
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Endothelialization of an ePTFE vessel prosthesis modified with an antithrombogenic fibrin/heparin coating enriched with bound growth factors†
Johanka Táborská a,
Zuzana Riedelováa,
Eduard Bryndaa,
Pavel Májekb and
Tomáš Riedel*a
a,
Zuzana Riedelováa,
Eduard Bryndaa,
Pavel Májekb and
Tomáš Riedel*a
aInstitute of Macromolecular Chemistry, Czech Academy of Sciences, Heyrovského Náměstí 2, 162 06 Prague 6, Czech Republic. E-mail: riedel@imc.cas.cz
bInstitute of Hematology and Blood Transfusion, U Nemocnice 1, 128 00 Prague 2, Czech Republic
First published on 3rd February 2021
Abstract
Early and late thrombosis remain the most frequent reasons for the failure of synthetic cardiovascular grafts. Long-term hemocompatibility of implanted synthetic grafts can be achieved if a natural living endothelium is formed over its blood-contacting surface. Here we present a modification of a standard expanded polytetrafluorethylene (ePTFE) vessel prosthesis by a controlled preparation of a fibrin mesh enriched with covalently bound heparin and noncovalently bound vascular endothelial growth factor (VEGF) and fibroblast growth factor (FGF). Compared to a bare prosthesis, the coated prosthesis showed excellent antithrombogenic properties after contact with heparinized fresh human blood. Human umbilical vein endothelial cells seeded on the inner surface of the coated prosthesis formed a confluent layer in 5 days, whereas only small colonies of cells were scattered on the bare prosthesis. Viability of the cells was promoted mainly by FGF immobilized on the coating. These findings suggest that the coating may prevent acute thrombus formation and support the self-endothelialization of an implanted ePTFE vascular graft in vivo.
Introduction
Cardiovascular diseases are the leading cause of death worldwide. Blood flow restriction due to the narrowing of the vessel lumen or vessel blockage leads to ischemia, which results in life-threatening conditions. To replace or bypass a damaged vessel, vascular grafts (VGs) are used. Currently, the best conduits for vascular grafting are autologous arteries or veins, typically the saphenous veins, internal mammary arteries, or radial arteries.1,2 Nevertheless, they have several drawbacks: limited availability; donor site morbidity and patient burden due to the requirement for additional surgery. As a result, synthetic polymer VGs made from expanded polytetrafluoroethylene (ePTFE), polyethylene terephthalate (PET, Dacron), and polyurethane (PU) are routinely used in current clinical practice as an alternative to autologous vessels.3 However, all synthetic grafts suffer from occasional acute, subacute, or late thrombotic complications. The risk of rapid thrombotic occlusion limits the application of synthetic VGs of less than 4 mm in diameter.4,5 The formation of a healthy natural endothelium over the luminal surface of an implanted VG seems to be a key step in reaching long-term hemocompatibility of synthetic VGs.6,7 The inability to form an endothelial lining leads to high failure rates in small and medium-sized synthetic VGs.4,5The endothelialization of the implanted synthetic VG's luminal surface includes the migration of vascular endothelial cells (ECs) from the anastomotic area (transanastomotic), ingrowth of ECs and microvessels from the perigraft tissue if the graft contains sufficiently large pores (transmural), and the adherence of circulating endothelial progenitor cells (EPCs) from peripheral blood (blood-borne). However, none of these processes are capable of spontaneously developing a neo-intima on VGs in humans, except for sporadically observed small islands of ECs.8 In vitro pre-seeding of VGs with autologous ECs may help the endothelialization after surgery. Nevertheless, the surgical harvesting of ECs from the patient, the in vitro ECs expansion and cultivation on the graft are time consuming and burdened with the risk of infection.8,9
On the other hand, in situ endothelialization is a promising method based on a graft modification that uses a coating which accelerates the self-endothelialization after the graft implantation while suppressing thrombosis until a functional neointima is formed.9,10 A design of the graft modification might be inspired by natural wound healing processes that lead to the regeneration of the endothelium after vascular injury. A blood clot, which is composed of a crosslinked fibrin mesh containing activated and aggregated platelets, erythrocytes, and leukocytes, is formed at sites of endothelium damage to stop bleeding and later serves as a temporary scaffold for ECs migrating from the neighbouring intact endothelium and for EPCs captured from peripheral blood.11–13 In addition, the repair of injured blood vessels is stimulated by various growth factors and cytokines released from activated platelets and leukocytes trapped in the fibrin mesh. The endothelium regeneration is particularly accelerated by vascular endothelial growth factor (VEGF) and fibroblast growth factors (FGF) accumulated in the blood clot by the attachment to specific binding sites in the fibrin structure.14,15 VEGF supports also adhesion, proliferation, differentiation and the maturation of various cells including EPCs and FGF supports the proliferation of smooth muscle cells and extracellular matrix remodelling in the injured vessel wall.16–19
Artificial fibrin meshes have been widely used for the modification of VGs as biodegradable substrates supporting adhesion and migration of ECs and EPCs.20–24 A significant improvement in the patency of ePTFE vascular grafts was observed when they were coated with a fibrin glue and endothelialized in vitro by the seeding of autologous endothelial cells before implantation.25 Fibrin glue is prepared by mixing a highly concentrated fibrinogen (Fbg) solution containing factor XIII with a thrombin solution immediately before use. In order to promote the in situ endothelialization, synthetic polymer VGs were modified with fibrin containing immobilized FGF and/or VEGF. A significant increase in luminal ECs proliferation and transmural capillary ingrowth was observed in ePTFE grafts modified by fibrin glue containing FGF factors in canine aortic models.26 On the other hand, an ePTFE graft modified with fibrin and genetically engineered recombinant VEGF showed effects deleterious to graft healing in a pig carotid artery interposition model, by increasing narrowing at the proximal anastomosis and neointimal hyperplasia, and by increased platelet deposition and thrombus formation in extracorporeal arteriovenous shunt perfusion.27
To reduce thrombogenicity in synthetic polymer VGs, the grafts are usually modified with immobilized anticoagulant heparin. ePTFE vascular grafts with covalently bound heparin significantly reduced platelet adhesion and thrombus formation both in vitro and in vivo.28 We have previously shown that a covalent binding of periodate activated heparin to a fibrin mesh prepared on PVC sheets prevented the formation of a blood clot on the sheets exposed to human blood in a Chandler loop model.29
The present work aims to develop a surface modification that might accelerate the in vitro and in situ endothelialization of cardiovascular grafts while suppressing acute thrombosis. The modification of standard ePTFE grafts with a fibrin mesh decorated by the covalently bound heparin and noncovalently bound FGF and VEGF presented in this work fulfils both requirements. The modification includes the controlled formation of a fibrin mesh (Fb) at a substrate surface using adapted techniques that we developed earlier29–31 followed by the covalent binding of chemically activated heparin to Fb and finally by the attachment of VEGF and FGF via their specific noncovalent binding to fibrin14,32 and heparin.33,34 The controlled formation of the fibrin mesh consists of three successive processes: (1) fibrinogen adsorption on the substrate, (2) thrombin attachment to the adsorbed fibrinogen monolayer, and (3) fibrin formation by the catalytic action of the bound thrombin on the fibrinogen in an ambient solution. Unlike the conventional mixing of fibrinogen solution with thrombin (e.g., fibrin glue), which leads to the spontaneous formation of bulky fibrin meshes and glues, the independent steps taken in our procedure make it possible to reproducibly control the structure and the thickness of the fibrin mesh formed at the surface of a substrate (including a surface of inner pores). The covalent attachment of heparin to the fibrin suppressed the blood clot formation on the prosthesis surface exposed to human blood. The further noncovalent binding of FGF and VEGF to the fibrin mesh did not affect the antithrombogenic properties of the prosthesis and accelerated the in vitro formation of a confluent monolayer from human umbilical vein endothelial cells (HUVECs) seeded on the luminal prosthesis surface.
Materials and methods
Materials and reagents
Fibrinogen (Fbg) and thrombin (Thr) from human plasma, heparin sodium salt from porcine intestinal mucosa, plasmin, Phalloidin – Atto 594, TWEEN® 20, Triton™ X-100, paraformaldehyde, bovine serum albumin (BSA), NaIO4, NaOH, NaCl, CaCl2, KCl, Na2HPO4·12H2O, KH2PO4, citric acid monohydrate, sodium citrate – tribasic dehydrate, Trizma© base, agarose, and glutaraldehyde were purchased from Sigma-Aldrich. Antithrombin III, and COATEST© Heparin KIT were purchased from Chromogenix. Human fibroblast growth factor-basic 154aa (FGF) and human vascular endothelial growth factor 165 (VEGF) were purchased from GenScript USA, Inc. Cell Counting Kit-8 (CCK-8 assay) was purchased from Dojindo. Blotting-Grade Blocker was purchased from Bio-Rad. Hoechst 33342, the VEGF Human ELISA Kit (KHG0111), the FGF-2 Human ELISA Kit (KHG0021), the Micro BCATM Protein Assay Kit, Fibrinogen Polyclonal Antibody (PA1-9526), and Goat anti-Chicken IgY Alexa Fluor 488 (A11039) were purchased from Thermo Fisher Scientific. Amicon Ultra-4 centrifugal filter devices were purchased from Merck Millipore. 24- and 96- glass bottom well plates were purchased from BIO-PORT Europe. Phosphate-buffered saline pH 7.4 (PBS), Tris–HCl buffer 0.05 M with 2 mM CaCl2 pH 7.4 (TB), and 0.01 M citrate buffer pH 4.0 (CB) were filtered through a Millipore 0.22 μm filter. Human umbilical vein endothelial cells (HUVECs) were purchased from Promocell and maintained in endothelial cell growth medium (EGM-2, Lonza). Expanded polytetrafluorethylene vascular prostheses (ePTFE vessel) were kindly provided by B. Braun Medical.Fibrin (Fb) and fibrin with covalently bound heparin (Fb/Hep) coatings
Characterization of coatings
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000) for 2 h in a 1% solution of Blotting-Grade Blocker (non-fat dry milk) in PBS-T (PBS + 0.02% Tween 20) and subsequent incubation with Goat anti-Chicken IgY Alexa Fluor 488 (1:4000) in PBS-T containing 1% Blotting-Grade Blocker for 1 h at RT. To visualize fibrin inside the ePTFE vessel wall, a flat sample of the Fb coated ePTFE vessel was fixed in 4% agarose gel and slices (80 μm thickness) were cut from the sample by a Vibratome Leica VT1200. The cut samples were subjected to immunofluorescence staining by incubation with a fibrinogen antibody (1:2000) for 72 h in a 1% solution of Blotting-Grade Blocker in PBS-T at 4 °C followed by incubation with Goat anti-Chicken IgY Alexa Fluor 488 antibody (1:4000) in PBS-T containing 1% Blotting-Grade Blocker for 2 h. Stained samples were observed using an Olympus IX83 confocal microscope.
:2000) for 2 h in a 1% solution of Blotting-Grade Blocker (non-fat dry milk) in PBS-T (PBS + 0.02% Tween 20) and subsequent incubation with Goat anti-Chicken IgY Alexa Fluor 488 (1:4000) in PBS-T containing 1% Blotting-Grade Blocker for 1 h at RT. To visualize fibrin inside the ePTFE vessel wall, a flat sample of the Fb coated ePTFE vessel was fixed in 4% agarose gel and slices (80 μm thickness) were cut from the sample by a Vibratome Leica VT1200. The cut samples were subjected to immunofluorescence staining by incubation with a fibrinogen antibody (1:2000) for 72 h in a 1% solution of Blotting-Grade Blocker in PBS-T at 4 °C followed by incubation with Goat anti-Chicken IgY Alexa Fluor 488 antibody (1:4000) in PBS-T containing 1% Blotting-Grade Blocker for 2 h. Stained samples were observed using an Olympus IX83 confocal microscope.GFs binding to and release from Fb/Hep coatings
Hemocompatibility of coatings
Viability and growth of HUVECs seeded on coated substrates
000 cells per cm2 in EGM-2 depleted from FGF and VEGF and supplemented with 1% FBS (Medium 2).:100) for F-actin and Hoechst (5 μg ml−1) for nuclei and observed under an Olympus IX83 confocal microscope.Statistical analysis
The quantitative data are presented as the mean with standard deviation (±S.D). The Student's one-tailed t-test was performed for statistical comparisons of cell viability results, and a value of p < 0.05 was considered statistically significant.Results and discussion
Characterization of Fb/Hep coating
Fibrin coating was grown on the surface of a glass bottom well in a 24-well glass bottom plate or ePTFE vessel prosthesis. The procedure is based on growing a controlled fibrin mesh on a material surface via the catalytic effect of a fibrin(ogen)-bound thrombin and the ability of antithrombin III to inhibit thrombin in solution but not fibrin(ogen)-bound thrombin.30,35The Scheme 1 illustrates successive modification of a substrate by a fibrin mesh (Fb), covalent binding of heparin (Fb/Hep) and noncovalent binding of GFs (Fb/Hep + VEGF, Fb/Hep + FGF, and Fb/Hep + FGF + VEGF). Fig. 1 shows an AFM image of a dried Fb coating on a glass surface (Fig. 1A) with a uniform fibrin mesh. The average thickness of the dried fibrin mesh measured by AFM was 40 ± 10 nm. The surface density of fibrin in the Fb coating on glass was 4.0 ± 0.7 μg cm−2 (evaluated by BCA assay). The activity of heparin covalently conjugated to the fibrin coating on glass (Fb/Hep) was 0.027 ± 0.02 U cm−2 as determined by an anti-FXa activity assay.
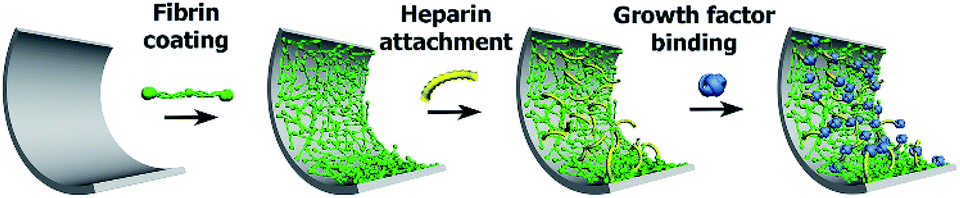 | ||
| Scheme 1 The scheme represents the preparation of Fb coating and its modification with heparin and subsequent attachment of growth factors. | ||
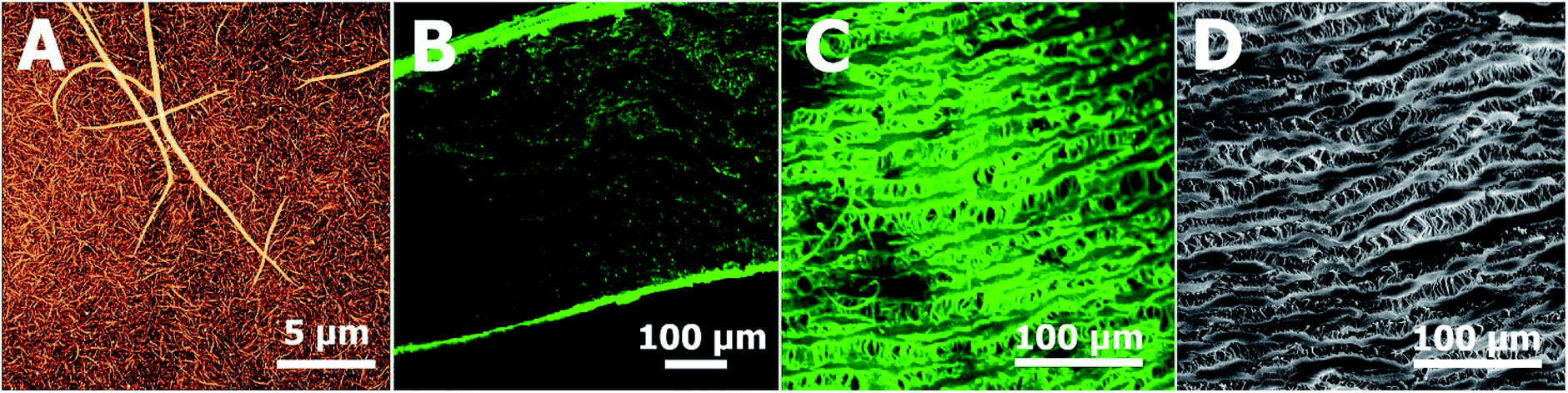 | ||
| Fig. 1 (A) Morphology of fibrin coating on glass observed using atomic force microscope. (B) Fibrin in a cross section of the coated ePTFE vessel wall (confocal microscope). (C) Fibrin on the inner surface of the coated ePTFE vessel wall (confocal microscope). (D) The inner surface of the uncoated ePTFE vessel wall (SEM). Fibrin was immunofluorescence stained with Alexa Fluor 488 (green). | ||
The confocal microscopic images of the surface and cross section of the Fb modified ePTFE vessel show both fibrin inside the vessel wall as well as fibrin homogenously decorating the surface of the ePTFE vessel (Fig. 1B and C), that copies the ePTFE morphology of inner surface on the unmodified prosthesis (Fig. 1D). SEM images in the ESI† show a homogeneous formation of the Fb localized at the surface of the ePTFE vessel prosthesis using the coating technology (Fig. S1†). The surface density of fibrin in the Fb/Hep coated ePTFE vessel was 40 ± 8 μg cm−2 and the heparin activity was 0.5 ± 0.1 U cm−2.
Several other coating techniques have been developed to cover ePTFE vascular grafts with fibrin, such as the incubation of a graft with fibrin and thrombin substances,27 the gelation of fibrin glue mixtures on ePTFE grafts in a vacuum,36 or the gelation of a fibrin–thrombin mixture under pressure in occluded grafts with the additional removal of excess fibrin on a surface.37,38 Unlike other techniques, our method makes it possible to reproducibly coat various types of surfaces, including the inner surface of porous materials, with fibrin meshes of thickness adjustable from nanometre to micrometre scale without the formation of a bulk fibrin gel.30,31
Binding of VEGF and FGF to Fb/Hep coatings
Amounts of VEGF or FGF attached to the Fb/Hep coatings prepared on the glass substrates are shown in Fig. 2A. The graph shows that the individual growth factors were attached to the Fb/Hep coatings in a dose-dependent manner. Furthermore, the amounts of the loaded VEGF were considerably higher than those of FGF (Fig. 2A). VEGF and FGF could bind to the Fb/Hep both via specific binding centers in fibrin and via specific interaction with immobilized heparin.32–34 | ||
| Fig. 2 (A) Dependence of the amount of VEGF (gray columns) and FGF (white columns) attached to Fb/Hep coatings on the concentration of the loaded protein in solution. (B) Fractions of VEGF (gray columns) and FGF (white columns) remaining in Fb/Hep + VEGF100 and Fb/Hep + FGF100 coatings, after 7 and 14 days in PBS. The GFs were loaded from 100 ng ml−1 solutions. The error bars represent standard deviation of the mean (n = 3). | ||
The amounts of bound FGF and VEGF did not depend on whether these were bound individually or simultaneously. The Fb/Hep + FGF100 + VEGF100 coating contained 972 ± 160 pg cm−2 of VEGF and 52 ± 4 pg cm−2 of FGF. These values did not significantly differ from the amounts of the individually attached VEGF (815 ± 378 pg cm−2) and FGF (67 ± 14 pg cm−2) loaded from the same concentration (100 ng ml−1), indicating that the binding of the GFs is independent. Indeed, it has been reported that binding of VEGF to fibrin is independent of FGF.14
The release of VEGF and FGF from Fb/Hep coatings
Release experiments were performed on the Fb/Hep coating loaded from 100 ng ml−1 solutions of VEGF or FGF. The ability of Fb/Hep coatings to retain GFs is depicted in Fig. 2B. After 7 days of incubation in PBS, 33 ± 3% of VEGF and 50 ± 3% of FGF remained bound in the Fb/Hep + VEGF100 and Fb/Hep + FGF100 coating, respectively. We did not observe any significant changes between 7 days and 14 days of incubation in PBS. After 14 days, 34 ± 6% of VEGF and 51 ± 8% of FGF remained bound in Fb/Hep + VEGF100 and Fb/Hep + FGF100, respectively. In addition, we have also measured the amount of released GFs during two weeks incubation in PBS (Fig. S2†). After a fast release during the first 24 h only a minimal release was detected for the next 14 days. The determined released amount is in line with the amount of GFs remaining on the Fb/Hep coatings after 14 days (Fig. 2B). The growth factors may be strongly immobilized by specific noncovalent binding to fibrin14,32 and heparin.33,34 In contrast to the long-term stability of covalent attachment of heparin in the Fb/Hep coatings incubated with PBS,29 some of the specifically bound or only nonspecifically adsorbed growth factors are only loosely bound in the coating.Hemocompatibility of the coatings
Hemocompatibility of the coatings was tested by contact with fresh human blood and by a plasma recalcification test (PRT). The prolongation of the recalcified plasma clotting time was measured to evaluate the coating's ability to resist the activation of the clotting cascade initiated when a biomaterial comes into contact with circulating blood.39 The activation of coagulation of freshly recalcified plasma was indicated by the increase in plasma turbidity on the coated glass surfaces in comparison with the uncoated glass. PRT of 16 ± 1 min on the unmodified glass was prolonged by the Fb coating by up to 20 ± 2 min. The attachment of heparin in Fb/Hep did not activate coagulation within 60 min when the test was terminated. Importantly, the binding of VEGF and FGF to the Fb/Hep coatings (Fb/Hep + VEGF100 and Fb/Hep + FGF100, respectively) also did not activate the plasma coagulation within 60 min when the test was terminated (Fig. 3B). This observation clearly indicates that heparin after binding of GFs remains active and is able to suppress blood plasma activation.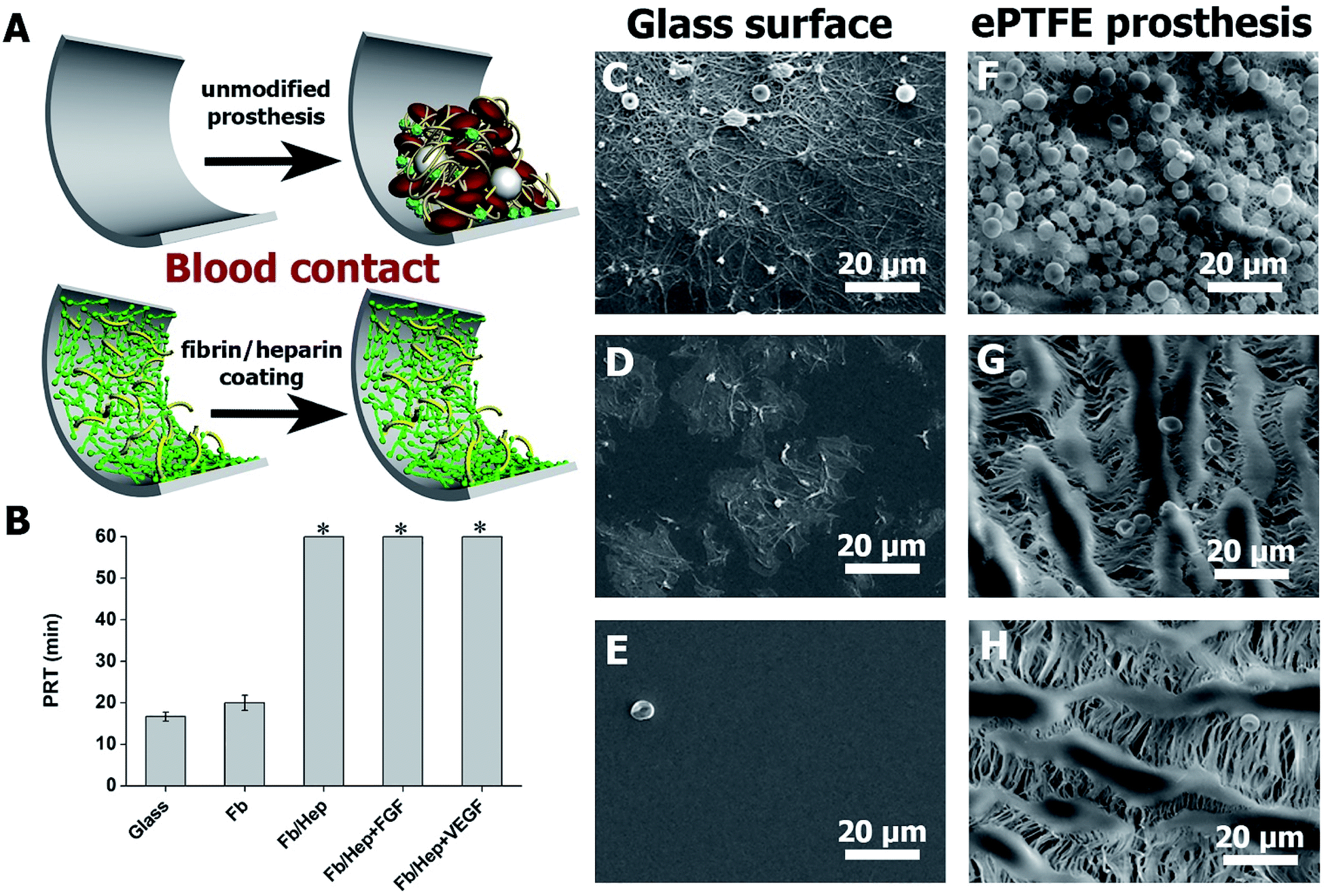 | ||
| Fig. 3 (A) Scheme showing a comparison of unmodified and fibrin/heparin modified prosthesis after blood contact. (B) Graph showing plasma recalcification times (PRT) after contact with various coatings on glass with human blood plasma recalcified with CaCl2 (0.02 M final concentration; mean ± S.D.; n = 3). * – an asterisk indicates samples where the plasma did not coagulate within 60 min. (C–E) SEM images of unmodified glass (C), Fb coated glass (D), and Fb/Hep coated glass(E), (F–H) SEM images of unmodified ePTFE vessels (F), Fb/Hep coated (G), Fb/Hep + FGF100 + VEGF100 coated (H) after 1 h incubation with fresh heparinized human blood at 37 °C. | ||
Additionally, in order to mimic initial thrombogenic processes activated after the vascular prosthesis implantation, the coated glass and ePTFE vessel samples were exposed to freshly drawn heparinized human blood and the resulting blood clot was analysed by SEM. Fig. 3C–E shows representative SEM images of bare and coated glass surfaces after their exposure to heparinized blood. Fig. 3C displays a blood clot composed of platelet aggregates, leukocytes, and red blood cells entrapped in a fibrin mesh on the bare glass. Fig. 3D shows patches of spread activated platelets, some platelet aggregates and nearly no leukocytes and red blood cells on the Fb coated glass. In contrast, no blood clot deposits were formed on the Fb/Hep coated glass (Fig. 3E). The images correlate with PRT data in Fig. 3A and indicate low thrombogenicity of the Fb/Hep surface modification. Fig. 3F–H shows SEM images of the bare ePTFE and the Fb/Hep and Fb/Hep + FGF100 + VEGF100 coated ePTFE vessel wall in contact with heparinized blood.
The images display an initial thrombus with platelet aggregates and many entrapped leukocytes and red blood cells on the unmodified ePTFE vessel (Fig. 3F) and only sparsely scattered red blood cells but no platelets or leukocytes on the inner side of the ePTFE vessel coated with Fb/Hep (Fig. 3G) and Fb/Hep + FGF100 + VEGF100 (Fig. 3H). Sparsely scattered red blood cells were also observed on the ePTFE vessels modified with the Fb/Hep + FGF100 and Fb/Hep + VEGF100 coatings containing immobilized heparin (Fig. S3†).
We have previously reported on the significantly improved in vitro hemocompatibility of PVC substrates29 and neurovascular stents40 that had been modified with Fb/Hep coatings by a similar technique as applied here. In contact with heparinized blood in a Chandler loop or a flow loop model mimicking human blood flow, the coatings suppressed thrombin generation, platelets and leukocyte activation, complement activation and blood clot formation.29,40 The results presented herein indicate that the binding of GFs preserve the excellent hemocompatibility of the Fb/Hep coating.
Viability of HUVECs on Fb coatings
The viability of HUVECs on days 3 and 7 after the seeding on the coated glass was characterized by measuring the cell metabolic activity. The results are presented in Fig. 4. The glass bottoms of wells were coated with Fb/Hep and loaded with FGF or/and VEGF at various concentrations (10 ng ml−1 to 10 μg ml−1, see Methods). Both Fb and Fb/Hep coating promoted the HUVEC viability compared to bare glass. This is in line with previously reported data that surface-attached fibrin improved adhesion and differentiation of endothelial cells.41 Moreover, a similar increase in HUVECs viability was obtained for the Fb or Fb/Hep coating thus indicating that bound heparin did not interfere with cell proliferation. A significant increase in the cell viability was reached if Fb/Hep coatings were loaded with FGF or VEGF from solutions containing the GFs at concentrations of 10 μg, 1 μg, or 100 ng per 1 ml (Fig. 4A). Relatively small differences in the cell viability on coatings loaded from these solutions contrasted with the huge differences in the initial amounts of VEGF or FGF bound in the respective coatings (Fig. 2A).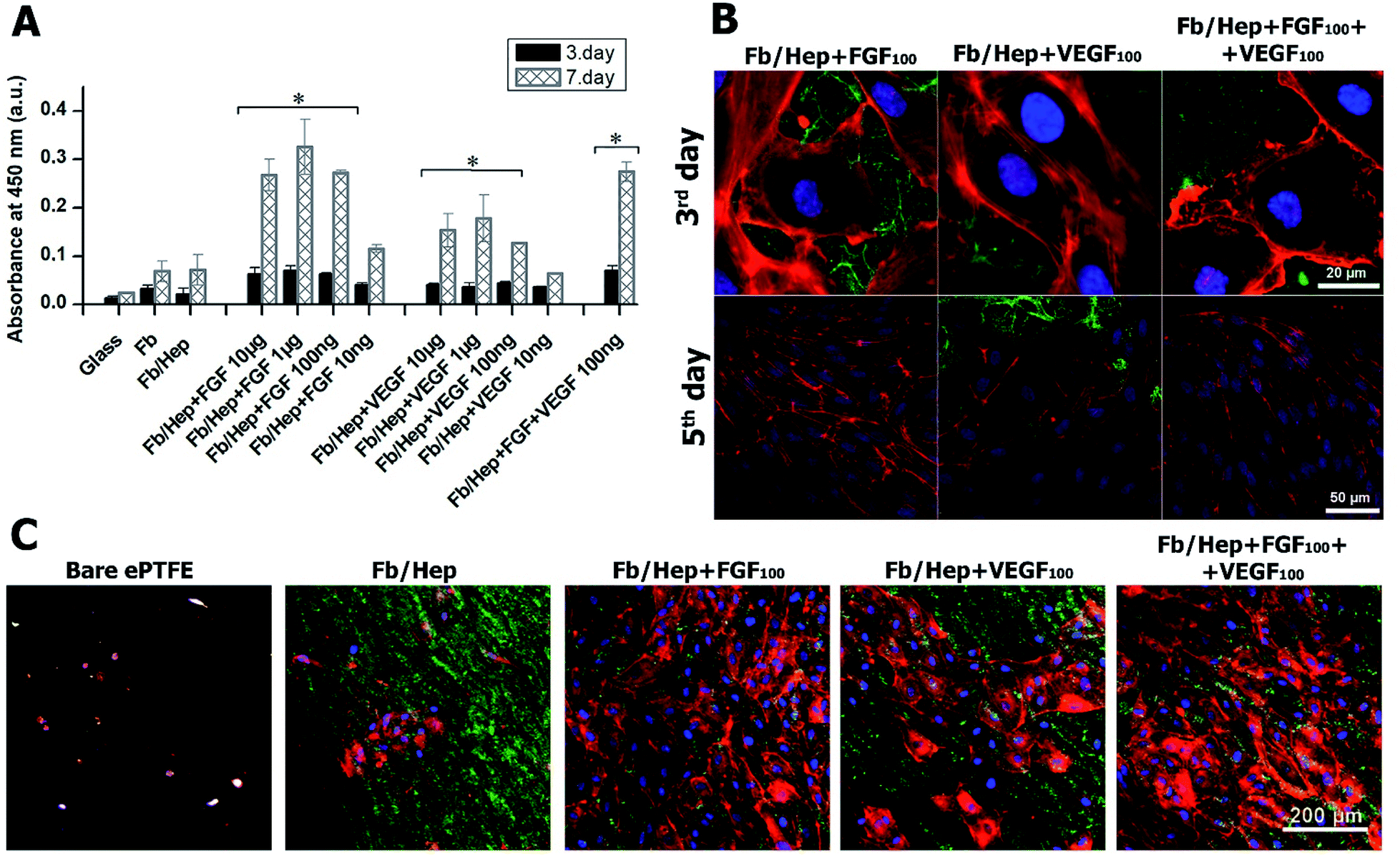 | ||
| Fig. 4 (A) The viability of HUVECs on days 3 and 7 after seeding on glass coated with Fb/Hep and modified by the attachment of growth factors from solutions containing various concentrations of FGF, VEGF, and a co-mixture of FGF (100 ng ml−1) and VEGF (100 ng ml−1). The data were obtained from four independent experiments, mean ± S.D, Student t-test (*p < 0.05). Representative confocal microscopy images of (B) HUVECs on day 3 and day 5 after seeding on the coated glass and (C) HUVECs on day 5 after seeding on the inner surface of the modified ePTFE vessel wall. Fb/Hep + FGF100, Fb/Hep + VEGF100, and Fb/Hep + FGF100 + VEGF100 were prepared by binding the GFs from solutions containing 100 ng ml−1 of the respective GF. The cells were stained with phalloidin for F-actin filaments (red) and with Hoechst for nuclei (blue); fibrin was immunofluorescence stained with Alexa Fluor 488 (green). | ||
Therefore, the Fb/Hep coatings loaded with the GFs from 100 ng ml−1 solutions (referred as Fb/Hep + VEGF100; Fb/Hep + FGF100; and Fb/Hep + VEGF100 + FGF100) were selected for further cell experiments because of their blood compatibility (Fig. 3) and the sufficient viability of HUVECs seeded on their surfaces (Fig. 4A). The release of GFs from the coatings (Fig. 2B, S2†) indicates that a considerable increase in the cell viability between day 3 and 7 may be due to the activity of a fraction of the GFs remaining immobilized in the coatings after the initial fast release. Remarkably, FGF promoted almost twice as much cell viability than VEGF, even if the initial FGF amounts in the coatings were considerably lower (Fig. 2A), e.g. Fb/Hep + FGF100 contained 67 pg cm−2 of FGF, while Fb/Hep + VEGF100 contained 815 pg cm−2 of VEGF. In accordance with our previous report, we did not observe any significant differences in cell viability between the Fb and Fb/Hep coating, indicating that the immobilized heparin did not have any undesirable effect on cell growth.25
Morphology of HUVECs on fibrin coatings
 | ||
| Fig. 5 Degradation of fibrin coating observed by confocal microscopy on day 5 after seeding HUVECs on coated glass (A) and on an ePTFE vessel (B). Upper row: (A) fibrin visualized on bare glass and glass coated with Fb, Fb/Hep, Fb/Hep + FGF100 + VEGF100 prepared by the attachment of the GFs from a solution containing 100 ng ml−1 FGF and 100 ng ml−1 VEGF. (B) Fibrin visualized in a cross section of an ePTFE vessel wall modified by Fb/Hep + FGF100 + VEGF100. Lower row: (A) merged image of fibrin and HUVECs visualized on bare and coated glass; (B) and on the inner surface of the ePTFE vessel wall. Fibrin was immunolabelled with Alexa Fluor 488 (green), and HUVECs were stained with phalloidin for actin (red) and with Hoechst for nuclei (blue). | ||
VEGF and FGF have been widely studied due to the essential role of growth factors in wound healing, angiogenesis and tissue engineering.43–45 The importance of FGF for HUVECs viability on the coated glass (Fig. 4A) and HUVECs growth on the ePTFE vessels (Fig. 4C) may correlate with the higher potency of FGF compared to VEGF to stimulate microvascular endothelial cells and HUVECs to form endothelial vessel-like tubes in a collagen matrix.44 The capacity of FGF to stimulate ECs proliferation in our experiments was probably further enhanced by the FGF attachment to fibrin.45 Contrary to the potent synergism between VEGF and FGF in the induction of the angiogenesis both in vitro,43,44 and in vivo,45 we did not find significant differences in the viability and growth of HUVECs seeded on Fb/Hep + FGF100 and on Fb/Hep + FGF100 + VEGF100. The VEGF effect on angiogenesis is associated with increased vascular permeability in endothelial cells.46 On the other hand, the presence of bound VEGF in Fb/Hep + FGF100 + VEGF100 coating may be important for capturing EPCs from peripheral blood, their homing and further differentiation into ECs.17,47 Recently it has been reported that immobilized VEGF also captures blood circulating monocytes and initiates their differentiation into a mixed endothelial (EC) and macrophage phenotype that further develops into mature EC in vivo.48
Moreover, ECs pre-seeded on fibrin-coated ePTFE grafts exhibited better resistance to shear stress than the control.38,49 Similarly, the coating of PET vascular grafts with fibrin using the technique described herein significantly increased the resistance of ECs seeded on the grafts to shear stress.50
Although the complete degradation of the Fb/Hep + FGF100 + VEGF100 coating below HUVECs was revealed on glass, some fibrin was still visible inside and on the surface of the ePTFE vessel prosthesis 5 days after seeding with HUVEC. The image of a cross section of the ePTFE vessel wall modified by Fb/Hep + FGF100 + VEGF100 (Fig. 5B, upper row) shows fibrin remaining inside the vessel wall and in a denser structure at the wall surface. An almost confluent layer of HUVECs with traces of fibrin was visible on the inner ePTFE vessel wall surface (Fig. 5B, lower row) and no cells were observed inside the wall. Evidently, HUVECs could not penetrate through a dense structure formed by PTFE nodes and fibrils at the inner wall surface (Fig. 1D).
The findings indicated that fibrinolysis occurred mainly in a close vicinity of the proliferating HUVECs. Such a fibrinolytic system contributes to the degradation of blood clots formed to stop bleeding after injury. During the process fibrin is lysed by serine protease plasmin. Plasmin is produced by the conversion of inactive zymogen plasminogen from circulating blood. The conversion is performed by tissue plasminogen activator (tPA) attachment of plasminogen and tPA to fibrin on the cell surfaces.51,52 In our in vitro experiments, fibrin in contact with proliferating HUVECs could have been cleaved by plasmin produced by the conversion of plasminogen from 1% serum contained in the culture medium. Alternatively, fibrin may be degraded by extracellular zinc-dependent endopeptidases responsible for the remodelling of the extracellular matrix, including fibrin (matrix metalloproteinases) secreted by HUVECs or that present in the serum.53–55 Additionally, the fibrinolysis may be potentially activated by VEGF stimulating the abluminal VEGFRs receptor of ECs.54 Regrettably, our experiments did not allow us to evaluate the decrease in the amount of fibrin under HUVECs growing on the inner surface of the ePTFE vessel wall as opposed to that on the coated glass. Nevertheless, we expect that a gradual degradation of the fibrin remaining inside the vessel wall might occur by a slow diffusion of fibrinolytic proteases from circulating blood and expressed by the cells growing on the ePTFE wall surface.
Therefore, covalently bound heparin and strongly noncovalently attached growth factors may be released from the coating remaining in the ePTFE vessel and prolong the capacity of the vessel to support endothelialization and suppress the thrombus formation after the implantation.14,56
Conclusions
In this study, we presented a new technique for surface modification of vascular prostheses that is based on the controlled growing of a fibrin mesh from fibrinogen solutions catalysed by thrombin immobilized on a substrate and the subsequent covalent binding of heparin and noncovalent binding of growth factors FGF and VEGF. This approach allows to coat reproducibly the surface and interior of a standard ePTFE vessel prosthesis with a controlled fibrin coating without the formation of a bulk fibrin gel. The coating completely prevents blood clot formation on the inner surface of the prosthesis exposed to heparinized fresh human blood in vitro and accelerates the creation of a monolayer of HUVECs seeded on the surface. Viability of the cells is promoted mainly by FGF immobilized in the Fb/Hep + FGF and Fb/Hep + FGF + VEGF coatings. The cells do not penetrate the ePTFE vessel wall and can degrade the coating of the wall surface by fibrinolytic processes localized at their surfaces. The coating remaining inside the vessel wall has probably been only slowly degraded by fibrinolytic agents diffusing from the wall surface. The obtained findings suggest that the developed coating, which is generally applicable to the modifications of various cardiovascular grafts, may prevent acute thrombus formation and support self-endothelialization of implanted ePTFE vessel prostheses and other grafts in situ. Additionally, the release of heparin and growth factors from the vessel wall may prevent thrombosis after the implantation of prostheses pre-seeded in vitro with autologous ECs and may support the transformation of the in vitro formed ECs layer to a functional endothelium.Author contributions
JT prepared the samples, performed cell experiments and wrote the manuscript. ZR designed and wrote the manuscript. EB wrote the manuscript. PM performed the hemocompatibility tests. TR performed the AFM and SEM measurements, performed the hemocompatibility tests, and designed and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Czech Science Foundation (project no. 18-01163S), the National Sustainability Program II (Biocev-FAR LQ1604) and the “BIOCEV” project (CZ.1.05/1.1.00/02.0109).Notes and references
- R. M. Maina, M. J. Barahona, M. Finotti, T. Lysyy, P. Geibel, F. D'Amico, D. Mulligan and J. P. Geibel, Innov. Surg. Sci., 2018, 3(3), 203–213 Search PubMed.
- M. A. Hiob, S. She, L. D. Muiznieks and A. S. Weiss, ACS Biomater. Sci. Eng., 2017, 3(5), 712–723 CrossRef CAS.
- D. S. Vara, H. J. Salacinski, R. Y. Kannan, L. Bordenave, G. Hamilton and A. M. Seifalian, Pathol. Biol., 2005, 53(10), 599–612 CrossRef.
- R. Y. Kannan, H. J. Salacinski, P. E. Butler, G. Hamilton and A. M. Seifalian, J. Biomed. Mater. Res., Part B, 2005, 74B(1), 570–581 CrossRef CAS.
- J. Chlupáč and B. Filová, Physiol. Res., 2009, 58(S2), S119–S139 Search PubMed.
- S. Pashneh-Tala, S. MacNeil and F. Claeyssens, Tissue Eng., Part B, 2016, 22(1), 68–100 CrossRef CAS.
- N. Nakamura, T. Kimura and A. Kishida, ACS Biomater. Sci. Eng., 2017, 3(7), 1236–1244 CrossRef CAS.
- P. Zilla, D. Bezuidenhout and P. Human, Biomaterials, 2007, 28(34), 5009–5027 CrossRef CAS.
- T. Liu, S. Liu, K. Zhang, J. Chen and N. Huang, J. Biomed. Mater. Res., Part A, 2014, 102(10), 3754–3772 CrossRef.
- P. M. Crapo, T. W. Gilbert and S. F. Badylak, Biomaterials, 2011, 32(12), 3233–3243 CrossRef CAS.
- P. M. Vanhoutte, Cardiovasc. Drugs Ther., 2010, 24, 299–303 CrossRef.
- R. A. F. Clark, Ann. N. Y. Acad. Sci., 2006, 936(1), 355–367 CrossRef.
- N. Laurens, P. Koolwijk and M. P. de Maat, J. Thromb. Haemostasis, 2006, 4(5), 932–939 CrossRef CAS.
- A. Sahni and C. W. Francis, Blood, 2000, 96(12), 3772–3778 CrossRef CAS.
- M. W. Mosesson, J. Thromb. Haemostasis, 2005, 3(8), 1894–1904 CrossRef CAS.
- C. Urbich and S. Dimmeler, Circ. Res., 2004, 95(4), 343–353 CrossRef CAS.
- M. Avci-Adali, G. Ziemer and H. P. Wendel, Biotechnol. Adv., 2010, 28(1), 119–129 CrossRef CAS.
- N. Turner and R. Grose, Nat. Rev. Cancer, 2010, 10(2), 116–129 CrossRef CAS.
- A. Sahni and C. W. Francis, Blood, 2004, 104(12), 3635–3641 CrossRef CAS.
- T. A. E. Ahmed, E. V. Dare and M. Hincke, Tissue Eng., Part B, 2008, 14(2), 199–215 CrossRef CAS.
- S. Ravi and E. L. Chaikof, Regener. Med., 2010, 5(1), 107–120 CrossRef CAS.
- N. Thottappillil and P. D. Nair, Vasc. Health Risk Manage., 2015, 11, 79–91 Search PubMed.
- J. P. Luyendyk, J. G. Schoenecker and M. J. Flick, Blood, 2019, 133(6), 511–520 CrossRef CAS.
- F. M. Shaikh, A. Callanan, E. G. Kavanagh, P. E. Burke, P. A. Grace and T. M. McGloughlin, Cells Tissues Organs, 2008, 188(4), 333–346 CrossRef CAS.
- M. Deutsch, J. Meinhart, P. Zilla, N. Howanietz, M. Gorlitzer, A. Froeschl, A. Stuempflen, D. Bezuidenhout and M. Grabenwoeger, J. Vasc. Surg., 2009, 49(2), 352–362 CrossRef.
- L. P. Brewster, D. Bufallino, A. Ucuzian and H. P. Greisler, Biomaterials, 2007, 28(34), 5028–5032 CrossRef CAS.
- B. H. Walpoth, P. Zammaretti, M. Cikirikcioglu, E. Khabiri, M. K. Djebaili, J. C. Pache, J. C. Tille, Y. Aggoun, D. Morel, A. Kalangos, J. A. Hubbell and A. H. Zisch, J. Thorac. Cardiovasc. Surg., 2007, 133(5), 1163–1170 CrossRef CAS.
- R. A. Hoshi, R. Van Lith, M. C. Jen, J. B. Allen, K. A. Lapidos and G. Ameer, Biomaterials, 2013, 34(1), 30–41 CrossRef CAS.
- O. Kaplan, T. Hierlemann, S. Krajewski, J. Kurz, M. Nevoralová, M. Houska, T. Riedel, Z. Riedelová, J. Zárubová, H. P. Wendel and E. Brynda, J. Biomed. Mater. Res., Part A, 2017, 105(11), 2995–3005 CrossRef CAS.
- T. Riedel, E. Brynda, J. E. Dyr and M. Houska, J. Biomed. Mater. Res., Part A, 2009, 88A(2), 437–447 CrossRef CAS.
- Z. Riedelová-Reicheltová, E. Brynda and T. Riedel, Physiol. Res., 2016, 65(S2), S263–S272 Search PubMed.
- A. Sahni, T. Odrljin and C. W. Francis, J. Biol. Chem., 1998, 273(13), 7554–7559 CrossRef CAS.
- W. Zhao, S. A. McCallum, Z. Xiao, F. Zhang and R. J. Linhardt, Biosci. Rep., 2012, 32(1), 71–81 CrossRef CAS.
- S. Guglieri, M. Hricovíni, R. Raman, L. Polito, G. Torri, B. Casu, R. Sasisekharan and M. Guerrini, Biochemistry, 2008, 47(52), 13862–13869 CrossRef CAS.
- J. I. Weitz, M. Hudoba, D. Massel, J. Maraganore and J. Hirsh, J. Clin. Invest., 1990, 86(2), 385–391 CrossRef CAS.
- E. Wijelath, T. R. Kohler, J. Murray, M. Namekata, M. Yagi and M. Sobel, J. Biomed. Mater. Res., Part A, 2010, 95A(2), 641–648 CrossRef CAS.
- C. Gosselin, D. A. Vorp, V. Warty, D. A. Severyn, E. K. Dick, H. S. Borovetz and H. P. Greisler, J. Surg. Res., 1996, 60, 327–332 CrossRef CAS.
- P. Fernandez, A. Deguette, L. Pothuaud, G. Belleannée, P. Coste and L. Bordenave, Biomaterials, 2005, 26(24), 5042–5047 CrossRef CAS.
- N. P. P. Rhodes and D. F. F. Williams, Plasma Recalcification as a Measure of Contact Phase Activation and Heparinization Efficacy after Contact with Biomaterials, Biomaterials, 1994, 15(1), 35–37 CrossRef CAS.
- A. Link, G. Cattaneo, E. Brynda, T. Riedel, J. Kucerova, C. Schlensak, H. P. Wendel, S. Krajewski and T. Michel, J. Visualized Exp., 2020, 2020(157), e60610 Search PubMed.
- E. Filová, E. Brynda, T. Riedel, J. Chlupáč, M. Vandrovcová, Z. Švindrych, V. Lisá, M. Houska, J. Pirk and L. Bačáková, J. Biomed. Mater. Res., Part A, 2014, 102(3), 698–712 CrossRef.
- A. Krüger-Genge, A. Blocki, R. P. Franke and F. Jung, Int. J. Mol. Sci., 2019, 20(18), 4411 CrossRef.
- M. R. Kano, Y. Morishita, C. Iwata, S. Iwasaka, T. Watabe, Y. Ouchi, K. Miyazono and K. Miyazawa, J. Cell Sci., 2005, 118(16), 3759–3768 CrossRef CAS.
- Y. Bai, Y. Leng, G. Yin, X. Pu, Z. Huang, X. Liao, X. Chen and Y. Yao, Cell Tissue Res., 2014, 356(1), 109–121 CrossRef CAS.
- P. Bao, A. Kodra, M. Tomic-Canic, M. S. Golinko, H. P. Ehrlich and H. Brem, J. Surg. Res., 2009, 153(2), 347–358 CrossRef CAS.
- M. O. Stefanini, F. T. H. Wu, F. M. Gabhann and A. S. Popel, PLoS Comput. Biol., 2009, 5(12), e1000622 CrossRef.
- L. Li, H. Liu, C. Xu, M. Deng, M. Song, X. Yu, S. Xu and X. Zhao, Stem Cell Res. Ther., 2017, 8(1), 237 CrossRef.
- R. J. Smith, B. Nasiri, J. Kann, D. Yergeau, J. E. Bard, D. D. Swartz and S. T. Andreadis, Nat. Commun., 2020, 11(1), 1622 CrossRef CAS.
- D. N. Coakley, F. M. Shaikh, K. O'Sullivan, E. G. Kavanagh, P. A. Grace, S. R. Walsh and T. M. McGloughlin, Int. J. Surg., 2016, 25, 31–37 CrossRef CAS.
- J. Chlupáč, E. Filová, T. Riedel, M. Houska, E. Brynda, M. Remy-Zolghadri, R. Bareille, P. Fernandez, R. Daculsi, C. Bourget, L. Bordenave and L. Bačáková, Physiol. Res., 2014, 63(2), 167–177 Search PubMed.
- G. Cesarman-Maus and K. A. Hajjar, Br. J. Haematol., 2005, 129(3), 307–321 CrossRef CAS.
- T. Urano and Y. Suzuki, J. Biomed. Biotechnol., 2012, 2012, 208108 Search PubMed.
- A. Mauro, M. Buscemi and A. Gerbino, J. Mol. Histol., 2010, 41(6), 367–377 CrossRef CAS.
- D. Ratel, S. Mihoubi, E. Beaulieu, Y. Durocher, G. E. Rivard, D. Gingras and R. Béliveau, Thromb. Res., 2007, 121(2), 203–212 CrossRef CAS.
- A. Bini, Y. Itoh, B. J. Kudryk and H. Nagase, Biochemistry, 1996, 35(40), 13056–13063 CrossRef CAS.
- A. Sahni and C. W. Francis, J. Thromb. Haemostasis, 2003, 1(6), 1271–1277 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional experimental data (Fig. S1–S6) of ePTFE modification with fibrin coating (SEM), GFs release from the Fb coating, hemocompatibility of various Fb coatings on ePTFE, and cell morphology images (PDF). See DOI: 10.1039/d1ra00053e |
| This journal is © The Royal Society of Chemistry 2021 |