
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cytotoxicity of targeted PLGA nanoparticles: a systematic review†
Hock Ing Chiua,
Nozlena Abdul Samada,
Lizhen Fangb and
Vuanghao Lim *a
*a
aIntegrative Medicine Cluster, Advanced Medical and Dental Institute, Universiti Sains Malaysia, Bertam, 13200 Kepala Batas, Penang, Malaysia. E-mail: vlim@usm.my; Tel: +604-5622427
bSchool of Pharmacy, Xinxiang Medical University, Xinxiang, Henan 453003, People's Republic of China
First published on 3rd March 2021
Abstract
Recent advances in nanotechnology have contributed tremendously to the development and revolutionizing of drug delivery systems in the field of nanomedicine. In particular, targeting nanoparticles based on biodegradable poly(lactic-co-glycolic acid) (PLGA) polymers have gained much interest. However, PLGA nanoparticles remain of concern for their effectiveness against cancer cells and their toxicity to normal cells. The aim of this systematic review is to identify a promising targeting PLGA nanoformulation based on the comparison study of their cytotoxicity potency in different cell lines. A literature search was conducted through the databases of Google Scholar, PubMed, ScienceDirect, Scopus and SpringerLink. The sources studied were published between 2009 and 2019, and a variety of keywords were utilized. In total, 81 manuscripts that met the inclusion and exclusion criteria were selected for analysis based on their cytotoxicity, size, zeta potential, year of publication, type of ligand, active compounds and cell line used. The half maximal inhibitory concentration (IC50) for cytotoxicity was the main measurement in this data extraction, and the SI units were standardized to μg mL−1 for a better view of comparison. This systematic review also identified that cytotoxicity potency was inversely proportional to nanoparticle size. The PLGA nanoparticles predominantly exhibited a size of less than 300 nm and absolute zeta potential ∼20 mV. In conclusion, more comprehensive and critical appraisals of pharmacokinetic, pharmacokinetic, toxicokinetic, in vivo and in vitro tests are required for the investigation of the full value of targeting PLGA nanoparticles for cancer treatment.
1. Introduction
Recent advances in nanotechnology have contributed tremendously to the development and revolutionizing of the drug delivery system in the nanomedicine field. The application of nanoparticles has long been recognized as a controlled release formulation for delivering a therapeutic agent to a specific targeted site. Nanoparticles provide a high therapeutic effect against cancers, which has earned them remarkable research interest among researchers. Nanoparticles offer a highly efficient targeted therapy compared to traditional cancer therapies. This targeted therapy can be done easily on nanoparticles using an ideal targeting ligand. Thus, targeting nanoparticles based on biodegradable polymers have gained much interest for treating cancer cells with minimal systemic side effects.1,2Poly(lactic-co-glycolic acid) (PLGA) is a biodegradable polymer that has attractive properties as a nanocarrier for cancer therapy. PLGA is a hydrophobic copolymer and mainly composed of two monomers: lactic acid and glycolic acid (Fig. 1). PLGA is approved by the European Medicine Agency and the US Food and Drug Administration (FDA) as an ideal material for designing a drug delivery system due to its biocompatibility and biodegradability. PLGA is widely adapted for preparing nanoparticles encapsulating hydrophilic and hydrophobic anti-therapeutic agents.3,4 PLGA offers an enhanced permeability and retention (EPR) effect, sustained and controlled drug delivery for cancer therapy, enhanced accumulation of drugs in tumor vasculature and targeted delivery by surface conjugation with targeting ligands.5
 | ||
| Fig. 1 Chemical structure of PLGA. | ||
Active and passive mechanisms are often practiced when targeting cancer cells using nanoparticles. Passive targeting is the application of polymeric nanoparticles owing to their size, shape and surface charge enabling them to be accumulated predominantly in the microenvironment of cancer cells via the EPR effect. This EPR effect is amplified based on the exclusive presence of leaky vasculature and impaired lymphatic drainage in tumors. Active targeting is the attachment or grafting of targeting/biorecognizable ligands on nanoparticles to target specific receptors/biomarkers that are overexpressed in cancer cells, excluding normal cells.6 Since targeting ligands are highly selective towards overexpressed receptors in cancer cells, in which can result in enhanced cellular uptake of nanoparticles as well as excluding harm to normal cells. Both passive and targeting mechanisms are considered a gold standard in designing a drug delivery system.
Recently, the use of PLGA nanoparticles for cancer therapy has received great interest due to the advantages offered and approval by the FDA. However, the safeness of PLGA nanoparticles containing cytotoxic therapeutic agent remains a huge concern. The anti-cancer drugs lack targeting specificity to cancer cells and could induce potent cytotoxic effects against both normal and cancer cells. In addition, the cellular concentration of the drugs is relatively low in cancer cells due to the low efficiency of non-targeting PLGA nanoparticles in delivering the drugs to the site of action. The cancer cell targeting of PLGA nanoparticles, enhanced cellular uptake of the drugs and low toxicity to normal cells are the most important criteria or measurements for chemotherapy. Therefore, it is vital for researchers to design a PLGA nanocarrier that, in addition to being biocompatible, biodegradable and cost-effective, can specifically release drugs at the target site with reduced systemic effects.
Hence, this systematic review is focused on a comparison study of the cytotoxicity potency of the targeting PLGA nanoparticles on the basis of published in vitro assessment findings from 2009 to 2019 in order to assess the PLGA nanoparticles with the ideal targeting ligands for specific cell lines – preferentially based on their IC50 cytotoxicity potency – and to correlate the size and zeta potential of nanoparticles with cytotoxicity potency.
The databases of Google Scholar, PubMed, ScienceDirect, Scopus and SpringerLink were searched for literature published between 2009 and 2019. Different combinations of keywords – including PLGA nanoparticles, cytotoxicity, targeting ligands and anti-cancer – were used for the literature search (Fig. 2). The methodology for the study was based on the Preferred Reporting Items for Systematic Review and Meta-Analysis Protocols (PRISMA-P) 2015.7–9 The inclusion criteria for our study were: (1) PLGA nanoparticles with different types of targeting ligands; (2) PLGA with nanoencapsulated active compounds and exhibited cytotoxic effects on normal and cancer cells; (3) PLGA with co-encapsulation of outer or inner polymers; (4) studies published between 2009 and 2019 (including in-press articles). Studies with the following criteria were omitted (exclusion criteria): (1) PLGA microparticles; (2) chemical conjugation of PLGA with other polymers to form nanoparticles; (2) all in vivo, ex vivo, in silico, clinical studies, and review articles; (4) studies without available cytotoxicity data; (5) articles that were not published in English. Based on the inclusion and exclusion criteria, the articles that fulfilled the requirements were selected for analysis. Screening of the articles was conducted by two independent reviewers. The data extraction involved analysis of the selected articles based on the types of cells, IC50 for cytotoxicity, year of publication, treatment duration, types of active compounds used, types of targeting ligands, types of studies (in vitro) and the size and zeta potential of the nanoparticles. The data were described and presented in a table. The IC50 for cytotoxicity was the main measurement in this data extraction. The SI units were standardized to μg mL−1 for a better view of comparison. The IC50 is the dose required to inhibit 50% of the cell viability. Based on the availability of the data of IC50, size and zeta potential of the nanoparticles, PLGA nanoparticles with the active targeting properties were selected for the purpose of studying the correlation between the particle size/zeta potential of PLGA nanoparticles and cytotoxicity potency.
 | ||
| Fig. 2 Flowchart of the selection of studies, using PRISMA guidelines. | ||
2. Cytotoxicity of PLGA nanoformulations
The databases of Google Scholar, PubMed, ScienceDirect, Scopus and SpringerLink were searched. These produced 113, 32, 113, 20 and 5 articles, respectively. From all the databases, a total of 266 articles were retrieved after 17 duplicates were removed. Following this, 170 articles that were not compliant with the inclusion criteria were identified and excluded from the study. The 96 articles left were thoroughly assessed according to the exclusion criteria defined in Introduction. After critical assessment, 15 articles were omitted due to the methodology and cytotoxicity data being insufficiently described. Hence, 81 studies have been integrated into the qualitative synthesis involving assessment of the data of in vitro studies.2.1 In vitro studies
The data of the cytotoxicity of PLGA nanoparticles conjugated with particular targeting ligands that deliver specific active compounds against different types of cells – such as brain, breast, lung, colon, stomach, gastric, liver, ovary, cervix, prostate, uterus, pancreas, skin, umbilical vein endothelial, esophagus, bladder, head, neck and kidney cells – are shown in Table 1. From the table, it can be seen that PLGA nanoparticles are time/dose-dependent on cytotoxicity.| Type of cell line | IC50 | Cell viability assay | Size (nm) | Zeta potential (mV) | Active compound | Exposure duration | Response relationship | Targeting ligand | Ref. | Year | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Brain | U87 | 1.03 μg mL−1 | MTT | ∼150 | −18.1 ± 0.5 | Doxorubicin and paclitaxel | 48 h | Dose-dependent | Transferrin | 10 | 2013 |
| 0.13 μg mL−1 | 96 h | Time-dependent | |||||||||
| Not available (NA) | SRB | ∼190 | +38.6 ± 4.1 | Doxorubicin and EGFR siRNA | 48 h | Dose-dependent | Angiopep-2 | 34 | 2015 | ||
| U87MG | NA | XTT | 181.9 ± 4.5 | +40.212 ± 2.844 | Etoposide | 6, 12, 24 and 48 h | Time-dependent | Lactoferrin and FA | 35 | 2015 | |
| C6 | NA | MTT | 182.8 ± 3.78 | −11.72 ± 2.27 | Paclitaxel | 24, 48 and 72 h | Time-dependent | Transferrin | 36 | 2009 | |
| 7.28 μg mL−1 | WST-1 | 138.5 ± 7.0 | −43.03 ± 0.38 | Paclitaxel | 24 h | Dose-dependent | Vitamin E TPGS | 37 | 2019 | ||
| GI-1 | NA | MTT | 85.5 | −26.5 ± 2.1 | Paclitaxel | 24, 48 and 72 h | Time-dependent | AS1411 | 38 | 2012 | |
| NA | MTT | ∼200 | NA | Paclitaxel | 24, 48 and 72 h | Time-dependent | AS1411 | 39 | 2012 | ||
| U251 | 0.185 μg mL−1 | CCK-8 | 137.3 ± 8.6 | −17.63 ± 3.10 | Doxorubicin | 48 h | Dose-dependent | Chondroitin sulphate | 40 | 2019 | |
| SH-SY5Y | NA | MTT | 257.10 ± 22.39 | −5.51 ± 0.73 | Doxorubicin | 26 h | Dose-dependent | Rabies virus-derived peptide | 41 | 2017 | |
| DKMG/EGFRvIII | NA | MTT | 251 ± 3 | −2.9 ± 1.2 | Curcumin | 24 h | Dose-dependent | Anti-EGFRvIII monoclonal antibody | 42 | 2018 | |
| Breast | D2F2 | >0.114 μg mL−1 | MTS | 124.2 ± 21.2 | +12 ± 7 | PE38KDEL | 48 h | Dose-dependent | Fab' fragments of a humanized anti-HER2 monoclonal antibody (rhu-MAbHER2) | 43 | 2009 |
| D2F2/E2 | 0.00078 ± 0.00024 μg mL−1 | ||||||||||
| SKBR-3 | 0.00497 ± 0.000176 μg mL−1 | MTS | 124.2 ± 21.2 | +12 ± 7 | PE38KDEL | 48 h | Dose-dependent | Fab' fragments of a humanized anti-HER2 monoclonal antibody (rhu-MAbHER2) | 43 | 2009 | |
| 0.00031 to 0.00049 μg mL−1 | MTS | 170.3 ± 7.6 | −18.9 ± 1.5 | Aromatase inhibitor, 7α-(4′-amino)phenylthio-1,4-androstadiene-3,17-dione (7α-APTADD) | 24 h | Dose-dependent | Transferrin | 11 | 2010 | ||
| NA | MTS | 180 ± 1 | −1.0 ± 0.1 | Paclitaxel | 24 h | Dose-dependent | Herceptin | 44 | 2013 | ||
| MCF-7 | 6.71 μg mL−1 | MTT | 110 | −32 | DDP prodrug (c,c, t-[Pt(NH3)2Cl2(O2CCH2CH2CH2CH2CH3)2]) | 24 h | Dose-dependent | Folate | 45 | 2012 | |
| NA | Alamar blue | 218 | −27 ± 3 | Paclitaxel | 120 h | Dose-dependent | AS1411 | 46 | 2013 | ||
| NA | MTS | 268.3 ± 23.4 | +24.21 ± 2.31 | Epirubicin | 24, 48, 72, 96 and 120 h | Time-dependent | Biotinylated chitosan | 47 | 2016 | ||
| ∼0.043 μg mL−1 | MTS | ∼180 | −22 | Paclitaxel and combretastatin A4 | 24 h | Dose-dependent | Cyclo-(Arg–Gly–Asp–D-Phe–Lys) | 48 | 2010 | ||
| NA | MTT | 85.5 | −26.5 ± 2.1 | Paclitaxel | 24, 48 and 72 h | Time-dependent | AS1411 | 38 | 2012 | ||
| NA | MTT | 222.7 | +10.2 | Epirubicin | 72 h | Dose-dependent | 5TR1 DNA aptamer (Apt) | 49 | 2017 | ||
| NA | MTT | 245.8 ± 7.8 | −8.57 ± 0.6 | Gold nanorods and doxorubicin | 28 h | Dose-dependent | Human serum albumin | 50 | 2019 | ||
| 0.02611 μg mL−1 | MTT | 274 ± 1.6 | −13.8 ± 5.1 | Rapamycin | 120 h | Dose-dependent | EGFR antibody | 51 | 2009 | ||
| 0.42 ± 0.15 μg mL−1 | MTT | 141 ± 58.41 | −2.61 ± 1.37 | Epirubicin | 24 h | Dose-dependent | Trastuzumab | 52 | 2019 | ||
| 1.84 ± 1.30 μg mL−1 | MTT | 153.41 ± 4.78 | +49.1 ± 1.2 | Salinomycin and paclitaxel | 24 h | Dose-dependent | Hyaluronic acid | 53 | 2016 | ||
| 0.053 ± 0.0065 μg mL−1 (48 h), 0.039 ± 0.0052 μg mL−1 (72 h) | MTT | 184.6 ± 4.1 | −28.6 ± 2.4 | Docetaxel, silibinin and SPIONs | 48 and 72 h | Time-dependent | LHRH decapeptide hormone | 54 | 2016 | ||
| NA | MTT | 210.4 ± 10.14 | +9.7 ± 0.2 | Antimir-21 and epirubicin | 72 h | Dose-dependent | MUC1 Apt | 55 | 2018 | ||
| 8.12 μg mL−1 | MTT | 151.7 ± 4.9 | −1.00 ± 0.11 | Paclitaxel | 48 h | Dose-dependent | Glutamate | 56 | 2016 | ||
| HER2-negative MCF7 | NA | MTT | 268.4 ± 6.6 | +11.80 ± 0.75 | Paclitaxel | 24, 48 and 72 h | Time-dependent | Herceptin | 57 | 2016 | |
| HER2-positive BT474 | NA | MTT | 268.4 ± 6.6 | +11.80 ± 0.75 | Paclitaxel | 24, 48 and 72 h | Time-dependent | Herceptin | 57 | 2016 | |
| BT-20 | 2.73 ± 0.28 μg mL−1 | MTT | 141 ± 58.41 | −2.61 ± 1.37 | Epirubicin | 24 h | Dose-dependent | Trastuzumab | 52 | 2019 | |
| MDA-MB-453 | 3.11 ± 0.19 μg mL−1 | MTT | 141 ± 58.41 | −2.61 ± 1.37 | Epirubicin | 24 h | Dose-dependent | Trastuzumab | 52 | 2019 | |
| MDA-MB-231 | NA | MTS | 286.20 ± 4.35 | −15.97 ± 1.28 | Docetaxel | 24, 48, 72 h | Time-dependent | Hyaluronic acid ceramide | 58 | 2014 | |
| 4 μg mL−1 | MTT | 150 ± 13.32 | −21.57 ± 2.93 | Quercetin | 24 h | Dose-dependent | Transferrin | 59 | 2018 | ||
| 0.048 ± 0.004 μg mL−1 | WST-1 | 91.2 ± 8.1 | −60.7 ± 1.0 | Doxorubicin and irinotecan | 240 h | Dose-dependent | Hyaluronic acid | 60 | 2015 | ||
| MBA-MD-231/ADR | NA | MTT | 198 ± 12 | −19.6 ± 1.5 | Doxorubicin | 48 h | Dose-dependent | FA | 61 | 2016 | |
| 4T1 mammary epithelial carcinoma cells | NA | MTT | 235.5 ± 71.30 | NA | Curcumin and bortezomib | 24 h | Dose-dependent | Alendronate | 62 | 2012 | |
| 0.37 μg mL−1 | MTT | 151.7 ± 4.9 | −1.00 ± 0.11 | Paclitaxel | 48 h | Dose-dependent | Glutamate | 56 | 2016 | ||
| MCF-7/ADR | 4.6 μg mL−1 | MTT | ∼210 | +27 | Doxorubicin | 48 h | Dose-dependent | Low-molecular weight protamine | 63 | 2014 | |
| NA | MTT | 245.8 ± 7.8 | −8.57 ± 0.6 | Gold nanorods and doxorubicin | 28 h | Dose-dependent | Human serum albumin | 50 | 2019 | ||
| JC | 0.034 ± 0.00085 μg mL−1 | MTS | 240 ± 1 | −19 ± 5 | Paclitaxel and tariquidar | 96 h | Dose-dependent | Biotin | 64 | 2009 | |
| NA | MTS | 200–250 | −12.1 ± 0.3 | Paclitaxel and P-gp-targeted siRNA | 24 h | Dose-dependent | Biotin | 65 | 2010 | ||
| T47D | NA | MTT | 210 ± 10 | −13 | Paclitaxel | 48 h | Dose-dependent | Human serum albumin | 66 | 2015 | |
| Lung | A549/T | 0.78 μg mL−1 | MTT | ∼210 | +27 | Doxorubicin | 48 h | Dose-dependent | Low-molecular weight protamine | 63 | 2014 |
| A549 | 0.34 μg mL−1 | MTT | ∼210 | +27 | Doxorubicin | 48 h | Dose-dependent | Low-molecular weight protamine | 63 | 2014 | |
| 30 μg mL−1 | MTS | 286 ± 10.3 | −45 ± 3.2 | Doxorubicin | 20 h | Dose-dependent | Cyclo-(1,12)-PenITDGEATDSGC (cLABL) peptide | 67 | 2009 | ||
| NA | MTT | 301 ± 10 | −37.4 ± 1.4 | Doxorubicin | 24 h | Dose-dependent | LFC131 peptide | 68 | 2014 | ||
| 0.00330 μg mL−1 | SRB | 108 ± 12.5 | −21.32 ± 1.91 | Doxorubicin | 48 h | Dose-dependent | Transferrin | 69 | 2015 | ||
| NA | MTS | 200 | 58.07 | Chlorin e6 | 24 h | Dose-dependent | Hyaluronic acid | 70 | 2017 | ||
| A549-Luc | 4.12 μg mL−1 | MTT | 58 | −29.4 | Docetaxel | 48 h | Dose-dependent | Hyaluronic acid | 71 | 2017 | |
| 0.91 μg mL−1 | MTT | 154 | −22.7 | Docetaxel | 48 h | Dose-dependent | Hyaluronic acid | 72 | 2016 | ||
| A549-luc-C8 | NA | MTT | 80 | −50 | Paclitaxel palmitate | 48 h | Dose-dependent | Cetuximab | 73 | 2013 | |
| TFR positive A549 | 0.01 μg mL−1 | CCK | 96–156 | NA | Hypocrellin A | 8 h | Dose-dependent | Transferrin | 74 | 2017 | |
| H1299, A549 and H1975 | A549 (∼0.011 μg mL−1), H1299 (∼0.024 μg mL−1), H1975 (∼0.0017 μg mL−1) | Trypan blue | 217 ± 13.54 | +29.5 ± 4.04 | Paclitaxel | 24 and 48 h | Time-dependent | RGD peptide | 12 | 2017 | |
| Colon | Caco-2 | 1.019 ± 0.233 μg mL−1 | MTT | 330 ± 3 | −3.9 ± 0.3 | Paclitaxel | 24 h | Dose-dependent | Wheat germ agglutinin | 13 | 2010 |
| 0.087 ± 0.020 μg mL−1 | 72 h | Time-dependent | |||||||||
| CCD-18Co | 1.007 ± 0.121 μg mL−1 | 24 h | Dose-dependent | ||||||||
| 0.137 ± 0.027 μg mL−1 | 72 h | Time-dependent | |||||||||
| Ht-29 | 0.061 ± 0.021 μg mL−1 | MTT | 330 ± 3 | −3.9 ± 0.3 | Paclitaxel | 24 h | Dose-dependent | Wheat germ agglutinin | 13 | 2010 | |
| 0.028 ± 0.008 μg mL−1 | MTT | 330 ± 3 | −3.9 ± 0.3 | Paclitaxel | 72 h | Time-dependent | Wheat germ agglutinin | 13 | 2010 | ||
| NA | MTT | 90 ± 1.9 | −36.3 ± 4.2 | Curcumin | 2 + 48 h | Dose-dependent | Ribonucleic acid (RNA) Apts against epithelial cell adhesion molecule (EpCAM) | 75 | 2014 | ||
| CT26 | NA | MTT | 245.8 ± 7.8 | −8.57 ± 0.6 | Gold nanorods and doxorubicin | 28 h | Dose-dependent | Human serum albumin | 50 | 2019 | |
| C26 | NA | MTT | 210.4 ± 10.14 | +9.7 ± 0.2 | Antimir-21 and epirubicin | 72 h | Dose-dependent | MUC1 Apt | 55 | 2018 | |
| 3.67 μg mL−1 | MTT | 102.3 ± 2.9 | −13.2 ± 2.4 | 10-Hydroxy camptothecin (HCPT) | 48 h | Dose-dependent | Chondroitin sulphate | 76 | 2019 | ||
| HCT116 | NA | MTT | 150 ± 12 | +0.2 ± 0.12 | Camptothecin | 72 h | Dose-dependent | Conatumumab (AMG 655) | 77 | 2011 | |
| Stomach | MKN74, MKN74-CD44v6+ and GP202 | NA | MTT | 293 ± 15 | −20.0 ± 0.4 | Paclitaxel | 24 and 48 h | Time-dependent | Human anti-human CD44v6 Fab, AbD15179 | 14 | 2018 |
| Gastric | HGC27 | 0.05 μg mL−1 | MTT | 406.6 ± 65.5 | −25.0 ± 2.0 | SN38 (7-ethyl-10-hydroxycamptothecin) | 48 h | Dose-dependent | AHNP | 39 | 2016 |
| MKN28 | NA | MTT | ∼200 | NA | Pheophorbide a | 2 h | Dose-dependent | Folate | 78 | 2018 | |
| Liver | HepG2 | 0.78 μg mL−1 | MTT | 138 ± 3.12 | NA | Epirubicin | 24 h | Dose-dependent | LFC131 peptide | 79 | 2016 |
| 0.38 μg mL−1 | 48 h | Time-dependent | |||||||||
| 5.4 ± 0.21 μg mL−1 | MTT | 204 | −5.6 | Docetaxel | 72 h | Dose-dependent | Human serum albumin | 80 | 2013 | ||
| 2.338 μg mL−1 | MTT | 111.3 ± 0.3 | −6.17 ± 0.41 | Doxorubicin | 24 h | Dose-dependent | Biotin | 81 | 2016 | ||
| 7.6 μg mL−1 | MTT | 209.4 | −16.7 | Paclitaxel | 48 h | Dose-dependent | Pullulan | 25 | 2016 | ||
| 3.1 μg mL−1 | MTT | 218.2 | −18.2 | Combretastatin A4 | 48 h | Dose-dependent | Pullulan | 25 | 2016 | ||
| ∼2 μg mL−1 | MTT | 288.4 ± 1.11 | −14.1 | Epirubicin and tocotrienols | 3 h | Dose-dependent | Asialofetuin | 82 | 2014 | ||
| JHH-7 | NA | MTT | 175.25 ± 1.82 | −19 | Sorafenib | 48 h | Dose-dependent | CXCR4 antagonist, AMD3100 | 83 | 2015 | |
| SMMC-7721 | NA | CCK-8 | 187.2 ± 10.6 | +28.9 ± 0.3 | Arsenic trioxide (As2O3) | 24, 48 and 72 h | Time-dependent | Lactobionic acid-modified chitosan | 84 | 2019 | |
| NA | CCK-8 | 249.1 ± 9.1 | −28.7 ± 1.1 | As2O3 | 24, 48 and 72 h | Time-dependent | Lactose acid | 85 | 2018 | ||
| Ovarian | NCI/ADR-RES | 0.044 ± 0.0026 μg mL−1 | MTS | 240 ± 1 | −19 ± 5 | Paclitaxel and tariquidar | 96 h | Dose-dependent | Biotin | 64 | 2009 |
| SKOV-3 | NA | SRB | 213.0 ± 3.5 | −1.3 ± 3.8 | Doxorubicin | 24 h | Dose-dependent | HER2 antibody | 22 | 2011 | |
| 0.062 μg mL−1 | MTT | 341.8 ± 86.8 | −3.5 ± 5.9 | Paclitaxel | 3 h | Dose-dependent | TAT peptide (RKKRRQRRR) | 86 | 2013 | ||
| 0.073 ± 0.002 μg mL−1 (48 h), 0.027 ± 0.001 μg mL−1 (72 h) | MTT | 184.6 ± 4.1 | −28.6 ± 2.4 | Docetaxel, silibinin and SPIONs | 48 and 72 h | Time-dependent | LHRH decapeptide hormone | 54 | 2016 | ||
| 4.91 μg mL−1 (24 h), 1.42 μg mL−1 (48 h) | CCK-8 | 165.61 ± 13.36 | +19.59 ± 1.74 | Cisplatin | 24 and 48 h | Time-dependent | Trastuzumab | 87 | 2019 | ||
| NA | SRB | 210 ± 7 | −1.0 ± 0.5 | Indocyanine green and doxorubicin | 24 h | Dose-dependent | Anti-HER2 | 88 | 2014 | ||
| OCSCs | 0.000754 μg mL−1 | MTT | 294.7 | NA | Paclitaxel | 24 h | Dose-dependent | FA | 18 | 2017 | |
| Cervical | Luciferase-expressing HeLa | 0.65 μg mL−1 | CellTiter-Glo® | 207 ± 4.461 | +5.29 ± 1.5 | Anti-luc siRNA | 72 h | Dose-dependent | Lipid | 89 | 2012 |
| HeLa | 1.86 ± 0.17 μg mL−1 | MTT | 190.91 ± 0.19 | −19.3 ± 0.59 | Doxorubicin | 24 h | Dose-dependent | Folate | 90 | 2018 | |
| 0.80 ± 0.06 μg mL−1 | 190.91 ± 0.19 | −19.3 ± 0.59 | 48 h | Time-dependent | Folate | 2018 | |||||
| 1.66 ± 0.16 μg mL−1 | 194.97 ± 1.33 | −15.4 ± 1.57 | 24 h | Dose-dependent | RGD peptide (Arg–Gly–Asp) | 2018 | |||||
| 0.79 ± 0.13 μg mL−1 | 194.97 ± 1.33 | −15.4 ± 1.57 | 48 h | Time-dependent | RGD peptide (Arg–Gly–Asp) | 2018 | |||||
| 0.61 μg mL−1 | MTT | 151.7 ± 4.9 | −1.00 ± 0.11 | Paclitaxel | 48 h | Dose-dependent | Glutamate | 56 | 2016 | ||
| KB-3-1 and KB-V1 | >9.95 μg mL−1 | MTT | 132.4 ± 1.5 | −40.3 ± 6.1 | Curcumin | 48 h | Dose-dependent | Anti-P-glycoprotein | 91 | 2012 | |
| KB-V1 | 0.051 μg mL−1 | MTT | 165.4 | −37.2 | Curcumin | 48 h | Dose-dependent | Mouse monoclonal anti-P-glycoprotein (P-gp) antibody (APgp; clone F4) | 92 | 2014 | |
| KB | NA | MTT | 198 ± 12 | −19.6 ± 1.5 | Doxorubicin | 48 h | Dose-dependent | FA | 61 | 2016 | |
| 0.0057 μg mL−1 | SRB | 200–250 | +9 | Docetaxel and SPIONs | 48 h | Dose-dependent | Folate–chitosan | 19 | 2017 | ||
| 2.7 μg mL−1 | MTT | 118 ± 3 | −8.5 ± 2.4 | Doxorubicin | 10 h | Dose-dependent | FA | 33 | 2015 | ||
| HCA-1 | NA | MTT | 175.25 ± 1.82 | −19 | Sorafenib | 48 h | Dose-dependent | CXCR4 antagonist AMD3100 | 83 | 2015 | |
| Prostate | PC3 | 0.48 ± 0.087 μg mL−1 | MTS | 123 ± 2 | −30.5 ± 1.4 | Doxorubicin | 72 h | Dose-dependent | Transferrin | 93 | 2017 |
| NA | CellTiter-Glo® | 207 ± 4.461 | +5.29 ± 1.5 | KIF11 siRNA | 72 h | Dose-dependent | Lipid | 89 | 2012 | ||
| 0.093 ± 0.016 μg mL−1 | WST-1 | 91.2 ± 8.1 | −60.7 ± 1.0 | Doxorubicin and irinotecan | 240 h | Dose-dependent | Hyaluronic acid | 60 | 2015 | ||
| 0.04 μg mL−1 | SRB | 200–250 | +9 | Docetaxel and SPIONs | 48 h | Dose-dependent | Folate–chitosan | 19 | 2017 | ||
| 0.01403 ± 0.00112 μg mL−1 (24 h), 0.00579 ± 0.00109 μg mL−1 (48 h), 0.00322 ± 0.00111 μg mL−1 (72 h) | MTT | 146.9 ± 8.6 | NA | Docetaxel and SPIONs | 24, 48 and 72 h | Dose- and time-dependent | Single-chain prostate stem cell antigen antibodies | 21 | 2011 | ||
| PC3M | NA | CCK-8 | 187.4 ± 32.7 | NA | Docetaxel and SPIONs | 24, 48 and 72 h | Dose- and time-dependent | Prostate stem cell antigen antibody | 94 | 2012 | |
| LNCaP | NA | CellTiter-Glo® | 207 ± 4.461 | +5.29 ± 1.5 | KIF11 siRNA | 72 h | Dose-dependent | Lipid | 89 | 2012 | |
| DU145 | NA | CellTiter-Glo® | 207 ± 4.461 | +5.29 ± 1.5 | KIF11 siRNA | 72 h | Dose-dependent | Lipid | 89 | 2012 | |
| <80 μg mL−1 | SRB | 206.9 | +21.7 | Bicalutamide | 120 h | Dose-dependent | FA | 95 | 2015 | ||
| Uterine | MES-SA/Dx5 | NA | SRB | 213.0 ± 3.5 | −1.3 ± 3.8 | Doxorubicin | 24 h | Dose-dependent | HER2 antibody | 22 | 2011 |
| Pancreas | PANC-1 | NA | Alamar blue | 150 | NA | Curcumin and SPIONs | 48, 72, 96 h | Dose- and time-dependent | AS1411 Apt | 96 | 2016 |
| NA | Alamar blue | 150 | NA | Curcumin and gemcitabine | 120 h | Dose-dependent | AS1411 Apt | 97 | 2013 | ||
| MIA PaCa-2 | NA | Alamar blue | 150 | NA | Curcumin and SPIONs | 48, 72, 96 h | Dose- and time-dependent | AS1411 Apt | 96 | 2016 | |
| NA | Alamar blue | 150 | NA | Curcumin and gemcitabine | 120 h | Dose-dependent | AS1411 Apt | 97 | 2013 | ||
| PK-1 | NA | Alamar blue | 150 | NA | Curcumin and gemcitabine | 120 h | Dose-dependent | AS1411 Apt | 97 | 2013 | |
| Normal hTERT-HPNE | NA | Presto blue and SRB | 200 ± 10 | −16 ± 1 | Bortezomib | 48 h | Dose-dependent | Human holo-transferrin | 98 | 2014 | |
| S2–013 | 0.0028 ± 0.00019 μg mL−1 | Presto blue and SRB | 200 ± 10 | −16 ± 1 | Bortezomib | 48 h | Dose-dependent | Human holo-transferrin | 98 | 2014 | |
| Skin | A375-luc | 0.09 μg mL−1 | MTS | 200 ± 10 | −16 ± 1 | Cisplatin and rapamycin | 24 h | Dose-dependent | Anisamide | 99 | 2014 |
| B16F10 | NA | Trypan blue | 244.1 ± 4.3 | −11.4 ± 2.3 | Paclitaxel and combretastatin A4 | 24 h | Dose-dependent | cRGDfK peptide | 100 | 2011 | |
| Umbilical vein endothelial | HUVECs | 0.0396 μg mL−1 | MTT | 209.4 | −16.7 | Paclitaxel | 48 h | Dose-dependent | Pullulan | 25 | 2016 |
| 0.0118 μg mL−1 | MTT | 218.2 | −18.2 | Combretastatin A4 | 48 h | Dose-dependent | Pullulan | 25 | 2016 | ||
| NA | MTT | 175.25 ± 1.82 | −19 | Sorafenib | 48 h | Dose-dependent | CXCR4 antagonist, AMD3100 | 83 | 2015 | ||
| NA | Trypan blue | 244.1 ± 4.3 | −11.4 ± 2.3 | Paclitaxel and combretastatin A4 | 24 h | Dose-dependent | cRGDfK peptide | 100 | 2011 | ||
| Oesophageal | OE21 | NA | MTT | 133 | −4.7 | Ruthenium-based DNA replication inhibitor and radiosensitizer (Ru(phen)2(tpphz)2+) | 24 h | Dose-dependent | hEGF | 27 | 2018 |
| OE33 | NA | MTT | 133 | −4.7 | Ruthenium-based DNA replication inhibitor and radiosensitizer (Ru(phen)2(tpphz)2+) | 24 h | Dose-dependent | hEGF | 27 | 2018 | |
| Bladder | UM-UC-3 and RT-4 | 0.509 μg mL−1 | WST-1 | 151 ± 32 | NA | Belinostat (NSC726630, PXD101) | 72 h | Dose-dependent | Poly(guanidinium oxanorbornene) | 101 | 2013 |
| Head and neck | UMSCC 22A | NA | MTT | 142 | −14.6 ± 0.5 | Doxorubicin | 3 h | Dose-dependent | Anti-EGFR antibody (cetuximab) | 29 | 2017 |
| Kidney | COS-7 | NA | MTT | 118 ± 3 | −8.5 ± 2.4 | Doxorubicin | 10 h | Dose-dependent | FA | 33 | 2015 |
2.2 Size and cytotoxicity
The cytotoxicity results show that PLGA nanoparticles induced an anti-proliferative effect as early as 3 h, taking 240 h at most. The particle size of PLGA nanoparticles ranged from 58 to 407 nm, with an average size of 189 nm. It was evident that particles smaller than 100 nm and larger than 300 nm were less commonly prepared, as a certain size is required to carry the load of the drugs and nor bigger size to promote the EPR effect in cancer cells. The IC50 of cytotoxicity of PLGA nanoparticles was plotted against particle size of the PLGA nanoparticles to assess the correlation between particle size and cytotoxicity (Fig. 3). It can be seen that as the particle size increased, the IC50 of cytotoxicity also increased, which indicates that smaller nanoparticles induced lower IC50 values and higher cytotoxicity potency. Although the application of corona materials such as hyaluronic acid increase the particle size, the efficacy of the encapsulated drugs to the desired site was enhanced. This is due to the cancer targeting properties provided by the binding of hyaluronic acid to the PLGA nanoparticles.53,58,60,70,72 Selective targeting enhances the cellular uptake of drugs in cancer cells and reduces the cytotoxic effect of drugs in normal cells. This further justifies that the ideal size and ability of PLGA nanoparticles to selectively deliver drugs to desired site are crucial in developing efficient and safe drug delivery systems. The IC50 values were mostly lower than 10 μg mL−1, but reached up to 80 μg mL−1 for FA-conjugated chitosan-functionalized PLGA nanoparticles (CPN) with particle size of 207 nm. This was due to the extremely low entrapment efficiency of bicalutamide in CPN (only about 1%).95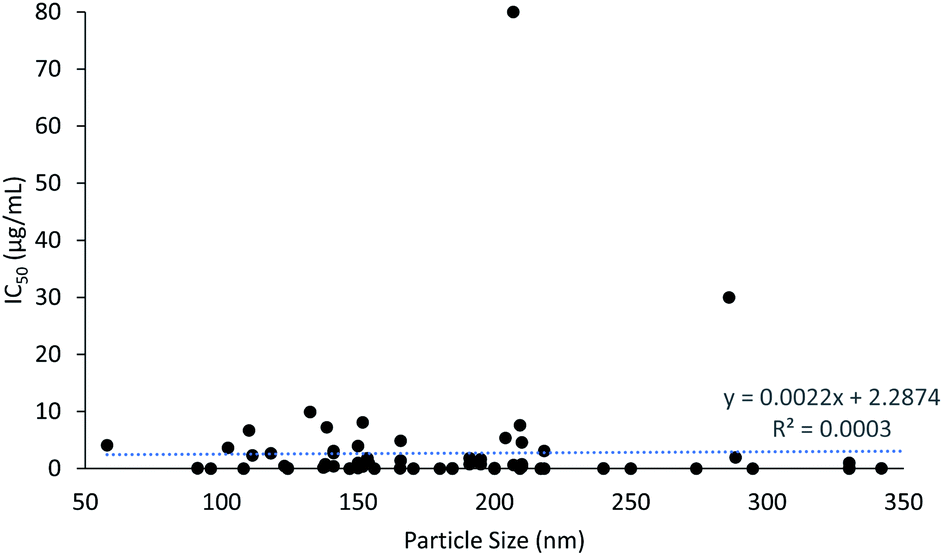 | ||
| Fig. 3 The correlation between particle size and cytotoxicity. | ||
2.3 Zeta potential and cytotoxicity
A plot of the zeta potential of the PLGA nanoparticles against their cytotoxicity IC50 values can be seen in Fig. 4. It is noticeable that PLGA nanoparticles were predominantly in negative charged because of its nature negatively charge of the carboxyl group end chain in PLGA. PLGA nanoparticles ranging from −13.2 to −19.3 mV showed the lowest IC50 value of 0.00031 μg mL−1 at −18.9 mV, with an average IC50 value of 1.15 μg mL−1 (n = 20) (ESI data†). Surface charge of the particle is defined by the absolute value of zeta potential. Zeta potential is a critical factor in designing a drug delivery system because it defines the stability of the nanoparticles. Interestingly, a former study reported that the cytotoxicity potency of PLGA nanoparticles is directly proportional to the absolute zeta potential.102 This is in agreement with a previous report that an absolute value of zeta potential 20 mV or much lower results in nanoparticles with adequate stability.103 The stability of nanoparticles is directly proportional to the absolute value of zeta potential. Since stronger repulsive forces were formed between the nanoparticles with high absolute values of zeta potential, stable nanoparticles with uniform size distributions were produced.104 PLGA is a hydrophobic polymer and can be stabilized by the hydrophilic corona materials such as chitosan.19,47,84 This is because of the absolute zeta potential of PLGA nanoparticles is increasing with the concentration of chitosan and indirectly resulted in increased stability of the PLGA nanoparticles. High stability in nanoparticles ensures that there is no early drug release along the route to the target site, which translates to a higher cytotoxic effect on the targeted site in cancer cells. | ||
| Fig. 4 The correlation between zeta potential and cytotoxicity. | ||
3. Discussion
This systematic review provides an overview of the application of different targeting ligands and active compounds/drugs used in PLGA nanoparticles to achieve active targeting for drug delivery to a particular cell line. This can give insight to researchers in regard to the designing of a potential drug delivery nanoparticle system for different types of cell lines. The potential of treating multiple cell lines using a single formulation makes the designation of a drug delivery system become more flexible in terms of its usage. For instance, a drug delivery nanoparticle system involving the encapsulation of paclitaxel and conjugation of transferrin as the targeting ligand can be used to treat several types of cancer cells, including breast and brain cancer cells. This can be explained by the fact that FR is a well-known biomarker that has a high affinity for FA in cancer cells due to its overexpression on several types of cancer cell lines, while paclitaxel has long been recognized as a mitotic inhibitor for treating various types of cancer cells. Both the drug and targeting ligand are suitable for treating breast and cervical cancers within a single formulation; this provides cost efficiency and compatibility to patients. The systematic review gives a summary of the IC50 of cytotoxicity of different types of cancer cells treated with various formulated PLGA nanoparticles, in which the overview of the cytotoxicity of PLGA nanoparticles is better understood. For instance, the exposure time and IC50 concentration for selected types of cells could serve as references for other researchers to use in planning and identifying their research interests and protocols, which would make these processes easier and save time.The cellular uptake of nanoparticles with the presence of targeting ligands provides a specific binding to the target or cancer site without causing any or less harms to the healthy cells. This phenomenon is attracting more interest from researchers due to its safeness and less toxicity being imposed as a result of its specific controlled release of anti-cancer drugs. These statements are in line with the findings of previous studies that reported that the surface conjugation of a targeting ligand in nanoparticles more greatly enhances the cellular uptake of drugs and cytotoxicity potency compared to non-conjugated nanoparticles.12,21,27 For instance, the cell viability for RGD antibody-conjugated nanoparticles (PTX-PLGA-CSNP-RGD, 35.2%) was significantly lower (p < 0.01) than for non-conjugated nanoparticles (PTX-PLGA-CSNP, 45.7%).12 On top of this, 24 h treatment of A549 cells with FluTax-PLGA-CSNP-RGD, on the basis of the fluorescence intensity of fluorescent paclitaxel (FluTax), revealed higher uptake of 28 pg/1 × 105 cells, compared to PTX-PLGA-CSNP by 28 pg/1 × 105 cells. The reduced cell viability and higher fluorescence intensity of FluTax in RGD antibody-conjugated nanoparticles demonstrates the selective absorption of paclitaxel through integrin receptor-mediated endocytosis compared to non-targeted delivery at the same dose.
Identifying the zeta potential and particle size of PLGA nanoparticles is crucial because an ideal nanoparticle size can provide an EPR effect for cancer cells' uptake of drugs, while proper zeta potential can provide nanoparticles with high stability. From this systematic review, it is evident that nanoparticles <300 nm in size and ∼20 mV in absolute zeta potential are favorable. This is because a decent size (∼200 nm) is required for nanoparticles to load the drugs, while high zeta potential provides a uniform and narrow size distribution, as well as high stability.105 Since nanoparticles are highly stable, the leakage of drugs is negligible during transportation along the target site, which means greater cytotoxicity potency and cellular uptake of nanoparticles.
Challenges are always present in designing a drug delivery system. Although active targeting of nanoparticles reduces toxicity to normal cells, the conjugation of targeting ligands to the nanoparticles might increase the particle size. This is time-consuming, as optimization of the preparation step in making nanoparticles of an ideal size is required. In addition, targeting ligand conjugation might also reduce the release of drugs from the nanoparticles due to the probability that the ligand may serve as an external barrier when releasing the drug from the nanoparticles. The process of conjugating targeting ligands onto the surface of nanoparticles often involves a two-step reaction. This two-step reaction may cause leakage or early release of drugs from the nanoparticles due to the involvement of a reaction incubation period as well as sonication to minimize the agglomeration of nanoparticles.
The studies analyzed in this systematic review involved different factors, including length of exposure time, active compounds and targeting ligands used in assessing cytotoxicity assays of PLGA nanoparticles in a single cell line. Comparisons among the available studies for a particular cell line are difficult to make due to the variations in the factors involved. Hence, there is still a lack of ideally designed PLGA nanoparticle drug delivery systems. Moreover, some in vitro studies lack proper descriptions for data, such as IC50 data for cytotoxicity assessment. Additionally, different in vitro cytotoxicity assays have been used (e.g. MTT, MTS, SRB), which may have resulted in variations in cytotoxicity data.106 Hence, it is difficult to obtain completely accurate results from comparison studies of cytotoxicity for PLGA nanoparticles.
Currently, only 19 drug formulations based on PLGA have been approved by the FDA.107 This negligible number displays that the development of PLGA formulations is very challenging. Thus far, none of the 19 FDA approved PLGA formulations are based on PLGA nanoparticles as they are mainly composed of PLGA microparticles, solid implant and in situ gel. This lacking availability of PLGA nanoformulations further indicates that more evaluations on its efficacy and safety requirements are needed before getting approved for clinical use. Poor drug entrapment efficiency and drug release kinetics from PLGA nanoformulations are the main challenges faced in order to deliver drugs effectively to the target site. For instances, initial burst drug release is the typical issue of low efficacy in designing PLGA nanoformulations. Although PLGA nanoformulations were reported to be safe and selective targeting to the cancer cells in in vitro and animal studies, they are insufficient to prove that the same outcomes will be observed in human trials. Safety concerns of a drug delivery system are the top priority when it comes to establish their application.
Nevertheless, challenges still arise along with opportunities. Despite the great challenges, PLGA nanoparticles are still of interest due to its biocompatibility and particularly, an FDA approved material for drug delivery. Over the past few decades, a significant advancement in the development of PLGA nanoparticles for the application of drug delivery has led to the revolution of pharmaceutical industry. The uniqueness of PLGA nanoformulations, specifically their sustained and controlled drug release as well as cancer targeting properties provide the assurance of great potentials as promising nanocarriers. There are still great potentials in PLGA nanoformulations by modifying their physicochemical properties with better understanding of the physiology of the cancer cells and pharmacokinetics of the drug delivery system. In terms of biocompatibility and toxicity in biological systems, the physicochemical properties of PLGA nanoformulations play an important role.108 Particle size, surface charge and selective targeting properties of PLGA nanoformulations are critical benchmarks to consider when it comes to efficacy. The potential of these state-of-the-art innovations and strategies to develop PLGA nanoformulations are contributing to the advancement of cancer treatment. Nevertheless, intensive evaluations for pharmacokinetics, biodistribution and toxicity are essential before progressing PLGA nanoformulations into clinical studies.
4. Conclusions
In conclusion, PLGA nanoparticles with active targeting properties have higher cytotoxicity and cellular uptake of drugs compared to non-targeting nanoparticles, regardless of the types of cells studied. The size and zeta potential of PLGA nanoparticles play a crucial role in defining the resulting cytotoxicity. Therefore, in future studies, greater focus should be placed on assessing the pharmacodynamic, pharmacokinetic and toxicokinetic profiles of the drug delivery system using cancer-targeting PLGA nanoparticles. In addition, although the listed PLGA nanoparticles exhibited the potency of pharmacological actions based on the in vitro data, there was a lack of in vivo data for many of them. This raises concerns regarding efficacy and safety of usage in the application of PLGA nanoparticles in human trials. Therefore, more relevant in vivo data on the efficacy and toxicity of PLGA nanoparticles are desired before human clinical trials should be commenced, as a pharmaceutical formulation can only be considered successful when both safety of usage and efficacy are guaranteed.Author contributions
Hock Ing Chiu: methodology, validation, formal analysis, investigation, writing – original draft, writing – review & editing, visualization, project administration. Nozlena Abdul Samad: data curation, formal analysis, investigation Lizhen Fang: data curation, formal analysis, investigation. Vuanghao Lim: conceptualization, methodology, validation, formal analysis, writing – review & editing, supervision, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Fundamental Research Grant Scheme (FRGS, 203.CIPPT.6711684) from the Ministry of Higher Education, Malaysia. Hock Ing Chiu would like to express his appreciation to USM for providing him the USM Fellowship Scheme.References
- A. D. Ayub, H. I. Chiu, S. N. A. Mat Yusuf, E. Abd Kadir, S. H. Ngalim and V. Lim, Artif. Cells, Nanomed., Biotechnol., 2019, 47, 353–369 CrossRef CAS PubMed.
- H. I. Chiu, A. D. Ayub, S. N. A. Mat Yusuf, N. Yahaya, E. Abd Kadir and V. Lim, Pharmaceutics, 2020, 12, 38 CrossRef CAS PubMed.
- A. Schoubben, M. Ricci and S. Giovagnoli, J. Pharm. Invest., 2019, 49, 381–404 CrossRef CAS.
- S. Rezvantalab and M. Keshavarz Moraveji, RSC Adv., 2019, 9, 2055–2072 RSC.
- K. T. Kim, J. Y. Lee, D. D. Kim, I. S. Yoon and H. J. Cho, Pharmaceutics, 2019, 11, 280 CrossRef CAS PubMed.
- J. Yoo, C. Park, G. Yi, D. Lee and H. Koo, Cancers, 2019, 11, 640 CrossRef CAS PubMed.
- D. Moher, L. Shamseer, M. Clarke, D. Ghersi, A. Liberati, M. Petticrew, P. Shekelle, L. A. Stewart and PRISMA-P Group, Syst. Rev., 2015, 4, 1–9 CrossRef PubMed.
- N. A. Hanan, H. I. Chiu, M. R. Ramachandran, W. H. Tung, N. N. Mohamad Zain, N. Yahaya and V. Lim, Int. J. Mol. Sci., 2018, 19, 1725 CrossRef PubMed.
- V. Lim, E. Schneider, H. Wu and I. H. Pang, Nutrients, 2018, 10, 1580 CrossRef PubMed.
- Y. Cui, Q. Xu, P. K. H. Chow, D. Wang and C. H. Wang, Biomaterials, 2013, 34, 8511–8520 CrossRef CAS PubMed.
- Y. Zheng, B. Yu, W. Weecharangsan, L. Piao, M. Darby, Y. Mao, R. Koynova, X. Yang, H. Li, S. Xu, L. J. Lee, Y. Sugimoto, R. W. Brueggemeier and R. J. Lee, Int. J. Pharm., 2010, 390, 234–241 CrossRef CAS PubMed.
- A. Babu, N. Amreddy, R. Muralidharan, G. Pathuri, H. Gali, A. Chen, Y. D. Zhao, A. Munshi and R. Ramesh, Sci. Rep., 2017, 7, 1–17 CrossRef CAS PubMed.
- C. Wang, P. C. Ho and L. Y. Lim, Int. J. Pharm., 2010, 400, 201–210 CrossRef CAS PubMed.
- P. J. Kennedy, F. Sousa, D. Ferreira, C. Pereira, M. Nestor, C. Oliveira, P. L. Granja and B. Sarmento, Acta Biomater., 2018, 81, 208–218 CrossRef CAS PubMed.
- Z. Yang, H. Luo, Z. Cao, Y. Chen, J. Gao, Y. Li, Q. Jiang, R. Xu and J. Liu, Nanoscale, 2016, 8, 11543–11558 RSC.
- Z. Zhang, S. Tan and S. S. Feng, Biomaterials, 2012, 33, 4889–4906 CrossRef CAS PubMed.
- S. Di-Wen, G. Z. Pan, L. Hao, J. Zhang, Q. Z. Xue, P. Wang and Q. Z. Yuan, Int. J. Pharm., 2016, 500, 54–61 CrossRef PubMed.
- A. Abou-ElNaga, G. Mutawa, I. M. El-Sherbiny, H. Abd-ElGhaffar, A. A. Allam, J. Ajarem and S. A. Mousa, Int. J. Mol. Sci., 2017, 18, 1–14 Search PubMed.
- A. Shanavas, S. Sasidharan, D. Bahadur and R. Srivastava, J. Colloid Interface Sci., 2017, 486, 112–120 CrossRef CAS PubMed.
- M. N. Wente, A. Jain, E. Kono, P. O. Berberat, T. Giese, H. A. Reber, H. Friess, M. W. Büchler, R. E. Reiter and O. J. Hines, Pancreas, 2005, 31, 119–125 CrossRef CAS PubMed.
- Y. Ling, K. Wei, Y. Luo, X. Gao and S. Zhong, Biomaterials, 2011, 32, 7139–7150 CrossRef CAS PubMed.
- T. Lei, S. Srinivasan, Y. Tang, R. Manchanda, A. Nagesetti, A. Fernandez-Fernandez and A. J. McGoron, Nanomedicine, 2011, 7, 324–332 CrossRef CAS PubMed.
- M. F. Frasco, G. M. Almeida, F. Santos-Silva, M. Do Carmo Pereira and M. A. N. Coelho, J. Biomed. Mater. Res., Part A, 2015, 103, 1476–1484 CrossRef PubMed.
- S. Guo, C. M. Lin, Z. Xu, L. Miao, Y. Wang and L. Huang, ACS Nano, 2014, 8, 4996–5009 CrossRef CAS PubMed.
- C. Zhang, T. An, D. Wang, G. Wan, M. Zhang, H. Wang, S. Zhang, R. Li, X. Yang and Y. Wang, J. Controlled Release, 2016, 226, 193–204 CrossRef CAS PubMed.
- Y. Kaneo, T. Tanaka, T. Nakano and Y. Yamaguchi, J. Controlled Release, 2001, 70, 365–373 CrossRef CAS PubMed.
- M. R. Gill, J. U. Menon, P. J. Jarman, J. Owen, I. Skaripa-Koukelli, S. Able, J. A. Thomas, R. Carlisle and K. A. Vallis, Nanoscale, 2018, 10, 10596–10608 RSC.
- D. T. Martin, C. J. Hoimes, H. Z. Kaimakliotis, C. J. Cheng, K. Zhang, J. Liu, M. A. Wheeler, W. K. Kelly, G. N. Tew, W. M. Saltzman and R. M. Weiss, Nanomedicine, 2013, 9, 1124–1134 CrossRef CAS PubMed.
- H. He, E. Markoutsa, Y. Zhan, J. Zhang and P. Xu, Acta Biomater., 2017, 59, 181–191 CrossRef CAS PubMed.
- V. Lim, K. Khiang Peh and S. Sahudin, Int. J. Mol. Sci., 2013, 14, 24670–24691 CrossRef PubMed.
- S. N. A. Mat Yusuf, Y. M. Ng, A. D. Ayub, S. H. Ngalim and V. Lim, Polymers, 2017, 9, 311 CrossRef PubMed.
- Y. M. Ng, S. N. A. Mat Yusuf, H. I. Chiu and V. Lim, Pharmaceutics, 2020, 12, 1–20 CrossRef PubMed.
- B. Wu, P. Yu, C. Cui, M. Wu, Y. Zhang, L. Liu, C. X. Wang, R. X. Zhuo and S. W. Huang, Biomater. Sci., 2015, 3, 655–664 RSC.
- L. Wang, Y. Hao, H. Li, Y. Zhao, D. Meng, D. Li, J. Shi, H. Zhang, Z. Zhang and Y. Zhang, J. Drug Targeting, 2015, 23, 832–846 CrossRef CAS PubMed.
- Y. C. Kuo and Y. C. Chen, Int. J. Pharm., 2015, 479, 138–149 CrossRef CAS PubMed.
- N. Shah, K. Chaudhari, P. Dantuluri, R. S. R. Murthy and S. Das, J. Drug Targeting, 2009, 17, 533–542 CrossRef CAS PubMed.
- C. Lei, P. Davoodi, W. Zhan, P. K. H. Chow and C. H. Wang, J. Pharm. Sci., 2019, 108, 1736–1745 CrossRef CAS PubMed.
- A. Aravind, P. Jeyamohan, R. Nair, S. Veeranarayanan, Y. Nagaoka, Y. Yoshida, T. Maekawa and D. S. Kumar, Biotechnol. Bioeng., 2012, 109, 2920–2931 CrossRef CAS PubMed.
- Z. Yang, H. Luo, Z. Cao, Y. Chen, J. Gao, Y. Li, Q. Jiang, R. Xu and J. Liu, Nanoscale, 2016, 8, 11543–11558 RSC.
- P. Liu, N. Chen, L. Yan, F. Gao, D. Ji, S. Zhang, L. Zhang, Y. Li and Y. Xiao, Carbohydr. Polym., 2019, 213, 17–26 CrossRef CAS PubMed.
- R. Huey, B. O'Hagan, P. McCarron and S. Hawthorne, Int. J. Pharm., 2017, 525, 12–20 CrossRef CAS PubMed.
- Z. Jamali, M. Khoobi, S. M. Hejazi, N. Eivazi, S. Abdolahpour, F. Imanparast, H. Moradi-Sardareh and M. Paknejad, Photodiagn. Photodyn. Ther., 2018, 23, 190–201 CrossRef CAS PubMed.
- J. Gao, G. Kou, H. Wang, H. Chen, B. Li, Y. Lu, D. Zhang, S. Wang, S. Hou, W. Qian, J. Dai, J. Zhao, Y. Zhong and Y. Guo, Breast Cancer Res. Treat., 2009, 115, 29–41 CrossRef CAS PubMed.
- C. Mattu, R. M. Pabari, M. Boffito, S. Sartori, G. Ciardelli and Z. Ramtoola, Eur. J. Pharm. Biopharm., 2013, 85, 463–472 CrossRef CAS PubMed.
- M. Zheng, P. Gong, D. Jia, C. Zheng, Y. Ma and L. Cai, Nano LIFE, 2011, 02, 1250002 CrossRef.
- A. Aravind, R. Nair, S. Raveendran, S. Veeranarayanan, Y. Nagaoka, T. Fukuda, T. Hasumura, H. Morimoto, Y. Yoshida, T. Maekawa and D. S. Kumar, J. Magn. Magn. Mater., 2013, 344, 116–123 CrossRef CAS.
- H. Chen, L. Q. Xie, J. Qin, Y. Jia, X. Cai, W. Bin Nan, W. Yang, F. Lv and Q. Q. Zhang, Colloids Surf., B, 2016, 138, 1–9 CrossRef CAS PubMed.
- Z. Wang and P. C. Ho, Small, 2010, 6, 2576–2583 CrossRef CAS PubMed.
- S. Taghavi, M. Ramezani, M. Alibolandi, K. Abnous and S. M. Taghdisi, Cancer Lett., 2017, 400, 1–8 CrossRef CAS PubMed.
- C. C. Chuang, C. C. Cheng, P. Y. Chen, C. Lo, Y. N. Chen, M. H. Shih and C. W. Chang, Int. J. Nanomed., 2019, 14, 181–193 CrossRef CAS PubMed.
- S. Acharya, F. Dilnawaz and S. K. Sahoo, Biomaterials, 2009, 30, 5737–5750 CrossRef CAS PubMed.
- F. Fathian kolahkaj, K. Derakhshandeh, F. Khaleseh, A. H. Azandaryani, K. Mansouri and M. Khazaei, J. Drug Delivery Sci. Technol., 2019, 53, 101136 CrossRef CAS.
- E. Muntimadugu, R. Kumar, S. Saladi, T. A. Rafeeqi and W. Khan, Colloids Surf., B, 2016, 143, 532–546 CrossRef CAS PubMed.
- P. Dadras, F. Atyabi, S. Irani, L. Ma’mani, A. Foroumadi, Z. H. Mirzaie, M. Ebrahimi and R. Dinarvand, Eur. J. Pharm. Sci., 2017, 97, 47–54 CrossRef CAS PubMed.
- A. Bahreyni, M. Alibolandi, M. Ramezani, A. Sarafan Sadeghi, K. Abnous and S. M. Taghdisi, Colloids Surf., B, 2019, 175, 231–238 CrossRef CAS PubMed.
- L. Li, X. Di, M. Wu, Z. Sun, L. Zhong, Y. Wang, Q. Fu, Q. Kan, J. Sun and Z. He, Nanomedicine, 2017, 13, 987–998 CrossRef CAS PubMed.
- K. Yu, J. Zhao, Z. Zhang, Y. Gao, Y. Zhou, L. Teng and Y. Li, Int. J. Pharm., 2016, 497, 78–87 CrossRef CAS PubMed.
- J. H. Park, J. Y. Lee, U. Termsarasab, I. S. Yoon, S. H. Ko, J. S. Shim, H. J. Cho and D. D. Kim, Int. J. Pharm., 2014, 473, 426–433 CrossRef CAS PubMed.
- A. Halder, P. Mukherjee, S. Ghosh, S. Mandal, U. Chatterji and A. Mukherjee, Mater. Today: Proc., 2018, 5, 9698–9705 CAS.
- H. Wang, P. Agarwal, S. Zhao, R. X. Xu, J. Yu, X. Lu and X. He, Biomaterials, 2015, 72, 74–89 CrossRef CAS PubMed.
- J. B. Du, Y. Cheng, Z. H. Teng, M. L. Huan, M. Liu, H. Cui, B. Le Zhang and S. Y. Zhou, Mol. Pharm., 2016, 13, 1711–1722 CrossRef CAS PubMed.
- S. I. Thamake, S. L. Raut, Z. Gryczynski, A. P. Ranjan and J. K. Vishwanatha, Biomaterials, 2012, 33, 7164–7173 CrossRef CAS PubMed.
- H. Wang, Y. Zhao, H. Wang, J. Gong, H. He, M. C. Shin, V. C. Yang and Y. Huang, J. Controlled Release, 2014, 192, 47–56 CrossRef CAS PubMed.
- Y. Patil, T. Sadhukha, L. Ma and J. Panyam, J. Controlled Release, 2009, 136, 21–29 CrossRef CAS PubMed.
- Y. B. Patil, S. K. Swaminathan, T. Sadhukha, L. Ma and J. Panyam, Biomaterials, 2010, 31, 358–365 CrossRef CAS PubMed.
- M. Esfandyari-Manesh, S. H. Mostafavi, R. F. Majidi, M. N. Koopaei, N. S. Ravari, M. Amini, B. Darvishi, S. N. Ostad, F. Atyabi and R. Dinarvand, Daru, J. Pharm. Sci., 2015, 23, 1–8 CrossRef CAS PubMed.
- C. Chittasupho, S. X. Xie, A. Baoum, T. Yakovleva, T. J. Siahaan and C. J. Berkland, Eur. J. Pharm. Sci., 2009, 37, 141–150 CrossRef CAS PubMed.
- C. Chittasupho, K. Lirdprapamongkol, P. Kewsuwan and N. Sarisuta, Eur. J. Pharm. Biopharm., 2014, 88, 529–538 CrossRef CAS PubMed.
- Y. Guo, L. Wang, P. Lv and P. Zhang, Oncol. Lett., 2015, 9, 1065–1072 CrossRef PubMed.
- K. E. Park, Y. W. Noh, A. Kim and Y. T. Lim, Carbohydr. Polym., 2017, 157, 476–483 CrossRef CAS PubMed.
- J. Wu, J. Zhang, C. Deng, F. Meng, R. Cheng and Z. Zhong, ACS Appl. Mater. Interfaces, 2017, 9, 3985–3994 CrossRef CAS PubMed.
- J. Wu, C. Deng, F. Meng, J. Zhang, H. Sun and Z. Zhong, J. Controlled Release, 2017, 259, 76–82 CrossRef CAS PubMed.
- N. Karra, T. Nassar, A. N. Ripin, O. Schwob, J. Borlak and S. Benita, Small, 2013, 9, 4221–4236 CrossRef CAS PubMed.
- X. Lin, S. Z. Yan, S. S. Qi, Q. Xu, S. S. Han, L. Y. Guo, N. Zhao, S. L. Chen and S. Q. Yu, Front. Pharmacol., 2017, 8, 1–16 Search PubMed.
- L. Li, D. Xiang, S. Shigdar, W. Yang, Q. Li, J. Lin, K. Liu and W. Duan, Int. J. Nanomed., 2014, 9, 1083–1096 CrossRef PubMed.
- Z. L. Liu, L. F. Li, S. Sen Xia, H. P. Tian, Z. H. Yan, G. J. Zhang, T. Zhou and Y. He, J. Drug Delivery Sci. Technol., 2019, 52, 1–7 CrossRef CAS.
- F. Fay, K. M. McLaughlin, D. M. Small, D. A. Fennell, P. G. Johnston, D. B. Longley and C. J. Scott, Biomaterials, 2011, 32, 8645–8653 CrossRef CAS PubMed.
- J. Son, S. M. Yang, G. Yi, Y. J. Roh, H. Park, J. M. Park, M. G. Choi and H. Koo, Biochem. Biophys. Res. Commun., 2018, 498, 523–528 CrossRef CAS PubMed.
- D.-W. Sun, G.-Z. Pan, L. Hao, J. Zhang, Q.-Z. Xue, P. Wang and Q.-Z. Yuan, Int. J. Pharm., 2016, 500, 54–61 CrossRef CAS PubMed.
- S. Manoochehri, B. Darvishi, G. Kamalinia, M. Amini, M. Fallah, S. N. Ostad, F. Atyabi and R. Dinarvand, Daru, J. Pharm. Sci., 2013, 21, 1–10 Search PubMed.
- Y. Dai, H. Xing, F. Song, Y. Yang, Z. Qiu, X. Lu, Q. Liu, S. Ren, X. Chen and N. Li, J. Pharm. Sci., 2016, 105, 2949–2958 CrossRef CAS.
- M. Nasr, N. Nafee, H. Saad and A. Kazem, Eur. J. Pharm. Biopharm., 2014, 88, 216–225 CrossRef CAS PubMed.
- D. Y. Gao, T. T. Lin, Y. C. Sung, Y. C. Liu, W. H. Chiang, C. C. Chang, J. Y. Liu and Y. Chen, Biomaterials, 2015, 67, 194–203 CrossRef CAS PubMed.
- X. Song, J. Wang, Y. Xu, H. Shao and J. Gu, Colloids Surf., B, 2019, 180, 110–117 CrossRef CAS PubMed.
- X. Song, J. You, H. Shao and C. Yan, Colloids Surf., B, 2018, 169, 289–297 CrossRef CAS PubMed.
- E. Gullotti, J. Park and Y. Yeo, Pharm. Res., 2013, 30, 1956–1967 CrossRef CAS PubMed.
- R. Domínguez-Ríos, D. R. Sánchez-Ramírez, K. Ruiz-Saray, P. E. Oceguera-Basurto, M. Almada, J. Juárez, A. Zepeda-Moreno, A. del Toro-Arreola, A. Topete and A. Daneri-Navarro, Colloids Surf., B, 2019, 178, 199–207 CrossRef.
- S. Srinivasan, R. Manchanda, T. Lei, A. Nagesetti, A. Fernandez-Fernandez and A. J. McGoron, J. Photochem. Photobiol., B, 2014, 136, 81–90 CrossRef CAS.
- W. Hasan, K. Chu, A. Gullapalli, S. S. Dunn, E. M. Enlow, J. C. Luft, S. Tian, M. E. Napier, P. D. Pohlhaus, J. P. Rolland and J. M. Desimone, Nano Lett., 2012, 12, 287–292 CrossRef CAS PubMed.
- D. Bi, L. Zhao, R. Yu, H. Li, Y. Guo, X. Wang and M. Hana, Drug Delivery, 2018, 25, 564–575 CrossRef CAS PubMed.
- W. Punfa, S. Yodkeeree, P. Pitchakarn, C. Ampasavate and P. Limtrakul, Acta Pharmacol. Sin., 2012, 33, 823–831 CrossRef CAS.
- W. Punfa, P. Pitchakarn, S. Yodkeeree, P. Limtrakul, S. Suzuki, T. Naiki and S. Takahashi, Asian Pac. J. Cancer Prev., 2014, 15, 9249–9258 CrossRef PubMed.
- A. M. Lopes, K. Y. Chen and D. T. Kamei, Mater. Sci. Eng., C, 2017, 73, 373–380 CrossRef CAS PubMed.
- X. Gao, Y. Luo, Y. Wang, J. Pang, C. Liao, H. Lu and Y. Fang, Int. J. Nanomed., 2012, 7, 4037–4051 CrossRef CAS PubMed.
- N. L. Dhas, P. P. Ige and R. R. Kudarha, Powder Technol., 2015, 283, 234–245 CrossRef CAS.
- B. Sivakumar, R. G. Aswathy, R. Romero-Aburto, T. Mitcham, K. A. Mitchel, Y. Nagaoka, R. R. Bouchard, P. M. Ajayan, T. Maekawa and D. N. Sakthikumar, Biomater. Sci., 2017, 5, 432–443 RSC.
- B. Sivakumar, R. G. Aswathy, Y. Nagaoka, S. Iwai, K. Venugopal, K. Kato, Y. Yoshida, T. Maekawa and D. N. Sakthi Kumar, RSC Adv., 2013, 3, 20579–20598 RSC.
- M. F. Frasco, G. M. Almeida, F. Santos-Silva, M. Do Carmo Pereira and M. A. N. Coelho, J. Biomed. Mater. Res., Part A, 2015, 103, 1476–1484 CrossRef PubMed.
- S. Guo, C. M. Lin, Z. Xu, L. Miao, Y. Wang, L. Huang, S. Guo, Z. Xu, Y. Wang and L. Miao, ACS Nano, 2014, 8, 4996–5009 CrossRef CAS PubMed.
- Z. Wang, W. K. Chui and P. C. Ho, Pharm. Res., 2011, 28, 585–596 CrossRef CAS PubMed.
- D. T. Martin, C. J. Hoimes, H. Z. Kaimakliotis, C. J. Cheng, K. Zhang, J. Liu, M. A. Wheeler, W. K. Kelly, G. N. Tew, W. M. Saltzman and R. M. Weiss, Nanomedicine, 2013, 9, 1124–1134 CrossRef CAS PubMed.
- W. Hasan, K. Chu, A. Gullapalli, S. S. Dunn, E. M. Enlow, J. C. Luft, S. Tian, M. E. Napier, P. D. Pohlhaus, J. P. Rolland and J. M. Desimone, Nano Lett., 2012, 12, 287–292 CrossRef CAS PubMed.
- S. Honary and F. Zahir, Trop. J. Pharm. Res., 2013, 12, 265–273 Search PubMed.
- C. Wen, Q. Yuan, H. Liang and F. Vriesekoop, Carbohydr. Polym., 2014, 112, 695–700 CrossRef CAS PubMed.
- D. Zukancic, E. J. A. Suys, E. H. Pilkington, A. Algarni, H. Al-Wassiti and N. P. Truong, Pharmaceutics, 2020, 12, 1–16 CrossRef PubMed.
- M. Mohd Narawi, H. I. Chiu, Y. K. Yong, N. N. Mohamad Zain, M. R. Ramachandran, C. L. Tham, S. F. Samsurrijal and V. Lim, Front. Pharmacol., 2020, 11, 1–15 CrossRef PubMed.
- K. Park, S. Skidmore, J. Hadar, J. Garner, H. Park, A. Otte, B. K. Soh, G. Yoon, D. Yu, Y. Yun, B. K. Lee, X. Jiang and Y. Wang, J. Controlled Release, 2019, 304, 125–134 CrossRef CAS PubMed.
- P. N. Navya, A. Kaphle, S. P. Srinivas, S. K. Bhargava, V. M. Rotello and H. K. Daima, Nano Convergence, 2019, 6, 1–30 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary data for Fig. 3 and 4. See DOI: 10.1039/d1ra00074h |
| This journal is © The Royal Society of Chemistry 2021 |