
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFullerene C60 promoted photochemical hydroamination reactions of an electron deficient alkyne with trimethylsilyl group containing tertiary N-alkylbenzylamines†
Suk Hyun Lim,
Hannara Jang and
Dae Won Cho *
*
Department of Chemistry, Yeungnam University, Gyeongsan, Gyeongbuk 38541, Korea. E-mail: dwcho00@yu.ac.kr
First published on 3rd February 2021
Abstract
C60-promoted photoaddition reactions of both trimethylsilyl- and a variety of alkyl group containing tertiary benzylamines (i.e., N-α-trimethylsilyl-N-alkylbenzylamines) with dimethyl acetylenedicarboxylate (DMAD) were carried out to explore the synthetic utility of trimethylsilyl group containing tertiary amines as a substrate in the photochemical hydroamination reactions with dimethyl acetylenedicarboxylate (DMAD). The results showed that photoreactions of all the trimethylsilyl containing N-alkylbenzylamines with DMAD, under an O2-purged environment, produced non-silyl containing enamines efficiently through a pathway involving addition of secondary amines to DMAD, the former of which are produced by hydrolytic cleavage of in situ formed iminium ions. Exceptionally, five-membered N-heterocyclic rings, pyrroles, could be produced competitively in photoreaction of bulky alkyl (i.e., tert-butyl) group substituted benzylamines through a pathway involving 1,3-dipolar cycloaddition of azomethine ylides to DMAD. Furthermore, C60-sensitized photochemical reactions of non-silyl containing benzylamines with DMAD under oxygenated conditions took place in a less efficient and non-regioselective manner to produce enamine photoadducts. The observations made in this study show that regioselectivity of C60-promoted photochemical reactions of N-α-trimethylsilyl-N-alkylbenzylamines, leading to formation of secondary amines, can be controlled by the presence of the trimethylsilyl group, and that these trimethylsilyl containing tertiary amines can serve as a precursor of secondary amines for hydroamination reactions with a variety of electron deficient acetylenes.
Introduction
The development of synthetic protocols for enamines possessing biologically interesting properties and their chemical manipulation have been a promising research topic in pharmaceutical and organic chemistry fields.1–3 In particular, because enamine moieties can be readily converted to valuable chiral amine moieties in asymmetric catalytic hydrogenation reactions of unsaturated compounds, enamines have served as useful intermediates in natural product synthesis.4 (Scheme 1). | ||
| Scheme 1 Chiral amine synthesis via asymmetric catalytic hydrogenation of enamines. | ||
Among the well-known enamine synthetic methods, inter- or intramolecular hydroamination reactions of alkynes with primary- or secondary amine substrates seem to be atom and step-economic transformations because of direct addition of the N–H moiety of amines to an alkyne substrate.5 Typically, hydroamination reactions through nucleophilic addition of amines to electron sufficient C–C multiple bonds are not readily available due to the repulsion between non-bonding electron on a nitrogen atom and π-electron on alkyne. To overcome these thermodynamic and kinetic constraint, a number of catalytic hydroamination reactions using a variety of metal catalysts5–8 and alkali bases9 have been employed, in which the use of metals facilitate amines to be readily added to electron-rich alkyne via metal–amine complex insertion to alkyne, while the alkali bases deprive proton to endow more nucleophilic ability. Although these metals and alkali base exhibit efficient catalytic activities for hydroamination reactions of alkynes, a cost-effective and toxic waste-free strategy amenable for the large scale applications are still remained.
In the studies exploring application of amine SET photochemistry for the synthesis of nitrogen-containing compounds, interesting observation was made in our recent studies aiming at exploring photochemical behaviors of both α-trimethylsilyl and α-alkoxycarbonyl group containing N-benzylamines (i.e., N-α-trimethylsilyl-N-benzylglycinates).10 The results showed that photoirradiation of N-α-trimethylsilyl-N-benzylglycinates in the presence of molecular oxygen (O2) and photosensitizer (PS) could convert starting tertiary amines to non-silyl containing secondary amines via hydrolytic cleavage of in situ formed iminium ions. This investigation suggested that α-trimethylsilyl group substituted tertiary amines can participate in photochemical hydroamination reactions with alkynes to produce enamine derivatives predominantly. To the best of knowledge, hydroamination reactions of alkynes with tertiary amines as a reaction substrates are not common.11
To explore substrate scope and synthetic utility of α-trimethylsilyl group containing tertiary aliphatic amines in the photochemical hydroamination reactions of C–C multiple bonds, in current study, we have prepared (trimethylsilyl)methyl and a variety of alkyl group substituted tertiary N-benzylamines (i.e., N-α-trimethylsilyl-N-alkylbenzylamines) and then, the photochemical reactions of them with dimethyl acetylenedicarboxylate (DMAD) in the presence of fullerence C60 were carried out. Since fullerene C60 is excellent electron acceptor owing to its high electron affinity and relatively high reduction potentials (for 3C60, 3Ered = 1.14 V vs. SCE),12,13 fullerene C60 as a photosensitizer can participate in rapid and efficient SET process with a variety of amine electron donors possess oxidation potentials lower than ca. 1.1 eV. The results of this study, presented below, showed that in contrast to those of non-silyl containing tertiary amines, aminium radicals generated by SET processes undergo H-atom abstraction regioselectively at α-carbons next to trimethylsilyl substituent to produce iminium ions, which are hydrolyzed to secondary amines. Then, generated secondary amines are added to alkyne to produce enamines. Especially, when the bulky alkyl group like tert-butyl is present on nitrogen atom, not only secondary amine but azomethine ylide intermediates could be formed.
Results and discussion
C60-promoted photoaddition reactions of N-α-trimethylsilyl-N-alkylbenzylamines and DMAD
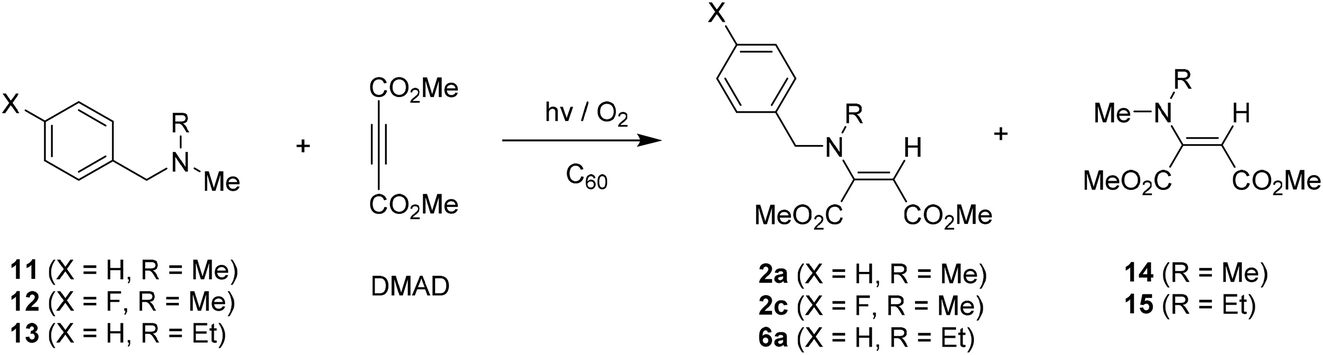
A variety of alkyl (i.e., methyl, ethyl, isopropyl, and tert-butyl) and (trimethylsilyl)methyl substituted N-benzylamines, where phenyl group within benzyl moiety possesses a substituent like H, Me and F at para-position, were readily prepared by using earlier reported synthetic procedure.14,15 With these tertiary benzylamines in hand, photoaddition reactions of toluene solutions (220 mL) containing benzylamines (0.7 mmol, 1 equiv.), dimethyl acetylenedicarboxylate (DMAD) (1.4 mmol, 2 equiv.) and fullerene (C60) photosensitizer (0.035 mmol, 0.05 equiv.) were performed by irradiation (450 W Hanovia medium pressure mercury lamp equipped with flint glass filter (>310 nm)) under oxygenated (O2-purged) conditions. The photolysates were then purified by column chromatography to determine photoproducts and their yields.First, C60-promoted photoaddition reactions of N-α-trimethylsilyl-N-methylbenzylamines 1a–1c with DMAD in the oxygenated toluene were conducted. As shown in Table 1, 10 min photoirradiation of toluene solutions containing 1a–1b and DMAD gave rise to formation of high yielding of enamines 2a–2b as the sole adduct. In the photoreaction of para-fluoro phenyl containing amine 1c, non-silyl enamine 2c was produced as a major product, along with silyl group containing enamine 3 as a minor adduct. Importantly, control experiments (entries 4 and 5 in Table 1) revealed that photoreaction did not take place in cases that the reactions were conducted under the either deoxygenated (freeze–pump–thaw degassing) condition or absence of C60 photosensitizer.
|
||||
|---|---|---|---|---|
| Entry | Amine | Irradiation time (min) | Conversiona (%) | Productb (%) |
| a Conversion was determined based on recovered amine.b Isolation yields.c Photoreaction in deoxygenated (using freeze–pump–thaw degassing) condition.d No reaction.e Photoreaction in the absence of C60. | ||||
| 1 | 1a | 10 | 100 | 2a (78) |
| 2 | 1b | 10 | 100 | 2b (73) |
| 3 | 1c | 10 | 93 | 2c (60), 3 (19) |
| 4c | 1a | 30 | —d | —d |
| 5e | 1a | 30 | —d | —d |
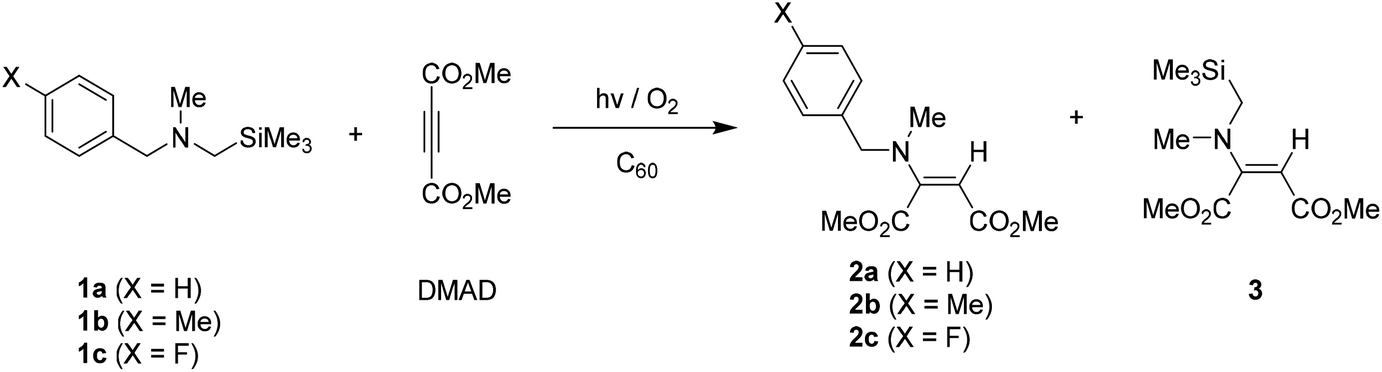
Similarly, photoreactions of N-α-trimethylsilyl-N-ethylbenzylamines 4a–4c with DMAD in the presence of C60 produced the same types of enamines 6a–6c as a single product even in the reaction of para-fluoro substituted amine 4c. (Table 2) When N-alkyl substituent of amine substrates was changed from ethyl (N-ethyl, 4a–4c) to isopropyl (N-isopropyl, 5a–5c) group, enamines 7a–7c were also produced. However, conversion yields of amine substrates 5a–5c and their photoproduct yields were much lower than those from 4a–4c. Especially, in case of photoreactions of para-fluoro phenyl tethered amine 5c, much longer irradiation time was required to bring about high conversion of starting amine 5c and high yielding of photoproduct 7c. (entries 8 and 9 in Table 2).
|
||||
|---|---|---|---|---|
| Entry | Amine | Irradiation time (min) | Conversiona (%) | Productb (%) |
| a Conversion was determined based on recovered amine.b Isolation yields. | ||||
| 1 | 4a | 10 | 100 | 6a (65) |
| 2 | 4b | 10 | 100 | 6b (76) |
| 3 | 4c | 10 | 90 | 6c (60) |
| 4 | 5a | 10 | 34 | 7a (18) |
| 5 | 5a | 30 | 96 | 7a (51) |
| 6 | 5b | 10 | 39 | 7b (21) |
| 7 | 5b | 30 | 100 | 7b (53) |
| 8 | 5c | 10 | 11 | 7c (6) |
| 9 | 5c | 50 | 100 | 7c (53) |
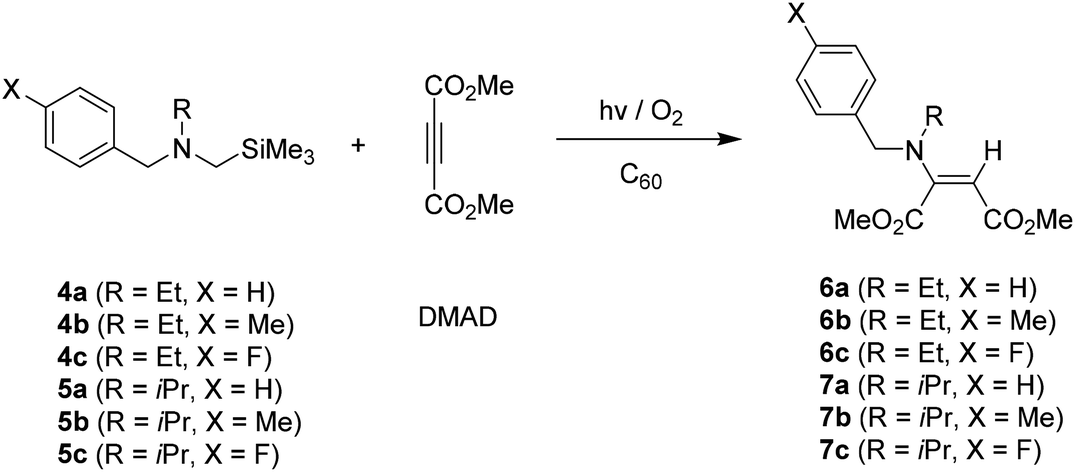
The results of photoreactions of tert-butyl group containing benzylamines 8a–8c with DMAD in the presence of C60 were quite different from those of less alkyl group containing benzylamines 1a–1c (N-methyl), 4a–4c (N-ethyl), 5a–5c (N-isopropyl) in terms of both photoreaction efficiency and photoproduct distribution pattern. As shown in Table 3, the photoaddition reactions of 8a–8c with DMAD took place much less efficiently and, moreover, the least reaction efficiency was made in the photoreaction using both tert-butyl and para-fluoro phenyl groups containing benzylamine 8c (see Tables 1 and 2). More interesting observation made in these reactions was that when much longer irradiation time was employed for the reactions, not only enamines 9a–9c but also the highly substituted pyrrole derivatives 10a–10c were produced, albeit relatively low yield. Considering our previous studies,10 it is likely that 1,3-dipolar cycloaddition reactions of in situ formed azomethine ylides from 8a–8c to DMAD are responsible for the formation of five membered N-heterocycles. In here, it is noteworthy to mention that the generation of non-stabilized azomethine ylides stimulated by sterically hindered N-substituent is quite unusual.
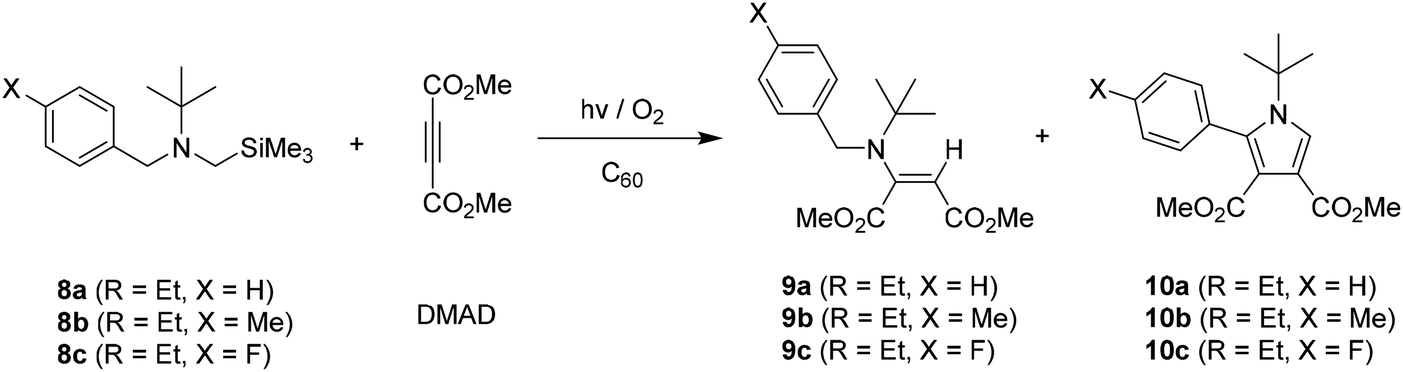
In order to evaluate how the trimethylsilyl group on the amine influence the reaction efficiencies and photoproduct distributions, we probed C60-promoted photoaddition reactions of non-silyl containing benzylamines 11–13. As described in Table 4, in contrast those of silyl group containing amine analogs (see Tables 1 and 2), photoreactions of non-silyl group tethered amines 11–13 with acetylene (DMAD) led to competitive formation of two types of enamine mixtures, 2a, 2c, 6a and 14–15 inefficiently. These observation reveal that silyl group resided on nitrogen atom is likely to be responsible for regioselective secondary amine formation. Especially, silyl group tethered aminium radicals undergo H-atom abstraction at α-carbons adjacent to silyl moiety regioselectively to form precursors of secondary amines (see reaction mechanism). In addition, considering irradiation time vs. conversion of amine substrates, silyl group on amines seem to be able to enhance the reaction efficiencies.
Visible light irradiated, C60-promoted photoaddition reactions of N-α-trimethylsilyl-N-alkylbenzylamines 1a–1c with DMAD
Visible-light promoted photochemical reactions have attracted great attention because of their wide application in organic synthesis and their use of sustainable and green energy sources. Especially, utilization of visible light is highly advantageous as most organic compounds do not absorb in this region and product decomposition and unwanted side products are readily avoided. Because fullerene C60 can absorbs light in the visible region as well as serve as an efficient electron acceptor, as a preliminary study, we explored viability of photoreactions of N-α-trimethylsilyl-N-alkylbenzylamines 1a–1c with DMAD using visible light. For this purpose, C60-promoted photoaddition reactions of benzylamines 1a–1c with DMAD under the O2-purged condition were performed by irradiation using commercially available 20 W compact fluorescent lamp (CFL) for fixed time periods (information about emitted wavelengths and their relative intensity from 20 W fluorescent lamp is available in ESI†). The results, given in Table 5, show that 20 h visible light irradiation of toluene solutions containing 1a–1c and DMAD led to production of the respective enamines 2a–2c, although reactions of para-fluorophenyl containing amine 1c gave rise to lower conversion and product yield. Definitely, employing much longer visible light irradiation time could enhance the conversion and photoproduct yields in the photoreactions of 1a–1c with DMAD.
Reaction mechanism
On the basis of observation made in earlier studies,10,16,17 plausible mechanistic pathways can be suggested. (Scheme 2) Enamine formation in these reactions seems to occur via Michael addition of formed secondary amines 19 to DMAD. Specifically, right after the excitation of ground state of fullerene C60 to singlet state of C60, followed rapid and efficient intersystem crossing (ΦISC = 1) processes, the SET process from benzylamines 16 to triplet state of fullerene C60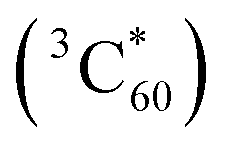 takes place initially to generate aminium radicals 17 and radical ions of C60 (C60˙−), latter of which are oxidized by molecular oxygen (O2) to ground state of C60 and radical anions of molecular oxygen (O2˙−). Among the several pathways open to aminium radicals 17, O2˙−-promoted regioselective H-atom abstraction of the aminium radicals seems to be the most feasible pathway.16 Then, generated iminium ions 18 undergo hydrolytic cleavage by hydrogen peroxide anions (HOO−) to produce non-silyl containing secondary benzylamines 19, which add to DMAD to form enamine adducts 20.5 When the sterically bulky N-substituent is present on the starting amine substrates, another photochemical process takes place competitively. A mechanistic pathway for this process, which is compatible with the results of photoreactions, is 1,3-dipolar cycloaddition reactions of in situ formed azomethine ylides to DMAD to produce N-heterocyclic pyrrole photoadducts. As shown in Scheme 2, the key step in this route is that aminium radicals 17 undergo desilylation to form α-amino radicals 21, which are consecutively converted to iminium ions 22 through a pathway involving second SET owing to the much lower oxidation potential of α-amino radicals in the range of −1 V (vs. SCE). Non-silyl containing iminium ions 22 formed in this manner then lose proton from benzyl position to produce 1,3-dipolar azomethine ylides 23 which serve as a precursor of pyrrole photoadducts 24.
takes place initially to generate aminium radicals 17 and radical ions of C60 (C60˙−), latter of which are oxidized by molecular oxygen (O2) to ground state of C60 and radical anions of molecular oxygen (O2˙−). Among the several pathways open to aminium radicals 17, O2˙−-promoted regioselective H-atom abstraction of the aminium radicals seems to be the most feasible pathway.16 Then, generated iminium ions 18 undergo hydrolytic cleavage by hydrogen peroxide anions (HOO−) to produce non-silyl containing secondary benzylamines 19, which add to DMAD to form enamine adducts 20.5 When the sterically bulky N-substituent is present on the starting amine substrates, another photochemical process takes place competitively. A mechanistic pathway for this process, which is compatible with the results of photoreactions, is 1,3-dipolar cycloaddition reactions of in situ formed azomethine ylides to DMAD to produce N-heterocyclic pyrrole photoadducts. As shown in Scheme 2, the key step in this route is that aminium radicals 17 undergo desilylation to form α-amino radicals 21, which are consecutively converted to iminium ions 22 through a pathway involving second SET owing to the much lower oxidation potential of α-amino radicals in the range of −1 V (vs. SCE). Non-silyl containing iminium ions 22 formed in this manner then lose proton from benzyl position to produce 1,3-dipolar azomethine ylides 23 which serve as a precursor of pyrrole photoadducts 24.
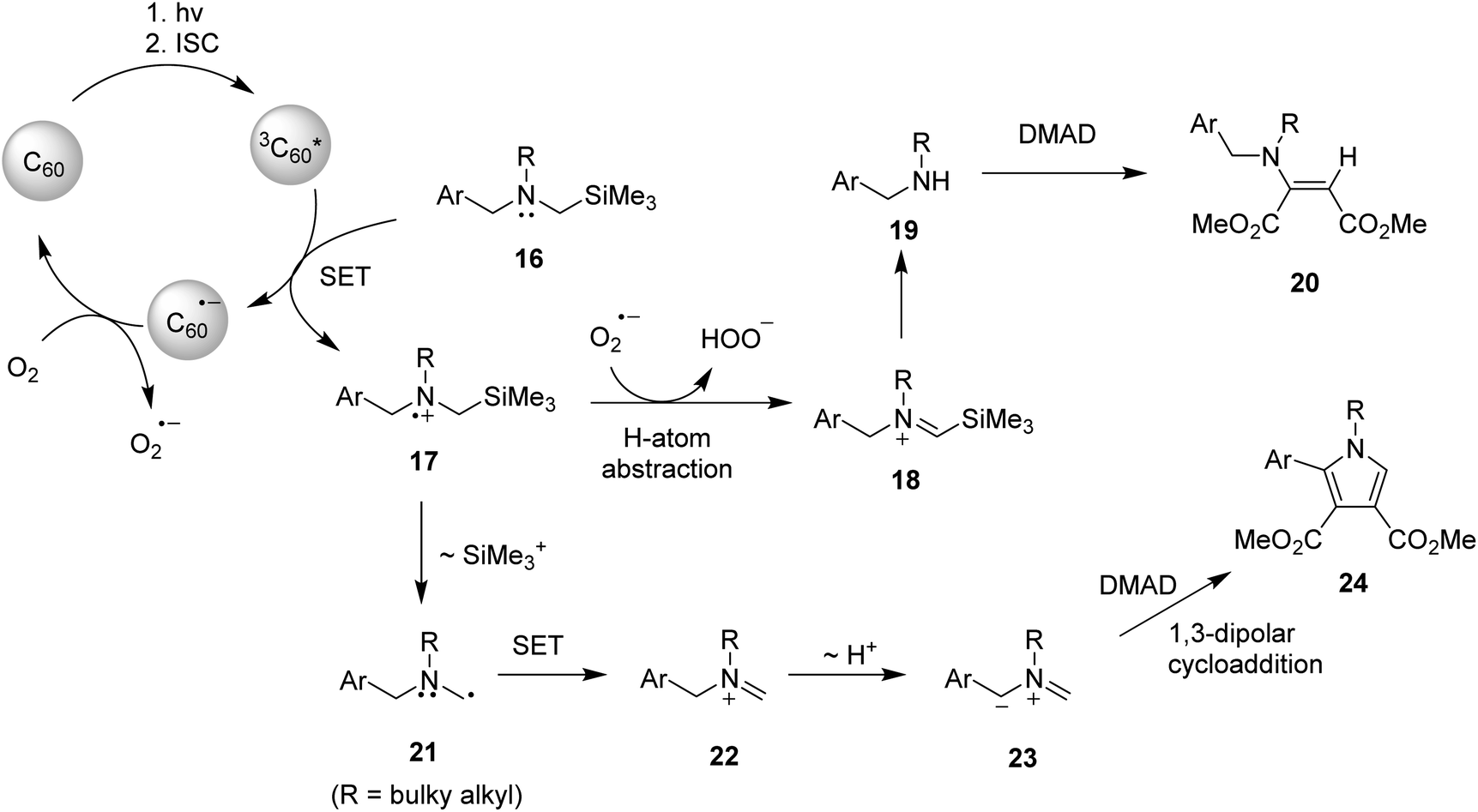 | ||
| Scheme 2 Plausible mechanistic pathways for the enamine and pyrrole photoadducts. | ||
Conclusion
In current study, C60-promoted photoaddition reactions of both trimethylsilyl- and a variety of alkyl group containing tertiary benzylamines (i.e., N-α-trimethylsilyl-N-alkylbenzylamines) with dimethyl acetylenedicarboxylate (DMAD) were carried out to explore synthetic utility of trimethylsilyl group containing tertiary amines as substrates in the photochemical hydroamination reactions with C–C multiple bonds. The results showed that photoreactions of all of trimethylsilyl containing N-alkylbenzylamines with DMAD under O2-purged environment, produced non-silyl containing enamines efficiently through a pathway involving addition of secondary amines to DMAD, former of which are produced by hydrolytic cleavage of in situ formed iminium ions. Exceptionally, five-membered N-heterocyclic ring, pyrroles, were also produced competitively in photoreaction of those of less bulky alkyl group (i.e., tert-butyl) substituted benzylamines through a pathway involving 1,3-dipolar cycloaddition of azomethine ylides to DMAD. Furthermore, photoreactions of non-silyl containing benzylamines with DMAD under oxygenated conditions took place less efficient and non-regioselective manner to produce enamine photoadducts.The observation made in this study show that regioselectivity of C60-promoted photochemical reactions of N-α-trimetylsilyl-N-alkylbenzylamines, leading to formation of secondary amines, can be controlled by the presence of trimethylsilyl group, and that this silyl containing tertiary amines can serve a precursor of secondary amines for the hydroamination reactions with electron deficient acetylenes.
Experimental
General
The 1H (300 MHz) and 13C NMR (75 MHz) spectra (obtained by using Bruker DPX300) were recorded on CDCl3, and chemical shifts were reported in parts per million (δ, ppm) relative to CHCl3 (7.24 ppm for 1H and 77.0 ppm for 13C) as an internal standard. High resolution (HRMS) mass spectra were obtained by using an EI or ESI. All starting materials used in the synthetic routes came from commercial sources. Photochemical reactions were conducted by using an immersion-well photochemical apparatus, consisting of a 450 W Hanovia medium pressure mercury vapor UV lamp (aceglass cat. # 7825-34), a power supply (aceglass cat. # 7830-61), a water-cooled quartz immersion well (aceglass cat # 7874-27), a borosilicate reaction vessel (agceglass cat. # 7841-03) and a flint glass filter (>310 nm). Detail information about UV lamps used in this study are provided on ESI.†General procedure of C60-promoted photoreactions of N-α-trimethylsilyl-N-alkylbenzylamines with DMAD.
The toluene solutions (220 mL) containing benzylamines (0.7 mmol, 1 equiv.), dimethyl acetylenedicarboxylate (1.4 mmol, 2 equiv.), and C60 (0.035 mmol, 0.05 equiv.) that were purged with O2 before and during irradiation were irradiated with a 450 W Hanovia medium pressure Hg lamp surrounded by a flint glass filter (>310 nm) in a water-cooled quartz immersion well for time periods given below. Then, photolysates were concentrated in vacuo and then the resulting mixture were separated by using silica gel column chromatographic isolation to give photoadducts.Photoreaction of 1a
10 min irradiation (100% conversion), column chromatography (EtOAc![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane = 1:5) to yield 2a10 (144 mg, 78%). 1H-NMR δ 2.72 (s, 3H), 3.61 (s, 3H), 3.90 (s, 3H), 4.27 (s, 2H), 4.65 (s, 1H), 7.19–7.34 (m, 5H); 13C-NMR δ 36.8, 50.7, 52.9, 56.3, 84.6, 127.3, 127.8, 128.7, 135.5, 154.9, 166.0, 168.0; HRMS (EI) m/z 263.1154 (M+, C14H17NO4 requires 263.1158).
:hexane = 1:5) to yield 2a10 (144 mg, 78%). 1H-NMR δ 2.72 (s, 3H), 3.61 (s, 3H), 3.90 (s, 3H), 4.27 (s, 2H), 4.65 (s, 1H), 7.19–7.34 (m, 5H); 13C-NMR δ 36.8, 50.7, 52.9, 56.3, 84.6, 127.3, 127.8, 128.7, 135.5, 154.9, 166.0, 168.0; HRMS (EI) m/z 263.1154 (M+, C14H17NO4 requires 263.1158).
Photoreaction of 1b
10 min irradiation (100% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2b (142 mg, 73%). 1H NMR δ 2.30 (s, 3H), 2.70 (s, 3H), 3.61 (s, 3H), 3.90 (s, 3H), 4.22 (s, 2H), 4.64 (s, 1H), 7.07–7.14 (m, 4H); 13C NMR δ 21.0, 36.7, 50.7, 52.9, 56.2, 84.6, 127.4, 129.4, 132.5, 137.6, 155.0, 166.1, 168.1; HRMS (EI) m/z 277.1313 (M+, C15H19NO4 requires 277.1314).
Photoreaction of 1c
10 min irradiation (93% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2c (119 mg, 78%) and 3 (34 mg, 19%).
2c: 1H NMR δ 2.68 (s, 3H), 3.58 (s, 3H), 3.88 (s, 3H), 4.2 (s, 2H), 4.62 (s, 1H), 6.94–7.0 (m, 2H), 7.15–7.19 (m, 2H); 13C NMR δ 36.5, 50.6, 52.8, 55.5, 84.7, 115.5 (d, JC–F = 21.5 Hz), 128.9 (d, JC–F = 8.1 Hz), 131.1 (d, JC–F = 3.1 Hz), 154.6, 160.5, 162.1 (d, JC–F = 245 Hz), 165.9, 167.8; HRMS (EI) m/z 28101066 (M+, C14H16FNO4 requires 281.1063).
3: 1H NMR δ 0.08 (s, 9H), 2.68 (s, 2H), 2.84 (s, 3H), 3.57 (s, 3H), 3.88 (s, 3H), 4.44 (s, 1H); 13C NMR δ −1.6, 50.5, 52.7, 82.5, 154.7, 166.1, 168.2; HRMS (EI) m/z 259.1242 (M+, C11H21NO4Si requires 259.1240).
Photoreaction of 4a
10 min irradiation (100% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 6a (127 mg, 78%). 1H NMR δ 1.05 (t, 3H, J = 7.2 Hz), 3.12 (q, 2H, J = 7.2 Hz), 3.55 (s, 3H), 3.87 (s, 3H), 4.26 (s, 2H), 4.62 (s, 1H), 7.18–7.28 (m, 5H); 13C NMR δ 50.4, 52.7, 77.2, 83.9, 126.9, 127.5, 128.5, 135.4, 154.0, 165.9, 168.0; HRMS (EI) m/z 277.1312 (M+, C15H19NO4 requires 277.1314).
Photoreaction of 4b
10 min irradiation (100% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 6b (156 mg, 76%). 1H NMR δ 1.06 (t, 3H, J = 7.2 Hz), 2.28 (s, 3H), 3.12 (q, 2H, J = 7.2 Hz), 3.57 (s, 3H), 3.88 (s, 3H), 4.23 (s, 2H), 4.63 (s, 1H), 7.09 (s, 4H); 13C NMR δ 20.9, 50.5, 52.7, 77.2, 83.8, 127.0, 129.2, 132.4, 137.2, 154.1, 166.0, 168.1; HRMS (EI) m/z 291.1474 (M+, C16H21NO4 requires 291.1471).
Photoreaction of 4c
10 min irradiation (90% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 6c (125 mg, 60%). 1H NMR δ 1.03 (t, 3H, J = 6.9 Hz), 3.09 (q, 2H, J = 6.9 Hz), 3.53 (s, 3H), 3.85 (s, 3H), 4.2 (s, 2H), 4.58 (s, 1H), 6.91–6.97 (m, 2H), 7.13–7.17 (m, 2H); 13C NMR δ 50.6, 52.8, 77.2, 84.2, 115.5 (d, JC–F = 21.4 Hz), 128.7 (d, JC–F = 8 Hz), 131.2, 153.9, 160.4, 162.1 (d, JC–F = 244.5 Hz), 165.9, 168.0; HRMS (EI) m/z 295.1221 (M+, C15H18FNO4 requires 295.1220).
Photoreaction of 5a
10 min irradiation (34% conversion), column chromatography (EtOAc:hexane = 1:6) to yield 7a (36 mg, 18%); 30 min irradiation (96% conversion), column chromatography (EtOAc:hexane = 1:6) to yield 7a (105 mg, 51%). 1H NMR δ 1.17 (d, 6H, J = 6.3 Hz), 3.53 (s, 3H), 3.79 (septet, 1H, J = 6.3 Hz), 3.92 (s, 3H), 4.31 (s, 2H), 4.47 (s, 1H), 7.18–7.24 (m, 3H), 7.27–7.32 (m, 2H); 13C NMR δ 20.9, 47.1, 50.7, 52.8, 52.9, 85.7, 126.0, 127.1, 128.6, 136.1, 154.5, 166.3, 168.2; HRMS (EI) m/z 291.1472 (M+, C16H21NO4 requires 291.1471).
Photoreaction of 5b
10 min irradiation (39% conversion), column chromatography (EtOAc:hexane = 1:6) to yield 7b (45 mg, 21%); 30 min irradiation (100% conversion), column chromatography (EtOAc:hexane = 1:6) to yield 7b (114 mg, 53%). 1H NMR δ 1.15 (d, 6H, J = 6.6 Hz), 2.28 (s, 3H), 3.52 (s, 3H), 3.75 (septet, 1H, J = 6.6 Hz), 3.91 (s, 3H), 4.26 (s, 2H), 4.46 (s, 1H), 7.04–7.11 (m, 4H); 13C NMR δ 20.8, 21.0, 46.9, 50.6, 52.7, 52.9, 85.4, 125.9, 129.2, 133.0, 136.6, 154.5, 166.3, 168.2; HRMS (EI) m/z 305.1624 (M+, C17H23NO4 requires 305.1627).
Photoreaction of 5c
10 min irradiation (11% conversion), column chromatography (EtOAc:hexane = 1:6) to yield 7c (12 mg, 6%); 50 min irradiation (100% conversion), column chromatography (EtOAc:hexane = 1:6) to yield 7c (116 mg, 53%). 1H NMR δ 1.14 (d, 6H, J = 6.6 Hz), 3.51 (s, 3H), 3.74 (septet, 1H, J = 6.6 Hz), 3.89 (s, 3H), 4.24 (s, 2H), 4.43 (s, 1H), 6.93–6.98 (m, 2H), 7.12–7.17 (m, 2H); 13C NMR δ 20.7, 46.4, 50.6, 52.7, 52.8, 85.9, 115.5 (d, JC–F = 21 Hz), 127.6 (d, JC–F = 8.3 Hz), 131.7 (d, JC–F = 3 Hz), 154.3, 160.1, 161.7 (d, JC–F = 244.5 Hz), 166.1, 167.9; HRMS (EI) m/z 309.1378 (M+, C16H20FNO4 requires 309.1376).
Photoreaction of 8a
10 min irradiation (19% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 9a (21 mg, 10%); 30 min irradiation (60% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 9a (41 mg, 73%) and 10a (41 mg, 19%).
9a: 1H NMR δ 1.37 (s, 9H), 3.59 (s, 3H), 3.80 (s, 3H), 4.43 (s, 2H), 4.96 (s, 1H), 7.18–7.33 (m, 5H); 13C NMR δ 28.8, 50.8, 52.8, 53.0, 59.5, 91.1, 126.5, 127.0, 128.5, 138.5, 154.1, 167.2, 167.8; HRMS (EI) m/z 305.1629 (M+, C17H23NO4 requires 305.1627).
10a: 1H NMR δ 1.38 (s, 9H), 3.52 (s, 3H), 3.78 (s, 3H), 7.32–7.38 (m, 5H), 7.46 (s, 1H); 13C NMR δ 31.3, 51.3, 51.5, 59.0, 112.3, 118.1, 124.4, 127.4, 128.6, 131.9, 132.9, 136.2, 164.2, 165.6; HRMS (EI) m/z 315.1471 (M+, C18H21NO4 requires 315.1471).
Photoreaction of 8b
10 min irradiation (15% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 9b (18 mg, 8%); 30 min irradiation (60% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 9b (55 mg, 25%) and 10b (18 mg, 8%).
9b: 1H NMR δ 1.36 (s, 9H), 2.30 (s, 3H), 3.59 (s, 3H), 3.80 (s, 3H), 4.38 (s, 2H), 4.95 (s, 1H), 7.08–7.15 (m, 4H); 13C NMR δ 28.8, 50.8, 52.8, 90.8, 126.4, 129.1, 135.4, 136.6, 154.1, 167.2, 167.9; HRMS (EI) m/z 319.1786 (M+, C18H25NO4 requires 319.1784).
10b: 1H NMR δ 1.39 (s, 9H), 2.37 (s, 3H), 3.55 (s, 3H), 3.78 (s, 3H), 7.14 (d, 2H, J = 7.8 Hz), 7.22 (d, 2H, J = 7.8 Hz), 7.45 (s, 1H); 13C NMR δ 21.4, 31.5, 51.3, 51.6, 59.0, 112.4, 118.2, 124.4, 128.3, 129.9, 131.9, 136.5, 138.6, 164.4, 165.9; HRMS (EI) m/z 329.1625 (M+, C19H23NO4 requires 329.1627).
Photoreaction of 8c
10 min irradiation (6% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 9c (7 mg, 3%); 90 min irradiation (73% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 9c (82 mg, 36%) and 10c (21 mg, 9%).
9c: 1H NMR δ 1.35 (s, 9H), 3.58 (s, 3H), 3.78 (s, 3H), 4.37 (s, 2H), 4.94 (s, 1H), 6.94–7.0 (m, 2H), 7.19–7.22 (m, 2H); 13C NMR δ 28.8, 50.8, 52.1, 52.8, 59.4, 91.9, 115.4 (d, JC–F = 21 Hz), 128.1 (d, JC–F = 8.3 Hz), 134.2 (d, JC–F = 3 Hz), 153.7, 161.8 (d, JC–F = 243.8 Hz), 167.1, 167.7; HRMS (EI) m/z 323.1530 (M+, C17H22FNO4 requires 323.1533).
10c: 1H NMR δ 1.39 (s, 9H), 3.55 (s, 3H), 3.79 (s, 3H), 7.01–7.07 (m, 2H), 7.3–7.34 (m, 2H), 7.46 (s, 1H); 13C NMR δ 31.5, 51.4, 51.6, 112.6, 114.7 (d, JC–F = 21.8 Hz), 118.6, 124.7, 129.0 (d, JC–F = 3.8 Hz), 133.9 (d, JC–F = 7.5 Hz), 135.1, 162.9 (d, JC–F = 247.5 Hz), 164.3, 165.6; HRMS (EI) m/z 333.1378 (M+, C18H20FNO4 requires 333.1376).
General procedure of C60-promoted photoreactions of non-silyl containing N-alkylbenzylamines with DMAD
Under the same reaction conditions employed above, the photoreactions of non-silyl containing each benzylamine 11–13 with DMAD were carried out. Then, photolysates were concentrated in vacuo and then the resulting mixture were separated by using silica gel column chromatographic isolation to give photoadducts.Photoreaction of 11 with DMAD
10 min irradiation (60% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2a (28 mg, 15%) and 14 (ref. 10) (29 mg, 22%); 20 min irradiation (100% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2a (52 mg, 28%) and 14 (55 mg, 42%).
Photoreaction of 12 with DMAD
. 10 min irradiation (35% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2c (28 mg, 14%) and 14 (12 mg, 9%); 20 min irradiation (79% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2c (69 mg, 35%) and 14 (28 mg, 21%).
14: 1H NMR δ 2.79 (s, 6H), 3.54 (s, 3H), 3.84 (s, 3H), 4.50 (s, 1H); 13C NMR δ 39.6, 50.5, 52.7, 84.2, 155.1, 165.9, 168.0; HRMS (EI) m/z 187.0845 (M+, C8H13NO4 requires 187.0845).
Photoreaction of 13 with DMAD
10 min irradiation (60% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 6a (31 mg, 16%) and 15 (21 mg, 15%); 20 min irradiation (100% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 6a (68 mg, 35%) and 15 (43 mg, 30%).
15: 1H NMR δ 1.09 (t, 3H, J = 7.2 Hz), 2.75 (s, 3H), 3.11 (q, d, J = 7.2 Hz), 3.55 (s, 3H), 3.86 (s, 3H), 4.51 (s, 1H); 13C NMR δ 36.6, 47.7, 50.6, 52.7, 83.6, 154.5, 166.0, 168.1; HRMS (EI) m/z 201.1002 (M+, C9H15NO4 requires 201.1001).
Visible light irradiated, C60-promoted photoreactions of N-α-trimethylsilyl-N-alkylbenzylamines 1a–1c with DMAD
The O2-purged toluene solutions (70 mL) containing each benzylamine 1a–1c (0.18 mmol, 1 equiv.), dimethyl acetylenedicarboxylate (0.35 mmol, 2 equiv.) and C60 (0.009 mmol, 0.05 equiv.) were simultaneously irradiated with a 20 W compact fluorescence lamp for fixed time periods (20, 40 and 60 h respectively). Then, photolysates were concentrated in vacuo and then the resulting mixture were separated by using silica gel column chromatographic isolation to give photoadducts.Photoreaction of 1a
20 h irradiation (47% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2a (15 mg, 32%); 40 h irradiation (100% conversion), column chromatography (EtOAc: hexane = 1:5) to yield 2a (32 mg, 70%).
Photoreaction of 1b
20 h irradiation (51% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2b (18 mg, 36%); 40 h irradiation (100% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2b (36 mg, 73%).
Photoreaction of 1c
20 h irradiation (20% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2b (5 mg, 10%); 40 h irradiation (48% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2c (14 mg, 28%); 60 h irradiation (80% conversion), column chromatography (EtOAc:hexane = 1:5) to yield 2c (23 mg, 45%).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was financially supported by Yeungnam University Research Grant (218A380049). The authors thanks the Core Research Center for Natural Products and Medical Materials (CRCNM) for technical support regarding NMR spectrometer.Notes and references
- (a) M. Joshi, M. Patel, R. Tiwari and A. K. Verma, J. Org. Chem., 2012, 77, 5633 CrossRef CAS; (b) T. J. Peelen, Y. Chi and S. H. Gellman, J. Am. Chem. Soc., 2005, 127, 11598 CrossRef CAS; (c) N. T. Patil, N. K. Pahadi and Y. Yamamoto, Tetrahedron Lett., 2005, 46, 2101 CrossRef CAS.
- (a) S. Mukherjee, L. W. Yang, S. Hoffman and B. List, Chem. Rev., 2007, 107, 5471 CrossRef CAS; (b) S. Kobayashi and H. Ishitani, Chem. Rev., 1999, 99, 1069 CrossRef CAS; (c) U. Kucklander, in The Chemistry of Enamines, ed. Z. Rappoport, Wiley-VCH, New York, 1994, pp. 523–636 Search PubMed; (d) K. D. Hesp and M. Stradiotto, J. Am. Chem. Soc., 2010, 132, 18026 CrossRef CAS.
- (a) M. Patel, R. K. Saunthwal and A. K. Verma, Acc. Chem. Res., 2017, 50, 240 CrossRef CAS; (b) B. Capon and Z.-P. Wu, J. Org. Chem., 1990, 55, 2317 CrossRef CAS; (c) Y.-Q. Zou, F. M. Hormann and T. Bach, Chem. Soc. Rev., 2018, 47, 278 RSC.
- (a) J. Xie, S. Zhu and Q. Zhou, Chem. Rev., 2011, 111, 1713 CrossRef CAS; (b) Z. Zhang, N. Butt and W. Zhang, Chem. Rev., 2016, 116, 14769 CrossRef CAS.
- (a) R. Severin and S. Doye, Chem. Soc. Rev., 2007, 36, 1407 RSC; (b) T. E. Muller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef.
- (a) F. Alonso, I. P. Beletskaya and M. Yus, Chem. Rev., 2004, 104, 3079 CrossRef CAS; (b) V. Lavallo, G. D. Frey, B. Donnadieu, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 5224 CrossRef CAS; (c) G. Kovas, A. Lledos and G. Ujaque, Angew. Chem., Int. Ed., 2011, 50, 11147 CrossRef; (d) R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2011, 50, 5560 CrossRef CAS.
- (a) M. J. Lopez-Gomez, D. Martin and G. Bertrand, Chem. Commun., 2013, 49, 4483 RSC; (b) E. Mizushima, T. Hayashi and M. Tanaka, Org. Lett., 2003, 5, 3350 CrossRef; (c) A. Leyva and A. Corma, Adv. Synth. Catal., 2009, 351, 2876 CrossRef CAS; (d) H. Duan, S. Sengupta, J. L. Petersen, N. G. Akhmedov and X. Shi, J. Am. Chem. Soc., 2009, 131, 12100 CrossRef CAS; (e) E. Alvarado, A. C. Badaj, T. G. Larocque and G. G. Lavoie, Chem.–Eur. J., 2012, 18, 12112 CrossRef CAS.
- (a) X. Zeng, G. D. Frey, S. Kousar and G. Bertrand, Chem.–Eur. J., 2009, 15, 3056 CrossRef CAS; (b) Y. Fukumoto, H. Asai, M. Shimizu and N. Chatani, J. Am. Chem. Soc., 2007, 129, 13792 CrossRef CAS; (c) C. Alonso-Moreno, F. Carrillo-Hermosilla, J. Romero-Fernandez, A. M. Rodriguez, A. Otero and A. Antinolo, Adv. Synth. Catal., 2009, 351, 881 CrossRef CAS; (d) G. V. Shanbhag, S. M. Kumbar, T. Joseph and S. B. Halligudi, Tetrahedron Lett., 2006, 47, 141 CrossRef CAS; (e) N. Pasha, N. S. Babu, K. T. V. Rao, P. S. S. Prasad and N. Lingaiah, Tetrahedron Lett., 2009, 50, 239 CrossRef CAS.
- (a) A. K. Verma, M. Joshi and V. P. Singh, Org. Lett., 2011, 13, 1630 CrossRef CAS; (b) M. Patel, R. K. Saunthawal and A. K. Verma, Tetrahedron Lett., 2014, 55, 1310 CrossRef CAS.
- S. H. Lim, A. B. Atar, G. Bae, K.-R. Wee and D. W. Cho, RSC Adv., 2019, 9, 5639 RSC.
- E. Baciocchi, T. D. Giacco and A. Lapi, Org. Lett., 2006, 8, 1783 CrossRef CAS.
- (a) D. Dubois, K. M. Kadish, S. Flanagan, R. E. Haufler, L. P. F. Chibante and L. J. Wilson, J. Am. Chem. Soc., 1991, 113, 4364 CrossRef CAS; (b) D. Dubois, K. M. Kadish, S. Flanagan and L. J. Wilson, J. Am. Chem. Soc., 1991, 113, 7773 CrossRef CAS; (c) J. W. Arbogast, C. S. Foote and M. Kao, J. Am. Chem. Soc., 1992, 114, 2277 CrossRef CAS.
- (a) J. W. Arbogast, A. O. Darmanyan, C. S. Foote, Y. Rubin, F. N. Diederich, M. M. Alvarez, S. J. Anz and R. L. Whetten, J. Phys. Chem., 1991, 95, 11 CrossRef CAS; (b) M. Terazima, N. Hirota, H. Shinohara and Y. Saito, J. Phys. Chem., 1991, 95, 9080 CrossRef CAS.
- (a) S. H. Lim, D. W. Cho and P. S. Mariano, Tetrahedron, 2018, 74, 383 CrossRef CAS; (b) S. H. Lim, J. Yi, G. M. Moon, C. S. Ra, K. Nahm, D. W. Cho, K. Kim, T. G. Hyung, U. C. Yoon, G. Y. Lee, S. Kim, J. Kim and P. S. Mariano, J. Org. Chem., 2014, 79, 6946 CrossRef CAS.
- (a) H. C. Jeong, S. H. Lim, Y. Sohn, Y.-I. Kim, H. Jang, D. W. Cho and P. S. Mariano, Tetrahedron Lett., 2017, 58, 949 CrossRef CAS; (b) S. H. Lim, H. C. Jeong, Y. Sohn, Y.-I. Kim, D. W. Cho, H.-J. Woo, I.-S. Shin, U. C. Yoon and P. S. Mariano, J. Org. Chem., 2016, 81, 2460 CrossRef CAS.
- (a) Y.-Q. Zou, L.-Q. Lu, L. Fu, N.-J. Chang, J. Rong, J.-R. Chen and W.-J. Xiao, Angew. Chem., Int. Ed., 2011, 50, 7171 CrossRef CAS; (b) M. Rueping, D. Leonori and T. Poisson, Chem. Commun., 2011, 47, 9615 RSC.
- (a) A. Fujiya, M. Tanaka, E. Yamaguchi, N. Tada and A. Itoh, J. Org. Chem., 2016, 81, 7262 CrossRef CAS; (b) L. Shi and W. Xia, Chem. Soc. Rev., 2012, 41, 7687 RSC; (c) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–5648 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00166c |
| This journal is © The Royal Society of Chemistry 2021 |