
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights into the C–H activation of methane mediated by the unsupported and silica-supported VO2OH and CrOOH: a DFT study†
Shidong Zhao‡
a,
Lishuang Ma‡a,
Yanyan Xibc,
Hongyan Shang*ac and
Xufeng Lin *ac
*ac
aDepartment of Chemistry, College of Science, China University of Petroleum (East China), Qingdao, 266580, P. R. China. E-mail: catagroupsh@163.com; hatrick2009@upc.edu.cn
bCollege of Chemical Engineering, China University of Petroleum (East China), Qingdao, 266580, P. R. China
cState Key Laboratory of Heavy Oil Processing, China University of Petroleum (East China), Qingdao, 266580, P. R. China
First published on 17th March 2021
Abstract
The direct activation and conversion of methane has been a topic of interest in both academia and industry for several decades. Deep understanding of the corresponding mechanism and reactivity mediated by diverse catalytic clusters, as well as the supporting materials, is still highly desired. In this work, the regulation mechanism of C–H bond activation of methane, mediated by the closed-shell VO2OH, the open-shell CrOOH, and their silica supported clusters, has been investigated by density functional theory (DFT) calculations. The hydrogen-atom transfer (HAT) reaction towards methane C–H bond activation is more feasible when mediated by the unsupported/silica-supported CrOOH clusters versus the VO2OH clusters, due to the intrinsic spin density located on the terminal Ot atom. The proton-coupled electron transfer (PCET) pathways are regulated by both the nucleophilicity of the Ot site and the electrophilicity of the metal center, which show no obvious difference in energy consumption among the four reactions examined. Moreover, the introduction of a silica support can lead to subtle influences on the intermolecular interaction between the CH4 molecule and the catalyst cluster, as well as the thermodynamics of the methane C–H activation.
1. Introduction
Methane (CH4), as the major component of natural gas, is not only an important clean fuel, but also an abundant hydrocarbon feedstock for chemical commodities.1,2 In the recent decades, with the development of modern catalytic technologies, the direct nonoxidative activation and conversion of methane to high value-added chemicals has received significant interest, from both industry and academia.3–6 Although a variety of strategies have been developed, C–H bond activation, the first and also critical step for methane conversion is still challenging. This challenge arises from the fact that the extremely symmetric nonpolar methane molecule exhibits a high C–H bond dissociation energy (439 kJ mol−1).7,8 To avoid the high temperature and harsh reaction conditions, efficient catalysis under mild conditions as well as the deep understanding of the underlying mechanism for direct methane activation has always been highly desirable.9–12Up to now, a great deal of effort has been devoted on the metal-cluster-mediated methane activation reactions, using both experimental and theoretical methods. These metal cluster species, including the metal oxide cluster,13,14 metal carbides,15,16 and metal cluster anions17,18 etc., can be regarded as simplified models of methane activation sites on heterogenous catalysts. Meanwhile, the knowledge of the fundamental reaction mechanism towards the methane activation and conversion is continuously being improved.13–18 For example, Sun et al. reported that the C–H bond activation may undergo a concerted pathway where the C and H atoms on the C–H bond attack two different sites, accompanied by formatting two new bonds (C–Ni and O–H), or a stepwise pathway where two separated products of Ni3O3H and ethyl radical can be obtained.19 Wan et al. found that the terminal oxygens [![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O] are more active than the bridge oxygens [–O–] for C–H bond activation of methane on molybdenum oxides.20 Interestingly, the adsorbed oxygen species on metal surfaces were demonstrated to exhibit promotional or inhibitory effects, which depends strongly on the nature of metals.21–26 In more recent years, a series of mechanistic scenarios were explored by Schwarz et al.,13–16,27–29 in which the classical hydrogen-atom transfer (HAT),27 proton-coupled electron transfer (PCET),28 and hydride transfer (HT)29 pathways were explored and distinguished in various reaction systems for methane C–H activation.
O] are more active than the bridge oxygens [–O–] for C–H bond activation of methane on molybdenum oxides.20 Interestingly, the adsorbed oxygen species on metal surfaces were demonstrated to exhibit promotional or inhibitory effects, which depends strongly on the nature of metals.21–26 In more recent years, a series of mechanistic scenarios were explored by Schwarz et al.,13–16,27–29 in which the classical hydrogen-atom transfer (HAT),27 proton-coupled electron transfer (PCET),28 and hydride transfer (HT)29 pathways were explored and distinguished in various reaction systems for methane C–H activation.
Although significant progress has been made in methane activation mediated by metal clusters, the reported gas-phase atom clusters obtained by state-of-the-art mass spectrometric are generally ionic species.13–20 While, researches on the catalytic activity of the neutral metal oxyhydroxide towards C–H bond activation of methane are still sparse. Typically, the earth abundant vanadium and chromium have been demonstrated good catalytic capability for C–H activation,30–32 but few studies have focused on their neutral oxyhydroxides, such as VO2OH and CrOOH. In addition, in industrial catalytic processes, catalyst supports are usually introduced to disperse and support active components, which can lead to a lower cost of the transition metal as well as a higher stability of the catalytic material compared with the unsupported ones.33 However, detailed understanding of the support effect on catalyst activity, and the interaction between support and active components is yet to be more thoroughly investigated, especially at the molecular level.33,34
Herein, we report a theoretical study on C–H bond activation of methane mediated by the silica-supported/unsupported VO2OH and CrOOH clusters, through the density functional theory (DFT) calculations. The HAT and PCET reaction channels were carefully examined to obtain a better understanding towards the catalytic activity of the closed-shell VO2OH and the open-shell CrOOH, as well as the effect of silica support.
2. Computational methods
In the present study, DFT calculations were performed using the Gaussian 09 program package,35 to investigate the reaction mechanism of methane activation catalyzed by the VO2OH and CrOOH catalysts, as well as their silica-supported complexes, as shown in the following equations:| CH4 + an unsupported VO2OH cluster | (i) |
| CH4 + a model silica supported VO2OH cluster | (ii) |
| CH4 + an unsupported CrOOH cluster | (iii) |
| CH4 + a model silica supported CrOOH cluster | (iv) |
In our calculations, a cluster containing Si4O6H3 was used to model the silica support surface, denoted as “Sili” in this paper. All Si centers have a tetrahedron local structure simulating the SiO4 tetrahedron moiety, and the H atoms were used to saturate the boundary dangling bonds of the Si centers. The VO3@Sili and CrO2@Sili were used to denote the complexes that the VO3− and CrO2− clusters supported on the silica model. In addition, a larger cluster models, Si16O30H3, denoted as “Sili2”, was used to investigate the cluster size effects (details can be seen from the Section 3.4), which showed that the smaller cluster model of Si4O6H3 used in this article are as reliable as that with the larger one.
All minima of the reactants and the corresponding products for Rxn (i)–(iv) were first obtained by full system optimization, using the hybrid density functional36–38 including empirical dispersion correction computed with Grimme's D3 formula (B3LYP-D3).39 Note that the open-shell configurations were calculated within the spin-unrestricted approximation, in which the broken-symmetry technique was introduced to treat the singlet (Rxn (i) and (ii)) and quartet (Rxn (iii) and (iv)) radical HAT pathway. The energy-consistent scalar-relativistic DF-adjusted 10-electron-core pseudopotential/ECP10MDF(8s7p6d1f/6s5p3d1f) basis set was used for V and Cr,40 while the def-TZVP41,42 basis set was applied for all other atoms. The transition states (TSs) for C–H bond activation of CH4 mediated by V and Cr catalysts were obtained by employing the conventional approach of transition state optimization. Based on the optimized geometries, vibrational frequency analyses were carried out at the same level of theory to ensure that each stationary point truly represented a minimum, or a saddle point, and to obtain the thermal enthalpy corrections at 298.15 K and 1 atm. Intrinsic reaction coordinate (IRC) computations43,44 were also conducted to confirm that the transition states connected to the appropriate reactants and products. Finally, the electronic energies were refined with the B3LYP-D3 functional and a larger basis set, in which the DF-adjusted 10-electron-core pseudopotential/ECP10MDF(8s7p6d2f1g)/[6s5p3d2f1g] was used for V and Cr,45 while the ma-def2-TZVPP46,47 basis set was used for all other atoms. To reduce the unaffordable computational cost on reactions mediated by the larger cluster model, Si16O30H3, the basis set for the reaction center was used as same as that smaller reaction models above, while a smaller def-SV basis set was applied to the remaining atoms away from the reaction center, the corresponding mixed basis set applied for the geometry optimization and frequency calculations is the denoted as BSI, and that used for the refined single-point-energy calculation is denoted as BSII (see Fig. S1†).
The charge and spin density populations analyses were performed by the Hirshfeld method.48 The spin-density isosurfaces (isovalue 0.02) were plotted using a multifunctional wavefunction analyzer (Multiwfn).49 The symmetry-adapted perturbation theory (SAPT) energy decomposition analysis was carried out at the SAPT0/def2-TZVPP level, by using PSI4 program package.50 The total SAPT0 interaction energy (ΔEint) between the CH4 and the catalyst cluster can be decomposed into contributions of the electrostatic (ΔEels), exchange-repulsion (ΔEexc), induction (ΔEind) and dispersion (ΔEdisp) terms:
| ΔEtot = ΔEels + ΔEexc + ΔEind + ΔEdisp |
The notations having the form of [R/TS/P]-n will be used in the later sections, where R/TS/P represents the reactant, transition state or product, and n represents one of (i)–(iv) for the Rxn numbers defined above. Meanwhile a superscript will be used before a species name to represent the spin multiplicity of this species. For example, the 4TSHAT-iv denotes the quartet transition state of Rxn (iv) along the HAT pathway.
3. Results and discussion
3.1 Structures and properties of the unsupported/silica-supported VO2OH and CrOOH clusters
The stable structures, charge and spin density properties of the unsupported and silica-supported VO2OH and CrOOH clusters were investigated at first. As shown in Fig. 1, the free VO2OH has two terminal oxygen atoms (Ot), which have the same V–Ot bond length of 1.59 Å and equivalent charge of −0.31e. The free CrOOH have one terminal Ot atom with −0.36e charge and an O–H bond of length 1.59 Å. These calculations results show that both VO2OH and CrOOH possess Lewis acid–base pair [Mn+(V5+/Cr3+)–O−] units,28 facilitating the C–H bond activation through the PCET pathway. For the HAT mechanism for C–H activation, the Ot-center spin is necessary as the precursor state to abstract H atom.51,52 Since the VO2OH is closed-shell singlet, zero spin density were found on the Ot atom. By contrast, obvious spin density of −0.21 was observed located on the Ot atom of the open-shell CrOOH cluster, which is reasonable due to the Cr3+ is initially a quartet, with three unpaired electrons in the 3d orbitals. In consequence, the Ot site on CrOOH is expected to be more advantageous for a radical HAT reaction; whereas, for the Ot site on the closed-shell VO2OH, an electron reorganization is needed to accumulate O-center spin density to trigger the radical HAT process, indicating a higher energy barrier should be overcome.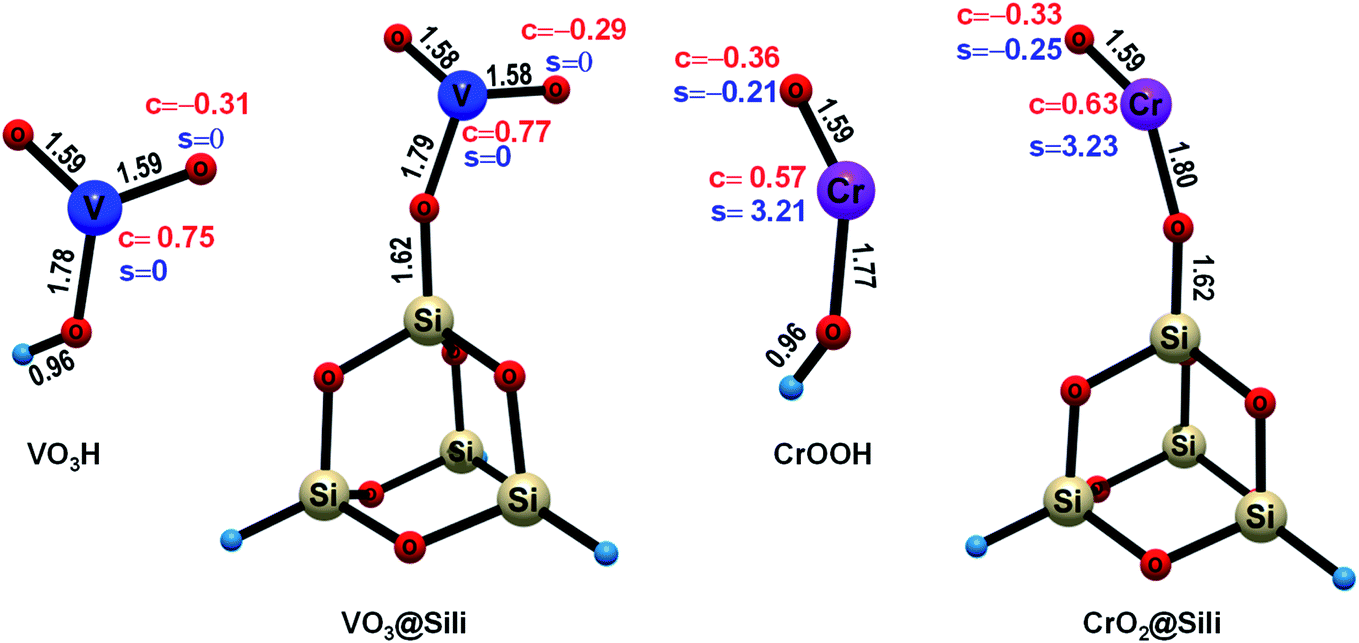 | ||
| Fig. 1 The diagrams of the key bond lengths (black), the Hirshfeld charge (c, red) and spin density (s, blue) distributions for the catalyst clusters studied in this paper (the property parameters on only one side Ot of VO2OH are labeled in this figure for clarity). | ||
In the presence of the silica support, the hydroxyl groups of both VO2OH and CrOOH have to deprotonate to combine with the Si atom through chemisorption, forming a new Si–O bond (1.62 Å). In this condition, slight structural change and charge reorganization were observed from our computational results. As shown in Fig. 1, the Ot atom is slightly less negatively charged upon silica support for both VO3@Sili and CrO2@Sili clusters [VO2OH (−0.31e) → VO3@Sili (−0.29e); CrOOH (−0.36e) → CrO2@Sili (−0.33e)]. Besides the charge-regulation effect, the spin density on the Ot atom of CrOOH is a little increased upon silica support [CrOOH (−0.21e) → CrO2@Sili (−0.25e)]. Since the methane C–H activation reaction is sensitive to the charge and spin density of the Ot atom, the silica support may lead to subtle influences on the potential energy profiles (PESs) of methane activation on the VO2OH and CrOOH clusters. Details about the mechanism of the reactions Rxn (i)–(iv) will be discussed in the Sections 3.2 and 3.3. It should be noted that, the free CrOOH in doublet state was calculated energetically 16.4 kcal mol−1 higher than that in quartet state. It suggests that the electronic ground state is a quartet state with three parallel-spin electrons occupying three 3d orbitals (t32ge0g). Thus, the doublet CrOOH were not considered in this work for the reactions Rxn (iii) and (iv).
3.2 Thermal activation of CH4 on VO2OH clusters
 | ||
| Fig. 2 (a) PESs (ΔH, kcal mol−1) for the reaction of CH4 with an unsupported VO2OH cluster, obtained at the B3LYP-D3/ma-def2-TZVPP//B3LYP-D3/TZVP level of theory. Critical structures are given with the key bond lengths in Å. (b) The evolution of the spin density distribution along the reaction pathway. | ||
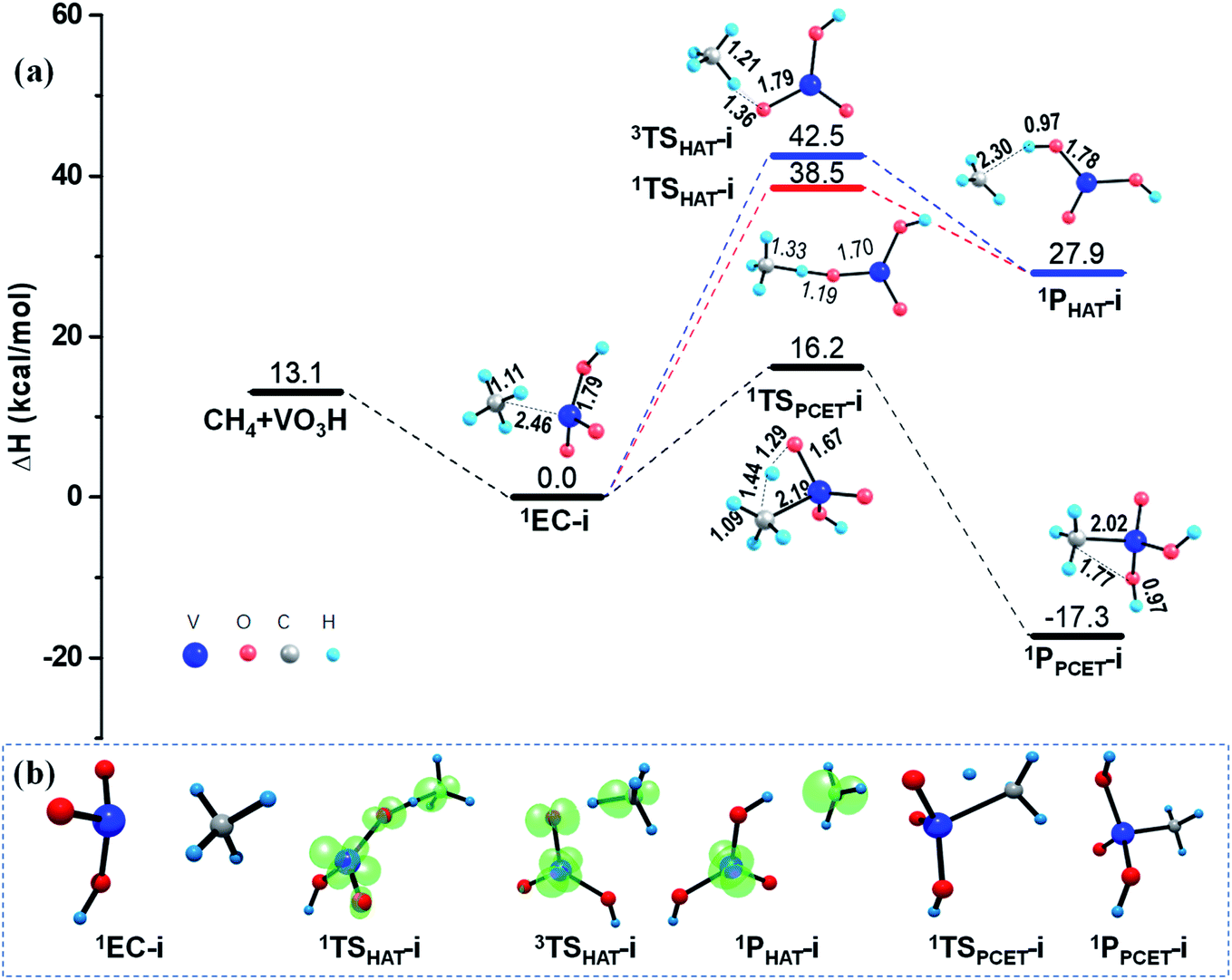
After 1EC-i is formed, one C–H bond of methane could be activated under two mechanisms in principle, that is, the HAT and the PCET mechanisms. As illustrated in Fig. 2, since the products of CH4 activation via a radical HAT mechanism are two separated radicals, the open-shell singlet channel and triplet channel were both examined for the HAT mechanism. Population analyses results reveal that the electron reorganization is necessary to access the transition states 1TSHAT-i or 3TSHAT-i, which finally generate the open-shell product 1PHAT-i, consisting of a tetravalent vanadium hydroxide [VO(OH)2] and a methyl radical CH3˙, through homolysis of C–H bond (see Fig. 2b and Table S2†). As discussed in Section 3.1, the Ot atom of VO2OH have zero spin density, which is hard to abstract a hydrogen atom from CH4 through the radical HAT mechanism. Consistently, noticeable energy consumptions (38.5–42.5 kcal mol−1) were found along the HAT pathways on both of the singlet and triplet PESs.
In contrast, the C–H activation of CH4 is relatively easy to proceed along the PCET pathway mediated by VO2OH, by overcoming a moderate barrier of 16.4 kcal mol−1, as shown in Fig. 2. In this case, the H atom is transferred as a proton from the CH4 moiety to the terminal Ot atom of VO2OH, forming a new O–H bond; meanwhile the generated methyl anion CH3− with an electron pair is transferred to the positive V center, forming a new V–C bond. This process eventually produces a relatively stable molecular structure (−17.3 kcal mol−1) of 1PPCET-i, without change of spin density. These calculated results suggest that the PCET mechanism is predominant for the Rxn (i) mediated by a VO2OH cluster.
 | ||
| Fig. 3 (a) PESs (ΔH, kcal mol−1) for the reaction of CH4 with the silica supported VO3− cluster, obtained at the B3LYP-D3/ma-def2-TZVPP//B3LYP-D3/def-TZVP level of theory. Critical structures are given with the key bond lengths in Å. (b) The evolution of the spin density distribution along the reaction pathway. | ||
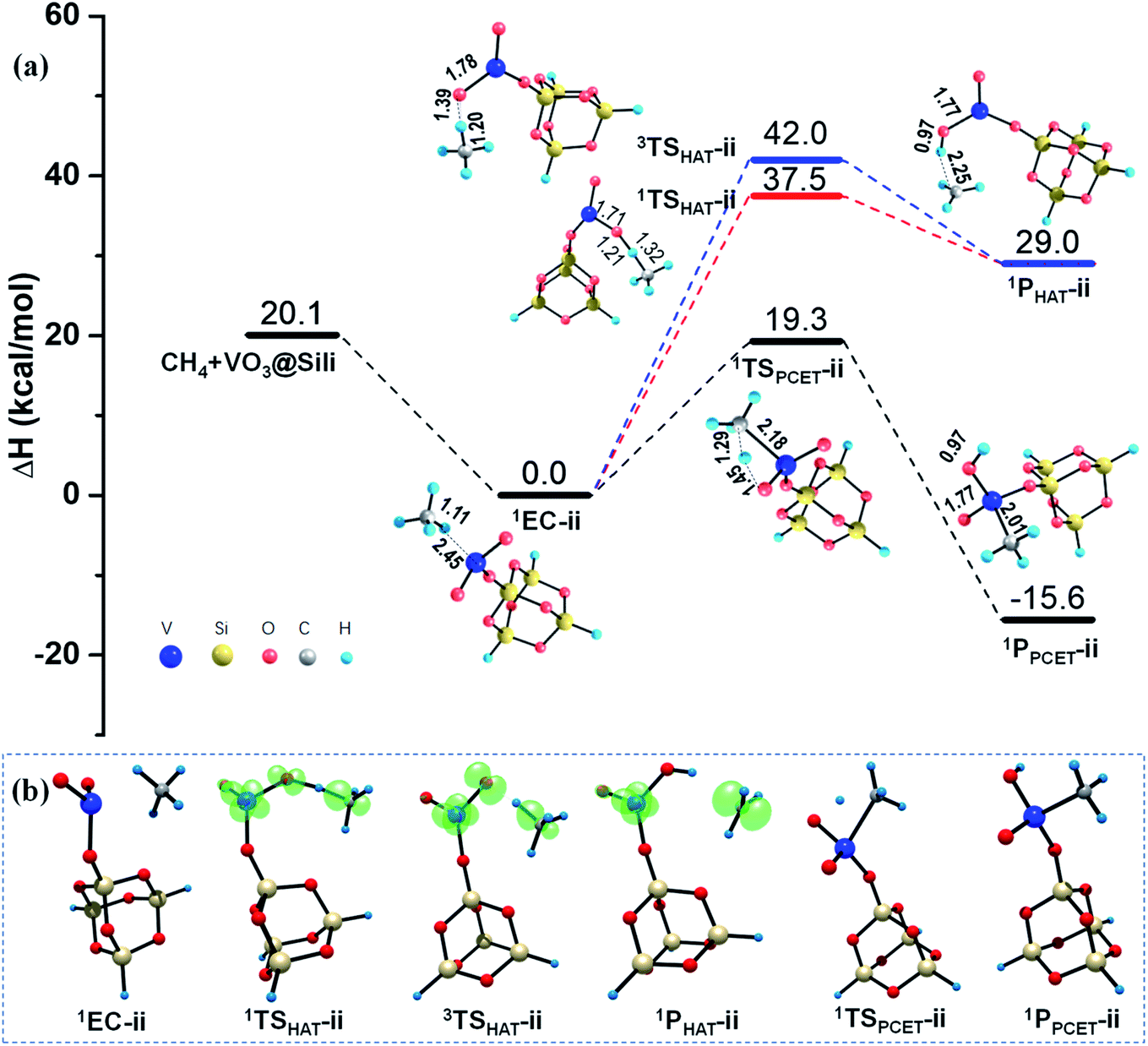
As illustrated in Fig. 3, the calculated results show negligible barrier changes of the HAT reaction paths in both singlet and triplet states, compared with the HAT reactions mediated by the free VO2OH cluster. This is reasonable since the VO2OH is intrinsically close-shell, and the spin density distribution is not affected in the presence of the silica support. While, for the PCET pathway, the silica support causes a larger energy barrier of 19.3 kcal mol−1, producing the 1PPCET-ii with −15.6 kcal mol−1 energy decrease. Though this cause a higher energy barrier than that in Rxn (i) from the point of view of kinetic features, both the HAT and PCET channels in Rxn (ii) are more advantageous thermodynamically due to the enhanced intermolecular interactions in the presence of silica support.
3.3 Thermal activation of CH4 on CrOOH clusters
 | ||
| Fig. 4 (a) PESs (ΔH, kcal mol−1) for the reaction of CH4 with an unsupported CrOOH cluster, obtained at the B3LYP-D3/ma-def2-TZVPP//B3LYP-D3/def-TZVP level of theory. Critical structures are given with the key bond lengths in Å. (b) The evolution of the spin density distribution along the reaction pathway. | ||
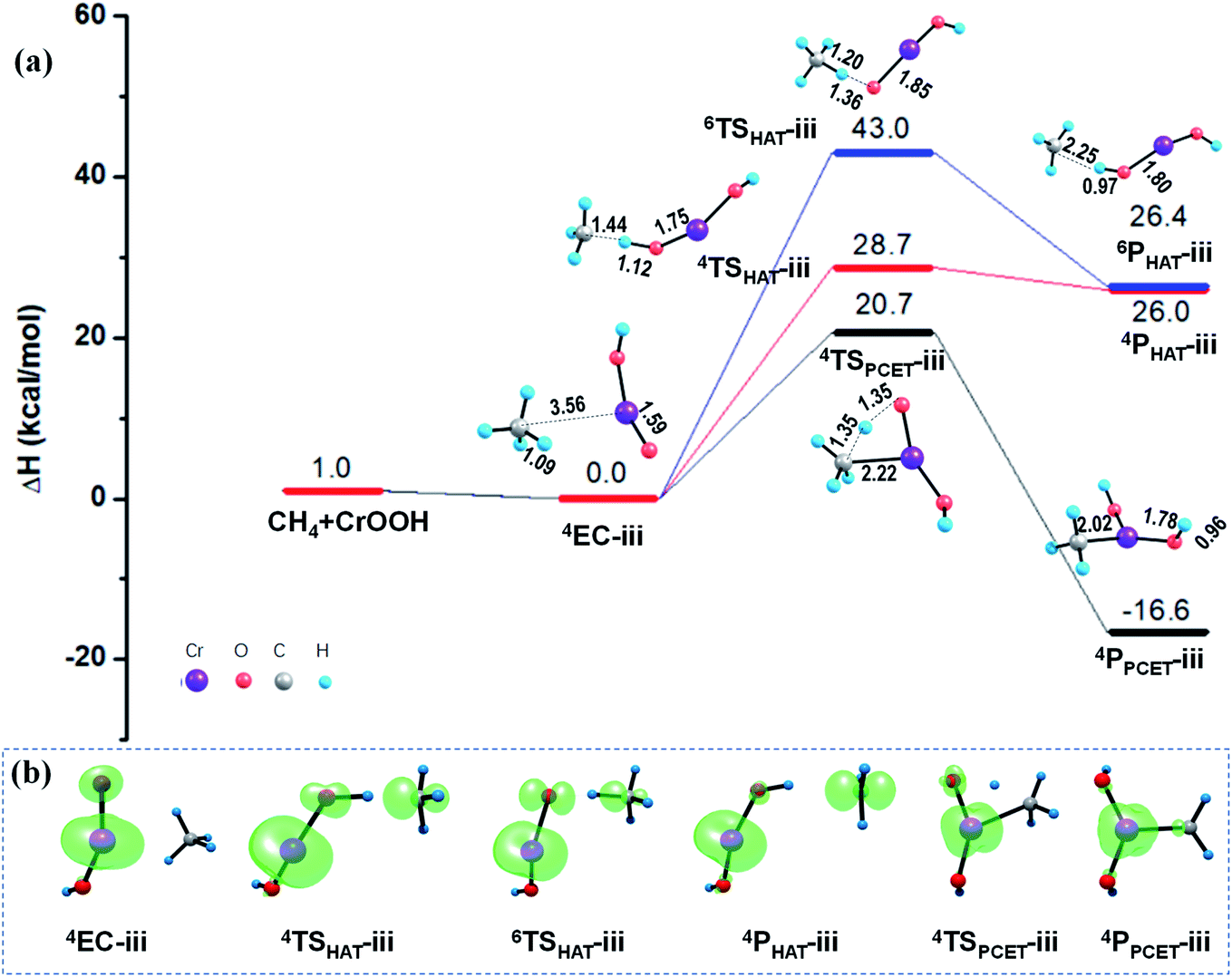
As mentioned in Section 3.1, unlike the PCET mechanism, the Ot-center spin is necessary as a prepared site to abstract the H atom in the HAT mechanism. Different from the close-shell VO2OH cluster, a spin density of −0.21 was found located on the terminal Ot atom of the quartet CrOOH cluster (see Fig. 4b and Table S2†). Correspondingly, the HAT reaction path in quartet state towards the methane C–H bond activation is more feasible with a smaller barrier of 28.7 kcal mol−1, compared with that in the Rxn (i) (38.5 kcal mol−1). The CH3˙ is then released, and the H atom is transferred to the Ot atom, forming a divalent chromium hydroxide [Cr(OH)2]. However, the HAT pathways on the sextet PESs in Rxn (iii) and (iv) are both suppressed severely by the energy consumption with more than 40.0 kcal mol−1 barriers, due to significant electronic reorganizations are need to access the sextet transition states 6TSHAT-iii and 6TSHAT-iv (see Fig. 4b, 5b and Table S2†).
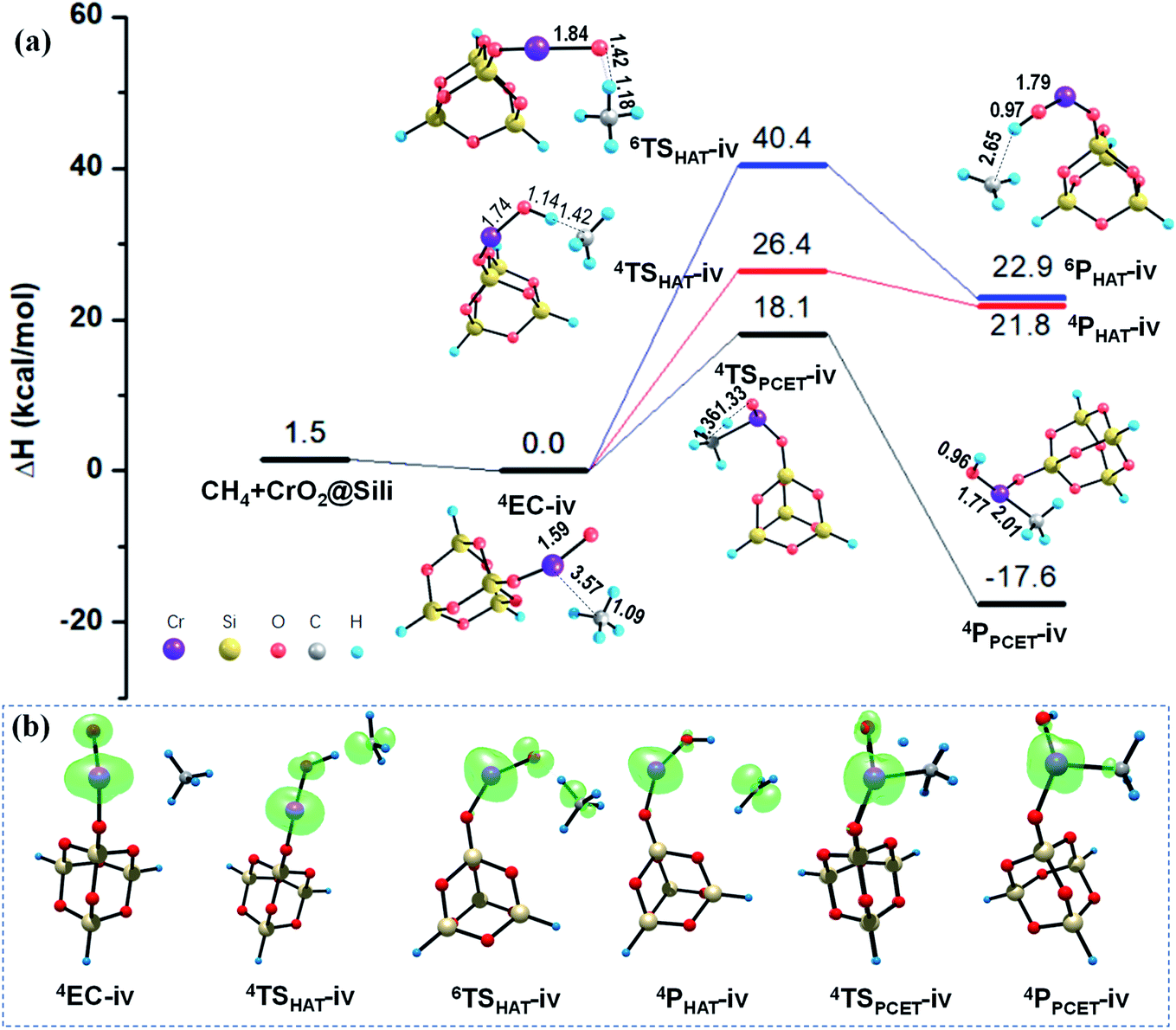 | ||
| Fig. 5 (a) PESs (ΔH, kcal mol−1) for the reaction of CH4 with the silica supported CrO2− cluster, obtained at the B3LYP-D3/ma-def2-TZVPP//B3LYP-D3/def-TZVP level of theory. Critical structures are given with the key bond lengths in Å. (b) The evolution of the spin density distribution along the reaction pathway. | ||
As described in Section 3.1, the spin density on the Ot atom is slightly increased once the CrO2@Sili is formed through chemisorption of the CrOOH cluster onto the silica support. The larger spin density on Ot atom means it is more facilitated to abstract a H atom from the CH4 molecule mediated by the supported CrO2− cluster versus that in the reaction Rxn (iii) mediated by the free CrOOH cluster. As expected, a smaller barrier (26.4 kcal mol−1) was found computationally along the singlet HAT pathway, which is 2.3 kcal mol−1 lower than that under the HAT pathway mediated by the free CrOOH in Rxn (iii). Moreover, the difference of the energy barriers along HAT paths are significantly smaller mediated by open-shell CrOOH and CrO2@Sili (26.4–28.7 kcal mol−1), than that mediated by the close-shell VO2OH and VO3@Sili clusters (37.5–38.5 kcal mol−1), which is in line with the previous findings that the spin density at the hydrogen-acceptor site plays a crucial role for C–H bond activation proceeding by HAT mechanism. It is worth mentioning that, though the PCET mechanism was found more active than that of HAT mechanism in the four methane activation reactions (Rxn (i)–(iv)), it is not always true for different reaction systems.51–54 For example, the HAT pathway was found more facilitated mediated by the cationic metal–carbon cluster FeC4+, induced by localized spin density generated in situ once the reactants encounter each other.53
3.4 Rxn (v) vs. Rxn (ii): investigation of silica cluster size effects
The results shown in the above sections about the silica support are based a small cluster model (Sili) containing 4 Si atoms. As is known, the metal sites are always resided on a rough surface since catalytic supports are always mesoporous materials. Furthermore, the particle size of metal or oxide clusters may possibly affect their catalytic activity, which depends on the localized or delocalized properties of the reactions toward the cluster surface.55,56 In order to know whether the small model of Sili is representative for a catalytic support of silica, and also to understand as well the cluster size effect of the silica model used in this work toward the C–H bond activation of methane, a larger Si16O30H3 cluster model was also examined for the reaction mediated by silica-supported VO3− cluster (VO3@Sili2 in Rxn (v)). Rxn (v) differs with Rxn (ii) only in the model of the silica support.As shown in Fig. 6, the differences of reaction barrier along the HAT reaction paths in both singlet and triplet states are within 0.6 kcal mol−1, predicted by using the two silica models (Si16O30H3 in Rxn (v) vs. Si4O6H3 in Rxn (ii)). The reaction barrier along the PCET pathways in Rxn (ii) and (v) show only a small difference of 0.1 kcal mol−1. The two products in Rxn (v) and (ii) also show similar thermodynamic stability with energy differences of 0.1–0.3 kcal mol−1. In addition, the changes of geometries along both the HAT and PCET pathways in Rxn (v) are very close to that in Rxn (ii), by using the larger and the smaller silica cluster models. In consequence, these calculated results show that the smaller cluster model of Si4O6H3 is as reliable as the larger one of Si16O30H3 at least for the current research in this work.
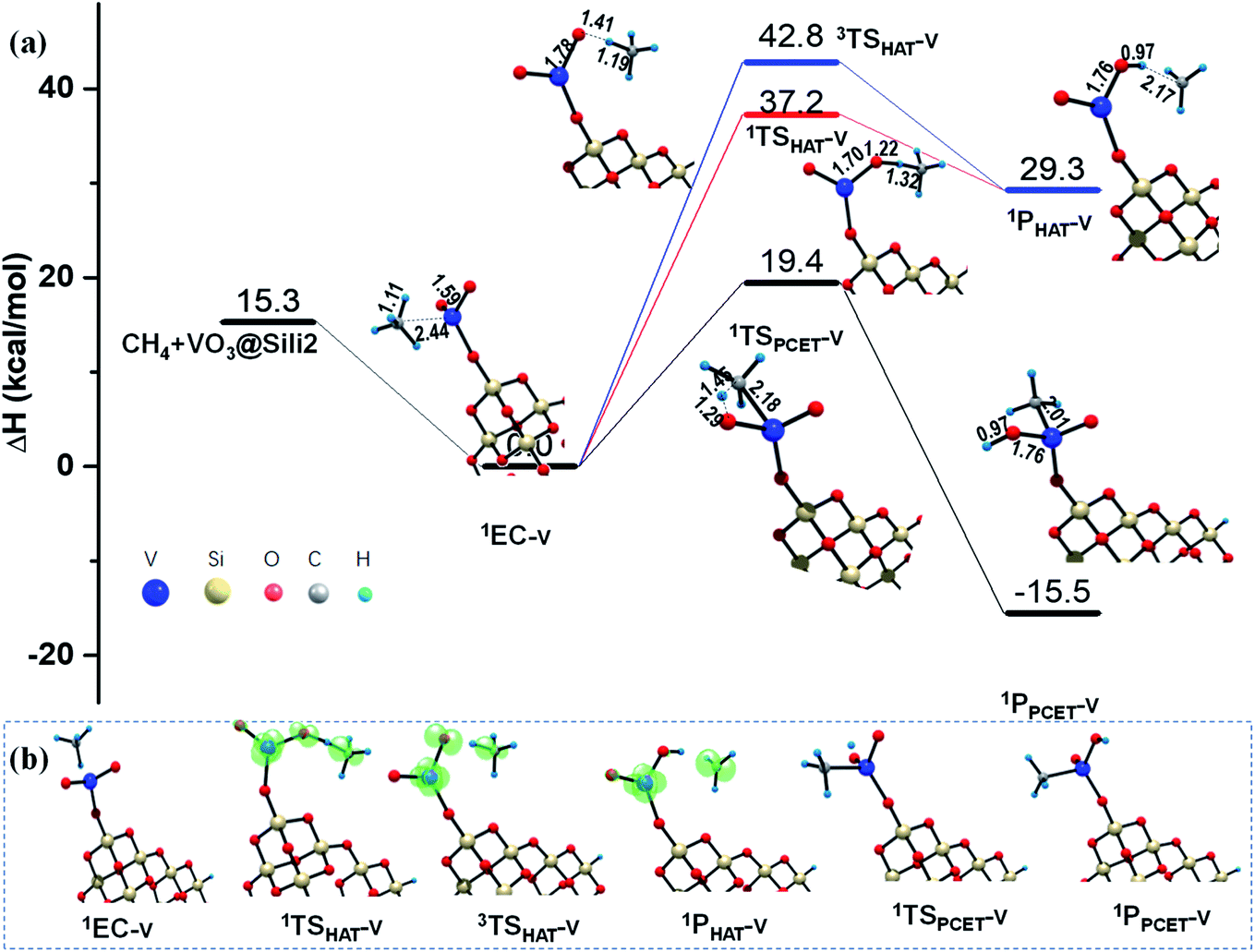 | ||
| Fig. 6 (a) PESs (ΔH, kcal mol−1) for the reaction of CH4 with VO3− cluster supported by the larger Si16O30H3 cluster, obtained at the B3LYP-D3/BSII//B3LYP-D3/BSI level of theory. Critical structures are given with the key bond lengths in Å. (b) The evolution of the spin density distribution along the reaction pathway. | ||
4. Conclusion
In this work, a systematic DFT computational work have been performed to provide the mechanism comparison regarding the C–H bond activation of methane through HAT and PCET pathways mediated by the close-shell VO2OH, the open-shell CrOOH, as well as their silica supported clusters.For the four reactions (Rxn (i)–(iv)) examined, the HAT reaction towards methane C–H bond activation is dominated by the spin density on the Ot site, and the PCET reaction is codetermined by the nucleophilicity of the Ot site and the electrophilicity of the metal center site. Benefiting from the obvious spin density on terminal Ot atom, the calculated HAT reactions in quartet state mediated by the open-shell CrOOH and CrO2@Sili clusters are demonstrated more favorable than that in singlet state mediated by the close-shell VO2OH and VO3@Sili clusters. In contrast, there are relatively smaller differences of the energy barriers through PCET mechanism among the four reactions (Rxn (i)–(iv)). Moreover, all the HAT reactions examined are disadvantaged compared with the PCET pathways, note that the products obtained under the PCET mechanism should suffer further M(V, Cr)–C bond cleavage to generate the free CH3˙ radical. In the presence of silica support, the intermolecular interaction between the CH4 molecule and the catalyst cluster is enhanced, especially for the reactions mediated by VO2OH cluster, leading to the C–H bond activation of methane being more thermodynamically feasible.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support from the National Natural Science Foundation of China (21576291), and Shandong Province Natural Science Foundation (ZR2014BM002) is gratefully acknowledged.References
- P. Tang, Q. Zhu, Z. Wu and D. Ma, Energy Environ. Sci., 2014, 7, 2580–2591 RSC.
- A. Caballero and P. J. Pérez, Chem. Soc. Rev., 2013, 42, 8809–8820 RSC.
- K. Otsuka and Y. Wang, Appl. Catal., A, 2001, 222, 145–161 CrossRef CAS.
- R. Horn and R. Schloegl, Catal. Lett., 2015, 145, 23–39 CrossRef CAS.
- C. Karakaya and R. J. Kee, Prog. Energy Combust. Sci., 2016, 55, 60–97 CrossRef.
- Y. Liu, D. Deng and X. Bao, Chem, 2020, 6, 2497–2514 CAS.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- X. Meng, X. Cui, N. P. Rajan, L. Yu, D. Deng and X. Bao, Chem, 2019, 5, 2296–2325 CAS.
- U. Zavyalova, M. Holena, R. Schloegl and M. Baerns, ChemCatChem, 2011, 3, 1935–1947 CrossRef CAS.
- H. Schwarz, Isr. J. Chem., 2015, 54, 1413–1431 CrossRef.
- M. G. Quesne, F. Silveri, N. H. D. Leeuw and R. R. A. Catlow, Front. Chem., 2019, 7, 182 CrossRef CAS PubMed.
- H. Schwarz, S. Shaik and J. Li, J. Am. Chem. Soc., 2017, 139, 17201–17212 CrossRef CAS PubMed.
- Y. X. Zhao, X. N. Li, Z. Yuan, Q. Y. Liu, Q. Shi and S. G. He, Chem. Sci., 2016, 7, 4730–4735 RSC.
- C. Geng, T. Weiske, J. Li, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 2018, 141, 599–610 CrossRef PubMed.
- H. F. Li, Z. Y. Li, Q. Y. Liu, X. N. Li, Y. X. Zhao and S. G. He, J. Phys. Chem. Lett., 2015, 6, 2287–2291 CrossRef CAS PubMed.
- B. A. Peter, Chem.–Eur. J., 2017, 23, 10–18 CrossRef PubMed.
- Y. X. Zhao, Z. Y. Li, Y. Yang and S. G. He, Acc. Chem. Res., 2018, 51, 2603–2610 CrossRef CAS PubMed.
- X. Lin, Y. Xi and J. Sun, J. Phys. Chem. C, 2012, 116, 3503–3516 CrossRef CAS.
- G. Fu, X. Xu, X. Lu and H. Wan, J. Am. Chem. Soc., 2005, 127, 3989 CrossRef CAS PubMed.
- B. Xing, X. Y. Pang and G. C. Wang, J. Catal., 2011, 282, 74–82 CrossRef CAS.
- B. Xing and G. C. Wang, Phys. Chem. Chem. Phys., 2014, 16, 2621–2629 RSC.
- Y. Q. Wang, C. Q. Lv and G. C. Wang, RSC Adv., 2015, 5, 66221–66230 RSC.
- J. J. Varghese, Q. T. Trinh and S. H. Mushrif, Catal. Sci. Technol., 2016, 6, 3984–3996 RSC.
- D. Hibbitts and M. Neurock, Surf. Sci., 2016, 650, 210–220 CrossRef CAS.
- J. Wang and G. C. Wang, J. Phys. Chem. C, 2018, 122, 17338–17346 CrossRef CAS.
- N. Dietl, M. Schlangen and H. Schwarz, Angew. Chem., Int. Ed., 2012, 51, 5544–5555 CrossRef CAS PubMed.
- J. Li, S. Zhou, J. Zhang, M. Schlangen, D. Usharani, S. Shaik and H. Schwarz, J. Am. Chem. Soc., 2016, 138, 11368–11377 CrossRef CAS PubMed.
- J. L. Li, S. D. Zhou, M. Schlangen, T. Weiske and H. Schwarz, Angew. Chem., Int. Ed., 2016, 128, 13266–13269 CrossRef.
- S. Feyel, J. Dobler, D. Schroder, J. Sauer and H. Schwarz, Angew. Chem., Int. Ed., 2006, 45, 4681–4685 CrossRef CAS PubMed.
- R. C. Bell and A. W. Castleman Jr, J. Phys. Chem. A, 2002, 106, 9893–9899 CrossRef CAS.
- X. Xu, F. Faglioni and W. A. Goddard III, J. Phys. Chem. A, 2002, 106, 7171–7176 CrossRef CAS.
- P. Serna and B. C. Gates, Acc. Chem. Res., 2014, 47, 2612–2620 CrossRef CAS PubMed.
- Y. Xi, B. Chen, X. Lin, H. Fu and C. Wang, Comput. Theor. Chem., 2016, 1076, 65–73 CrossRef CAS.
- M. J. Frisch, et al.Gaussian 09, Revision B. 01, Gaussian, Inc., Wallingford, 2010 Search PubMed.
- A. D. Becke and D. Axel, J. Chem. Phys., 1993, 98, 1372–1377 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 247–257 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- M. Dolg, U. Wedig, H. Stoll and H. Preuss, J. Chem. Phys., 1987, 86, 866–872 CrossRef CAS.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- K. Eichkorn, F. Weigend, O. Treutler and R. Ahlrichs, Theor. Chem. Acc., 1997, 97, 119–124 Search PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- H. P. Hratchian and H. B. Schlegel, J. Chem. Phys., 2004, 120, 9918–9924 CrossRef CAS PubMed.
- J. M. L. Martin and A. Sundermann, J. Chem. Phys., 2001, 114, 3408 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- J. Zheng, X. Xu and D. G. Truhlar, Theor. Chem. Acc., 2011, 128, 295–305 Search PubMed.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- J. M. Turney, A. C. Simmonett, R. M. Parrish, E. G. Hohenstein, F. A. Evangelista, J. T. Fermann, B. J. Mintz, L. A. Burns, J. J. Wilke and M. L. Abrams, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 556–565 CAS.
- W. Lai, C. Li, P. D. H. Chen and P. D. S. Shaik, Angew. Chem., Int. Ed., 2012, 51, 5556–5578 CrossRef CAS PubMed.
- S. Ye and F. Neese, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1228–1233 CrossRef CAS PubMed.
- C. Geng, J. Li, T. Weiske and H. Schwarz, Chem.–Eur. J., 2019, 25, 12940–12945 CrossRef CAS PubMed.
- X.-L. Ding, X.-N. Wu, Y.-X. Zhao and S.-G. He, Acc. Chem. Res., 2012, 45, 382–390 CrossRef CAS PubMed.
- C. S. Cooper, R. J. Oldmana and C. R. A. Catlowb, Chem. Commun., 2015, 51, 5856–5859 RSC.
- Z. Hu and C. H. Turner, J. Am. Chem. Soc., 2007, 129, 3863–3878 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: The symmetry-adapted perturbation theory (SAPT) energy decomposition and Hirshfeld spin density and charge population analyses, and Cartesian coordinates of all optimized structures. See DOI: 10.1039/d0ra10785a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2021 |