
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLigand substituent effect on the cytotoxicity activity of two new copper(II) complexes bearing 8-hydroxyquinoline derivatives: validated by MTT assay and apoptosis in MCF-7 cancer cell line (human breast cancer)†
Arif Alia,
Somesh Banerjeeb,
Saima Kamaala,
Mohammad Usmanc,
Neeladrisingha Dasb,
Mohd Afzal d,
Abdullah Alarifid,
Nayim Sepaye,
Partha Royb and
Musheer Ahmad*a
d,
Abdullah Alarifid,
Nayim Sepaye,
Partha Royb and
Musheer Ahmad*a
aDepartment of Applied Chemistry, ZHCET, Faculty of Engineering and Technology, Aligarh Muslim University, Aligarh–202002, India. E-mail: amusheer4@gmail.com
bMolecular Endocrinology Laboratory, Department of Biotechnology, Indian Institute of Technology Roorkee, Roorkee, Uttarakhand–247667, India
cDepartment of Chemistry, IIT Kanpur, Uttar Pradesh 208016, India
dDepartment of Chemistry, College of Science, King Saud University, Riyadh–11451, Saudi Arabia
eDepartment of Chemistry, Lady Brabourne College, Kolkata–700 017, India
First published on 16th April 2021
Abstract
In this study, we have examined the effect of ligand substituent on the structure–cytotoxicity relationships of the MCF-7 cancer cell line (human breast cancer), by two copper(II) complexes {[Cu(qmbn)(Hqmba)(q)]·NO3·2H2O} (1) and {[Cu(Hqmba)2(q)]·NO3·2H2O} (2) (where, qmbn = 2-(quinolin-8-yloxy)(methyl) benzonitrile (L1); Hqmba = 2-((quinolin-8-yloxy)methyl)benzoic acid (L2) and q = quinolin-8-olate). The structural analysis reveals that both the complexes exhibit distorted octahedral (CuN3O3) configuration which is further corroborated by density functional theory (DFT) calculations. The cytotoxicity impact of ligands (L1 and L2) and complexes (1 and 2) was screened against the MCF-7 cell line (human breast cancer). The MTT assay uptake indicated that the presence of –COOH functionality in complex 2 leads to higher cytotoxicity (lower IC50) than that observed for complex 1 containing a –CN group. This could be due to the strong H-bonding forming propensity of the carboxylic acids. Incubation of MCF-7 cancer cells with IC50 concentrations of 1 and 2 promoted cellular detachments via nuclear condensation and membrane destabilization followed by apoptosis as a result of metal-assisted generation of reactive oxygen species. Flow cytometry analysis showed that 1 and 2 might prompt early apoptosis in MCF-7 cells as the maximum percentage of cells appeared in the LR quadrant. Furthermore, mRNA expression analysis confirmed that both the complexes induced apoptosis in MCF-7 cells. Comparative mRNA expression analysis of complexes with their respective ligands also confirmed the enhanced apoptotic behavior of complexes. Furthermore, molecular docking studies of the complexes have also been performed with the active site of EGFR kinase receptors (major target for any cancer causing agent) due to similar analogues with FDA-approved EGFR inhibitors in order to rationalize its promising cytotoxicity activity.
Introduction
Quinoline and its analogues (both natural and synthetic) are the basic constituents of various bioactive compounds.1 The quinoline framework is a useful structural motif for the improvement of therapeutic preparations such as NSC3852 (potency against human breast cancer),2 idelalisib (PI3Kδ inhibitor) and omipalisib (dual PI3K/mTOR inhibitor).3 The mechanism of action includes inhibition of several cell growth promoting factors such as tyrosine kinases, proteasome, topoisomerase and DNA repair; they are in high demand for their use as precursors in metal-based drug therapeutic regimes.4 Furthermore, these derivatives successfully show a range of biological activity, such as antimalarial, antibacterial, anti HIV, cardiotonic, anti-inflammatory, and analgesic activity.5–8 Several studies have deciphered that the natural/synthetic quinoline based metal complexes exhibited enormous potential as anticancer agents with minimal side effects and maximal function.3,7–11 Recently, quinoline-based frameworks such as azolyalkyl quinolines with diverse azole groups (benzothiazole (SRA-HX-1), tetrazole (SRA-HX-2), and 1,2,4-triazole (SRAHX-3)) have been designated as potent antitumor agents for breast cancer cells in vitro.12 Zhang et al. reported a series of quinoline-3-carbonitrile derivatives which were selective cytotoxic candidates toward SMMC-7721 cell line in comparison to Gefitinb.13 The aim to obtain such complexes is the design and selection of bioactive ligands, because the synergistic effect of copper and the ligand will endow them with excellent drug features.14–16Cancer remains the second leading cause of misery worldwide after the cardiovascular disease.17–19 Breast cancer is the most prevalent noncutaneous form of solid tumor among women. Although the incidence of breast cancer increases with age,20 due to an elevated level of estrogen metabolites which generate reactive oxygen species (ROS), induces uncontrolled DNA production, increased kinases phosphorylation, play a pivotal role on the development of breast cancer risk.20 In this direction, one of the rapidly growing candidates of cancer treatment is the exploitation of metal complexes as drugs and as a regulators of gene expression.21 Thus, the design and fabrication of metal based anticancer agents of high efficacy, good selectivity and diverse mechanism of action to overcome the drawbacks of platinum based drugs.22,23 In contrast to platinum, copper has been found to be an essential cofactor in tumor angiogenesis and elevated copper levels (both in tissue or serum) have been detected in breast, prostate, colon, lung, and brain cancers.14 Therefore, it is necessary to chelate excess intracellular copper accumulation by using clioquinol and 8HQs and thereby accelerating cancer cell death is of prime concern. Many metal complexes have been extensively studied to acts as potential growth inhibitors of human breast cancer cells by in vitro and in vivo.24–28 More efforts are being made to synthesize efficacious copper(II) complexes which exhibited remarkable anticancer activity, with lower toxicity than platinum drugs.18,19,27,29 In the recent past, Zhai and his team demonstrated that both 8-hydroxyquinoline (8-OHQ) and CQ/copper complexes are able to inhibit the proteasome activity, resulting in proliferation suppression and apoptosis in cultured breast cancer cells.30
The role of the ligands design in the activity of the metal complexes is obviously crucial, since they can modulate important aspects such as; (i) modulate the permeability through the cell membranes by tuning their lipophilic character, (ii) direct the toxicities of the metals toward specific intracellular targets, (iii) exhibit an intrinsic cytotoxic activity when they dissociate from the center metals, and (iv) ability to chelate excess intracellular metal concentration.7,31,32 One of encouraging approach in ligand design is the covalent grafting of another pharmacophores to create a single molecule with potential therapeutic properties and has ability to counterbalance the side effects caused by another part.33–35
In view of aforementioned facts, the efforts were being made to develop new 8-hydroxyquinoline derivatives by incorporating, 2-(bromomethyl) benzonitrile moiety and further acidification of nitrile group results terminal carboxylate group at ortho position of the quinolinic system. The copper(II) complexes {[Cu(qmbn)(Hqmba)(q)]·NO3·2H2O} (1) and {[Cu(Hqmba)2(q)]·NO3·2H2O} (2) have been synthesized and thoroughly characterized. The comparative cytotoxic potential of the complexes 1 and 2 were explored on the human breast cancer cell line MCF-7 and cellular uptake was monitored by fluorescence microscopy, in order to identify a correlation between the structural modifications of the ligands and the cytotoxicity of their copper (II) complexes. It seems that the results of this study can help us to find new copper-based agent in inhibiting the expansion and development of breast cancer cell type, therefore could represent a new strategy for the research of anticancer agents.
Experimental section
Materials
Reagent grade 2-(bromomethyl) benzonitrile and 8-hydroxyquinoline were purchased from Sigma Aldrich. Solvents, K2CO3, potassium iodide, NaOH and Cu(NO3)2·3H2O were procured from ThermoFisher scientific, India.Physical measurements
Microanalyses were performed using a CE–440 elemental analyzer (Exeter Analytical Inc.). Infrared spectra were obtained (KBr disk, 400–4000 cm−1) on Thermo Scientific iS50 FTIR. UV-visible and photoluminescence spectra were carried out with Thermo Scientific Evolution 201 UV-visible spectrophotometer and PerkinElmer LS55 fluorescence spectrophotometer, respectively.Single crystal X-ray studies
The diffraction data was collected with a Bruker SMART APEX CCD diffractometer using monochromatic Mo-Kα radiation (λ = 0.71073 Å) at 100(2) K. Using Olex2, the structure was solved with the olex2.solve36 structure solution program using Charge Flipping and refined with the olex2.refine36 refinement package using Gauss–Newton minimisation. All hydrogen atoms were located in difference Fourier maps in the structures and refined isotropically. All non-H atoms were refined anisotropically. The crystal and refinement data for complexes 1 and 2 are given in Table S1,† while selected bond distances are summarized in Table 2.Synthesis of ligands (L1 and L2) and complexes (1 and 2)
The ligands (L1 and L2) and complexes (1 and 2) were synthesized according to the earlier reported protocols with slight modifications.37,38 The detailed synthetic procedure of ligands L1 and L2 have been provided in ESI.†Synthesis of copper(II) complexes
Cell culture
The human estrogen receptor positive breast adenocarcinoma adherent cell line, MCF-7, was purchased from National Centre for Cell Science (NCCS), Pune, India. The cells were maintained in Dulbecco's Modified Eagles' Medium (DMEM; Himedia, Mumbai, India) supplemented with 10% heat sterilized fetal bovine serum, 100 U mL−1 penicillin–streptomycin and 100 mg mL−1 ampicillin antibiotic mixture (both from Gibco, Thermo Fischer Scientific, Waltham, MA, USA). The cells were propagated in humidified incubator supplied with 5% CO2 and maintained at 37 °C.Evaluation of cytotoxicity
The cytotoxicity impact of ligands (L1 and L2) and complexes (1 and 2) on MCF-7 cell line was evaluated by performing MTT assay39 and the inhibitory concentration (IC50) was evaluated. Briefly, 5000 cells were seeded in each well of 96-wells plate with 200 μL of DMEM media. After 24 h, the media was replaced with fresh media supplemented with varying concentration of synthesized complexes. DMSO (Himedia, Mumbai, India) treated cells were considered as vehicle control for evaluation. After 24 h, the media was replaced with fresh media supplemented with varying concentration of both the synthesized ligands and complexes were also evaluated. Then the media from each well was replaced with 200 μL of DMSO to dissolve the formed formazon crystal in each well. The absorbance of the formazon dissolved solution was measured at 570 nm in a microplate reader (FLUOstar Omega, BMG Labtech, Ortenberg, Germany). The cytotoxicity values were evaluated using absorbance values implementing formula:| Percentage cytotoxicity = 100 − [(absorbance of molecule treated cells/absorbance of vehicle treated cells) × 100] |
The inhibitory concentration (IC50) was evaluated using Graph Pad prism software 6.01 (San Diego, CA, USA) using the absorbance values.
Fluorescence microscopy analysis of apoptosis and nuclear fragmentation in cells
The visualization of cellular morphological change and formation of apoptotic bodies in cells was performed by acridine orange/ethidium bromide (AO/EB) staining.40 Briefly, 1 × 106 cells were seeded in 6-well plate with 2 mL of DMEM media. After 24 h, the cells were treated with evaluated IC50 concentration of ligands (L1 and L2) and complexes 1 and 2 for another 24 h. Then the cells were stained with 1 μL of AO/EB mixture (each 100 μg mL−1 in PBS) (Sigma, St. Louis, MO, USA) and the images were captured immediately using fluorescent microscope (EvosFloid cell imaging station, Thermo Fischer, Waltham, MA, USA).The nuclear morphology of complex treated cells was observed by performing DAPI staining.41 Briefly, 1 × 106 cells were seeded with 2 mL DMEM media in 6-well plate. After 24 h, the cells were treated with evaluated IC50 concentration of complexes 1 and 2 for another 24 h. Then the cells were fixed with 3.7% formaldehyde solution and washed gently with PBS twice. The fixed cells were permeabilized with 0.2% triton X-100 for 5 min at room temperature and incubated for another 3 min with DAPI labeling solution (0.1% 4,6-diamidino-2-phenylindole in 5% DMSO) (Sigma, St. Louis, MO, USA). The labeling solution was aspirated and rinsed with PBS twice. The images of stained cells were captured using fluorescence microscope at excitation wavelength of 359 nm and emission wavelength of 461 nm using fluorescent microscope (EvosFloid cell imaging station, Thermo Fischer, Waltham, MA, USA).
Flow cytometry for analysis of apoptotic cells
The induction of apoptosis in complexes 1 and 2 treated cells were determined quantitatively by performing Annexin V- conjugated Alexa Fluor 488 apoptosis assay kit (Thermo Fischer Scientific, Waltham, MA, USA) following the manufacturer's instruction. Briefly, after 24 h treatment of cells with IC50 concentration of complexes 1 and 2, the cells were harvested and pelleted. The cells were then incubated with Annexin V AlexaFluor 488 and propidium iodide (PI) in binding buffer in dark conditions at room temperature for 15 min. Then the cells were analyzed using flow cytometry (FACS Calibur, BD Biosciences, San Jose, CA, USA) and the data was analyzed using Cell Quest 3.3 software.Gene expression analysis
Total RNA of MCF-7 cells treated at IC50 concentration of complexes 1 and 2 and with DMSO (control) was isolated using RNA isolation reagent (RNA X-Press Reagent, Himedia, Mumbai, India) as per the manufacturer's instruction. The RNA samples were first quantified to synthesized first-strand cDNA using 2 μg of each RNA samples. Primers for caspase-3, caspase-8, caspase-9 and β-actin were designed using Primer3 software (Whitehead Institute for Biomedical Research, Cambridge, MA, USA) (Table 1). The effect of ligands L1 and L2 on the expression of caspase-3 were also evaluated. The semi-quantitative polymerase chain reaction was performed to evaluate the expression pattern and obtained PCR products were separated on 2% agarose gel and visualized in a gel documentation system (BioRad, Hercules, CA, USA). The band intensity was analyzed using ImageJ 1.52a software (NIH, Bethesda, MD, USA).| Name of gene | Forward primer sequence (5′–3′) | Reverse primer sequence (5′–3′) | Product size (bp) | Annealing temperature (°C) |
|---|---|---|---|---|
| Caspase-3 | GGTTCATCCAGTCGCTTTGTG | GCGTCAAAGGAAAAGGACTCA | 121 | 60 |
| Caspase-8 | CTGCCTACAGGGTCATGCTCTA | AGCAGGTTCATGTCATCATCCA | 124 | 60 |
| Caspase-9 | AGGCCCCATATGATCGAGGA | GGCCTGTGTCCTCTAAGCAG | 139 | 60.3 |
| β-Actin | TCACCCACACTGTGCCCCATCTACGA | CAGCGGAACCGCTCATTGCCAATGG | 298 | 65 |
Statistical analysis
All data were represented as mean ± SEM of three independent experiment. The difference among set conditions was evaluated by performing a one-way analysis of variance (ANOVA) test followed by the Bonferroniposthoc test using Graph Pad Prism 6.01 software (San Diego, CA, USA). The analyzed data with p-value < 0.05 were considered statistically significant.DFT methodology
All reported computations were performed using ORCA computational package.42 Experimental geometry was obtained from single crystal X-ray data. The geometry optimization calculations were carried out by hybrid B3LYP functional43 using Aldrich's def2–TZVP basis set for copper atom and def2–SVP basis set for C, H, O, N atoms.44–46 To speed up the calculations we employed the resolution of identity (RI) approximation with the decontracted auxiliary def2-SVP/J and def2–TZV/J Coulomb fitting basis sets and the chain-of-spheres (RIJCOSX) approximation to exact exchange as executed in ORCA.47,48 DFT calculation utilizes the atom-pairwise dispersion correction with the Becke–Johnson damping scheme (D3BJ).47,49Molecular docking methodology
The geometrical coordinates of 1 and 2 obtained from single crystal X-ray crystallography have been utilized for docking studies. For other small molecules, the three dimensional structures were obtained from ChemDraw 3D and energy minimized structure were performed using AMBER force field. All these obtained structures were converted into pdbqt files (required for docking) using MGL Tools after adding hydrogens and fixing formal charges.50 In the same way, the EGFR protein structure (PDB ID: 1M14)51 was prepared for the studies. The docking of the protein and all the compounds individually was performed in the 60 × 60 × 60 grid box through Lamarkial Genetic Algorithm with the help of AutoDock 4.2 application.52Results and discussion
To establish the structure–cytotoxicity relationships on MCF-7 cancer cell line (human breast cancer), we synthesized copper(II) complexes {[Cu(qmbn)(Hqmba)(q)]·NO3·2H2O} (1) and {[Cu(Hqmba)2(q)]·NO3·2H2O} (2) bearing ligands with different functional groups. We have performed the comparative biological evaluations of both the ligands 2-(quinolin-8-yloxy)(methyl)benzonitrile (qmbn) and 2-((quinolin-8-yloxy)methyl)benzoic acid (Hqmba) and their copper(II) complexes 1 and 2 respectively. Both the ligands and complexes were well characterized by different analytical and diffraction techniques. Single crystal X-ray analysis revealed the distorted octahedral environment around Cu(II) metal centers (Scheme 1). The MTT analysis indicated that the complex 2 induces higher cytotoxicity potential in MCF-7 cells as compared to complex 1 and the respective ligands.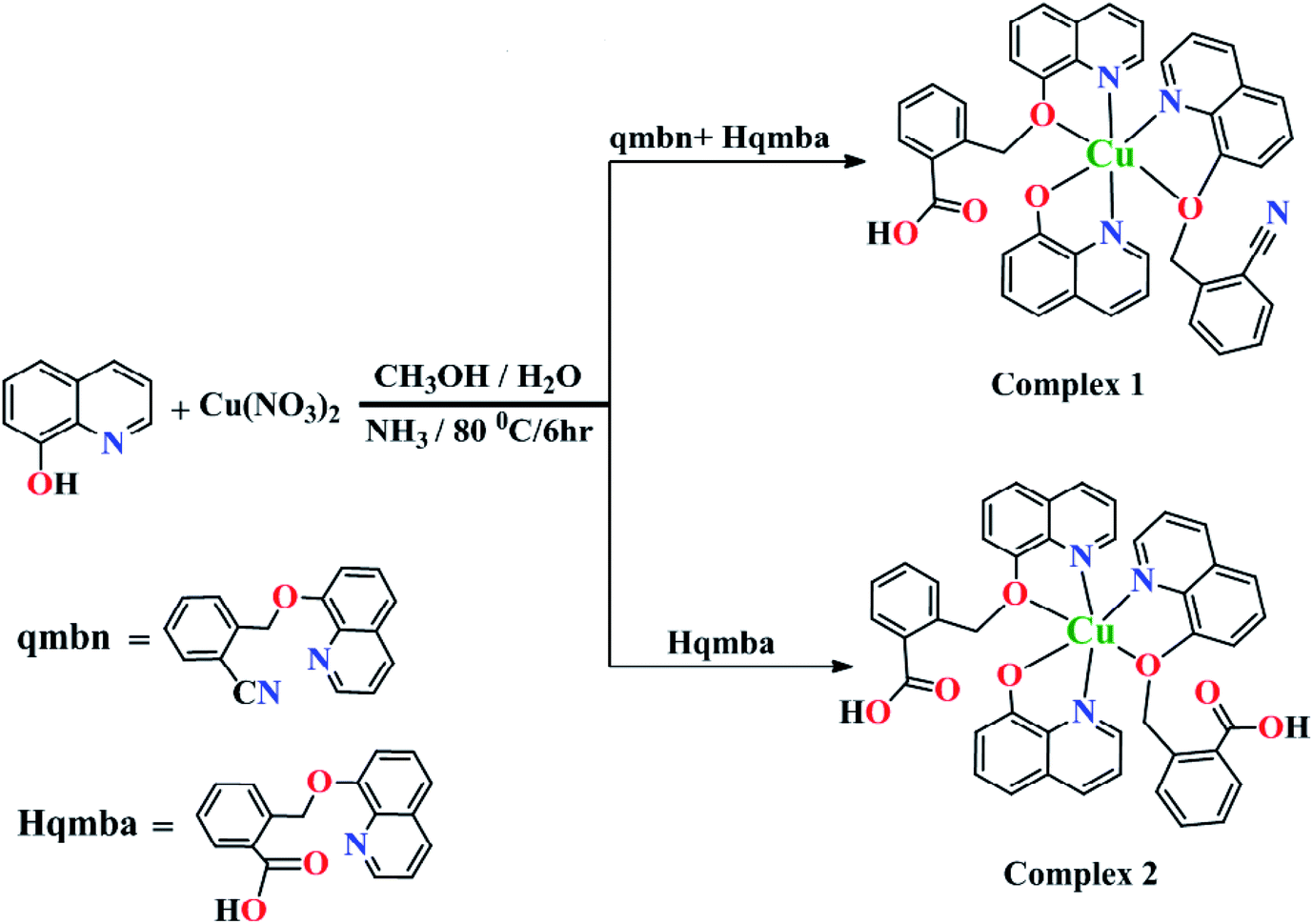 | ||
| Scheme 1 Synthetic route for complexes 1 and 2. | ||
Structural description
Complexes 1 and 2 were crystallized in triclinic crystal system with P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group. The asymmetric unit of both the complexes are similar: complex 1 consists of one Cu(II) ion, one q−1, one qmbn and Hqmba ligands, whereas complex 2 contains one Cu(II) ion, one q−1, two Hqmba ligands. Crystallized lattice molecules (one nitrate and two water) are same in both the complexes as shown in Fig. 1a and b. In both the complexes, metal ion exhibits distorted octahedral CuN3O3 configuration (Fig. 1c and d).
space group. The asymmetric unit of both the complexes are similar: complex 1 consists of one Cu(II) ion, one q−1, one qmbn and Hqmba ligands, whereas complex 2 contains one Cu(II) ion, one q−1, two Hqmba ligands. Crystallized lattice molecules (one nitrate and two water) are same in both the complexes as shown in Fig. 1a and b. In both the complexes, metal ion exhibits distorted octahedral CuN3O3 configuration (Fig. 1c and d).
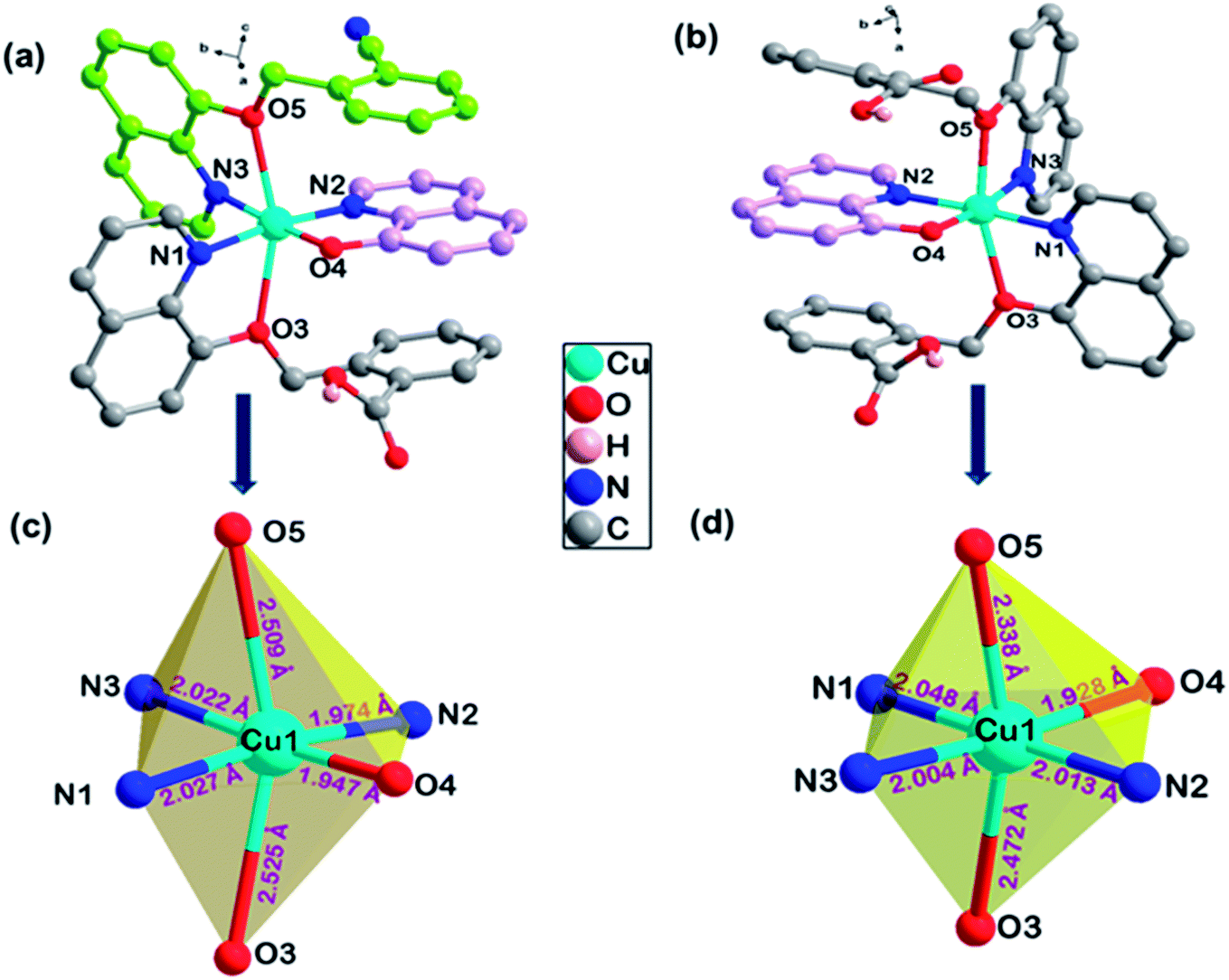 | ||
| Fig. 1 Representation of molecular structure (a) complex 1, (b) complex 2 and coordination environment around metals (c and d). | ||
In the complex 1, each independent complex unit is connected through C–N⋯H (C43–N4⋯H31), O–H⋯O (O2–H2⋯O4) contacts and π–π interactions between phenyl ring of Hqmba ligand along with bc plane forming 2D chain. Further the presence of water and nitrate molecules in the lattice leads to formation of 3D supramolecular network connected via C–H⋯O, O–H⋯O interactions (Fig. 2a). Similarly, in complex 2 each independent complex unit is connected through π–π contacts between Hqmba and O–H⋯O contact formed by the carboxylic acid group of the Hqmba ligand. One COOH group is forming the classical H-bonding with O6–H6⋯O7 contact with the neighbouring complex unit and other COOH group is forming O2–H2⋯O4 contact with the O-atom of the q ligand and O9–H9a⋯O1 contact with the O-atom of the water molecule. Moreover, water and nitrate molecules in the lattice forming a 3D supramolecular network connected via C–H⋯O, O–H⋯O interactions (Fig. 2b).
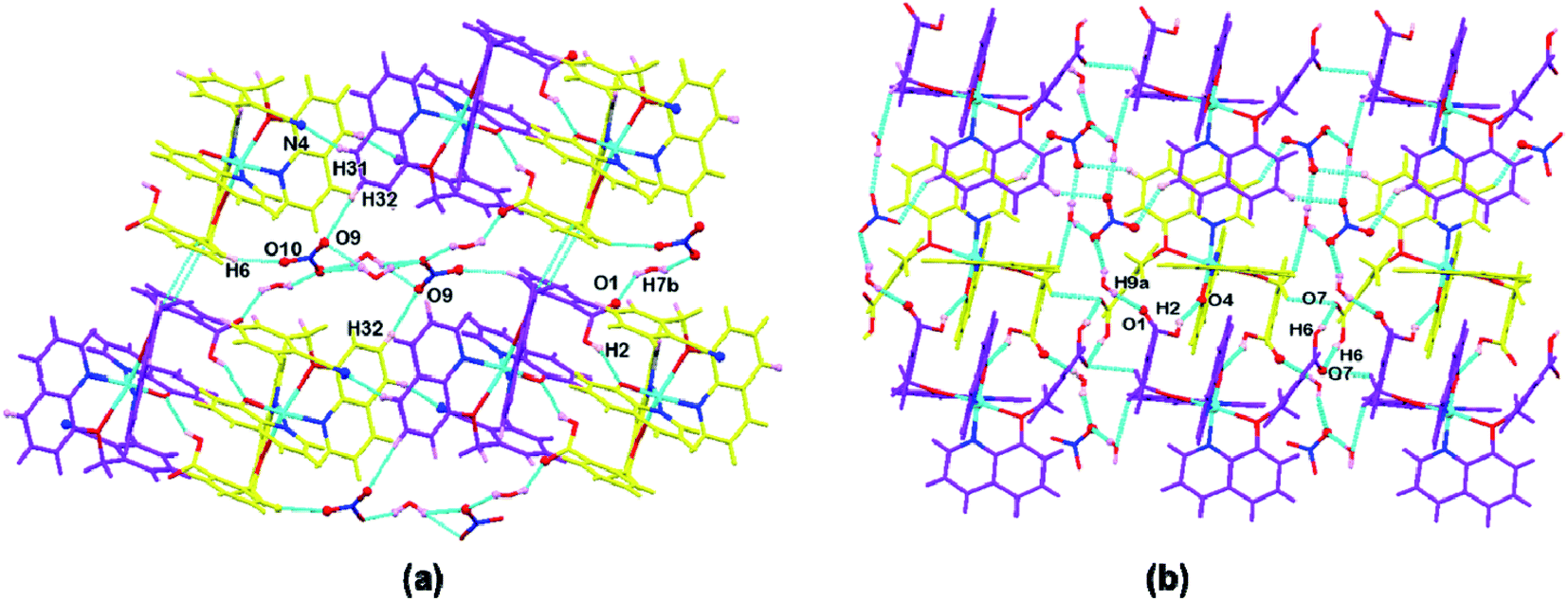 | ||
| Fig. 2 Representation of supramolecular interactions in (a) complex 1 and (b) complex 2. | ||
Crystal structure of the complex 1 comprises hydrogen-bonded chains running along [011] directions and consisting of the complex cations connected by means of the nitrate anions and water molecules. More precisely, the Cu(q)(qmbn)(Hqmba) cations form hydrogen bonded dimers, which are in turn hydrogen-bonded to the clusters consisting of four water molecules and two nitrate anions (Fig. 3).
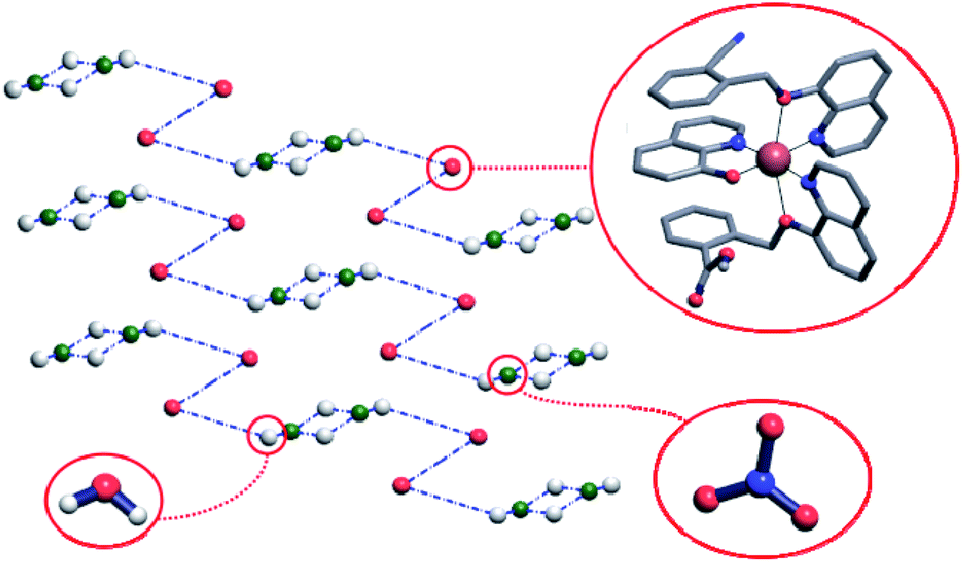 | ||
| Fig. 3 Standard representation of the crystal structure of complex 1 with hydrogen-bonded chains. In the simplified net each sphere represents the center of mass of corresponding molecule, while blue dashed-dotted lines represent hydrogen bonds between the molecules or molecular ions. Hydrogen atoms that do not participate in hydrogen-bonding are omitted for clarity. | ||
Crystal structure of the complex 2 comprises hydrogen-bonded layers lying in plane (011) and consisting of the complex cations connected by means of the nitrate anions and water molecules. The Cu(q)(Hqmba)2 cations form hydrogen bonded dimers, which are in turn hydrogen-bonded to the clusters consisting of four water molecules and two nitrate anions, structurally analogous to those found in crystal structure of complex 1 (Fig. 4).
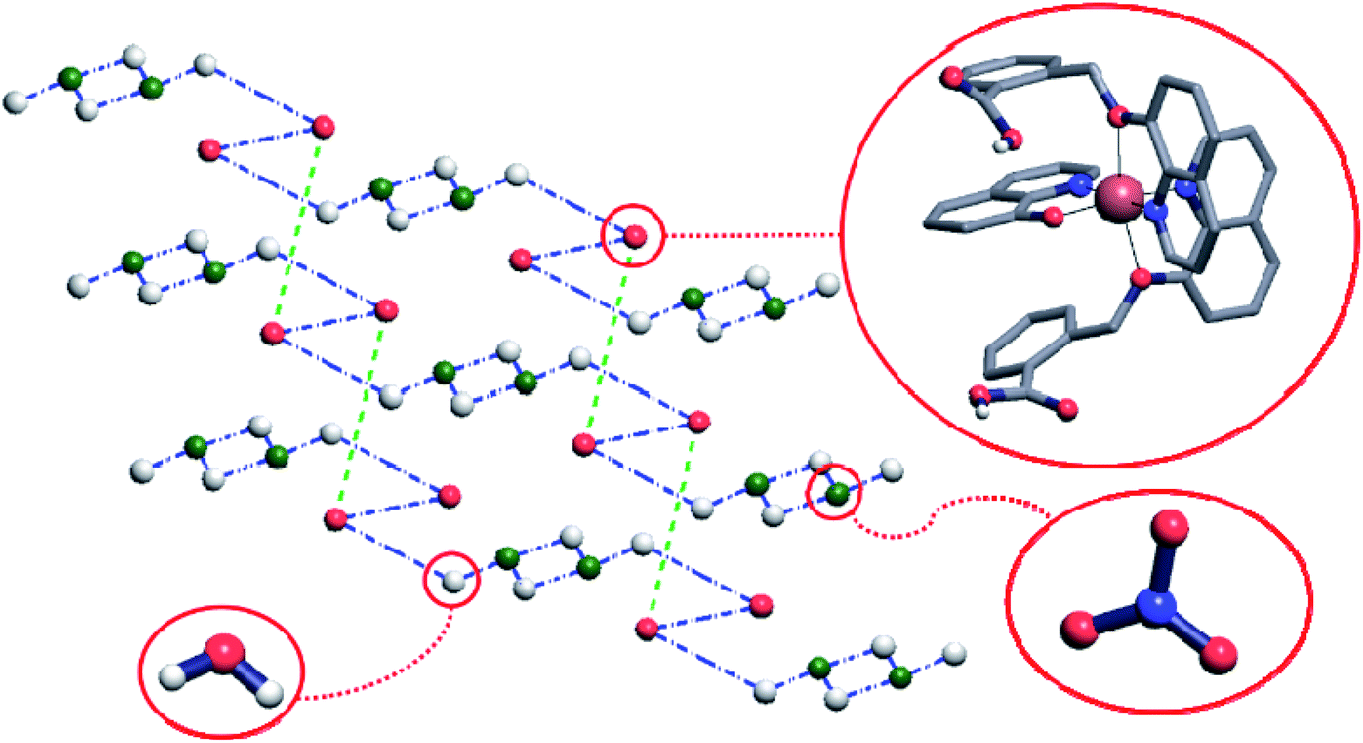 | ||
| Fig. 4 The simplified net corresponding to crystal structure of complex 2. Each ball represents the center of mass of corresponding molecule, while blue and green dashed-dotted lines represent hydrogen bonds between the molecules or molecular ions. Hydrogen atoms that do not participate in hydrogen-bonding are omitted for clarity. | ||
To have better insight of 1 and 2, we performed deep structural-interrelation between the complexes under consideration. The increase in dimensionality of the hydrogen-bonded network in 2 is attributed to the formation of the additional hydrogen bonded link between Cu(q)(Hqmba)2 cations (shown as a green dashed line between red spheres in Fig. 4). This link formed by carboxylic acid dimer synthon is enabled by the second Hqmba ligand in Cu(q)(Hqmba)2 cation as shown in details in Fig. 5. Obviously, the qmbn ligand of complex cation in structure 1 is devoid of the formation of such hydrogen bonded link.
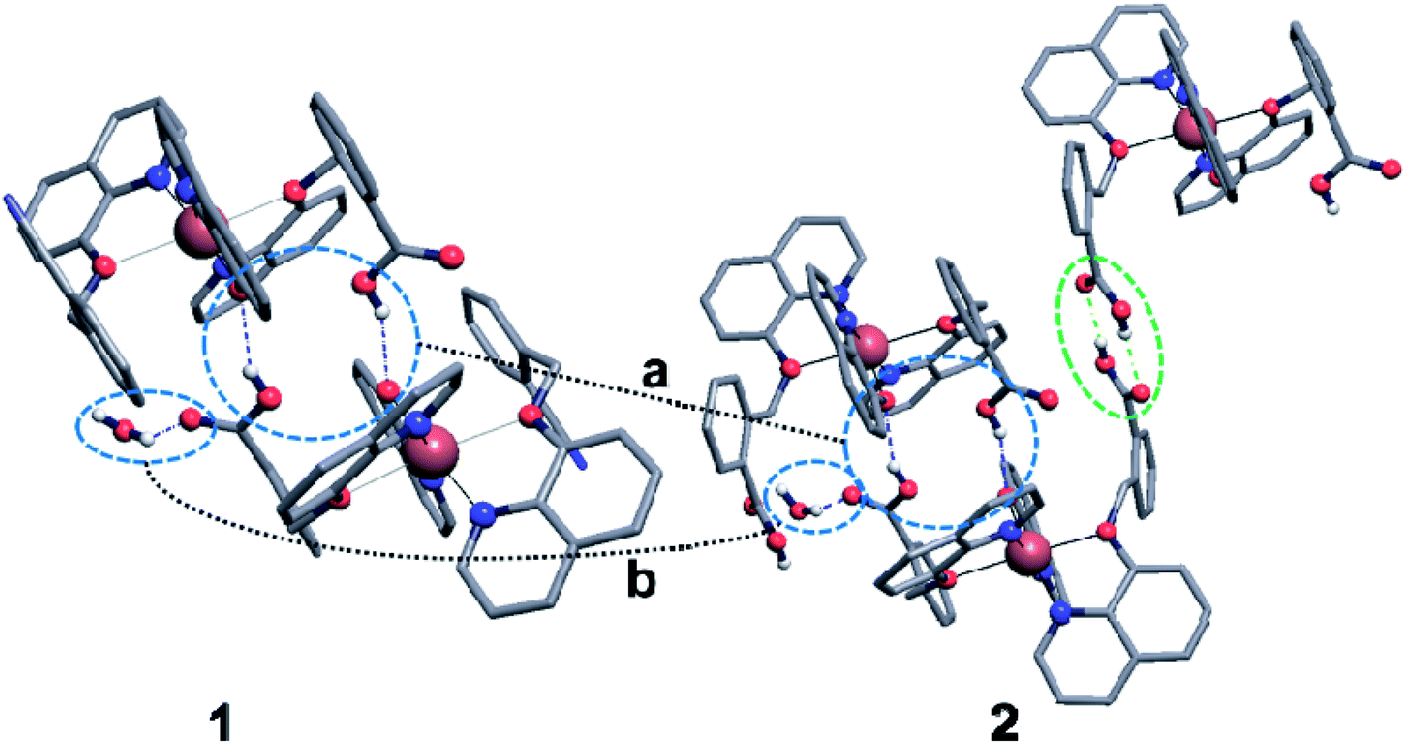 | ||
| Fig. 5 Hydrogen bonds between complex cations Cu(q)(qmbn)(Hqmba) (left) and Cu(q)(Hqmba)2 (right) in crystal structures of 1 and 2. Blue circles highlight connections between cations and neighbour molecules analogous in the two structures under consideration: a – hydrogen-bonded dimers of complex cations; b – hydrogen bond between water molecule and carboxylic acid group of Hqmba ligand. Green circle highlights the additional link between complex cations, which is absent in crystal structure of 1. | ||
DFT studies
To gain more insight into the electronic distribution and structure of 1 and 2, theoretical calculations were executed. Geometry optimizations of 1 and 2 were carried out with the actual compounds, without symmetry restrictions, and the resulting optimized structures are shown in Fig. 6. Counter anion has not been included for the geometry optimization calculations involving 1 and 2 giving an overall charge of +1 to the complexes. Complexes 1 and 2 have been optimized in doublet state. The structural parameters of the final structures are in good match with obtained by X-ray diffraction (Table 2).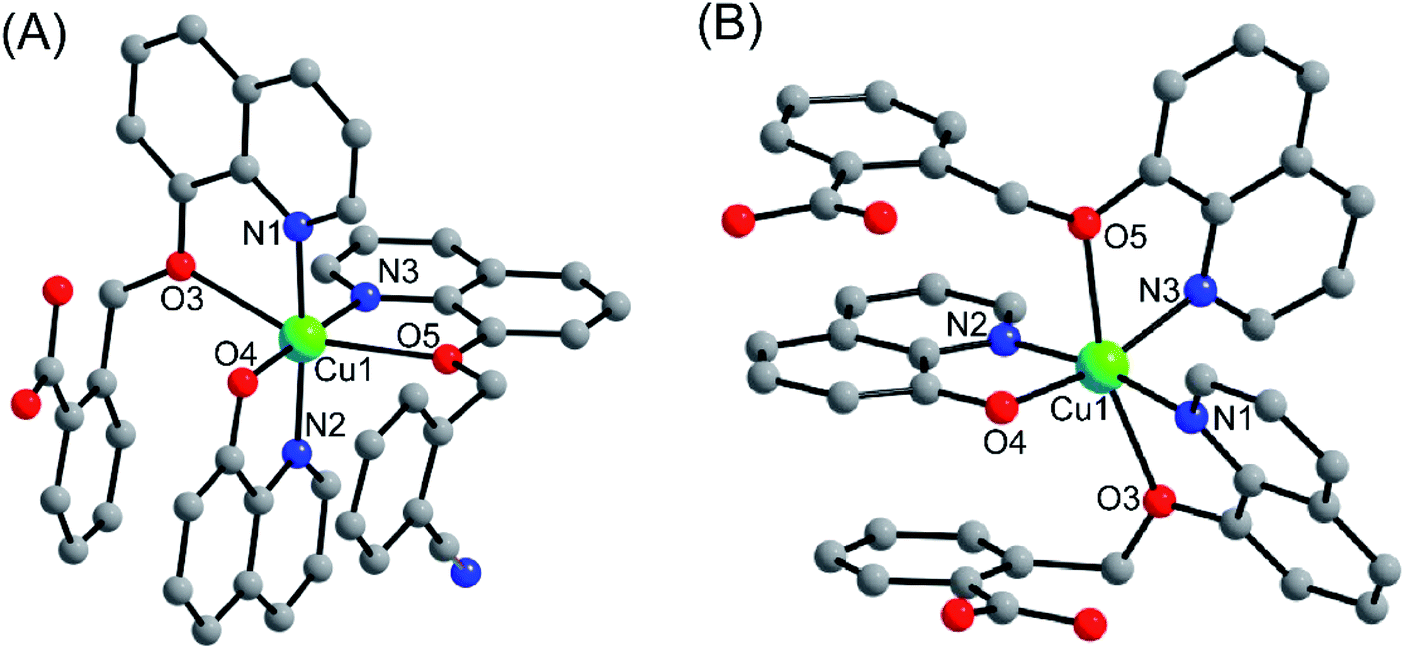 | ||
| Fig. 6 DFT/B3LYP optimized structures of complexes 1 and 2. | ||
| Bond distances | 1 | 2 | ||
|---|---|---|---|---|
| X-Ray | DFT | X-Ray | DFT | |
| Cu1–N1 | 2.024(4) | 2.091 | 2.048(18) | 2.118 |
| Cu1–N2 | 1.976(4) | 2.014 | 2.013(18) | 2.091 |
| Cu1–N3 | 2.024(4) | 2.089 | 2.004(17) | 2.087 |
| Cu1–O3 | 2.254(3) | 2.301 | 2.472(15) | 2.541 |
| Cu1–O4 | 1.947(3) | 2.007 | 1.928(15) | 2.010 |
| Cu1–O5 | 2.508(3) | 2.596 | 2.338(16) | 2.410 |
The frontier molecular orbitals (HOMOs and LUMOs) play an important role in demonstrating the molecule's chemical reactivity, kinetic stability and electrical transport properties. Metal complexes with a small energy gap (ΔE) between the HOMO and LUMO are more polarizable, and thereby act as soft molecules with higher chemical and biological reactivity. However, complexes with a large energy gap often offer greater stability and highly stable in terms of their lower reaction to chemical and biochemical responses. The slight degree of intramolecular charge transfer (ICT) from the electron-donor groups to electron-acceptors was correlated with a small energy gap between HOMO and LUMO. Molecular systems with high EHOMO are good donors of electron whereas systems with lower ELUMO are good acceptors of electron. Therefore, calculated energy gap between HOMO and LUMO of 1 and 2 were found to be 2.91 and 2.82 eV, respectively. Furthermore, major part of LUMO was mainly distributed on coordinated ligand moieties while HOMO has mixed distribution on Cu2+ ion and ligand (Fig. 7). The FMO analysis clearly suggested that the peripheral functional group (CN and COOH) substitution on aromatic ring of pharmacophore ligand scaffold affects the chemical and as well biological activity of its copper complexes. Hence, the greater magnitude of anticancer activity of 2 compared to 1 has been justified by studying the effects of frontier molecular orbital (HOMO and LUMO) analysis obtained from DFT/B3LYP/D3BJ calculations (Fig. 7).
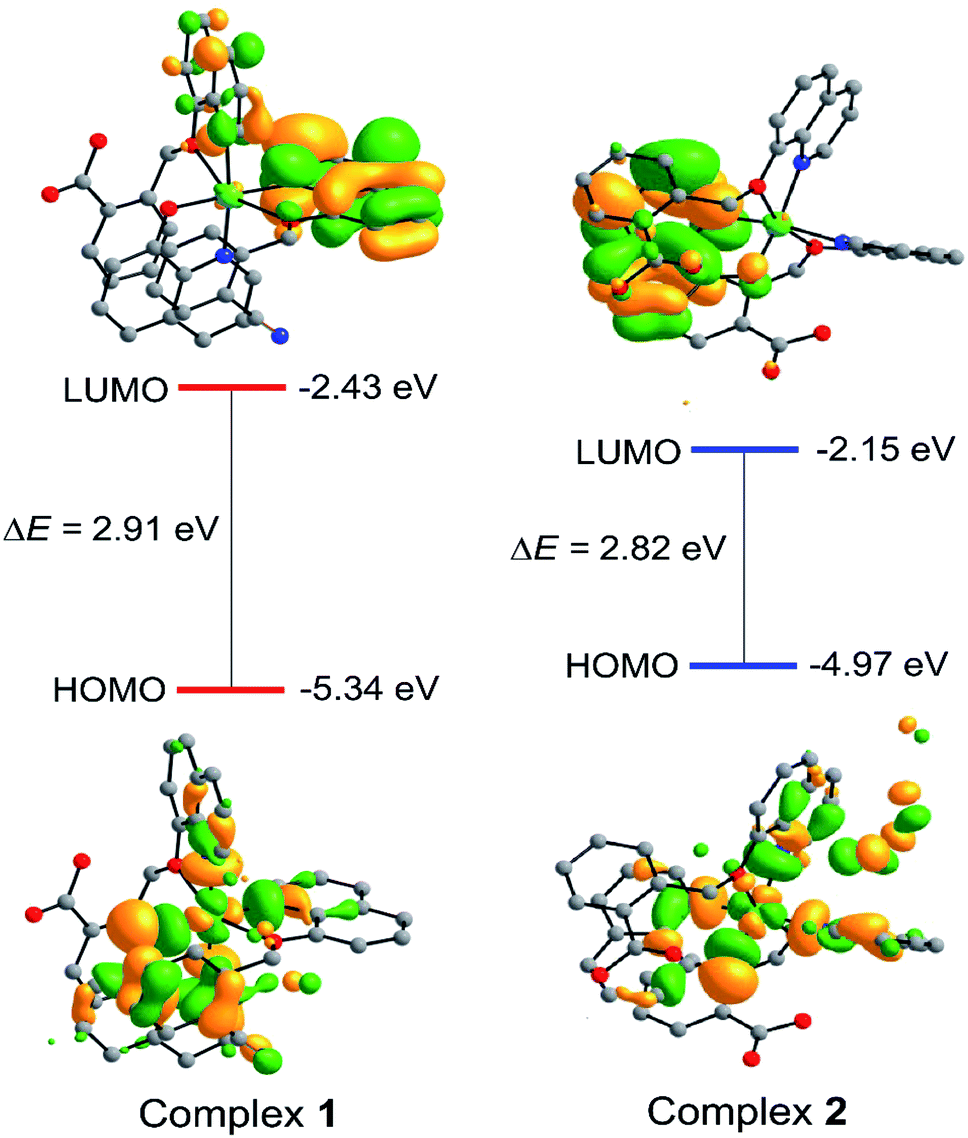 | ||
| Fig. 7 Isodensity surfaces (isovalue 0.03) for the FMOs of complexes 1 and 2 were generated from the Kohn–Sham orbitals. | ||
Estimation of cytotoxicity
The cytotoxicity impact of ligands (L1 and L2) and complexes (1 and 2) were evaluated on MCF-7 cell line by well-established MTT assay in order to examine the role of ligand functionality. The cells were treated with four different concentrations of ligands L1 and L2 (base structural analogue of complex 1 and 2) and our designed complexes 1 and 2 (0.5, 1.0, 2.0 and 4.0 μM) towards the MCF-7 cell line for 24 h. Dose-dependent inhibition of cell growth and cell death were observed in MCF-7 cells after treatment with ligands and complexes to a different extent. The observed IC50 values for L1 and L2 were 7.9 ± 1.1 and 5.6 ± 0.7 μM, respectively while for complexes 1 and 2 were found to be 1.86 ± 0.27 and 0.77 ± 0.12 μM, respectively. MCF-7 cells retained around 64% and 55% cell survivability at maximum evaluated 4 μM concentration of L1 and L2, respectively (Fig. S1a†). However, the cells survivability were reduced by 50% around 2 μM and 1 μM concentration of complex 1 and 2 respectively (p < 0.05) (Fig. 8). These IC50 values are about 5.1- and 7.3-fold higher compared to IC50 values of our complexes 1 and 2, respectively, which confirmed that the cytotoxic potential of ligands are lower compared to complexes. Thus, we conclude that the structural modulation in the ligand framework leads to better the cytotoxic effect on MCF-7 cancer cell lines.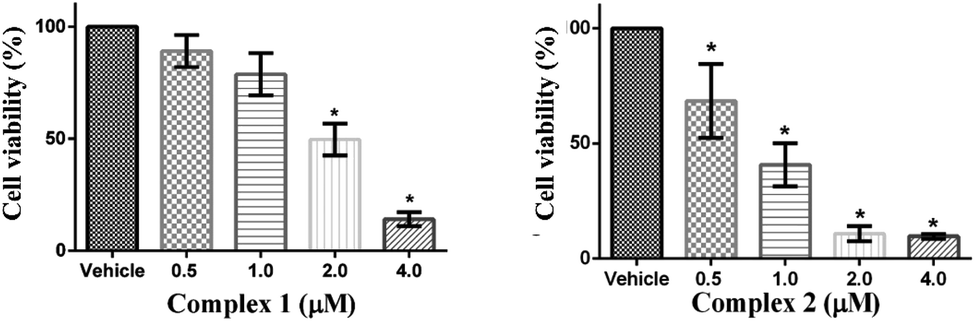 | ||
| Fig. 8 Evaluation of cell survivability of MCF-7 in presence of complexes 1 and 2. MCF-7 cells were treated with varying concentration of complexes and the cell survivability was evaluated by performing MTT assay. * represents p < 0.05 as compared to the vehicle treated control group. | ||
Apoptosis in cytotoxic MCF-7 cells
At first, we have evaluated the potential of L1 and L2 for inducing apoptosis in MCF-7 cells by determining the expression of caspase-3 (effector caspase) at their respective IC50 values. For the purpose, we have opted the caspase-3 because of its central role in any caspase dependent pathway. It was observed that both the ligands L1 and L2 enhanced the caspase-3 expression by 2.2- and 2.5-fold compared to vehicle treated cells (Fig. S1b†). However, detailed investigations on the apoptotic activity of complexes 1 and 2 on MCF-7 cells were examined by performing AO/EB fluorescence staining assay. In general, the vital AO dye stains both live and dead cells green, whereas, EB dye stains only those cells red which are dead due to loss of cell membrane integrity.40 As shown in Fig. 9, the vehicle treated control cells had all viable green cells with regular cellular morphology and almost none red stained cells. On the other side, the complex 1 and 2 treated cells showed a significant increase in EB stained cells, confirming the cells to be in early and/or late apoptosis. The appearance of bright green dots in cells confirmed the cells in early apoptosis, whereas orange stained cells confirmed to be in late apoptosis stage (Fig. 9).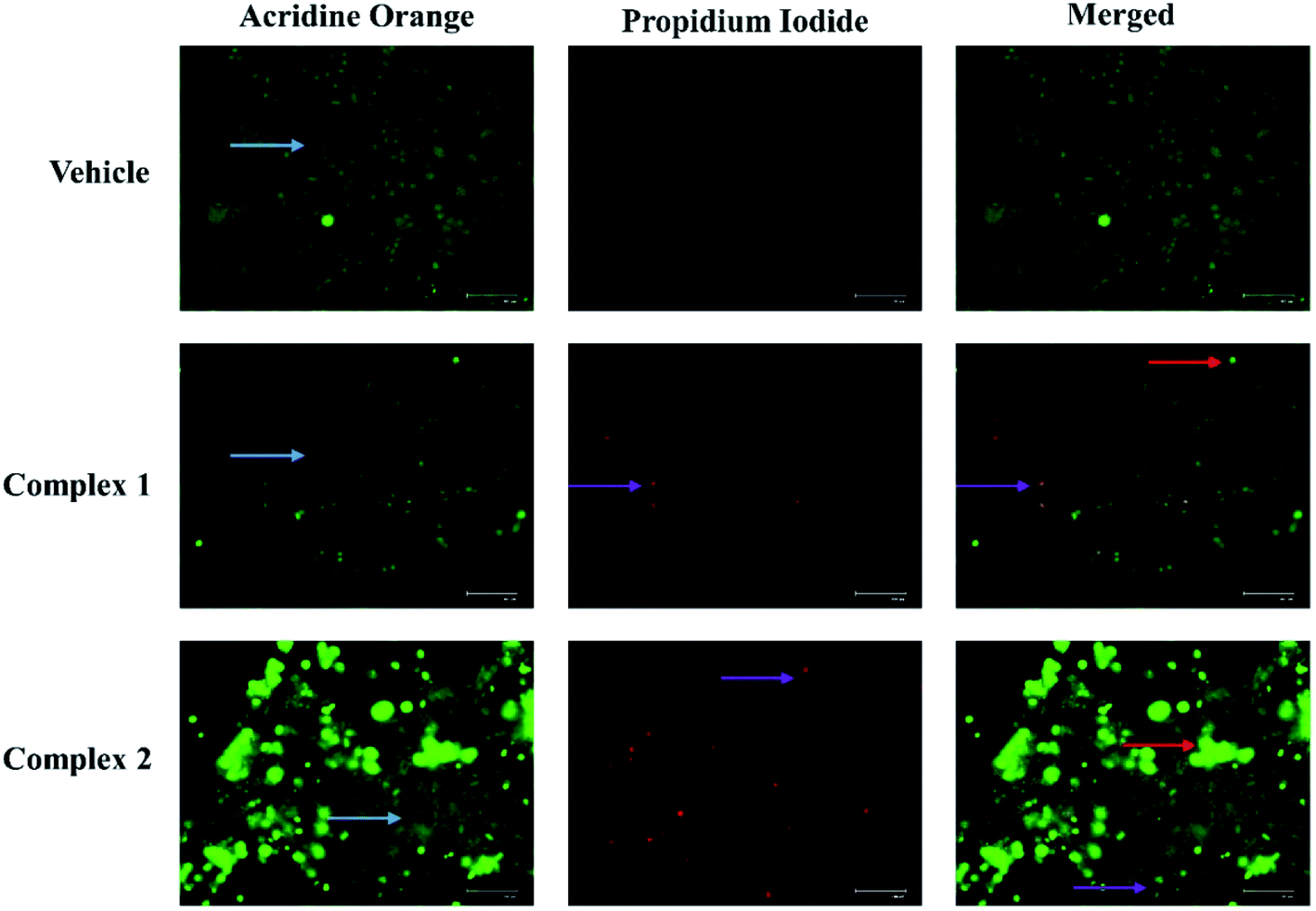 | ||
| Fig. 9 Evaluation of apoptosis in MCF-7 cells. Representative acridine orange/ethidium bromide staining in MCF-7 cells treated at IC50 concentration of complexes 1 and 2. The light blue colored arrow represents live cells, red and purple colored arrow represents cells in early and late apoptosis respectively. | ||
The designed complexes 1 and 2 induce cytotoxicity in MCF-7 cells with high potential compared to the respective ligands as evident in the MTT analysis. Further, the evaluation of expression of apoptotic marker, caspase-3, revealed that complexes 1 and 2 executed the apoptosis in the breast cancer MCF-7 cells more significantly, at much lower concentration compared to the ligand.
The loss of nuclear integrity and induction of nuclear fragmentation was further confirmed by DAPI staining. DAPI is a blue fluorescent DNA labeling dye.41 Fluorescent microscopy images of MCF-7 cells after 24 treatments with complexes 1 and 2, and DMSO treated cells (control) are represented in Fig. 10. It is evident from Fig. 10 that, complex treated cells showed increased blue fluorescence as compared to control condition. The morphological changes associated with apoptosis such as chromatin condensation, and nuclear fragmentation are evident in the cells upon treatment as visualized.
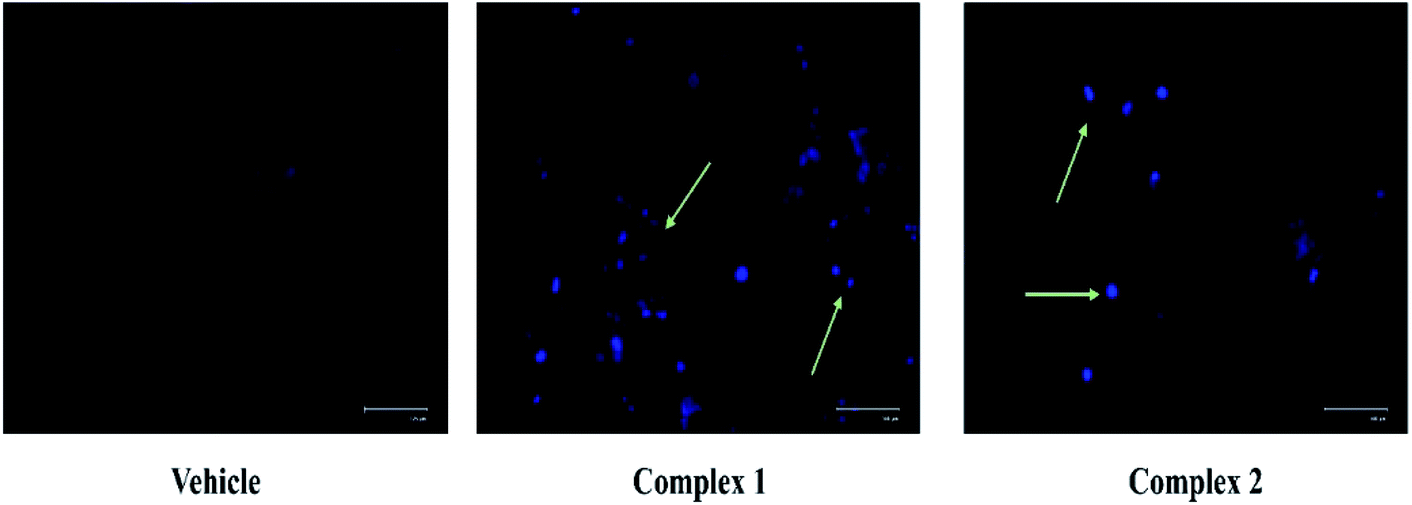 | ||
| Fig. 10 Evaluation of nuclear condensation in MCF-7 cells by DAPI staining. Representative DAPI staining images in MCF-7 cells treated at IC50 concentration of complexes 1 and 2. The light green colored arrow represents the dead cells with nuclear condensation and fragmentation. | ||
The quantitative estimation of complexes 1 and 2 treated MCF-7 was evaluated by flow cytometry analysis. The cell population in fluorescence cell sorting histograms were divided into four quadrants with live cells in lower left (LL), dead cells in upper left (UL), early apoptotic cells in lower right (LR) and late apoptotic cells in upper right (UR) quadrants. MCF-7 cells treated at IC50 concentration of complexes 1 and 2 showed distinct apoptosis after 24 h of incubation. As shown in Fig. 11, the total percentages of apoptotic cells were found to be 50 and 45% in complexes 1 and 2 treated cells at IC50 concentration respectively (p < 0.05). It was also evident that both the complexes prompted early apoptosis in MCF-7 cells as maximum percentage of cells appeared in LR quadrant.
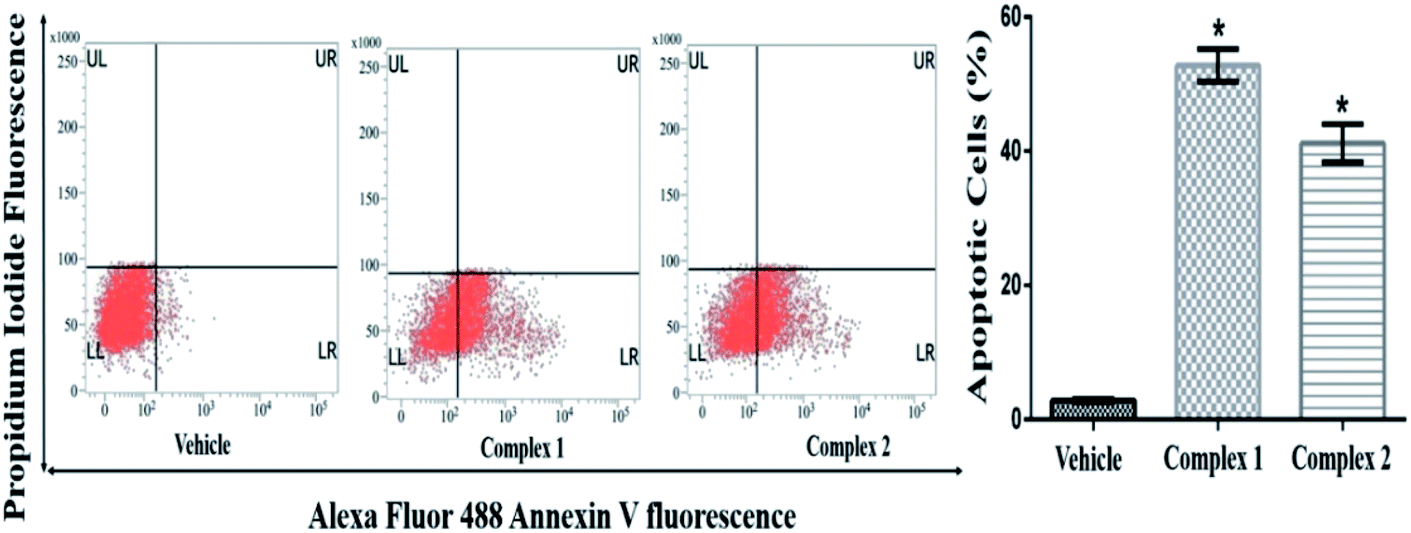 | ||
| Fig. 11 Evaluation of apoptosis in MCF-7 cells by flow cytometry. Representative FACS analysis of the cells performed using Annexin V as a marker. Each bar of histograms indicates a mean ± SEM of three independent experiments and expressed as percent apoptosis rate. * represents p < 0.05 as compared to the vehicle treated control group. The cells were treated at IC50 concentration of both complexes 1 and 2. | ||
The inductions of apoptosis in complexes 1 and 2 treated MCF-7 cells were further examined to determine the expression pattern of initiator and effector caspases at transcriptional level. In MCF-7 cells treated with IC50 concentration of complexes 1 and 2 for 24 h, the expression of caspase-3 (effector caspase) was found to increase by about 4.7- and 4.8-fold respectively as compared to vehicle treated condition (Fig. 12) (p < 0.05).53,54 These data further provided an additional clue about differential activation of caspase-dependent pathways by complexes 1 and 2.
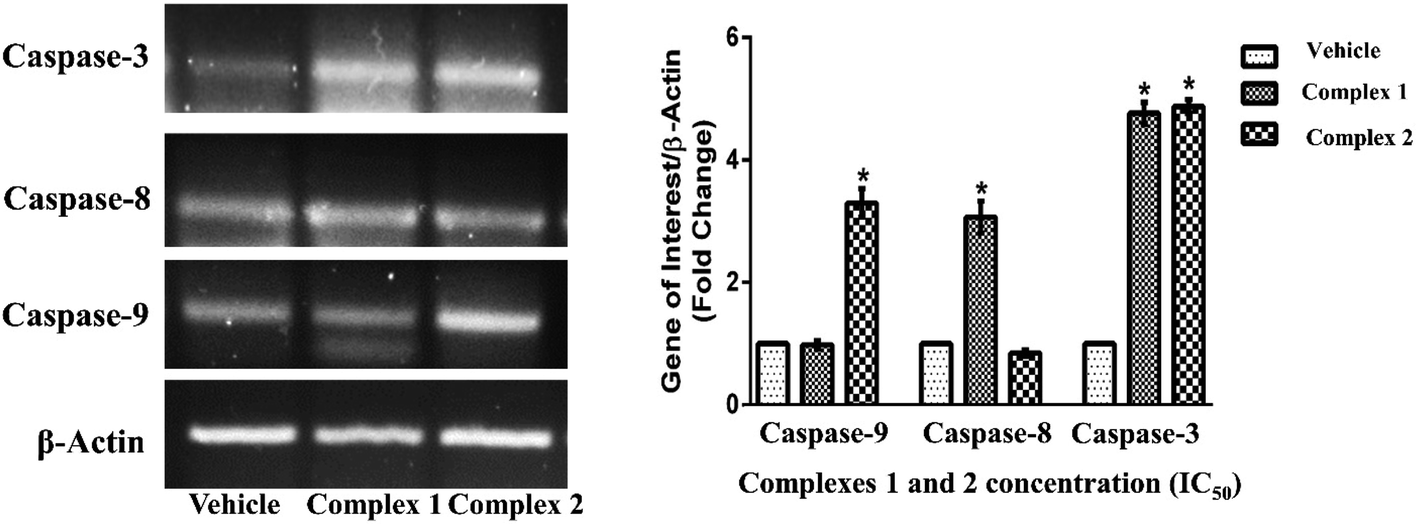 | ||
| Fig. 12 Evaluation of expression pattern of apoptotic genes at transcriptional level. Representative PCR images indicating the transcriptional pattern of β-actin, capase-3, -8 and -9 in response to complexes 1 and 2 treated MCF-7 cell. β-Actin gene was used as housekeeping control. Histogram in the panel represents the fold change in the expression pattern of genes in treatment with IC50 concentration of complex 1 and 2 as compared to vehicle treated control cells condition. Results are the mean ± SEM of three independent experiments. * indicates p < 0.05 as compared to the vehicle treated control group. | ||
Molecular docking
In humans, a transmembrane receptor protein, epidermal growth factor receptor (EGFR) is of extracellular protein. The epidermal growth factor protein binds with its receptor protein (EGFR) and dimerizes the EGFR subunits. The result of the process is the autophosphorylation of intracellular tyrosine residues which opens the receptor to bind the intracellular effector proteins. Such binding led to cellular proliferation, cellular invasion, angiogenesis, metastasis, apoptosis resistance and DNA repairing process. Mutation, rearrangement, EGFR overexpression or amplification of EGFR gene are responsible for a number of cancer in different organs and malignancies.The compounds erlotinib, gefitinib and lapatinib are FDA-approved EGFR inhibitors and they belong to TKIs inhibitors group (Fig. 13a). All the three compounds are structurally N-phenylquinazolin-4-amine having one phenyl ring attached at the 4-position of quinazolin. Furthermore, the moiety has two rotatable bonds and three hetero-atoms for hydrogen bonding. The structure of erlotinib, gefitinib and lapatinib are also obvious that the increase of hetero atoms do not hamper their EGFR binding which depends on the structural feature of N-phenylquinazolin-4-amine (Fig. 13a). The quinolin-8-ol is structurally similar with quinazolin-4-amine. However, the moiety contain less number of hetero atom. Therefore, we have designed 2-((quinolin-8-yloxy)methyl)benzoic acid to address the aforesaid hetero atom shortage problem. The presence of acid group can increase its water solubility and molecular flexibility. Such structural similarity between ligand and FDA-approved EGFR inhibitors indicates that the complexes (1 and 2) can show anticancer activities through EGFR inhibition.
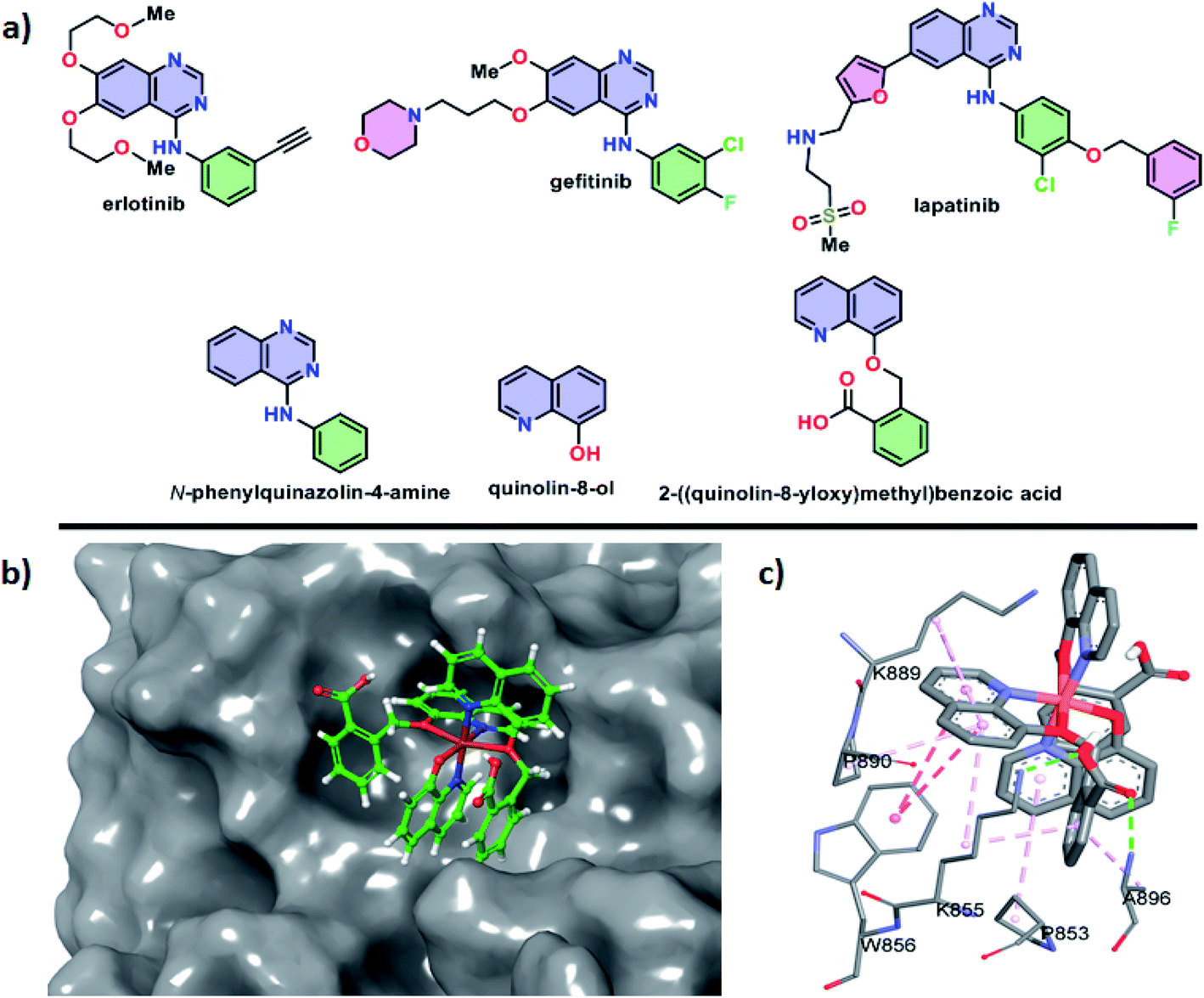 | ||
| Fig. 13 (a) Structural features of FDA-approved EGFR inhibitors with designed ligands, (b) docking pose of the complex 2 inside the active site of the EGFR protein, (c) non-covalent interactions between active site amino acid residues of EGFR with complex 2. | ||
Docking studies of small molecules and protein give opportunity to understand their binding affinity and mechanism. Here, the study was utilized to understand the behavior of complexes 1 and 2 with the EGFR. The study showed that the negative values of the change of Gibbs free energy for the interaction between these complexes (ΔG° = −5.53 kcal mol−1 for 1 and −5.83 kcal mol−1 for 2) and EGFR are higher than that of erlotinib (ΔG° = −4.26 kcal mol−1), gefitinib (ΔG° = −4.02 kcal mol−1) and lapatinib (ΔG° = −3.86 kcal mol−1) and the free ligands of the complexes (ΔG° = −3.58 kcal mol−1 for quinolin-8-ol and −3.91 kcal mol−1 for 2-((quinolin-8-yloxy)methyl)benzoic acid). Therefore, the complex 2 was found to be best in the binding with EGFR and the compound showed the best anti-cancer activities.
The docking pose showed that the complex 2 binds at the active site of the EGFR where erlotinib, gefitinib and lapatinib compounds bind (Fig. 13b). The complex binds through both polar and hydrophobic interactions. The acid group of 2-((quinolin-8-yloxy)methyl)benzoic acid of the complex 2 involved in hydrogen bonding with K855 and A896 residues (Fig. 13c). The amino acid residues P853, K855, W856, K889, P890 and A896 are interact through hydrophobic and CH–π interactions. Therefore, the complex 2 preferred hydrophobic interactions over hydrophilic interactions during its binding with EGFR.
Conclusions
The work presented in this manuscript provides a comprehensive insight of copper(II) complexes namely, {[Cu(qmbn)(Hqmba)(q)]·NO3·2H2O} (1) and {[Cu(Hqmba)2(q)]·NO3·2H2O} (2) which exhibited potential in vitro cytotoxicity as compared to 8-hydroxyquinoline derivatives against MCF-7 cell line. Single crystal study reveals distorted octahedral (CuN3O3) geometry around the metal centers. In order to determine the impact of substituents incorporated into the 8-hydroxyquinoline core, a comprehensive study of complexes were accomplished on the antiproliferative efficiency towards MCF-7 cancer cell lines. Dose-dependent inhibition of cell growth and cell death induced by complexes 1 and 2 in MCF-7 cells revealed that the presence of –COOH functionality in complex 2 leads to higher cytotoxicity (lower IC50 = 0.77 ± 0.12 μM) than that observed for complex 1 containing –CN group (higher IC50 = 1.86 ± 0.27 μM). The AO/EB/DAPI, annexin-V-FITC and propidium iodide nuclear staining assays displayed an apoptotic mechanism of cell death, possibly through membrane destabilization, and triggering apoptosis and necrosis owing to the metal-assisted generation of reactive oxygen species. Gene expression analysis at transcriptional level induced by complexes 1 and 2 revealed different responses to caspases-3/8 and 9 proteins. The expression of caspase-3 prompted by complexes 1 and 2 were found to increase by 4.7- and 4.8-fold, respectively as compared to vehicle treated condition. However, the complex 1 increased the expression of caspase-8 by about 3.6-fold while complex 2 increased the expression of caspase-9 by about 3.5-fold as compared to vehicle treated conditions, thus both the complexes induced apoptotic cell death by activating pro-apoptotic genes. However, further detailed studies are needed to get a mechanistic overview about the differential caspase activation by complexes 1 and 2. Additionally the molecular docking studies of copper(II) complexes 1 and 2 with EGFR kinase receptor, which were in agreement with the results obtained from cytotoxicity assay.Conflicts of interest
There are no conflicts of interest.Acknowledgements
The Department of Applied Chemistry, Faculty of Engineering and Technology, Aligarh Muslim University, UP, India, and Researchers Supporting Project number (RSP-2020/288), King Saud University, Riyadh, Saudi Arabia are gratefully acknowledged for financial support and laboratory facilities. UGC Start-up grant is thanked for extensive help to procure chemicals.Notes and references
- A. Weyesa and E. Mulugeta, RSC Adv., 2020, 10, 20784–20793 RSC.
- A. R. Martirosyan, R. Rahim-Bata, A. B. Freeman, C. D. Clarke, R. L. Howard and J. S. Strobl, Biochem. Pharmacol., 2004, 68, 1729–1738 CrossRef CAS PubMed.
- S. H. Abbas, A. A. Abd El-Hafeez, M. E. Shoman, M. M. Montano and H. A. Hassan, Bioorg. Chem., 2019, 82, 360–377 CrossRef CAS PubMed.
- S. Jain, V. Chandra, P. Kumar Jain, K. Pathak, D. Pathak and A. Vaidya, Arabian J. Chem., 2019, 12, 4920–4946 CrossRef CAS.
- Q.-P. Qin, Z.-F. Chen, J.-L. Qin, X.-J. He, Y.-L. Li, Y.-C. Liu, K.-B. Huang and H. Liang, Eur. J. Med. Chem., 2015, 92, 302–313 CrossRef CAS PubMed.
- V. Oliveri and G. Vecchio, Eur. J. Med. Chem., 2016, 120, 252–274 CrossRef CAS PubMed.
- B. Deka, T. Sarkar, S. Banerjee, A. Kumar, S. Mukherjee, S. Deka, K. K. Saikia and A. Hussain, Dalton Trans., 2017, 46, 396–409 RSC.
- Y.-Q. Hu, C. Gao, S. Zhang, L. Xu, Z. Xu, L.-S. Feng, X. Wu and F. Zhao, Eur. J. Med. Chem., 2017, 139, 22–47 CrossRef CAS PubMed.
- S. Tardito, A. Barilli, I. Bassanetti, M. Tegoni, O. Bussolati, R. Franchi-Gazzola, C. Mucchino and L. Marchiò, J. Med. Chem., 2012, 55, 10448–10459 CrossRef CAS PubMed.
- L. B. de O. Freitas, T. F. Borgati, R. P. de Freitas, A. L. T. G. Ruiz, G. M. Marchetti, J. E. de Carvalho, E. F. F. da Cunha, T. C. Ramalho and R. B. Alves, Eur. J. Med. Chem., 2014, 84, 595–604 CrossRef CAS PubMed.
- X. Jia, F.-F. Yang, J. Li, J.-Y. Liu and J.-P. Xue, J. Med. Chem., 2013, 56, 5797–5805 CrossRef CAS PubMed.
- S. Rasoul-Amini, A. Khalaj, A. Shafiee, M. Daneshtalab, A. Madadkar-Sobhani, S. Fouladdel and E. Azizi, Int. J. Cancer Res., 2006, 2, 102–108 CrossRef CAS.
- S. L. Zhang, X. Zhai, S. J. Zhang, H. H. Yu and P. Gong, Chin. Chem. Lett., 2010, 21, 939–942 CrossRef CAS.
- A. Barilli, C. Atzeri, I. Bassanetti, F. Ingoglia, V. Dall'Asta, O. Bussolati, M. Maffini, C. Mucchino and L. Marchiò, Mol. Pharm., 2014, 11, 1151–1163 CrossRef CAS PubMed.
- M. Carcelli, M. Tegoni, J. Bartoli, C. Marzano, G. Pelosi, M. Salvalaio, D. Rogolino and V. Gandin, Eur. J. Med. Chem., 2020, 194, 112266 CrossRef CAS PubMed.
- A. Paul, R. K. Gupta, M. Dubey, G. Sharma, B. Koch, G. Hundal, M. S. Hundal and D. S. Pandey, RSC Adv., 2014, 4, 41228–41236 RSC.
- P. P. Kattimani, R. R. Kamble, M. Y. Kariduraganavar, A. Dorababu and R. K. Hunnur, Eur. J. Med. Chem., 2013, 62, 232–240 CrossRef CAS PubMed.
- M. Mohamed Subarkhan, R. N. Prabhu, R. Raj Kumar and R. Ramesh, RSC Adv., 2016, 6, 25082–25093 RSC.
- V. Oliveri, M. Viale, G. Caron, C. Aiello, R. Gangemi and G. Vecchio, Dalton Trans., 2013, 42, 2023–2034 RSC.
- S. Modi, C. Saura, C. Henderson, N. U. Lin, R. Mahtani, J. Goddard, E. Rodenas, C. Hudis, J. O'Shaughnessy and J. Baselga, Breast Cancer Res. Treat., 2013, 139, 107–113 CrossRef CAS PubMed.
- C. Santini, M. Pellei, V. Gandin, M. Porchia, F. Tisato and C. Marzano, Chem. Rev., 2014, 114, 815–862 CrossRef CAS PubMed.
- D. Mahendiran, S. Amuthakala, N. S. P. Bhuvanesh, R. S. Kumar and A. K. Rahiman, RSC Adv., 2018, 8, 16973–16990 RSC.
- S. Mukherjee, S. Chowdhury, A. Ghorai, U. Ghosh and H. Stoeckli-Evans, Polyhedron, 2013, 51, 228–234 CrossRef CAS.
- M. Frezza, S. Hindo, D. Chen, A. Davenport, S. Schmitt, D. Tomco and Q. Ping Dou, Curr. Pharm. Des., 2010, 16, 1813–1825 CrossRef CAS PubMed.
- V. Milacic and Q. P. Dou, Coord. Chem. Rev., 2009, 253, 1649–1660 CrossRef CAS PubMed.
- C. K. Adokoh, RSC Adv., 2020, 10, 2975–2988 RSC.
- L. Cattaruzza, D. Fregona, M. Mongiat, L. Ronconi, A. Fassina, A. Colombatti and D. Aldinucci, Int. J. Cancer, 2011, 128, 206–215 CrossRef CAS PubMed.
- K. Choroba, L. R. Raposo, J. Palion-Gazda, E. Malicka, K. Erfurt, B. Machura and A. R. Fernandes, Dalton Trans., 2020, 49, 6596–6606 RSC.
- A. N. Kate, A. A. Kumbhar, A. A. Khan, P. V. Joshi and V. G. Puranik, Bioconjugate Chem., 2014, 25, 102–114 CrossRef CAS PubMed.
- S. Zhai, L. Yang, Q. C. Cui, Y. Sun, Q. P. Dou and B. Yan, JBIC, J. Biol. Inorg. Chem., 2010, 15, 259–269 CrossRef CAS PubMed.
- B.-L. Fei, S. Tu, Z. Wei, P. Wang, C. Qiao and Z.-F. Chen, Eur. J. Med. Chem., 2019, 176, 175–186 CrossRef CAS PubMed.
- Y. Song, H. Xu, W. Chen, P. Zhan and X. Liu, Medchemcomm, 2015, 6, 61–74 RSC.
- Y. Wang, P. J. Jones, T. W. Batts, V. Landry, M. K. Patel and M. L. Brown, Bioorg. Med. Chem., 2009, 17, 7064–7072 CrossRef CAS PubMed.
- M. Wichert, N. Krall, W. Decurtins, R. M. Franzini, F. Pretto, P. Schneider, D. Neri and J. Scheuermann, Nat. Chem., 2015, 7, 241–249 CrossRef CAS PubMed.
- D. Moser, J. M. Wisniewska, S. Hahn, J. Achenbach, E. Buscató, F.-M. Klingler, B. Hofmann, D. Steinhilber and E. Proschak, ACS Med. Chem. Lett., 2012, 3, 155–158 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- A. Ali, S. Mishra, S. Kamaal, A. Alarifi, M. Afzal, K. Das Saha and M. Ahmad, Bioorg. Chem., 2021, 106, 104479 CrossRef CAS PubMed.
- A. Ali, N. Sepay, M. Afzal, N. Sepay, A. Alarifi, M. Shahid and M. Ahmad, Bioorg. Chem., 2021, 110, 104772 CrossRef CAS PubMed.
- P. Kumar, A. Nagarajan and P. D. Uchil, Cold Spring Harb. Protoc., 2018, 2018, pdb.prot095505 CrossRef PubMed.
- S. Kasibhatla, Cold Spring Harb. Protoc., 2006, 2006, pdb.prot4493-pdb.prot4493 Search PubMed.
- B. Chazotte, Cold Spring Harb. Protoc., 2011, 2011, pdb.prot5556-pdb.prot5556 Search PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- A. Schäfer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829–5835 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- C. Steffen, K. Thomas, U. Huniar, A. Hellweg, O. Rubner and A. Schroer, J. Comput. Chem., 2010, 2967–2970 CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- N. Sepay, C. Guha, S. Maity and A. K. Mallik, Eur. J. Org. Chem., 2017, 2017, 6013–6022 CrossRef CAS.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, J. Biol. Chem., 2002, 277, 46265–46272 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- B. Tummers and D. R. Green, Immunol. Rev., 2017, 277, 76–89 CrossRef CAS PubMed.
- S. Fulda, Cancer Lett., 2009, 281, 128–133 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedure for ligand L1 and L2, effect of ligands L1 and L2 on the MCF-7 cells and X-ray crystallographic data in CIF format. CCDC reference number 2040551 (1) and 2040552 (2). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00172h |
| This journal is © The Royal Society of Chemistry 2021 |