13), Li(2)–O(4) 1.944(14); O(2B)–Li(1)–O(3) 104.3(7), O(1)–Li(2)–O(2C) 138.7(7), Li(1B)–O(4)–Li(2) 130.3(6). | |
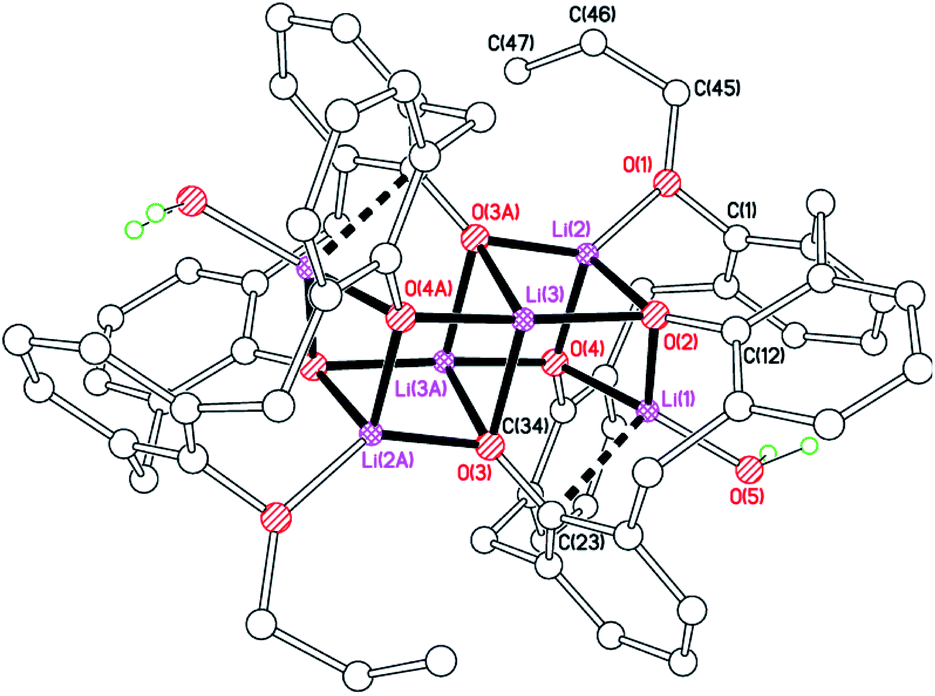
For our catalytic studies, we were also interested in evaluating the effect of the presence of alkyl chains at the lower rim. With this in mind, 1,3-di-n-propyloxycalix[4]arene, 1,3-L(OH)2(nPrO)2, which we abbreviate as LPr2H2, was treated with nBuLi in hexane. Following work-up, small crystals suitable for X-ray diffraction using synchrotron radiation were obtained. The molecular structure (Fig. 9) revealed the complex to be [Li6(LPr)2(H2O)2]·hexane (9 hexane). Half of the complex and half a solvent molecule are unique, both on i. The core comprises two face-sharing distorted cubes. The distortion arises because Li(1) and O(3) are not directly bound, and instead Li(1) makes a π-interaction with C(23), bound to phenolate oxygen O(3). Additionally, Li(3) binds to the water molecule which resides in the calixarene cavity. Each calixarene adopts a pinched conformation with rings attached to O(2) and O(4) splayed out, whilst those at O(1) and O(4) are pinched together.
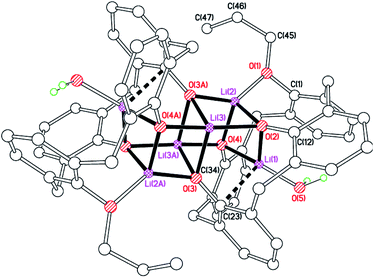 |
| | Fig. 9 Molecular structure of [Li6(LPr)2(H2O)2]·hexane (9 hexane). Most H atoms and hexane of crystallization omitted for clarity. Selected bond lengths (Å) and angles (°): Li(1)–O(2) 1.913(3), Li(1)–O(4) 1.935(3), Li(1)–O(5) 1.885(3), Li(2)–O(1) 1.883(3), Li(2)–O(2) 1.963(3), Li(3)–O(3A) 1.948(3), Li(2)–O(4) 1.973(3); Li(1)–O(2)–Li(2) 78.64(13), Li(1)–O(4)–Li(3A) 102.41(13), O(2)–Li(3)–O(4A) 166.43(16). | |
ROP studies
ε-caprolactone (ε-CL). We have examined the ability of the complexes prepared herein (not 3) to act as catalysts for the ROP of ε-CL (Table 1). When conducting the reaction in toluene at 130 °C, poor activity was observed upon using a monomer to catalyst ratio of 100:1 and in the presence of one equiv./Li of BnOH as co-activator. In fact, the conversions observed were in the range of 10–20%. The reaction was then repeated under solvent-free conditions in the absence of a co-activator (Table 2). While no reactivity was obtained after 24 h for compound 1 and in the presence of the Na-containing species 4 (runs 1 and 4), complete monomer conversion was achieved within 10 minutes in the presence of the dimethyleneoxa-derivative 6 (run 7). Moreover, interesting reactivity was exhibited by the SO2-bridged congener 5 and the methylene-bridged compound 7. Indeed, the formation of polymer blocking the stirring of the mixture took place within 1 h, affording 40 and 80% conversion for 5 and 7, respectively. Good activity was also shown by 2 and 9 (45 and 77% conversion, respectively), albeit requiring longer reaction times. The activity trend (6 > 7 >8 > 5 > 9 > 2) indicated a positive effect of the dimethyleneoxa bridge systems, compared with its –CH2– and S-containing congeners. Interestingly, similar behaviour has been previously observed for ethylene polymerization promoted by V-based catalysts.25 Such higher activity could be attributed to the greater flexibility of the dimethyleneoxa bridge (over –CH2– and –S–) allowing the monomer better access to active centres and/or to the presence of oxygen atoms which help stabilize the active species. With respect to the CH2-bridged complexes, the higher activity of complex 7 over its de-butylated analogue 8 could be attributed to its higher solubility in the reaction medium. Finally, the lower activity of 9 could be due to the steric encumbrance of its propoxy groups. In all cases, Mn higher than the calculated values were observed, suggesting a concentration of the active species lower than expected. Moreover, the rather broad Mw/Mn values indicated poor control over the polymerization, possibly due to the forcing reaction conditions as well as to the heterogenization of the reaction medium. These factors impeded the rigorous study of the kinetics aspects of the reaction. The 1H NMR spectrum of the PCL synthesized with 6 showed the presence of a triplet at δ 3.6 ppm, compatible with linear –CH2OR (R = H or tBu) terminated polymers (Fig. S6, ESI†). This was confirmed by mass spectrometry. In fact, the MALDI-ToF spectrum of the sample (Fig. S7†) displayed one population of peaks (separated by 114 m/z units) compatible with an α-tBuO-ω-OH terminated linear PCL. This suggested that the tert-butoxy group of 6 serves as initiating group of the ROP process. Upon performing the reaction in air, low conversion (14–17%) was obtained in the presence of 5, 6, and 7 (runs 5, 7, and 9) while no reaction was observed for 1, 8 and 9 (runs 2, 11, and 13). This significant drop of activity was attributed to the hydrolysis of the complexes affording catalytically inactive parental calixarenes; separate runs using the parent calixarenes failed to produce any PCL.
Table 1 ROP of ε-CL catalysed by the Li complexes
| Run |
Catalyst |
ε-CL:Li:BnOH |
T (°C) |
Time (h) |
Conversiona (%) |
| Determined by 1H NMR spectroscopy on crude reaction mixture. |
| 1 |
1 |
100:1:1 |
130 |
24 |
16 |
| 2 |
2 |
100:1:1 |
0 |
| 3 |
4 |
100:1:1 |
0 |
| 4 |
5 |
100:1:1 |
18 |
| 5 |
6 |
100:1:1 |
10 |
| 6 |
7 |
100:1:1 |
20 |
| 7 |
8 |
100:1:1 |
11 |
| 8 |
9 |
100:1:1 |
7 |
Table 2 ROP of ε-CL catalysed by the Li complexes under solvent-free conditions
| Run |
Catalyst |
ε-CL:Li |
T (°C) |
Time (h) |
Conversiona (%) |
Mnb,c |
Mncalcd |
PDIb |
| Determined by 1H NMR spectroscopy on crude reaction mixture. From GPC. Values corrected considering Mark–Houwink factor (0.56) from polystyrene standards in THF. Calculated from ([Monomer]0/[Cat.]0) × conv. (%) × monomer molecular weight. Reaction performed in air. Stirring stopped due to the polymer formation. |
| 1 |
1 |
100:1 |
130 |
24 |
None |
— |
— |
— |
| 2e |
24 |
None |
— |
— |
— |
| 3 |
2 |
24 |
45 |
1800 |
5130 |
1.25 |
| 4 |
4 |
24 |
None |
— |
— |
— |
| 5 |
5 |
1f |
40 |
14640 |
4560 |
2.01 |
| 6e |
24 |
17% |
nd |
|
nd |
| 7 |
6 |
10 min |
>99 |
27000 |
11290 |
1.67 |
| 8e |
24 |
17 |
— |
— |
— |
| 9 |
7 |
1f |
80 |
16130 |
9130 |
1.78 |
| 10e |
24 |
14 |
— |
— |
— |
| 11 |
8 |
15 minf |
72 |
27520 |
8210 |
1.76 |
| 12e |
24 |
None |
— |
— |
— |
| 13 |
9 |
6 |
77 |
17040 |
8780 |
2.05 |
| 14e |
24 |
None |
— |
— |
— |
δ-valerolactone (δ-VL). The ROP of δ-valerolactone (δ-VL) was also investigated. By performing the reaction in toluene and in the presence of BnOH as co-activator, none of the catalysts proved active, affording either nil or trace polymer after 24 h at 130 °C. Furthermore, the tests were repeated under solvent-free conditions (Table 3). Due to the disappointing results achieved in the CL case, the Na-containing species 4 was not further tested. Poor activity was exhibited by complexes 1 and 2, allowing for 18 and 10% conversion, respectively, after 24 h (runs 1 and 2). Good conversion (58%) was obtained in the presence of the SO2-bridged derivative 5 after 10 minutes (run 3); however, further reaction was impeded by the formation of a highly viscous mixture. A similar outcome was observed in the presence of complexes 7–9, albeit with longer reaction times (30 min to 12 h, runs 4–6). Finally, complete conversion was attained after 12 h with the dimethyleneoxa-derivative 6, affording a PVL with Mn lower than the expected values and rather broad polydispersity (1.57, run 4). This indicated the occurrence of extensive transesterification. Noteworthy, the Mw/Mn values observed for the PCLs and PVLs do not present significant differences (with respect to the uncertainty of the GPC measurements). Thus, the same extent of transesterification can be assumed for both monomers. This was primarily attributed to the lack of homogeneity of the reaction medium, as the formation of polymer often impeded efficient stirring of the mixture. On the other hand, Mn higher than the expected values were obtained in the presence of complexes 5 and 7–9, indicating partial catalyst activation. Similar to the ε-CL case, the 1H NMR spectrum of the PVL synthesized with 6 suggested the formation of linear –CH2OH capped polymer chains (Fig. S8†). The MALDI-ToF spectrum of the sample highlighted the presence of a main set of peaks (Fig. S9†) compatible with an α-hydroxyl-ω-(carboxylic acid)-terminated PVL, suggesting that, unlike the PCL case, the polymerization was initiated by adventitious water. Two minor distributions accountable to cyclic species and Na-doped linear adducts were also observed (Fig. S10 and S11,† respectively).
Table 3 ROP of δ-VL catalysed by the Li complexes under solvent-free conditions
| Run |
Catalyst |
δ-VL:Li |
T (°C) |
Time (h) |
Conversiona (%) |
Mnb,c |
Mncalcd |
PDIb |
| Determined by 1H NMR spectroscopy on crude reaction mixture. From GPC. Values corrected considering Mark–Houwink factor (0.56) from polystyrene standards in THF. Calculated from ([Monomer]0/[Cat.]0) × conv. (%) × monomer molecular weight. Stirring stopped due to the polymer formation. |
| 1 |
1 |
100:1 |
130 |
24 |
18 |
nd |
1800 |
nd |
| 2 |
2 |
24 |
10 |
1320 |
1000 |
1.29 |
| 3 |
5 |
10 mine |
58 |
30100 |
5800 |
2.12 |
| 4 |
6 |
12 |
>99 |
2990 |
9900 |
1.57 |
| 5 |
7 |
1e |
46 |
16360 |
4600 |
1.29 |
| 6 |
8 |
30 mine |
60 |
21680 |
6000 |
1.52 |
| 7 |
9 |
12e |
64 |
31650 |
6400 |
2.08 |
rac-Lactide (r-LA). The complexes were then employed as catalysts in the ROP of r-LA under solvent-free conditions at 150 °C (Table 4). While low molecular weight oligomers were obtained with 2, 4, 5, 7, and 9, wax-like materials were isolated in the presence of 1, 6, and 8. Mn higher than the calculated values and polydispersities of ca. 1.7 suggested partial catalyst activation as well as the occurrence of transesterification. Again, here the dimethyleneoxa-bridged complex 6 performed best. The samples showed high heterotactic enrichment (Pr 0.88–0.91), as observed by 2D J-resolved 1H NMR spectroscopy (Fig. S12–S14†).26 However, due to the poor activity of the catalysts in solution, it has not been possible to perform mechanistic investigations by NMR spectroscopy in order to assess the type of stereocontrol involved. The MALDI-ToF spectrum of the PLA prepared in the presence of 6 (Fig. S15†) highlighted the presence of only one population accountable to cyclic species. In addition, the fact that the peaks are separate by 72 m/z units (mono-lactyl group) suggested the occurrence of transesterification.
Table 4 ROP of r-LA catalysed by the Li complexes
| Run |
Catalyst |
r-LA:Li |
T (°C) |
Time (h) |
Conversiona (%) |
Mnb,c |
Mncalcd |
PDIb |
Pre |
| Determined by 1H NMR spectroscopy on crude reaction mixture. From GPC. Values corrected considering Mark–Houwink factor (0.56) from polystyrene standards in THF. Calculated from ([Monomer]0/[Cat.]0) × conv. (%) × monomer molecular weight. Determined 2D J-resolved 1H NMR spectroscopy. |
| 1 |
1 |
100:1 |
150 |
24 |
57 |
17340 |
8210 |
1.73 |
0.91 |
| 2 |
2 |
100:1 |
18 |
|
Liquid oligomers |
|
|
| 3 |
3 |
100:1 |
25 |
nd |
3600 |
nd |
nd |
| 4 |
5 |
100:1 |
69 |
|
Liquid oligomers |
|
|
| 5 |
6 |
100:1 |
87 |
18470 |
12530 |
1.72 |
0.90 |
| 6 |
7 |
100:1 |
18 |
nd |
1150 |
nd |
nd |
| 7 |
8 |
100:1 |
57 |
22650 |
8210 |
1.71 |
0.88 |
| 8 |
9 |
100:1 |
66 |
|
Liquid oligomers |
|
|
ω-pentadecalactone (ω-PDL). All complexes were found to be inactive in the ROP of the more challenging 15-membered ring monomer ω-pentadecalactone, both in solution and under solvent-free conditions.
Conclusions
In conclusion, the use of LiOtBu (or LiOH or nBuLi in the case of 4 and 7) on interaction with a series of calixarenes containing a range of bridging groups, namely –CH2–, –S–, –SO–, –SO2– or –CH2OCH2– allowed access to a number of lithiated calix[3 and 4]arenes (see Chart 1). In the case of the –SO–, adventitious oxidation can result in the formation of mixed –SO–/–SO2– bridging.
All complexes (except 1 and 4) proved active in the ROP of ε-CL, δ-VL and r-LA under solvent-free conditions and in the case of ε-CL, the activity trend was found to be 6 > 7 > 8 > 5 > 9. Similar results were also observed in the case of the other monomers, with the dimethyleneoxa-bridged derivative outperforming the other systems. The higher activity of 6 was thought to be due to the higher flexibility of the –CH2OCH2– bridge allowing better access to the active centre(s) and/or to the stabilization of the active species by the oxygen atoms of said bridge. In most of the cases, incomplete catalyst activation and lack of control were observed. The NMR spectroscopy characterization of selected samples suggested the formation of linear –CH2OH capped PCLs and PVLs and highly heterotactic PLAs. None of the catalysts proved active in the ROP of the larger monomer ω-pentadecalactone, while the conversion of only r-LA was observed during ε-CL/r-LA co-polymerization.
Experimental
General
All manipulations were carried out under an atmosphere of nitrogen using standard Schlenk and Cannula techniques or in a conventional nitrogen-filled glove-box. Solvents were refluxed over an appropriate drying agent, and distilled and degassed prior to use. THF-d8 was stirred over CaH2 for 48 h, vacuum transferred and then stored over 3A molecular sieves. Elemental analyses were performed by the microanalytical services at the London Metropolitan University or the Chemistry Department at the University of Hull. NMR spectra were recorded on a Varian VXR 400 S spectrometer at 400 MHz; chemical shifts are referenced to the residual protio impurity of the deuterated solvent. IR spectra (nujol mulls, KBr windows) were recorded on PerkinElmer 577 and 457 grating spectrophotometers. The compounds 1,3-di-n-propyloxycalix[4]arene and p-tert-butylhexahomotrioxacalix[3]arene were prepared from using the literature methods.27 The compound p-tert-butylthiacalix[4]arene was purchased from TCI UK, whilst the ligands p-tert-butylsulfinylcalix[4]arene and p-tert-butylsulfonylcalix[4]arene were gifts from Dr Hitoshi Kumagaya of the Cosmo oil company. The complex [LS4Li5(OH)(THF)4] was prepared by the method of Zeller and Radius.18 All other chemicals were obtained commercially and used as received unless stated otherwise.
Preparation of [Li8(L4S)2(THF)4]·5THF (1·5THF)
A solution of lithium tert-butoxide (7.21 ml, 1 M in THF, 7.21 mmol) was added to LSH4 (1.00 g, 1.39 mmol) in THF (30 ml) at ambient temperature. After stirring for 1 h, the orange solution was concentrated to about 20 ml, and was left to stand for 48 h at room temperature to afford colourless crystals of 1·5THF suitable for X-ray diffraction analysis in 65% yield (0.90 mmol, 1.90 g). Anal. cald. for C56H76S8Li8O8: C, 56.58; H, 6.44%; found C, 56.49; H, 6.28%. IR: 1587 w, 1357 m, 1306 w, 1260 s, 1199 s, 1093 bs, 1022 bs, 911 w, 885 m, 865 m, 800 bs, 767 m, 733 m, 702 w, 668 w, 620 w, 546 m. MS (M+): 1488.6 m/z. 1H NMR (THF-d8, 400 MHz, 298 K) δ: 7.61 (s, 16H, ArH), 1.31 (s, 72H, (CH3)3C). 7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 1.50 (s).
Preparation of [Li6(L4SOH)2(THF)2]·5(THF) (2·5THF)
A solution of lithium tert-butoxide (2.01 ml, 1 M in THF, 2.01 mmol) was added to LSOH4 (0.51 g, 0.65 mmol) in THF (10 ml) at ambient temperature. After stirring for 1 h, the solution was concentrated to about 10 ml, and was left to stand for 48 h at room temperature, to afford colourless crystals of 2·8THF suitable for X-ray diffraction analysis in 31% yield (0.20 mmol, 0.22 g). Anal. cald. for C88H106Li6O18S8 (sample dried in vacuo for 12 h, –5THF) requires C 60.40, H 6.11% Found C 59.87, H 6.39%. IR: 2727 w, 1772 w, 1602 m, 1305 w, 1093 s, 1019 s, 878 w, 801 s, 660 w, 643 w, 600 w, 491 w. 7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 3.24 (s).
Preparation of [Li8(calix[4]arene(SO)(SO2)(SO1.68)2)2(THF)6]·8(THF) (3·8THF)
A solution of lithium tert-butoxide (6.62 ml, 1 M in THF, 6.62 mmol) was added to LSOH4 (1.00 g, 1.27 mmol) in THF (30 ml) at ambient temperature. After stirring for 1 h, the solution was concentrated to about 20 ml, and was left to stand for 48 h at room temperature to afford colourless crystals of 3·8THF suitable for X-ray diffraction analysis in 70% yield (0.89 mmol, 2.40 g). Anal. cald. for C136H200Li8O34.72S8 requires C 60.34, H 7.45%. Found C 60.19, H 7.39%. IR: 1606 w, 1462 m, 1377 m, 1260 m, 1087 m, 1020 m, 799 m, 722 w, 566 w. 1H NMR (THF-d8, 400 MHz, 298 K) δ: 7.98–7.70 (m, 16H, ArH), 1.32–1.29 (m, 72H, (CH3)3C). 7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 1.37 (bs).
Preparation of [Li5Na(LSO/3SO2H)2(THF)5]·7.5(THF) (4·7.5THF)
To L4SOH4 (1.00 g, 1.27 mmol) and LiOH (0.16 g, 6.68 mmol) in THF (30 ml) at ambient temperature. After stirring for 12 h, the system was filtered, concentrated to about 20 ml, and was left to stand for 48 h at room temperature affording colourless prisms of 4·7.5THF suitable for X-ray diffraction analysis in 26% yield (0.33 mmol, 0.45 g). Anal. cald. for C106H142Li5NaO35.28S8 (–7THF, sample dried in vacuo for 3 h) requires C 58.32, H 6.56%. Found C 58.57, H 6.92%. IR: 3530 bs, 2826 w, 2360 m, 2341 w, 1604 m, 1261 s, 1220 w, 1092 s, 1020 s, 866 w, 799 s, 723 m, 678 w, 634 w, 563 w. 1H NMR (THF-d8, 400 MHz, 298 K) δ: 8.82 (bs, 2H, –OH, 7.97–7.72 (m, 16H, ArH), 1.25–1.03 (m, 72H, (CH3)3C). 7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 0.14 (bs).
Preparation of [Li5L4SO2(OH)(THF)4]·2THF (5·2THF)
A solution of lithium tert-butoxide (6.12 ml, 1 M in THF, 6.12 mmol) was added to LSO2H4 (1.00 g, 1.18 mmol) in THF (30 ml) at ambient temperature. After heating at 70 °C for 1 h, the solution was filtered hot under nitrogen atmosphere on a POR4 frit, and was left to stand for 48 h at room temperature affording colourless crystals of 5·2THF suitable for X-ray diffraction analysis in 60% yield (0.71 mmol, 0.89 g). Anal. cald. for C60H85S4Li5O18: C, 57.32; H, 6.82%; found C, 57.21; H, 6.85%. IR: 3182 bw, 1609 m, 1500 m, 1396 w, 1365 m, 1328 w, 1292 m, 1261 s, 1214 m, 1197 m, 1155 m, 1127 s, 1080 bs, 1019 bs, 962 m, 902 w, 866 w, 796 s, 735 w, 722 w, 685 w, 660 w, 631 m, 562 m, 523 w, 495 w, 478 w. 1H NMR (THF-d8, 400 MHz, 298 K) δ: 7.99 (s, 8H, ArH), 1.34 (s, 36H, (CH3)3C). OH signal not detected. 7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 1.23 (s).
Preparation of [Li6(LCOC)2(HOtBu)2]·0.78THF·1.22hexane (6·0.78THF·1.22hexane)
A solution of lithium tert-butoxide (9.02 ml, 1 M in THF, 9.02 mmol) was added to LCOCH4 (1.00 g, 1.73 mmol) in THF (30 ml) at ambient temperature. After stirring for 1 h, the volatiles were removed in vacuo, and the residue was taken up in hexane (30 ml). Filtration of the solution under nitrogen atmosphere on a POR4 frit and cooling at 0 °C afforded colourless crystals of 6·0.78THF·1.22hexane suitable for X-ray diffraction analysis in 11% yield (0.19 mmol, 0.25 g). Further crops of crystals can be obtained by concentration of the mother liqueur to 10 ml and standing at −40 °C for 10 days (total isolated yield 45%, 0.78 mmol, 1.02 g). C90.45H133.36Li6O14.77 requires C 72.48, H 8.97%. Found C, 71.96; H, 8.49%. IR: 3662 (bs), 2727 w, 1636 m, 1304 w, 1260 w, 1260 w, 1211 m, 1094 m, 1018 m, 975 m, 875 w, 802 m, 722 m, 530w. 1H NMR (THF-d8, 400 MHz, 298 K) δ: 7.08–6.70 (m, 12H, ArH), 5.16–4.81 (m, 12H, endo-CH2), 4.53–4.40 (m, 12H, exo-CH2), 2.27–2.03 (bs, 2H, (CH3)3COH), 1.30–1.19 (m, 54H, (CH3)3C)), 1.10 (s, 18H, (CH3)3COH). 7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 0.14 (bs), 0.13 (s).
Preparation of [LH2Li2(thf)(OH2)2]·3THF (7·3THF)
A solution of lithium tert-butoxide (4.33 ml, 1 M in THF, 4.33 mmol) was added to LH4 (1.00 g, 1.73 mmol) in THF (30 ml) at ambient temperature. After stirring for 4 h, the solution was concentrated to about 20 ml, and was left to stand for 48 h at room temperature to afford colourless crystals of 7 suitable for X-ray diffraction analysis in 45% yield (0.59 g, 0.78 mmol). C60H90Li2O10 requires C 73.14, H 9.21%. Found C, 71.48; H, 10.35%.28 IR: 3611 s, 3476 w, 2727 w, 2360 w, 1744 w, 1603 w, 1577 w, 1303 s, 1155 w, 1130 m, 1104 w, 1040 s, 967 w, 910 m, 871 s, 821 m, 795 m, 736 s, 668 w, 606 m. 1H NMR (THF-d8, 400 MHz, 298 K) δ: 7.13–6.76 (m, 16H, ArH), 5.08 (d, 2H, J = 12 Hz, endo-CH2), 4.66 (d, 2H, J = 12 Hz, endo-CH2), 4.57 (d, 2H, J = 12 Hz, endo-CH2), 4.14 (d, 2H, J = 12 Hz, endo-CH2), 3.40–3.02 (m, 8H, exo-CH2), 1.43–1.11 (m, 72H, (CH3)3C). OH signal not detected. 7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 2.44 (bs), 1.77 (bs).
Preparation of {[Li5(de-BuL)(OH)(NCMe)3]·2MeCN}n (8·2MeCN)
A solution of lithium tert-butoxide (12.25 ml, 1 M in THF, 12.25 mmol) was added to de-BuLH4 (1.00 g, 2.36 mmol) in THF (30 ml) at ambient temperature. After stirring for 4 h the volatiles were removed in vacuo, and the residue was taken up in MeCN (30 ml). Filtration under nitrogen atmosphere on a POR4 frit and cooling at 0 °C afforded, after 1 day, colourless crystals of 8·2MeCN suitable for X-ray diffraction analysis in 55% yield (1.30 mmol, 0.88 g). Cald. for C38H36Li5N5O5 (–MeCN, sample dried in vacuo for 2 h) C 67.94, H 5.23, N 8.81%. Found C 69.31, H 5.77, N 8.84%.28 Found C 65.76, H 5.86, N 2.04%. IR: 3609 w, 1587 w, 1301 m, 1288 m, 1261 s, 1218 w, 1092 bs, 1046 s, 1020 bs, 944 w, 909 w, 860 w, 847 m, 800 s, 752 m, 723 w, 705 w, 678 w. 1H NMR (THF-d8, 400 MHz, 298 K) δ: 7.85–7.76 (m, 8H, ArH), 7.04–6.64 (m, 8H, ArH), 6.24–5.99 (m, 8H, ArH), 5.10 (d, 2H, J = 13 Hz, endo-CH2), 4.94 (d, 2H, J = 13 Hz, endo-CH2), 4.64 (d, 2H, J = 13 Hz, endo-CH2), 4.17 (d, 2H, J = 13 Hz, endo-CH2), 3.61 (d, 2H, J = 12 Hz, exo-CH2), 3.41–3.03 (m, 6H, exo-CH2), 2.08 (s, 12H, MeCN), 1.29 (s, 6H, MeCN).7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 3.46 (s), 0.46 (bs), −2.36 (bs).
Preparation of [Li6(LPr)2(H2O)2]·hexane (9·hexane)
To a solution of LPr2H2 (1.00 g, 1.36 mmol) in THF (30 ml) at −78 °C was added nBuLi (4.35 ml, 1.6 M, 2.75 mmol). After stirring for 12 h at ambient temperature, the volatiles were removed in vacuo, and the residue was taken up in hexane (30 ml). Filtration of the solution under nitrogen atmosphere on a POR4 frit and cooling to 0 °C afforded, after 3 days, small colourless crystals of 9·hexane suitable for X-ray diffraction analysis in 25% yield (0.34 mmol, 0.52 g). Anal. cald. for C100H136Li6O10: C, 78.00; H, 8.90%; found C, 77.67; H, 8.78%. IR: 3376 bm, 3182 w, 1747 w, 1671 w, 1598 s, 1337 m, 1301 s, 1261 s, 1236 m, 1194 s, 1096 s, 1055 bs, 969 s, 917w, 871 s, 799 s, 755 w, 723 m, 675 w, 633 w. 1H NMR (THF-d8, 400 MHz, 298 K) δ: 7.03–6.95 (m, 8H, ArH), 6.90–6.45 (m, 8H, ArH), 6.60–6.33 (m, 8H, ArH), 4.31 (d, 4H, J = 12 Hz, endo-CH2), 4.12 (d, 4H, J = 13 Hz, endo-CH2), 3.92 (t, 4H, J = 7 Hz, –OCH2CH2CH3), 3.30 (d, 4H, J = 13 Hz, exo-CH2), 3.95 (d, 4H, J = 12 Hz, exo-CH2), 2.06 (m, 4H, –OCH2CH2CH3), 1.32 (t, 6H, –OCH2CH2CH3). 7Li NMR (THF-d8, 194.3 MHz, 298 K) δ: 2.17 (s), 1.55 (bs).
Ring open polymerization (ROP) procedures
Reaction in toluene. Under inert atmosphere, a THF solution of the complex (1 mM) was added into a Schlenk tube and the solvent was removed under reduced pressure at room temperature. Toluene (2 ml) was added along with the monomer (4.5 mmol) and the required amount of BnOH (as a toluene solution). The reaction mixture was then placed into an oil bath pre-heated to the required temperature, and the solution was stirred for the prescribed time. The polymerization mixture was then quenched by addition of an excess of glacial acetic acid (0.2 ml) into the solution, and the resultant solution was then poured into methanol (200 ml). The resultant polymer was then collected on filter paper and dried in air at room temperature.
Solvent-free conditions. Under an inert atmosphere, a THF solution of the complex (1 mM) was added into a Schlenk tube and the solvent was removed under reduced pressure at room temperature. The monomer (9.0 mmol) was then added and the reaction was stirred at 130 °C for 12–24 h, or until a mass of polymer blocking the stirring formed. The mixture was then taken up in CH2Cl2 and quenched with acidified methanol (200 ml). The resultant polymer was then collected on filter paper and dried in air at room temperature.
Crystal structure determinations
Diffraction data for 1·5THF, 5·2THF, and 8·2MeCN were collected on a Bruker Apex 2 CCD diffractometer using a sealed tube source; for 2·5THF & 7·3THF at the Diamond Light Source synchrotron, station I19, equipped with Rigaku Saturn724+ CCD or Crystal Logic diffractometers; for 3·8THF, 4·7.5THF, and 6·0.78THF·1.22hexane, on Rigaku Saturn 724+ CCD diffractometers equipped with sealed tube (3·8THF and 4·7.5THF) or rotating anode (6·0.78THF·1.22hexane) sources; and for 9·hexane at the Advanced Light Source synchrotron, Station 11.3.1, equipped with a Bruker Apex 2 CCD diffractometer. Further details are given in Table 6 and in the deposited cif files. All crystal structures were collected at low temperature. Data were corrected for absorption and Lp effects. The structures were solved using direct methods29 or a dual-space, charge-flipping algorithm and refined on F2.30,31 This set of lithium-containing calixarene structures were challenging due to often weak diffracting power caused by their light-atom make-up, and the often-encountered tBu group and solvent of crystallization disorder. Some tBu groups needed to be modelled with the methyl groups or the whole moiety split over two sets of positions. Solvent molecules of crystallization were often disordered and needed to be modelled with split positions, or as diffuse electron density via the Platon Squeeze procedure (for 1·5THF, 2·5THF, 4·7.5THF, 5·2THF and 6·0.78THF·1.22hexane).32 The ‘squeezed’ solvent contribution is included in the chemical formula in each case. The number of solvent molecules of crystallization should only be taken as approximate. Where disorder has been modelled, restraints were applied to anisotropic displacement parameters and the geometry of the affected and immediately adjacent atoms. For 2·5THF there was some evidence of partial disorder in the O atom on each –SO– bridging group, with the O sometimes positioned in the other tetrahedral site on the S; this was modelled and the major components at O(5), O(6), O(7), and O(8) were 88.4(8), 91.4(7), 81.6(8), and 81.9(9)%, respectively. For 3·8THF the occupancies of O(6) and O(11) were refined as 68.1(10) and 68.0(10)%, respectively. For 4·7.5THF O(20) was only 78.0(18)% occupied. For 5·2THF both THFs are disordered along the 4-fold axis and both were modelled with ‘Squeeze’. One resides in the calixarene cavity, the other above the coordinated THFs. In 6·0.78THF·1.22hexane the solvent of crystallization site was provisionally modelled with point atoms as having part hexane and part THF in a 61.2:38.8(4)% ratio, but due to the severity of the disorder was subsequently modelled with the Platon Squeeze procedure.32 Atoms O(7)/H(7) were modelled a two-fold disordered with major site occupancy of 54.9(12)% while atoms O(6)/C(36) and the associated hydrogens were also modelled as two-fold disordered with major site occupancy 85.2(4)%. The diffraction data for 8·2MeCN were non-merohedrally twinned via a 180° rotation about real axis 0 0 1 with a twin component ratio of 52.2:47.8(4)%. Hydrogen atoms were placed in geometrically determined positions, except for those on hetero atoms in structures with good data, where coordinates were refined.
Table 6 Crystallographic data for complexes 1–9
| Compound |
1·5THF |
2·5THF |
3·8THF |
4·7.5THF |
5·2THF |
| Formula |
C116H160S8Li8O17 |
C108H146Li6O23S8 |
C136H200Li8O34.72S8 |
C134H198Li5NaO35.28S8 |
C60H85S4Li5O18 |
| Formula weight |
2138.43 |
2110.36 |
2702.48 |
2687.55 |
1257.21 |
| Crystal system |
Monoclinic |
Triclinic |
Monoclinic |
Monoclinic |
Tetragonal |
| Space group |
I2/a |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
P21/c |
C2 |
P4/n |
| Unit cell dimensions |
|
|
|
|
|
| a (Å) |
25.323(13) |
13.354(12) |
17.8795(13) |
38.167(3) |
13.306(2) |
| b (Å) |
17.655(9) |
13.959(9) |
23.8479(17) |
20.3517(14) |
13.306(2) |
| c (Å) |
26.894(14) |
17.735(14) |
17.9950(13) |
18.4867(13) |
19.079(3) |
| α (°) |
90 |
70.39(4) |
90 |
90 |
90 |
| β (°) |
105.218(5) |
84.17(5) |
111.9050(11) |
99.9979(5) |
90 |
| γ (°) |
90 |
62.02(3) |
90 |
90 |
90 |
| V (Å3) |
11602(10) |
2744(4) |
7118.9(9) |
14141.7(18) |
3377.9(9) |
| Z |
4 |
1 |
2 |
4 |
2 |
| Temperature (K) |
150(2) |
100(2) |
100(2) |
100(2) |
150(2) |
| Wavelength (Å) |
0.71073 |
0.6889 |
0.71073 |
0.71073 |
0.71073 |
| Calculated density (g cm−3) |
1.224 |
1.277 |
1.261 |
1.262 |
1.236 |
| Absorption coefficient (mm−1) |
0.216 |
0.231 |
0.199 |
0.203 |
0.205 |
| Transmission factors (min./max.) |
0.950 and 0.981 |
0.984 and 0.998 |
0.965 and 0.982 |
0.672 and 1.000 |
0.941 and 0.984 |
| Crystal size (mm3) |
0.24 × 0.11 × 0.09 |
0.07 × 0.05 × 0.01 |
0.18 × 0.17 × 0.09 |
0.25 × 0.20 × 0.12 |
0.30 × 0.22 × 0.08 |
| θ(max) (°) |
22.5 |
26.6 |
27.6 |
27.5 |
25.0 |
| Reflections measured |
35870 |
27343 |
66538 |
98566 |
26220 |
| Unique reflections |
7584 |
12071 |
16117 |
32341 |
2987 |
| Rint |
0.230 |
0.183 |
0.110 |
0.047 |
0.081 |
| Reflections with F2 > 2σ(F2) |
2884 |
3704 |
13043 |
30584 |
2071 |
| Number of parameters |
748 |
707 |
965 |
1657 |
191 |
| R1 [F2 > 2σ(F2)] |
0.098 |
0.109 |
0.0973 |
0.063 |
0.064 |
| wR2 (all data) |
0.272 |
0.329 |
0.280 |
0.176 |
0.187 |
| GOOF, S |
0.93 |
0.88 |
1.07 |
1.05 |
1.08 |
| Largest difference peak and hole (e Å−3) |
0.41 and −0.32 |
0.53 and −0.31 |
1.08 and −0.88 |
1.17 and −0.40 |
0.67 and −0.43 |
| Compound |
6·0.78THF·1.22hexane |
7·3THF |
8·2MeCN |
9·hexane |
| Formula |
C90.45H133.36Li6O14.77 |
C60H90Li2O10 |
C38H36Li5N5O5 |
C100H136Li6O10 |
| Formula weight |
1498.68 |
985.19 |
677.42 |
1539.72 |
| Crystal system |
Triclinic |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P |
P21 |
P21/m |
P |
| Unit cell dimensions |
|
|
|
|
| a (Å) |
13.350(8) |
11.763(14) |
13.011(7) |
10.5688(9) |
| b (Å) |
13.868(9) |
36.82(4) |
10.480(6) |
13.1163(11) |
| c (Å) |
14.564(9) |
13.119(16) |
13.837(8) |
17.2651(14) |
| α (°) |
114.022(7) |
90 |
90 |
102.6019(11) |
| β (°) |
95.245(6) |
96.406(11) |
112.332(8) |
91.2345(12) |
| γ (°) |
111.486(6) |
90 |
90 |
93.5123(12) |
| V (Å3) |
2198(2) |
5647(11) |
1745.2(17) |
2329.8(3) |
| Z |
1 |
4 |
2 |
1 |
| Temperature (K) |
100(2) |
100(2) |
150(2) |
150(2) |
| Wavelength (Å) |
0.71073 |
0.6889 |
0.71073 |
0.7749 |
| Calculated density (g cm−3) |
1.132 |
1.159 |
1.289 |
1.097 |
| Absorption coefficient (mm−1) |
0.074 |
0.075 |
0.084 |
0.080 |
| Transmission factors (min./max.) |
0.980 and 0.993 |
0.340 and 1.000 |
0.974 and 0.997 |
0.990 and 0.993 |
| Crystal size (mm3) |
0.28 × 0.16 × 0.09 |
0.06 × 0.05 × 0.02 |
0.31 × 0.24 × 0.04 |
0.15 × 0.10 × 0.10 |
| θ(max) (°) |
31.5 |
22.5 |
26.5 |
33.6 |
| Reflections measured |
29498 |
37790 |
20601 |
34077 |
| Unique reflections |
12781 |
15742 |
6965 |
13962 |
| Rint |
0.039 |
0.133 |
0.199 |
0.061 |
| Reflections with F2 > 2σ(F2) |
8911 |
9255 |
2595 |
9552 |
| Number of parameters |
493 |
1333 |
298 |
561 |
| R1 [F2 > 2σ(F2)] |
0.071 |
0.150 |
0.115 |
0.072 |
| wR2 (all data) |
0.215 |
0.451 |
0.276 |
0.228 |
| GOOF, S |
1.05 |
1.05 |
1.04 |
1.06 |
| Largest difference peak and hole (e Å−3) |
0.44 and −0.36 |
0.56 and −0.49 |
0.40 and −0.52 |
0.66 and −0.36 |
CCDC 2038615-23† contain the crystal data for the structures reported herein.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We thank the EPSRC for funding (EP/R023816/1 and EP/S025537/1), and the EPSRC National Crystallography Service at Southampton (UK) and the EPSRC Mass Spectrometry Service, Swansea (UK) for data collection. This research used resources of the Advanced Light Source, which is a DOE Office of Science User Facility under contract no. DE-AC02-05CH11231.
Notes and references
-
(a) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS;
(b) C. K. Williams and M. A. Hillmyer, Polym. Rev., 2008, 48, 1–10 CrossRef CAS;
(c) M. A. Woodruff and D. W. Hutmacher, Prog. Polym. Sci., 2010, 35, 1217–1256 CrossRef CAS;
(d) A. L. Sisson, D. Ekinci and A. Lendlein, Polymer, 2013, 54, 4333–4350 CrossRef CAS.
-
(a) A. Arbaoui and C. Redshaw, Polym. Chem., 2010, 1, 801–826 RSC;
(b) I. Nifant'ev and P. Ivchenko, Molecules, 2019, 24, 4117 CrossRef PubMed;
(c) O. Santoro, X. Zhang and C. Redshaw, Catalysts, 2020, 10, 800 CrossRef CAS.
- D. Homden and C. Redshaw, Chem. Rev., 2008, 108, 5086–5130 CrossRef CAS PubMed.
- O. Santoro and C. Redshaw, Catalysts, 2020, 10, 210 CrossRef CAS.
- C. Floriani and R. Floriani-Moro, Adv. Organomet. Chem., 2001, 47, 167–233 CrossRef CAS.
- See for example
(a) B. Masci, in Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield, and J. Vicens, Kluwer Academic Publishers, 2001, ch. 12 Search PubMed;
(b) N. Morohashi, F. Narumi, N. Iki, T. Hattori and S. Miyano, Chem. Rev., 2006, 106, 5291–5316 CrossRef CAS PubMed;
(c) K. Cottet, P. M. Marcos and P. J. Cragg, Beilstein J. Org. Chem., 2012, 8, 201–226 CrossRef CAS PubMed.
-
(a) A. K. Sutar, T. Maharana, S. Dutta, C.-T. Chen and C. Lin, Chem. Soc. Rev., 2010, 39, 1724–1746 RSC;
(b) J. Gao, D. Zhu, W. Zhang, G. A. Solan, Y. Ma and W.-H. Sun, Inorg. Chem. Front., 2019, 6, 2619–2652 RSC.
-
(a) C. D. Gutsche, M. Iqbal, K. S. Nam, K. See and I. Alam, Pure Appl. Chem., 1988, 60, 483–488 CAS;
(b) R. Abidi, M. V. Baker, J. M. Harrowfield, D. S.-C. Ho, W. R. Richmond, B. W. Skelton, A. H. White, A. Varnek and G. Wipff, Inorg. Chim. Acta, 1996, 246, 275–286 CrossRef CAS;
(c) K. C. Nam, D. S. Kim and J. M. Kim, Bull. Korean Chem. Soc., 1997, 18, 636–640 CAS;
(d) For FTIR studies see B. Brzezinski, F. Bartl and G. Zundel, J. Phys. Chem. B, 1997, 101, 5611–5653 CrossRef CAS.
- H. Bock, A. John, C. Nather and Z. Havlas, J. Am. Chem. Soc., 1995, 117, 9367–9368 CrossRef CAS.
- M. G. Davidson, J. A. K. Howard, S. Lamb and C. W. Lehmann, Chem. Commun., 1997, 1607–1608 RSC.
- G. Guillemot, E. Solari, C. Rizzoli and C. Floriani, Chem.–Eur. J., 2002, 8, 2072–2080 CrossRef CAS.
- E. D. Gueneau, K. M. Fromm and H. Goesmann, Chem.–Eur. J., 2003, 9, 509–514 CrossRef CAS PubMed.
- T. A. Hanna, L. Liu, A. M. Angeles-Boza, X. Kou, C. D. Gutsche, K. Ejsmont, W. H. Watson, L. N. Zakharov, C. D. Incarvito and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 6228–6238 CrossRef CAS PubMed.
- D. Mendoza-Espinosa, B. A. Martinez-Ortega, M. Quiroz-Guzman, J. A. Golen, A. L. Rheingold and T. A. Hanna, J. Organomet. Chem., 2009, 694, 1509–1523 CrossRef CAS.
- L. Liu, L. N. Zakharov, J. A. Golen, A. L. Rheingold, W. H. Watson and T. A. Hanna, Inorg. Chem., 2006, 45, 4247–4260 CrossRef CAS PubMed.
- N. P. Clague, J. D. Crane, D. J. Moreton, E. Sinn, S. J. Teat and N. A. Young, J. Chem. Soc., Dalton Trans., 1999, 3535–3536 RSC.
- D. S. Lee, M. R. J. Elsegood, C. Redshaw and S. Zhan, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2009, 65, m291–m295 CrossRef CAS PubMed.
- J. Zeller and U. Radius, Inorg. Chem., 2006, 45, 9487–9492 CrossRef CAS PubMed.
- C. Redshaw, O. Rowe, D. L. Hughes, A. M. Fuller, I. Alvarado Ibarra and S. M. Humphrey, Dalton Trans., 2013, 42, 1983–1986 RSC.
- M. Yamada, M. R. Gandhi and F. Hamada, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2018, 74, 575–579 CrossRef CAS PubMed.
-
(a) B.-T. Ko and C.-C. Lin, J. Am. Chem. Soc., 2001, 123, 7973–7977 CrossRef CAS PubMed;
(b) M. H. Chisholm, C.-C. Lin, J. C. Gallucci and B.-T. Ko, Dalton Trans., 2003, 406–412 RSC;
(c) C.-A. Huang and C.-T. Chen, Dalton Trans., 2007, 5561–5566 RSC;
(d) W. Clegg, M. G. Davidson, D. V. Graham, G. Griffen, M. D. Jones, A. R. Kennedy, C. T. O'Hara, L. Russo and C. M. Thomson, Dalton Trans., 2008, 1295–1301 RSC;
(e) Y. Huang, Y.-H. Tsai, W.-C. Hung, C.-S. Lin, W. Wang, J.-H. Huang, S. Dutta and C.-C. Lin, Inorg. Chem., 2010, 49, 9416–9435 CrossRef CAS PubMed;
(f) W.-Y. Lu, M.-W. Hsiao, S. C. N. Hsu, W.-T. Peng, Y.-J. Chang, Y.-C. Tsou, T.-Y. Wu, Y.-C. Lai, Y. Chen and H.-Y. Chen, Dalton Trans., 2012, 41, 3659–3667 RSC;
(g) N. Ikpo, C. Hoffmann, L. N. Dawe and F. M. Kerton, Dalton Trans., 2012, 41, 6651–6660 RSC;
(h) R. K. Dean, A. M. Reckling, H. Chen, L. N. Dawe, C. M. Schneider and C. M. Kozak, Dalton Trans., 2013, 42, 3504–3520 RSC;
(i) J. Char, O. G. Kulyk, E. Brulé, F. de Montigny, V. Guérineau, T. Roisnel, M. J.-L. Tschan and C. M. Thomas, C. R. Chim., 2016, 19, 167–172 CrossRef CAS;
(j) D. Alhashmialameer, N. Ikpo, J. Collins, L. N. Dawe, K. Hattenhauer and F. M. Kerton, Dalton Trans., 2015, 44, 20216–20231 RSC;
(k) Y. F. Al-Khafaji, T. J. Prior, L. Horsburgh, M. R. J. Elsegood and C. Redshaw, ChemistrySelect, 2017, 2, 759–768 CrossRef.
- C. Redshaw, Dalton Trans., 2016, 45, 9018–9030 RSC.
-
(a) P. v. d. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194 CrossRef;
(b) A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- O. Santoro, M. R. J. Elsegood, E. V. Bedwell, J. A. Pryce and C. Redshaw, Dalton Trans., 2020, 49, 11978–11996 RSC.
- C. Redshaw, M. A. Rowan, L. Warford, D. M. Homden, A. Arbaoui, M. R. J. Elsegood, S. H. Dale, T. Yamato, C. Pérez-Casas, S. Matsui and S. Matsuura, Chem.–Eur. J., 2007, 13, 1090 CrossRef CAS PubMed.
-
(a) C. Ludwig and M. R. Viant, Phytochem. Anal., 2010, 21, 22 CrossRef CAS PubMed;
(b) M. J. Walton, S. J. Lancaster and C. Redshaw, ChemCatChem, 2014, 6, 1892 CrossRef CAS.
-
(a) A. Arduini and A. Casnati, in Macrocycle Synthesis, ed. D. Parker, Oxford University Press, New York, 1996, ch. 7 Search PubMed;
(b) P. D. Hampton, Z. Bencze, W. Tomg and C. E. Daitch, J. Org. Chem., 1994, 59, 4838–4843 CrossRef CAS.
- Problems with obtaining satisfactory elemental analysis due to rapid decomposition of compounds of this type were noted by Fromm et al., see ref. 12.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
-
(a) A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9–18 CrossRef CAS PubMed;
(b) P. v. d. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Alternative views of 1, 2, 3 and 6; polymer 1H and 2D-resolved NMR spectra, MALDI-TOF spectra. CCDC 2038615–2038623. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00175b |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Simon J. Teat
b,
Simon J. Teat