
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a thermally stable Pt catalyst by redispersion between CeO2 and Al2O3
Li Lanab,
Xin Huangc,
Weiqi Zhouc,
Hongmei Lib,
Junhuai Xiang a,
Shanhu Chen*d and
Yaoqiang Chene
a,
Shanhu Chen*d and
Yaoqiang Chene
aCollege of Materials and Mechatronics, Jiangxi Science and Technology Normal University, Nanchang, 330013, P.R. China
bCollege of Food and Bioengineering, Chengdu University, Chengdu, 610064, P.R. China
cCollege of Mechanical Engineering, Chengdu University, Chengdu, 610064, P.R. China
dCollege of Chemistry and Chemical Engineering, Jiangxi Science and Technology Normal University, Nanchang, 330013, P.R. China. E-mail: cshscu@163.com
eCollege of Chemistry, Sichuan University, Chengdu, 610064, P.R. China
First published on 10th February 2021
Abstract
For catalytic systems consisting of Pt as the active component and CeO2–Al2O3 as the support material, the metal–support interaction between the Pt and CeO2 components is widely applied to inhibit aggregation of Pt species and thus enhance the thermal stability of the catalyst. In this work, a highly thermostable Pt catalyst was prepared by modifying the synthesis procedure for conventional Pt/CeO2/Al2O3 (Pt/Ce/Al) catalyst, that is, the CeO2 component was introduced after deposition of Pt on Al2O3. The obtained CeO2/Pt/Al2O3 (Ce/Pt/Al) catalyst exhibits significantly different aging behavior. During the hydrothermal aging process, redispersion of Pt species from the surface of Al2O3 to the surface of CeO2 occurs, resulting in a stronger metal–support interaction between Pt and CeO2. Thus, the formed Pt–O–Ce bond could act as an anchor to retard aggregation of Pt species and help Pt species stay at a more oxidative state. Consequently, excellent reduction capability and superior three-way catalytic performance are acquired by Ce/Pt/Al-a after hydrothermal aging treatment.
Introduction
Supported noble metals are some of the most important and widely applied nanoparticles in the field of catalysis, especially in exhaust gas after-treatment systems.1,2 It is widely accepted that the catalytic performance generally relies on the dispersion, configuration, and valence state of the noble metals, such as three-way catalytic (TWC) performance on supported platinum-group metals (Pt, Pd, Rh).3–5 In practical applications of TWC, Pt and Pd are usually used as active components due to their outstanding low-temperature oxidation activity of CO/HC, while Rh usually accompanies them owing to its prominent NO purification efficiency.6,7 Recently, persistent increases in the price of noble metals have resulted in increasingly heavy cost pressures for TWCs. Therefore, without sacrificing the catalytic performance, the development of catalysts with a higher proportion of Pt is crucial for cost reduction, because of the comparatively lower price of Pt.Besides the properties of noble metals, the support material is equally important in the catalytic performance of TWCs.1 As is well known, Al2O3 with high surface area is a commonly used support material to achieve favorable noble metal dispersion of the catalyst.8–12 In most cases γ-Al2O3 is employed. However, phase transformation of γ-Al2O3 to other transitional phases (θ/δ) or even α-Al2O3 takes place under high temperature conditions, leading to a significantly reduced surface area. Hence, stabilizing agents, such as La2O3, BaO, CeO2, ZrO2, etc., are often added to enhance the thermal stability of Al2O3, as well as its supported catalyst system.6
On the other hand, in exhaust-gas purification catalysts, CeO2 usually serves as an oxygen buffer, as demonstrated by in situ monitoring of a catalyst bed based on a microwave cavity perturbation method.13,14 Furthermore, CeO2-containing components are also reported to have the ability to promote the noble metal dispersion and enhance the thermal stability of the Al2O3 support.6,15–19 What is more, to the best of our knowledge, CeO2 is recognized as a favorable stabilizer of Pt owing to the strong metal–support interaction, that is, the formation of the Pt–O–Ce bond could act as an anchor to inhibit agglomeration of Pt particles.20–23 In addition, some researchers have reported that redispersion of Pt on CeO2 may take place when submitted to high-temperature treatment, which could also promote the thermal stability of Pt species.24–27 However, as we know, pure CeO2 would undergo severe sintering at temperatures higher than 800 °C, which consequently leads to a dramatically reduced surface area and deterioration in oxygen mobility, accompanied by serious aggregation and even encapsulation of the noble metal species.28–30 Nevertheless, when CeO2 is highly dispersed on the surface of Al2O3, remarkably enhanced thermal stability can be obtained owing to the synergistic interaction between CeO2 and Al2O3.31–34 In addition, it is found that when Pt is first loaded onto Al2O3 and subsequently physically mixed with the CeO2 component, migration of Pt from Al2O3 to CeO2 could be observed at elevated temperatures (800 °C), which could promote the dispersion and anti-aging resistance of Pt.15 Accordingly, it can be inferred that the combination of CeO2 and Al2O3 would be a more desirable candidate to support the Pt catalyst.
Therefore, to take full advantage of the favorable metal–support interaction, along with the synergistic effect between CeO2 and Al2O3, a modified synthesis route was employed to prepare Pt catalyst in this work. By successively loading a salt solution of Pt and precursor of CeO2 onto Al2O3, an advanced CeO2–Al2O3-supported Pt catalyst is developed.
Experimental section
Catalyst preparation
Pt/CeO2/Al2O3 catalyst was synthesized by first impregnating a precursor of CeO2 on Al2O3 powders, followed by loading Pt species on the CeO2/Al2O3 support. The weight ratio of CeO2/Al2O3 was controlled at 1/5 and the loading content of Pt was 2 wt%. The details were as follows. First, 6.43 g of Ce(NH4)2(NO3)6 was dissolved in distilled water, then 10 g of γ-Al2O3 powders (provided by Solvay Company, the specific surface area was measured as 187 m2 g−1) was introduced under vigorous stirring. Then, the mixture was dried at 100 °C for 3 h in an oven and calcined at 550 °C for 3 h in a muffle furnace. Subsequently, 2.4 g of Pt(NO3)2 salt solution (purchased from Shanghai Jiuyue Chemical Co. Ltd., the concentration of Pt was 10 wt%) was impregnated on the obtained CeO2/Al2O3 powders, followed by drying at 100 °C for 3 h and calcining at 550 °C for 3 h to obtain Pt/CeO2/Al2O3 powders, which were then added into distilled water and the resulting slurry was coated onto cordierite monoliths (400 cells per in2, Corning, USA) to obtain the monolithic catalyst Pt/CeO2/Al2O3 (labeled as Pt/Ce/Al).The advanced CeO2/Pt/Al2O3 (labeled as Ce/Pt/Al) catalyst with the same composition was prepared by modifying the synthesis procedure for Pt/Ce/Al. Specifically, the impregnation sequence of Pt(NO3)2 solution and CeO2 precursor was exchanged, that is, Pt(NO3)2 solution was first loaded onto γ-Al2O3 powders, and then Ce(NH4)2(NO3)6 solution was introduced to proceed with the impregnation on the Pt/Al2O3 powders. The drying and calcining processes, together with the coating process, were all the same as for Pt/Ce/Al.
In addition, to evaluate the anti-aging resistance of the catalysts, the above two catalysts were further submitted to hydrothermal treatment in an atmosphere consisting of 10 vol% of H2O in air at 900 °C for 10 h, and the catalysts obtained are hereafter referred to as Pt/Ce/Al-a and Ce/Pt/Al-a, respectively. In addition, for comparison purposes, Pt/Al and Pt/Al-a catalysts were also synthesized.
Catalyst characterization
Phase identification of the catalysts was performed using a Rigaku DX-2500 diffractometer and CuKα was applied as the radiation source. The experiment was conducted under conditions of 40 mV and 100 mA and X-ray diffraction (XRD) patterns within the 2θ range 10–90° were obtained, with a step size of 0.02°.Raman spectra of the catalysts were collected on a Renishaw inVia reflex Raman spectrometer, using a Nd:YAG laser with an excitation wavelength of 532 nm. During the whole testing process, the laser power was fixed at 20 mW, and the resolution of the equipment was 1 cm−1.
The surface elemental information was analyzed by an ESCALAB 250 XI electron spectrometer using monochromatic AlKα (binding energy (BE) = 1486.6 eV) as the radiation. The data for Ce 3d, Al 2p, O 1s and Pt 4d were collected, and, during the experimental process, the binding energies were calibrated by fixing the C 1s binding energy at 284.8 eV.
The morphology of the samples was observed on a ZEISS Libra 200 FE transmission electron microscope. Before the measurement, the powder specimen was introduced into ethanol and dispersed by ultrasonic agitation. Both the transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM) images were obtained at an accelerating voltage of 200 kV.
The CO Fourier transform-infrared (FTIR) experiment was carried out using a Nicolet 6700 FTIR spectrometer (ThermoFisher Scientific). Prior to the measurement, the samples were reduced in H2 atmosphere (25 mL min−1) at 400 °C for 1 h and then cooled to room temperature. Afterwards, the atmosphere was switched to N2 (25 mL min−1) and was held for 30 min. Subsequently, CO (25 mL min−1) adsorption proceeded, followed by sweeping with N2 (25 mL min−1) for 30 min, and eventually the CO adsorption spectra were collected.
Hydrogen temperature-programmed reduction (H2-TPR) was conducted to estimate the reduction features of the various catalysts. Before the experiment, the samples were purged in N2 (25 mL min−1) at 400 °C for 1 h and then cooled to room temperature. Thereafter, a mixture of H2 in N2 (5 vol% H2, 25 mL min−1) was passed through, and the reduction experiment proceeded by heating the samples at a constant rate of 5 °C min−1. In this process the H2 consumption was continuously monitored by a thermal conductivity detector.
Evaluation of the three-way catalytic performance
The evaluation of the three-way catalytic performance was conducted in a continuous flow reactor. A simulated exhaust gas mixture, which was composed of CO (4600 ppm), C3H6 (220 ppm), C3H8 (110 ppm), NO (1250 ppm), H2 (1533 ppm), H2O (10%), CO2 (12%), O2 (adjustable) and N2 as the balance, was passed through the monolithic catalyst. The conversions of CO, HC and NO in a temperature range of 100–500 °C over fresh and aged catalysts were evaluated by fixing the O2 concentration at 3600 ppm to ensure a so-called stoichiometric air-to-fuel circumstance. Before the measurement, the catalysts were pretreated under the simulated exhaust gas mixture at 550 °C for at least 1 h. The compositions of the inlet and outlet gas mixtures were analyzed by FTIR spectrometry (Antaris IGS, ThermoFisher Scientific).Results and discussion
XRD analysis results
Fig. 1 presents the XRD patterns of the fresh and aged catalysts. From Fig. 1(a) no obvious differences can be observed for fresh Pt/Ce/Al and Ce/Pt/Al, and only two phases, including cubic CeO2 and γ-Al2O3 phases, are identified for both samples. Because CeO2 can scatter X-rays, it causes a decrease in the intensities of the γ-Al2O3 peaks.15 In addition, it is worth noting that no peaks related to Pt species are found, implying that Pt species exist in a highly dispersed state or the particle size of Pt is comparatively small, and so beyond the detection limit of the XRD technique.21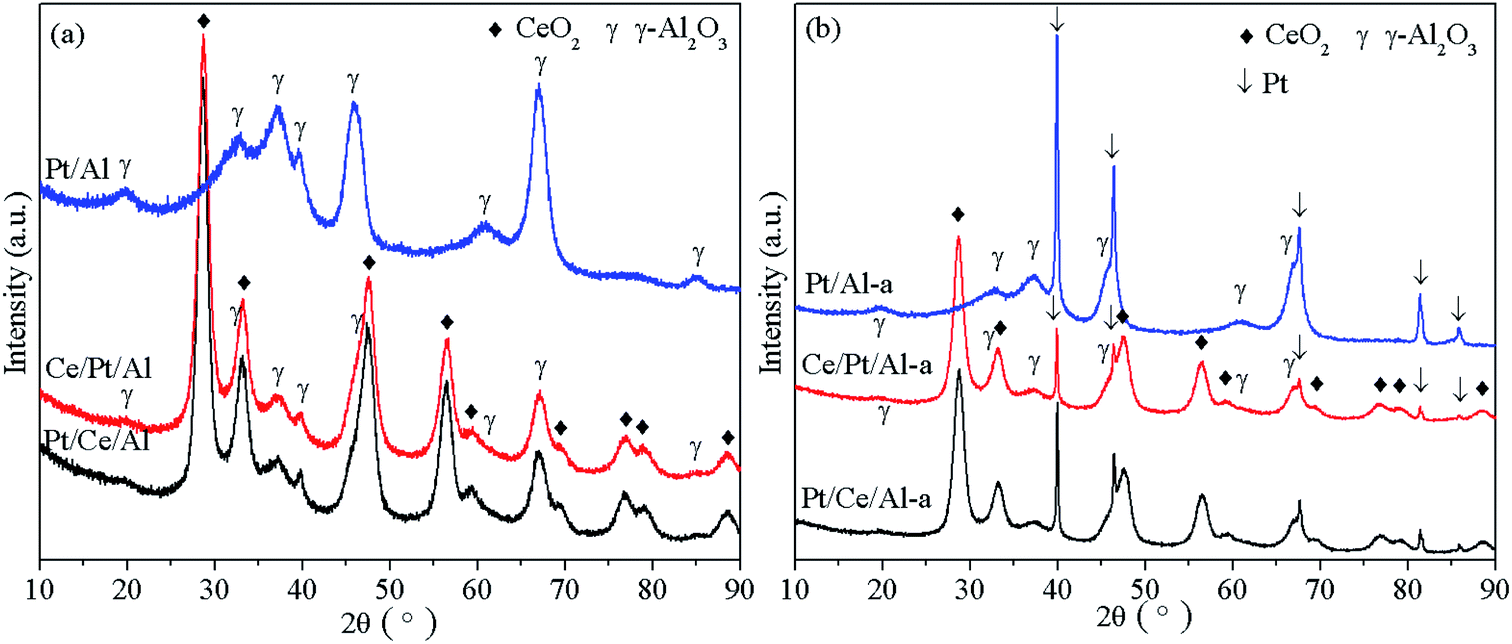 | ||
| Fig. 1 XRD patterns of the (a) fresh and (b) aged catalysts. | ||
After aging treatment, significant peak sharpening is found for all samples, as seen in Fig. 1(b), indicating that grain growth has taken place during the hydrothermal aging treatment process. However, it should be pointed out that besides the characteristic peaks of cubic CeO2 and γ-Al2O3 phases, appreciable peaks corresponding to (111), (200), (220), (311) and (222) facets of metallic Pt are clearly observed at 2θ values of 39.8°, 46.2°, 67.4°, 81.3° and 85.7° for all samples.24 However, it is also noticeable that the intensities of the Pt peaks show an obvious decreasing tendency of Pt/Al-a > Pt/Ce/Al-a > Ce/Pt/Al-a, which can be ascribed to the strong metal–support interaction in the CeO2-containing samples. This demonstrates that an even stronger metal–support interaction existed in Ce/Pt/Al-a compared to Pt/Ce/Al-a, which consequently leads to higher dispersion and smaller particle size for Pt in Ce/Pt/Al-a.
Raman characterization
Fig. 2 displays the Raman spectra of the fresh and aged Pt/Ce/Al and Ce/Pt/A catalysts. The most dominant Raman band appears at ca. 460 cm−1, which is generally attributed to the triply degenerate F2g mode of cubic fluorite CeO2, while other signals appearing at ca. 153, 196, 550 and 680 cm−1 are all associated with Pt species. According to the literature,1,35,36 the band located at 153 cm−1 is ascribed to Pt2+ ion square planar-coordinated on the CeO2 facet, and the feature at 196 cm−1 can be assigned to the Raman-active Ag(w1) mode of PtOx. Moreover, the two peaks centered at around 550 and 680 cm−1 are mainly associated with highly dispersed PtOx species on the surface of CeO2, and are tentatively assigned to the vibration of Pt–O–Ce species and the vibration of Pt–O species, respectively, which are indicative of the existence of strong metal–support interaction between Pt and CeO2 species.1,21,22,37 In addition, it is worth mentioning that metallic Pt is not Raman active.35 Therefore, metallic Pt species cannot be detected by the Raman technique, although the presence of such species is clearly verified in the XRD measurements.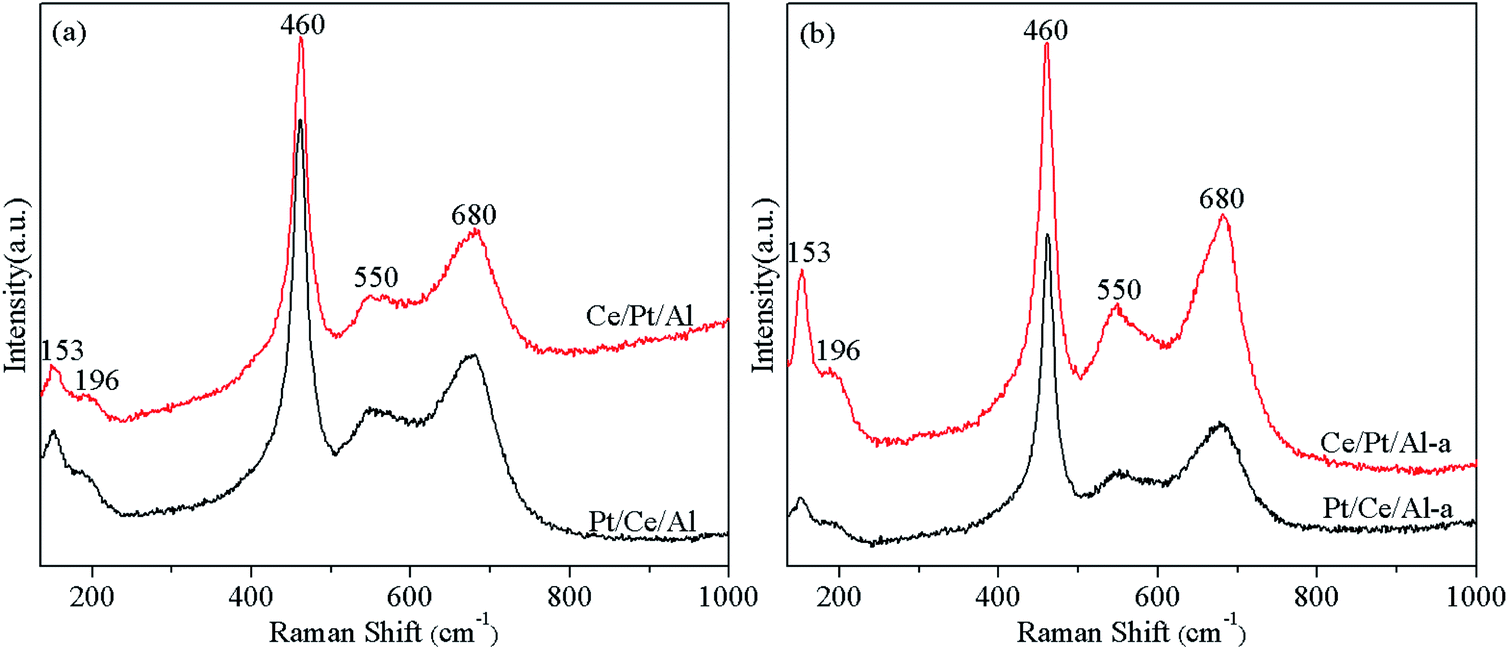 | ||
| Fig. 2 Raman spectra of the (a) fresh and (b) aged catalysts. | ||
The Raman results in Fig. 2 reveal that, while the two fresh samples Pt/Ce/Al and Ce/Pt/Al have similar Raman features, after hydrothermal aging treatment the Raman characteristics associated with the Pt species are significantly different. As shown in Fig. 2(b), the Pt-related peaks of Ce/Pt/Al-a exhibit considerably higher intensities compared with those of Pt/Ce/Al-a, demonstrating that for Ce/Pt/Al-a more Pt species are anchored on the surface of CeO2 and stronger interaction between Pt and CeO2 is obtained on account of the formation of the Pt–O–Ce bond, which could thus facilitate improvement of the hydrothermal stability of the Ce/Pt/Al-a catalyst.
XPS analysis results
The surface elemental information was analyzed using X-ray photoelectron spectroscopy (XPS), and the results obtained are summarized in Table 1. It can be seen from Table 1 that, for fresh samples, Pt/Ce/Al shows a higher surface concentration of Pt than Ce/Pt/Al, which, considering the difference in their synthesis routes, is rational and understandable. After hydrothermal aging treatment, the surface content of Pt is somewhat declined for Pt/Ce/Al-a, indicating that aggregation of Pt species has occurred. However, it is worth pointing out that Ce/Pt/Al-a exhibits an inversely higher surface concentration of Pt compared with fresh Ce/Pt/Al, implying that migration of Pt species from the surface of Al2O3 to the surface of CeO2, that is, redispersion of Pt species, might have taken place during the hydrothermal aging process.| Samples | Relative amount (%) | Ce3+ (%) | Ptδ+ (%) | Oβ (%) | |||
|---|---|---|---|---|---|---|---|
| Ce | Al | O | Pt | ||||
| Pt/Ce/Al | 0.52 | 39.99 | 59.33 | 0.16 | 16.43 | 55.34 | 45.62 |
| Ce/Pt/Al | 0.58 | 40.02 | 59.29 | 0.11 | 16.74 | 43.75 | 34.66 |
| Pt/Ce/Al-a | 0.56 | 38.73 | 60.59 | 0.12 | 17.14 | 30.92 | 39.75 |
| Ce/Pt/Al-a | 0.52 | 39.23 | 60.07 | 0.18 | 18.43 | 49.46 | 48.75 |
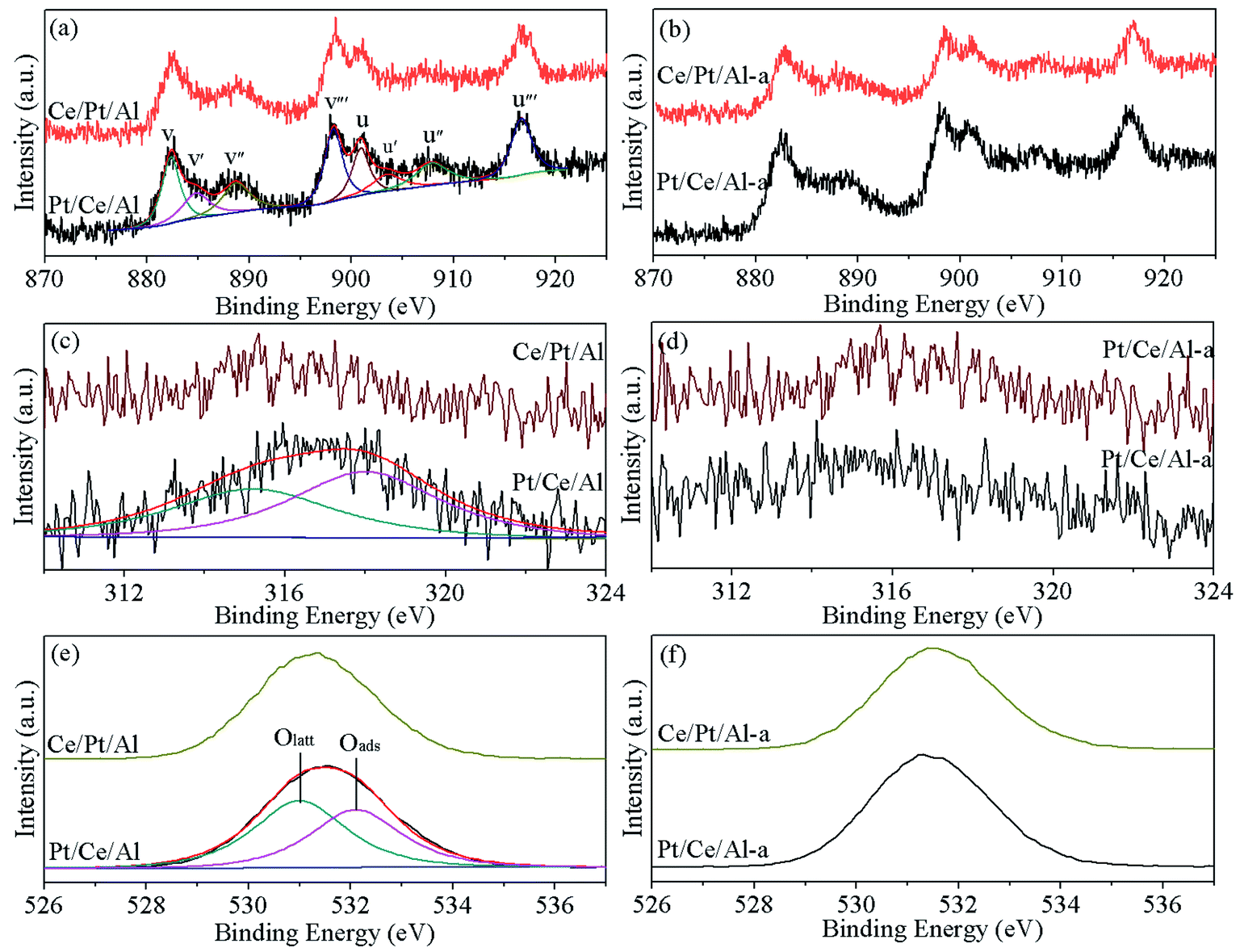
In addition, to better analyze the valence state of the surface elements, the Ce 3d, Pt 4d and O 1s XPS spectra of the various catalysts are displayed in Fig. 3. From Fig. 3(a) and (b), it can be seen that the Ce 3d XPS curves of the CeO2-containing samples can be fitted into eight peaks, in which the peaks marked as v, v′′ and v′′′ originate from Ce4+ 3d5/2, those labeled u, u′′ and u′′′ are associated with Ce4+ 3d3/2, and the doublet v′ and u′ are attributed to Ce3+ 3d5/2 and Ce3+ 3d3/2, respectively.38,39 Accordingly, the ratio of Ce3+ in the total Ce species is calculated based on the corresponding peak areas, which is reported to be important for the formation of oxygen vacancies.40,41 As listed in Table 1, Ce/Pt/Al possesses a slightly higher ratio of Ce3+ compared with Pt/Ce/Al, especially after hydrothermal aging treatment, which could facilitate the formation of more oxygen vacancies. Consequently, higher oxygen mobility associated with better reduction capability can be achieved for Ce/Pt/Al-a.
 | ||
| Fig. 3 Ce 3d, Pt 4d and O 1s XPS spectra of the (a, c and e) fresh and (b, d and f) aged catalysts. | ||
For Pt species, the binding energies of the Pt 4f peaks overlap with those of Al 2p, making them indistinguishable. Thus, Pt 4d XPS peaks were collected as an alternative. As shown in Fig. 3(c) and (d), the Pt 4d3/2 XPS peak can be deconvoluted into two peaks. The one with lower binding energy is ascribed to metallic Pt0 species, and the other, located at higher binding energy, is derived from oxidative Ptδ+ species.9,10,24 From Table 1, it is found that for the fresh samples Pt/Ce/Al has a higher proportion of oxidative Ptδ+ species, which may be due to the majority of Pt species closely interacting with CeO2. On the other hand, for Ce/Pt/Al, the Pt species are predominately located on the surface of Al2O3, leading to the formation of more metallic Pt0 species, which is detrimental to its reduction property and catalytic behavior.42 However, after hydrothermal aging treatment, it can be seen that, owing to the severe aggregation of Pt species, the proportion of Ptδ+ species is clearly reduced for Pt/Ce/Al-a, whereas for Ce/Pt/Al-a, owing to redispersion of Pt, the Pt species more closely interact with CeO2, leading to the formation of more oxidative Ptδ+ species.
In addition, from Fig. 3(e) and (f), two types of oxygen species are observed for all samples. The peak labeled as Oα is characteristic of lattice oxygen, and the peak denoted as Oβ is attributed to surface-adsorbed oxygen species.10,43 As claimed in the literature,44 the relative concentration of Oβ plays a significant role in the catalytic reaction process on account of the higher mobility of the surface-adsorbed oxygen species (Oβ) than that of lattice oxygen (Oα). From Table 1, it can be seen that Pt/Ce/Al possesses a higher proportion of Oβ, which is advantageous to its oxygen mobility. Upon aging treatment, some loss of Oβ is observed for Pt/Ce/Al-a, while Ce/Pt/Al-a exhibits an inversely larger proportion of Oβ than its fresh counterpart, and the value is also larger than for Pt/Ce/Al-a, indicating that oxygen on the surface of Ce/Pt/Al-a may be more active. This is, without doubt, beneficial to its catalytic reaction process.
TEM and HRTEM measurements
Fig. 4 presents the TEM images of the fresh and aged catalysts. As seen therein, the rod-like particles are identified as Al2O3 particles, while CeO2 particles are mainly spherical in shape. For the three fresh samples, it is found that Pt-related particles are difficult to distinguish owing to their considerably smaller particle size, implying that the Pt species are all in a highly dispersed state, which is in good agreement with the XRD observations. When the catalysts suffer hydrothermal aging treatment, aggregation of the noble metal species is clearly observed. Especially for the comparison sample Pt/Al-a, large agglomerates of Pt species are generated, as shown by the arrows in Fig. 4(f). Fortunately, the CeO2-containing samples exhibit comparatively better anti-aging resistance, and the particle sizes of Pt species are much smaller than those of Pt/Al-a. Furthermore, to better compare the particle size of the Pt species in the three aged samples, the corresponding particle size distributions are displayed in the insets of Fig. 4(d–f). It is found that the particle size of Pt species in Pt/Al-a is indeed distributed over a considerably larger range, and the average value is as much as 61.2 nm. In addition, the average particle size of Pt species for Ce/Pt/Al-a (4.7 nm) is smaller than for Pt/Ce/Al-a (6.2 nm). In other words, higher dispersion of Pt species is achieved for Ce/Pt/Al-a, which can probably be ascribed to the redispersion behavior of Pt during the hydrothermal aging treatment process. | ||
| Fig. 4 TEM images of (a) Pt/Ce/Al, (b) Ce/Pt/Al, (c) Pt/Al, (d) Pt/Ce/Al-a, (e) Ce/Pt/Al-a, (f) Pt/Al-a, and the corresponding particle size distributions of Pt species (insets). | ||
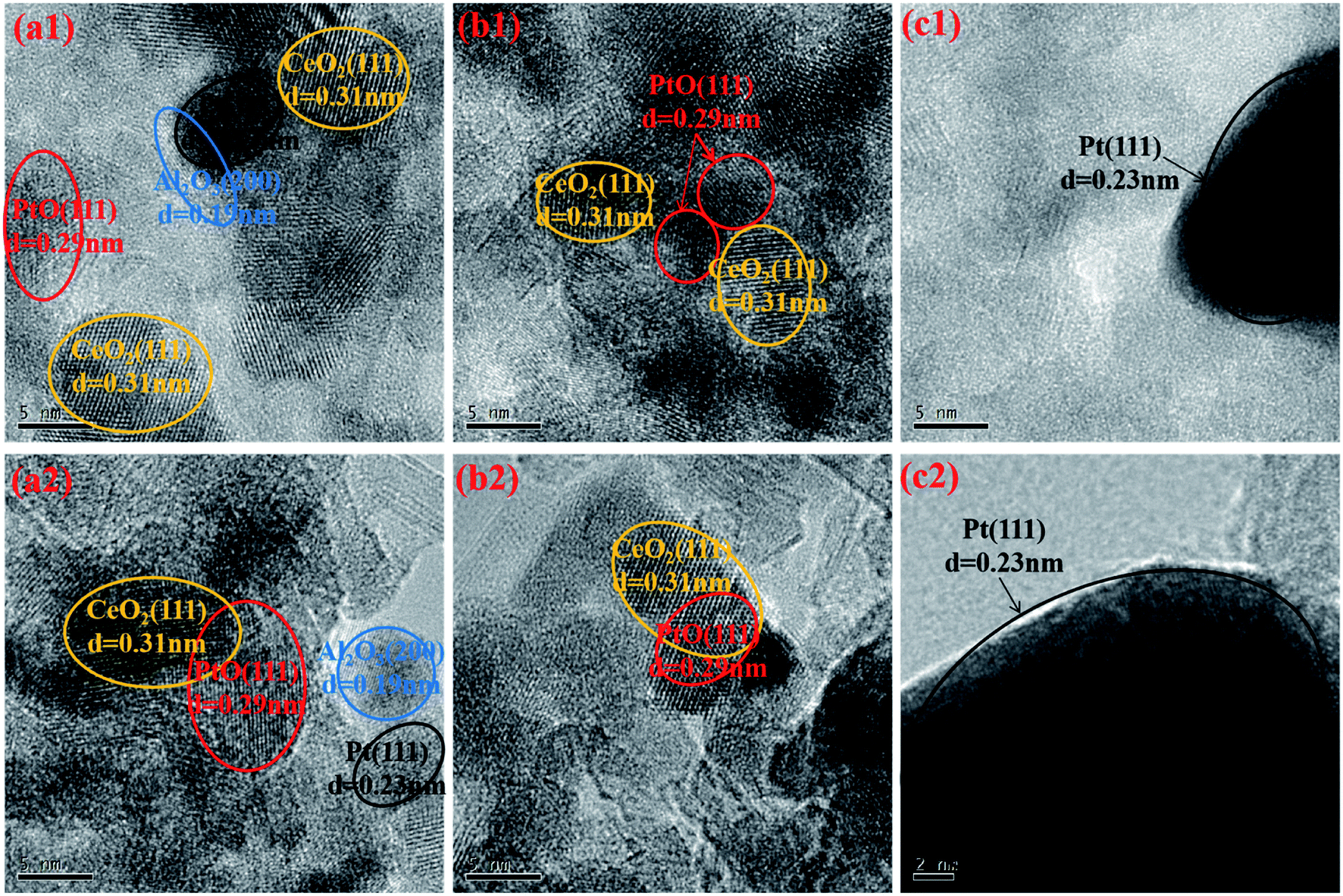
To better analyze the microstructural characteristics of the aged catalysts, HRTEM measurements were conducted and the images are displayed in Fig. 5. The identification of different particles is based on the measured interplanar spacing.1,10,45 From Fig. 5(c1) and (c2), large metallic Pt particles are easily recognized for Pt/Al-a, which again confirms the serious aggregation of Pt species, accompanied by its reduction into the more inactive Pt0 state. Particular attention should be paid to the differences between Pt/Ce/Al-a and Ce/Pt/Al-a. By carefully comparing Fig. 5(a1) and (a2) with Fig. 5(b1) and (b2), it can be seen that the particle sizes of Pt species for Pt/Ce/Al-a are obviously larger than those for Ce/Pt/Al-a, and metallic Pt0 particles are easily observed for Pt/Ce/Al-a. Accordingly, it can be inferred that during the hydrothermal aging process the interaction between Pt and CeO2 in Pt/Ce/Al-a deteriorates, and so Pt species would undergo severe aggregation and transformation into a more reductive state. Fortunately, due to the special form of distribution in Ce/Pt/Al, migration of Pt species from the surface of Al2O3 to the surface of CeO2 happens during aging treatment, which gives rise to favorable interaction between Pt and CeO2, and, consequently, the formation of the Pt–O–Ce bond may act as an anchor to inhibit the agglomeration of Pt species. Meanwhile, a higher oxidation state could be achieved for Pt species owing to electron transfer from Pt to CeO2.
 | ||
| Fig. 5 HRTEM images of (a1 and a2) Pt/Ce/Al-a, (b1 and b2) Ce/Pt/Al-a and (c1 and c2) Pt/Al-a. | ||
CO-FTIR measurement
The CO adsorption experiment was conducted on an FTIR spectrometer, and the spectra obtained are shown in Fig. 6. As can be seen, the band located at 2175 cm−1 originated from CO in the gaseous phase,25,46 and other peaks, including two intense bands at ca. 2085 and 2065 cm−1 along with a weak band centered at around 1830 cm−1, are all associated with Pt-related species. According to the literature,9,15,47 the two bands at 2085 cm−1 and 2065 cm−1 are associated with CO species linearly bonded to one Pt atom; the former is attributed to CO linearly adsorbed on Pt terraces, and the latter to linearly adsorbed CO species on open sites of Pt particles. In addition, the weak band centered at around 1830 cm−1 is assigned to CO bonded on two surface Pt atoms, i.e., bridge-adsorbed CO species. | ||
| Fig. 6 CO-FTIR spectra of the (a) fresh and (b) aged catalysts. | ||
For the fresh catalysts, as observed in Fig. 6(a), the Pt-related peaks for Pt/Ce/Al show higher intensities compared with Ce/Pt/Al, indicating that Pt/Ce/Al possesses higher dispersion of Pt, which is consistent with the XPS analysis results. After hydrothermal aging treatment, the CO adsorption peaks of Pt/Ce/Al-a exhibit obviously reduced intensities, which is indicative of the aggregation of Pt particles and corresponds to the lower dispersion of Pt. On the other hand, Ce/Pt/Al-a displays prominent hydrothermal stability, as can be found in Fig. 6(b). The Pt-related peaks maintain relatively high intensities, which is in line with the XPS and TEM results that higher dispersion and lower particle size of Pt are maintained for Ce/Pt/Al-a, and could well contribute to its superior three-way catalytic performance.
Discussion of the hydrothermal aging behaviors of the Pt catalysts
Based on the above analysis results, it can be deduced that the migration and sintering behaviors of the three Pt catalysts are undoubtedly different from each other. Thus the hydrothermal aging behavior of the three Pt catalysts is schematically described in Fig. 7. As can be seen, without participation of the CeO2 component, Pt/Al shows the worst anti-aging resistance. Due to the absence of the favorable metal–support interaction, severe aggregation of Pt species into large metallic Pt particles inevitably occurs, which leads to a drastically declined reduction ability and three-way catalytic activity for Pt/Al-a. When CeO2 is introduced prior to the impregnation of Pt species, as shown in Fig. 7(a), Pt species are mainly located on the surface of the CeO2 component and so the Pt/Ce/Al catalyst exhibits higher hydrothermal stability than Pt/Al owing to the metal–support interaction between the Pt and CeO2 components. Nevertheless, upon aging treatment at such high temperature aggregation of Pt species is the predominantly occurring aging behavior, that is, Pt species would still experience a considerable degree of aggregation. Furthermore, it should be pointed out that if the CeO2 component is introduced after the deposition of Pt species on Al2O3, the hydrothermal aging behavior is significantly different from that of Ce/Pt/Al. As described in Fig. 7(b), redispersion of Pt species is probably the predominate aging behavior. This is because Pt species are mainly distributed on the surface of Al2O3, and the subsequently impregnated CeO2 component may act as a diffusion barrier or protective layer to retard the aggregation of Pt species upon aging treatment. On the other hand, owing to the much stronger Pt–CeO2 interaction compared with Pt–Al2O3,24,37 CeO2 can effectively trap the mobile Pt species and, hence, a large portion of Pt species originally located on the surface of Al2O3 may undergo migration towards the surface of the CeO2 component. In this way, strong metal–support interaction is achieved by the formation of the Pt–O–Ce bond, which can further act as an anchor to inhibit aggregation of Pt species, while the higher oxidation state of Pt could be preserved. As a consequence, Ce/Pt/Al-a, with higher dispersion and a higher oxidative state for Pt, may be predicted to present a more desirable three-way catalytic performance. | ||
| Fig. 7 Schematics describing the hydrothermal aging behaviors of the supported Pt catalysts: (a) Pt/Ce/Al, (b) Ce/Pt/Al and (c) Pt/Al. | ||
H2-TPR studies
The reduction features of the fresh and aged catalysts were characterized using the H2-TPR technique, and the profiles obtained are depicted in Fig. 8. As displayed in Fig. 8(a), the reduction profiles of the fresh samples can be categorized into two regions. The portion at temperatures below 300 °C is mainly attributed to reduction of Ptδ+ species accompanied by reduction of some active oxygen species in the vicinity of Pt, while the peak located at the temperature range 300–500 °C originated from the reduction of oxygen species distant from Pt.24,25,48,49 By comparing the reduction characteristics of Pt/Ce/Al and Ce/Pt/Al, it is found that the reduction of Pt/Ce/Al begins at comparatively lower temperatures, which could be ascribed to its higher dispersion of Pt on the surface as well as the presence of more reducible Ptδ+ species. Considering that Pt species loaded on Al2O3 may inevitably be covered by the subsequently introduced CeO2 component, it appears reasonable that the reduction of Pt species on Ce/Pt/Al is more difficult than that on Pt/Ce/Al.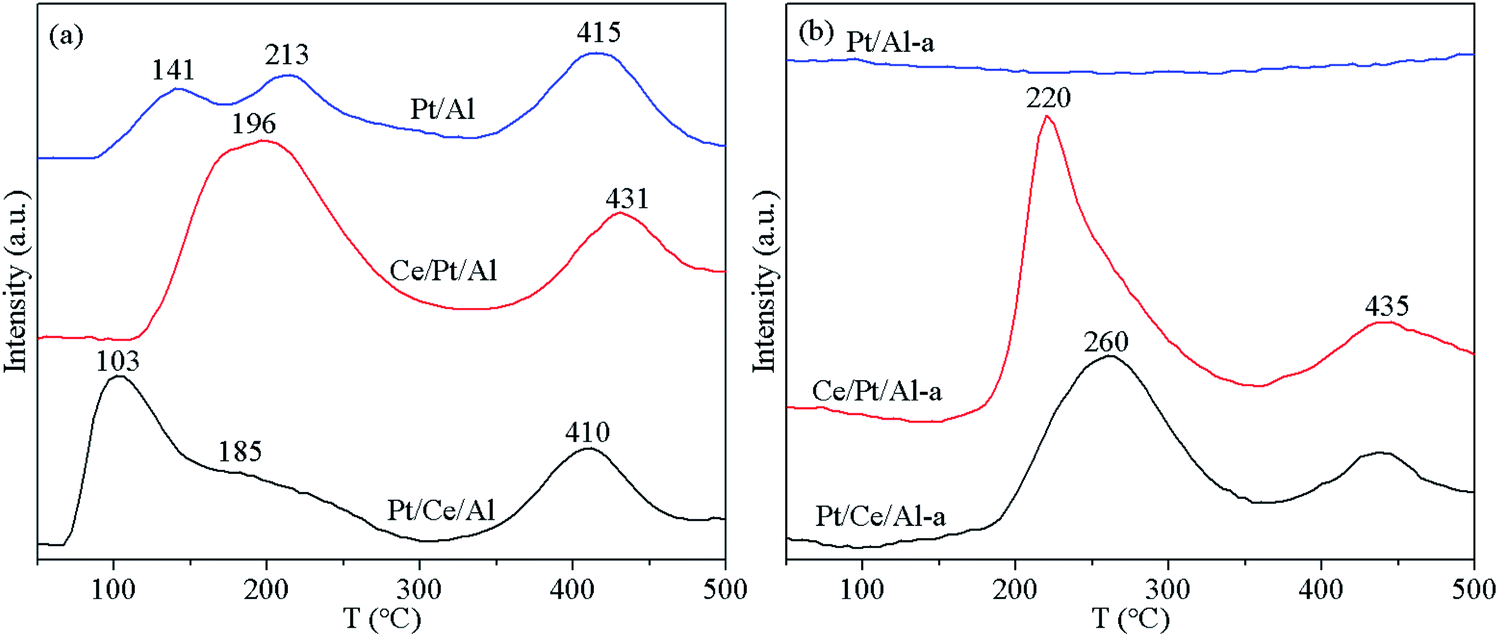 | ||
| Fig. 8 H2-TPR profiles of the (a) fresh and (b) aged catalysts. | ||
As is well known, the metal–support interaction between Pt and Al2O3 is comparatively weaker compared with that between Pt and CeO2.24,37 Thus, completely different behavior is observed on H2-TPR curves, especially for the aged samples. As shown in Fig. 8(b), the reduction peaks for Pt/Al-a almost disappear, since Pt species are aggregated to form large metallic Pt particles upon hydrothermal aging treatment, as confirmed by the TEM observations. In addition, after aging treatment, the reduction capability of Pt/Ce/Al-a and Ce/Pt/Al-a also exhibits deterioration to some extent. Fortunately, through redispersion of Pt species, Ce/Pt/Al-a maintains better reduction performance than Pt/Ce/Al-a, benefiting from the higher dispersion and more oxidative state of Pt species associated with the formation of the favorable Pt–O–Ce bond.
Three-way catalytic performance
The three-way catalytic performance, in terms of the conversion curves of CO, HC and NO as a function of temperature for the fresh and aged catalysts, is displayed in Fig. 9. This shows that the catalytic activity of the fresh catalysts roughly follows a sequence of Pt/Ce/Al > Ce/Pt/Al > Pt/Al. So, Pt/Al without the participation of oxygen buffering component (CeO2) shows the worst catalytic performance, while Pt/Ce/Al appears to be more active than Ce/Pt/Al owing to its higher surface dispersion and more oxidative state of Pt species, as well as its better reduction property.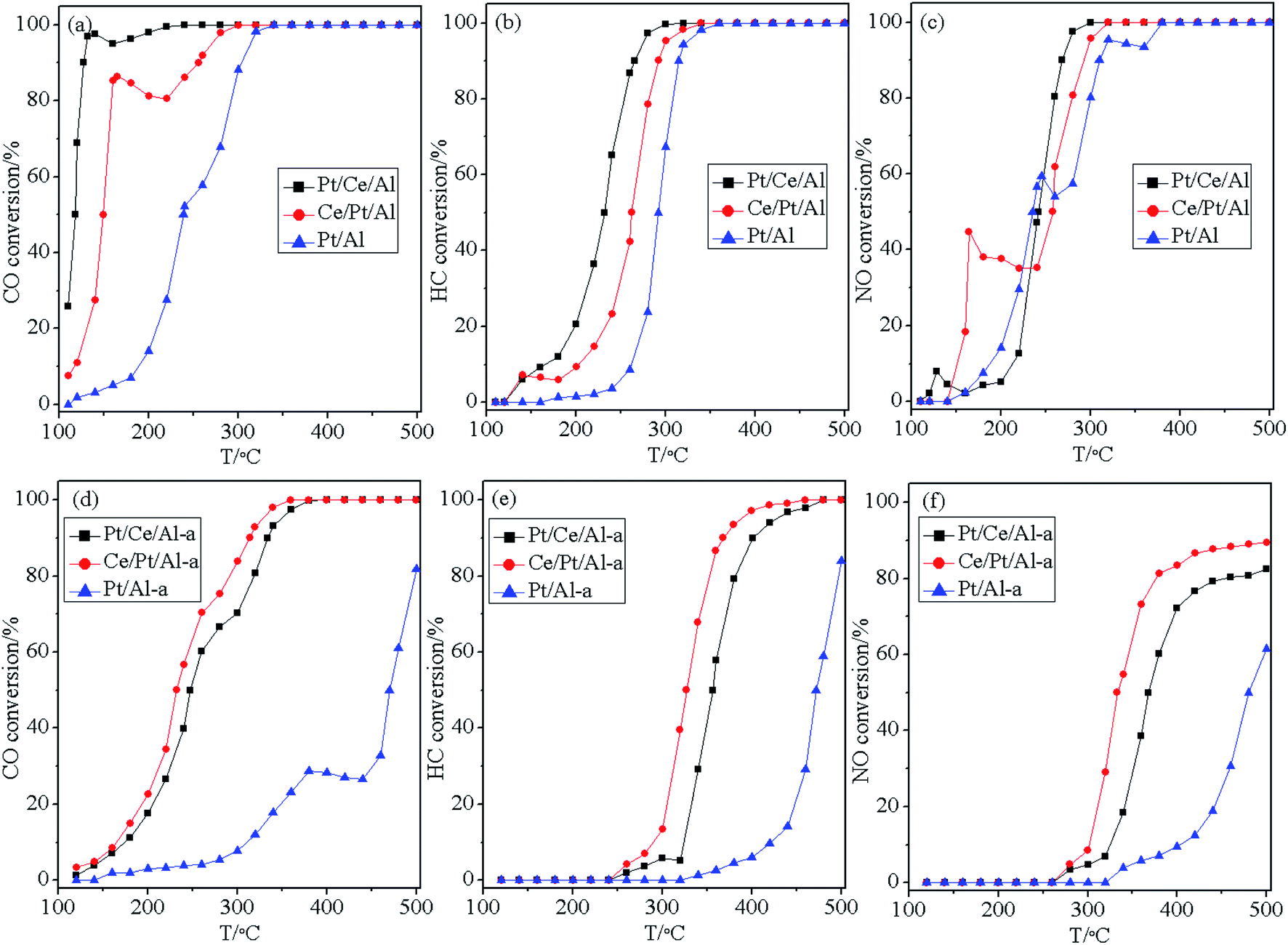 | ||
| Fig. 9 Conversion curves of CO, HC and NO over the supported (a–c) fresh and (d–f) aged catalysts. | ||
As displayed in Fig. 9(b), the different hydrothermal aging behaviors of the catalysts bring about deactivation of the catalysts to differing extents. With the absence of strong metal–support interaction, Pt species on Al2O3 undergo severe aggregation to large metallic Pt particles. Thus, it is not surprising to find that Pt/Al-a has extremely low anti-aging resistance, and its catalytic performance decreases dramatically to the largest extent. In addition, owing to the aggregation and transformation of the Pt species to a more reductive state, Pt/Ce/Al-a still exhibits undesirable hydrothermal stability. However, it is worth noting that although the three-way catalytic performance over fresh Ce/Pt/Al is inferior to that over Pt/Ce/Al, after hydrothermal aging treatment the Ce/Pt/Al-a behaves even better than Pt/Ce/Al-a. This phenomenon is closely related to the redispersion of Pt species during the hydrothermal aging process. Namely, migration of Pt species from the surface of Al2O3 to the surface of CeO2 occurs, which gives rise to strong metal–support interaction between Pt and CeO2, and, as a consequence, the formation of the Pt–O–Ce bond could act as an anchor to retard the aggregation of Pt species. Therefore, a higher dispersion and more oxidative state for the Pt species could be maintained for Ce/Pt/Al-a, which contributes to its prominent reduction property and superior three-way catalytic performance.
In addition, as is well known, Pd–Rh bimetallic catalyst is currently the most common commercial TWC in industry. Therefore, to further verify the application potential of the designed Pt catalyst, the Ce/Pt/Al catalyst was physically mixed with a Rh catalyst supported on a CeO2–ZrO2-based material prior to aging treatment. A commercial Pd–Rh catalyst with almost the same composition and treated in the same manner was used as the reference sample. Evaluation of the three-way catalytic activity results for the commercial Pd–Rh and the as-prepared Pt–Rh catalysts are depicted in Fig. 10. As can be seen in Fig. 10, the two catalysts exhibit quite similar three-way catalytic performance in terms of the similar tendency in variation of the conversion curves for CO, HC and NO. Taking into account the comparatively lower price of Pt compared with that of Pd, the Pt-containing catalyst therefore possesses practical merits compared with commercial Pd–Rh catalyst.
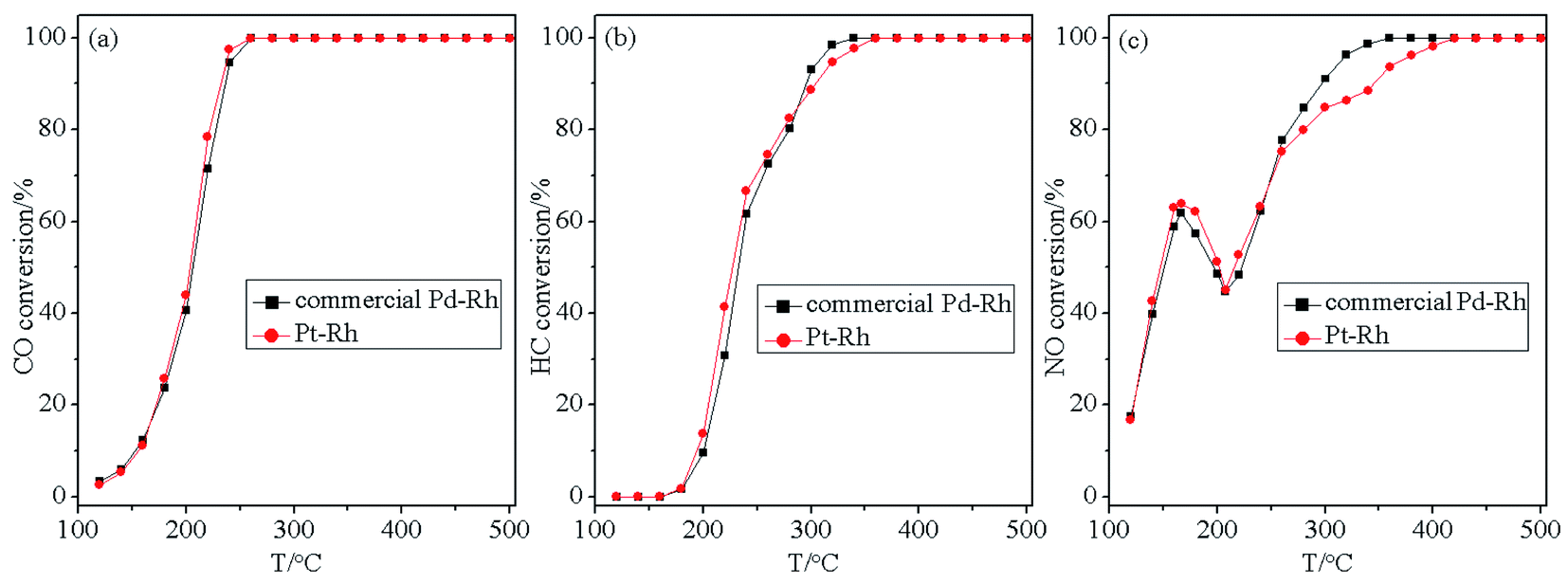 | ||
| Fig. 10 Conversion curves of (a) CO, (b) HC and (c) NO over commercial Pd–Rh and the as-prepared Pt–Rh catalysts. | ||
Conclusions
In this work, an advanced CeO2/Pt/Al2O3 (Ce/Pt/Al) catalyst with enhanced hydrothermal stability was prepared by modifying the synthesis route of a conventional Pt/CeO2/Al2O3 (Pt/Ce/Al) catalyst. Based on the above analysis, it can be concluded that the two catalysts undergo different hydrothermal aging behaviors. Upon hydrothermal aging treatment, conventional Pt/Ce/Al predominately suffers from inevitable aggregation of Pt species, and a large amount of Pt species transform into the more inactive metallic Pt0 state due to the weakened metal–support interaction between Pt and CeO2. Fortunately, when the CeO2 component is introduced after the deposition process of Pt onto Al2O3, the obtained Ce/Pt/Al catalyst exhibits more desirable hydrothermal stability. During hydrothermal aging treatment, redispersion of Pt species takes place, that is, migration of Pt species from the surface of Al2O3 to the surface of CeO2 occurs, resulting in stronger metal–support interaction between Pt and CeO2, and, as a consequence, the Pt–O–Ce bond formed could further act as an anchor to inhibit the aggregation of Pt species. Therefore, higher dispersion of Pt species is achieved for Ce/Pt/Al-a, which could provide more active sites for the catalytic reaction process. On the other hand, the strong metal–support interaction between Pt and CeO2 could help Pt species stay at a more oxidative state. Hence, an improved reduction property and superior three-way catalytic performance are acquired for Ce/Pt/Al-a.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Key Research and Development Program of Sichuan Science and Technology Department (2019YFS0513).References
- A. M. Gänzler, M. Casapu, F. Maurer, H. Störmer, D. Gerthsen, G. Ferré, P. Vernoux, B. Bornmann, R. Frahm, V. Murzin, M. Nachtegaal, M. Votsmeier and J. Grunwaldt, ACS Catal., 2018, 8, 4800–4811 CrossRef.
- G. Ertl, Angew. Chem., Int. Ed., 2008, 47, 3524–3535 CrossRef CAS.
- S. Parres-Esclapez, M. J. Illán-Gómez, C. Salinas-Martínez de Lecea and A. Bueno-López, Appl. Catal., B, 2010, 96, 370–378 CrossRef CAS.
- A. Iglesias-Juez, A. Martínez-Arias, M. A. Newton, S. G. Fiddy and M. Fernández-García, Chem. Commun., 2005, 4092–4094 RSC.
- A. M. Gänzler, M. Casapu, D. E. Doronkin, F. Maurer, P. Lott, P. Glatzel, M. Votsmeier, O. Deutschmann and J. Grunwaldt, J. Phys. Chem. Lett., 2019, 10, 7698–7705 CrossRef.
- J. Kašpar, P. Fornasiero and N. Hickey, Catal. Today, 2003, 77, 419–449 CrossRef.
- R. Polvinen, M. Vippola, M. Valden, T. Lepistö, A. Suopanki and M. Härkönen, J. Catal., 2004, 226, 372–381 CrossRef CAS.
- Z. Z. Zhu, G. Z. Lu, Y. Guo, Y. L. Guo, Z. G. Zhang and Y. Q. Wang, J. Ind. Eng. Chem., 2012, 18, 2135–2140 CrossRef CAS.
- B. A. Riguetto, S. Damyanova, G. Gouliev, C. M. P. Marques, L. Petrov and J. M. C. Bueno, J. Phys. Chem. B, 2004, 108, 5349–5358 CrossRef CAS.
- Y. Chen, Y. X. Feng, L. Li, J. Y. Liu, X. L. Pan, W. Liu, F. F. Wei, Y. T. Cui, B. T. Qiao, X. C. Sun, X. Y. Li, J. Lin, S. Lin, X. D. Wang and T. Zhang, ACS Catal., 2020, 10, 8815–8824 CrossRef CAS.
- N. H. An, X. L. Yuan, B. Pan, Q. L. Li, S. Y. Li and W. X. Zhang, RSC Adv., 2014, 4, 38250–38257 RSC.
- A. Russell, C. Henry, N. W. Currier, A. Yezerets and W. S. Epling, Appl. Catal., A, 2011, 397, 272–284 CrossRef CAS.
- G. Beulertz, M. Fritsch, G. Fischerauer, F. Herbst, J. Gieshoff, M. Votsmeier, G. Hagen and R. Moos, Top. Catal., 2013, 56, 405–409 CrossRef CAS.
- G. Beulertz, M. Votsmeier and R. Moos, Appl. Catal., B, 2015, 165, 369–377 CrossRef CAS.
- J. Jones, H. F. Xiong, A. T. DeLaRiva, E. J. Peterson, H. Pham, S. R. Challa, G. Qi, S. Oh, M. H. Wiebenga, X. I. P. Hernández, Y. Wang and A. K. Datye, Science, 2016, 353, 150–154 CrossRef CAS.
- A. M. Gänzler, M. Casapu, P. Vernoux, S. Loridant, F. J. C. S. Aires, T. Epicier, B. Betz, R. Hoyer and J. Grunwaldt, Angew. Chem., 2017, 129, 13258–13262 CrossRef.
- A. Bruix, Y. Lykhach, I. Matolínová, A. Neitzel, T. Skála, N. Tsud, M. Vorokhta, V. Stetsovych, K. Ševčíková, J. Mysliveček, R. Fiala, M. Václavů, K. C. Prince, S. Bruyère, V. Potin, F. Illas, V. Matolín, J. Libuda and K. M. Neyman, Angew. Chem., Int. Ed., 2014, 53, 10525–10530 CrossRef CAS.
- M. Hatanaka, N. Takahashi, N. Takahashi, T. Tanabe, Y. Nagai, A. Suda and H. Shinjoh, J. Catal., 2009, 266, 182–190 CrossRef CAS.
- A. P. Ferreira, D. Zanchet, R. Rinaldi, U. Schuchardt, S. Damyanova and J. M. C. Bueno, Appl. Catal., A, 2010, 388, 45–56 CrossRef CAS.
- Y. Nagai, T. Hirabayashi, K. Dohmae, N. Takagi, T. Minami, H. Shinjoh and S. Matsumoto, J. Catal., 2006, 242, 103–109 CrossRef CAS.
- J. Lee, Y. Ryou, X. Chan, T. J. Kim and D. H. Kim, J. Phys. Chem. C, 2016, 120, 25870–25879 CrossRef CAS.
- D. M. F. Alves, F. S. Alves, D. G. Ribeiro, S. S. Montani, F. R. L. Faulstich, M. J. B. Cardoso, C. A. Henriques and F. M. Z. Zotin, Chem. Eng. J., 2013, 223, 239–245 CrossRef CAS.
- M. Hatanaka, N. Takahashi, T. Tanabe, Y. Nagai, K. Dohmae, Y. Aoki, T. Yoshida and H. Shinjoh, Appl. Catal., B, 2010, 99, 336–342 CrossRef CAS.
- J. Lee, Y. Ryou, J. Kim, X. Chan, T. J. Kim and D. H. Kim, J. Phys. Chem. C, 2018, 122, 4972–4983 CrossRef CAS.
- L. Nie, D. H. Mei, H. F. Xiong, B. Peng, Z. B. Ren, X. I. P. Hernandez, A. DeLaRiva, M. Wang, M. H. Engelhard, L. Kovarik, A. K. Datye and Y. Wang, Science, 2017, 358, 1419–1423 CrossRef CAS.
- T. X. Wu, X. Q. Pan, Y. B. Zhang, Z. Z. Miao, B. Zhang, J. W. Li and X. G. Yang, J. Phys. Chem. Lett., 2014, 5, 2479–2483 CrossRef CAS.
- Y. Nagai, K. Dohmae, Y. Ikeda, N. Takagi, T. Tanabe, N. Hara, G. Guilera, S. Pascarelli, M. A. Newton, O. Kuno, H. Jiang, H. Shinjoh and S. Matsumoto, Angew. Chem., Int. Ed., 2008, 47, 9303–9306 CrossRef CAS.
- G. W. Graham, H. W. Jen, W. Chun and R. W. McCabe, J. Catal., 1999, 182, 228–233 CrossRef CAS.
- Y. H. Zhou, J. M. Perket and J. Zhou, J. Phys. Chem. C, 2010, 114, 11853–11860 CrossRef CAS.
- A. Bruix, K. M. Neyman and F. Illas, J. Phys. Chem. C, 2010, 114, 14202–14207 CrossRef CAS.
- N. Kakuta, N. Morishima, M. Kotobuki, T. Iwase, T. Mizushima, Y. Sato and S. Matsuura, Appl. Surf. Sci., 1997, 121/122, 408–412 CrossRef CAS.
- S. Roy and A. K. Saroha, RSC Adv., 2014, 4, 56838–56847 RSC.
- P. J. S. Prieto, A. P. Ferreira, P. S. Haddad, D. Zanchet and J. M. C. Bueno, J. Catal., 2010, 276, 351–359 CrossRef CAS.
- M. Ozawa, T. Okouchi and M. Haneda, Catal. Today, 2015, 242, 329–337 CrossRef CAS.
- Z. Say, E. I. Vovk, V. I. Bukhtiyarov and E. Ozensoy, Appl. Catal., B, 2013, 142–143, 89–100 CrossRef CAS.
- E. A. Derevyannikova, T. Y. Kardash, A. I. Stadnichenko, O. A. Stonkus, E. M. Slavinskaya, V. A. Svetlichnyi and A. I. Boronin, J. Phys. Chem. C, 2019, 123, 1320–1334 CrossRef CAS.
- W. Y. Lin, A. A. Herzing, C. J. Kiely and I. E. Wachs, J. Phys. Chem. C, 2008, 112, 5942–5951 CrossRef CAS.
- X. D. Wu, L. H. Xu and D. Weng, Appl. Surf. Sci., 2004, 221, 375–383 CrossRef CAS.
- G. J. Zhang, Z. R. Shen, M. Liu, C. H. Guo, P. C. Sun, Z. Y. Yuan, B. H. Li, D. T. Ding and T. H. Chen, J. Phys. Chem. B, 2006, 110, 25782–25790 CrossRef CAS.
- J. Fan, X. D. Wu, X. D. Wu, Q. Liang, R. Ran and D. Weng, Appl. Catal., B, 2008, 81, 38–48 CrossRef CAS.
- C. Bozo, N. Guilhaume and J. Herrmann, J. Catal., 2001, 203, 393–406 CrossRef CAS.
- W. Tang, Z. P. Hu, M. J. Wang, G. D. Stucky, H. Metiu and E. W. McFarland, J. Catal., 2010, 273, 125–137 CrossRef CAS.
- T. Q. Cheng, J. L. Wang, S. N. Wang, Y. J. Cui, H. L. Zhang, S. Yan, S. D. Yuan and Y. Q. Chen, Appl. Surf. Sci., 2017, 426, 745–754 CrossRef CAS.
- H. Li, K. Li, X. Zhu, Y. Du, Y. Wei, K. Zhai and H. Wang, J. Ind. Eng. Chem., 2017, 54, 126–136 CrossRef CAS.
- M. M. Sun, J. Y. Liu, C. Song, Y. Ogata, H. Rao, X. Q. Zhao, H. D. Xu and Y. Q. Chen, ACS Appl. Mater. Interfaces, 2019, 11, 23102–23111 CrossRef CAS.
- M. Kottwitz, Y. Y. Li, R. M. Palomino, Z. Y. Liu, G. J. Wang, M. Balasubramanian, D. Lu, R. G. Nuzzo and A. I. Frenkel, ACS Catal., 2019, 9, 8738–8747 CrossRef CAS.
- A. Martínez-Arias, J. M. Coronado, R. Cataluǹa, J. C. Conesa and J. Soria, J. Phys. Chem. B, 1998, 102, 4357–4365 CrossRef.
- Z. Y. Chen, J. X. Mao and R. X. Zhou, Appl. Surf. Sci., 2019, 465, 15–22 CrossRef CAS.
- M. Q. Shen, L. F. Lv, J. Q. Wang, J. X. Zhu, Y. Huang and J. Wang, Chem. Eng. J., 2014, 255, 40–48 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2021 |