
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photocatalytic performance of rhodamine B and enrofloxacin by Pt loaded Bi4V2O11: boosted separation of charge carriers, additional superoxide radical production, and the photocatalytic mechanism†
Yanjun Zhaoa,
Xintong Liub,
Shaonan Guc and
Jiemin Liu *a
*a
aSchool of Chemistry and Biological Engineering, University of Science and Technology Beijing, No. 30 Xueyuan Road, Haidian District, Beijing 100083, People's Republic of China. E-mail: liujm@ustb.edu.cn; Fax: +86-10-6233-2281; Tel: +86-10-8237-6678
bSchool of Light Industry, Beijing Technology and Business University, No. 33 Fucheng Road, Haidian District, Beijing 100048, People's Republic of China
cKey Laboratory of Fine Chemicals in Universities of Shandong, School of Chemistry and Pharmaceutical Engineering, Qilu University of Technology, Shandong Academy of Sciences, No. 3501 Daxue Road, Changqing District, Jinan 250353, Shandong Province, People's Republic of China
First published on 5th March 2021
Abstract
Photocatalytic performance is influenced by two contradictory factors, which are light absorption range and separation of charge carriers. Loading noble metals with nanosized interfacial contact is expected to improve the separation and transfer of photo-excited charge carriers while enlarging the light absorption range of the semiconductor photocatalyst. Therefore, it should be possible to improve the photocatalytic performance of pristine nontypical stoichiometric semiconductor photocatalysts by loading a specific noble metal. Herein, a series of novel Pt–Bi4V2O11 photocatalysts have been successfully prepared via a surface reduction technique. The crystal structure, morphology, and photocatalytic performance, as well as photo-electron properties of the as-synthesized samples were fully characterized. Moreover, the series of Pt–Bi4V2O11 samples were evaluated to remove typical organic pollutants, rhodamine B and enrofloxacin, from aqueous solutions. The photoluminescence, quenching experiments and the electron spin resonance technique were utilized to identify the effective radicals during the photocatalytic process and understand the photocatalytic mechanism. The photocatalytic performance of Pt–Bi4V2O11 was tremendously enhanced compared with pristine Bi4V2O11, and there was additional ˙O2− produced during the photocatalytic process. This study deeply investigated the relation between the separation of charge carriers and the light harvesting, and revealed a promising strategy for fabricating efficient photocatalysts for both dyes and antibiotics.
1. Introduction
Organic dyes and antibiotics are closely related to human society due to their wide field of application.1,2 However, their emission into aquatic ecosystems poses severe threats to environment, and causes detrimental effects on human health as well. For example, rhodamine B (RhB) is a widely used cationic dye in textiles, dyeing, and leather industries and biomedical laboratories.3 However, RhB is also toxic and carcinogenic for humans and animals.4,5 In addition, enrofloxacin (ENR) is a common fluoroquinolone antibiotic, which is widely used in veterinary and human medicine as well as a feed additive in animal husbandry. However, the residual ENR in environment can promote antibiotic-resistance of bacteria and cause potential threats to ecosystems and human health.6,7 Although the hazards of organic dyes and antibiotics were different, the degradation of them was difficult because of their complex and stable structures. Thus, the development of effective and environmentally friendly methods to degrade these organic pollutants became significant and necessary.Among the various water treatment methods, semiconductor photocatalysis is considered as a green and promising alternative to remove organic pollutants from aqueous solutions due to its environmental friendliness and high efficiency.8–10 It is known that light absorption range and separation of charge carriers are two decisive factors in the photocatalytic process.11,12 However, these two factors are contradictory: enlarging the light absorption range requires narrowing down the band gap, but the narrowed band gap may promote the recombination of charge carriers.13 Therefore, one of the main emphasis in semiconductor photocatalysis is to improve the light absorption capacities of the photocatalysts while facilitate the separation of charge carriers.14–16
Compared with typical stoichiometric semiconductor photocatalysts, the corresponding nontypical stoichiometric semiconductor photocatalysts showed great potential in photocatalysis due to their distinct band structures, efficient separation and mobility of photogenerated charge carriers.17 Therefore, otherwise than typical stoichiometric semiconductors, the nontypical stoichiometric semiconductors were also ideal candidates as efficient photocatalysts to remove organic pollutants, such as W18O49,18,19 Bi3O4Cl,20 Bi2VO5.5,21 Bi5O7I,22 Bi24O31Cl10,23,24 Bi24O31Br10,25,26 Bi4V2O11.27,28 Among these nontypical stoichiometric semiconductor photocatalysts, Bi4V2O11 has attracted continuous attentions because of the consistent element composition and similar crystal structure relative to BiVO4.29 Moreover, the light absorption range of Bi4V2O11 is much larger than that of BiVO4. However, the recombination of charge carriers of Bi4V2O11 is also much higher than that of BiVO4, which restricted its photocatalytic performance. Notably, promoting the separation of charge carriers is essential for enhancing the photocatalytic performance of Bi4V2O11. In addition, due to the limitation of energy band potential, Bi4V2O11 could only produce specific radicals during the process of photocatalytic.30 Hence, the induction of Bi4V2O11 for producing additional radicals is also beneficial for enhancing the photocatalytic performance.31
Loading noble metals with nanosized interfacial contact was one of the effective methods to improve the separation and transportation of photo-excited charge carriers, which resulted from the favorable interfacial contact and short transfer pathway.32–34 Besides, some noble metals could also enlarge the light absorption range of the semiconductor photocatalyst.35,36 Recently, several novel noble metal loaded visible light driven photocatalysts have been reported to show enhanced catalytic performance.37–39 Besides, loading noble metal might also produce some additional superoxide radicals by directly utilizing the photogenerated electrons.31 Hence, it is of great importance to employ appropriate noble metal on the surface of the Bi4V2O11 to enhance the photocatalytic performance.
Therefore, in this paper, novel Pt loaded Bi4V2O11 catalysts were successfully prepared via a surface reduction method, and were evaluated as photocatalysts to degrade typical organic dyes and antibiotics, namely rhodamine B (RhB) and enrofloxacin (ENR). The photocatalytic experiments illustrated that the photocatalytic performance of Pt–Bi4V2O11 was tremendously enhanced. The photoluminescence (PL) and quenching experiments results demonstrated that the improvement of photocatalytic performance could mainly attributed to the rapid transmission of photo-excited charge carriers and the additional production of superoxide radicals. This study not only provided a promising strategy for fabricating efficient photocatalysts for organic dyes and antibiotics, but also carried forward the photocatalytic theory.
2. Experimental
2.1. Synthesis of samples
The chemicals used in this work were supplied by Sinopharm Chemical Reagent Corporation (Shanghai, China), and the chemicals were of analytical grade. The preparation of Bi4V2O11 photocatalyst was through one-pot facile solvothermal method.40 First, 70 mL ethylene glycol (EG) was used to dissolve 1 g urea and 2.43 g Bi(NO3)3·5H2O under vigorous stirring. Then 0.30 g NaVO3 was added, and the pH of the above mixture was modulated to about 7.5 by adding diluent ammonia. Subsequently, the as-prepared precursor suspension was added into a 100 mL Teflon-lined stainless autoclave and kept at 478 K for 24 h. Afterwards, the reactor was cool naturally. Afterwards, the prepared Bi4V2O11 was first washed with ultra-pure water three times then washed with ethanol three times. Finally, Bi4V2O11 was dried in an oven at 373 K for 10 h.The preparation method of 2%, 4%, 6%, and 8% Pt loaded Bi4V2O11 samples (where 2%, 4%, 6% and 8% are the atomic ratio of Pt/Bi) was described as follows. 0.5 mmol as-synthesized pure Bi4V2O11 was added into the certain amount of H2PtCl6 solutions under ultrasonic-assistance for 1 h. Next, the sample was dried for 12 h at 373 K and cooled naturally. Then the obtained sample was impregnated within 30 mL methanol by ultrasonication, and the obtained dark brown suspension was stirred for another 1 h. At last, the suspension was centrifuged, and washed by ultra-pure water and ethanol, then the synthesized Pt loaded Bi4V2O11 was dried at 333 K for 10 h.
2.2. Characterization of the photocatalysts
The crystal structure of pure Bi4V2O11 and Pt–Bi4V2O11 series samples were examined by X-ray diffraction (XRD) (D/MAX-RB; Rigaku, Japan). Scanning electron microscopy (SEM) (S-4800; Hitachi, Japan) was used to investigate the surface morphologies of the as-synthesized photocatalysts. Transmission electron microscope (F-20, FEI; USA) was examined to further identify the formation of Pt–Bi4V2O11. The Brunauer–Emmett–Teller (BET) surface areas were determined using an automatic volumetric sorption analyzer (Micromeritics model ASAP 2000) at 77 K. Moreover, the UV-Vis diffuse reflectance spectra (DRS) of the as-synthesized photocatalysts were determined by a T9s UV-Vis spectrophotometer (Persee, China). Besides, the X-ray photoelectron spectroscopies (XPS) of Pt–Bi4V2O11 were analyzed by an ESCALAB 250Xi X-ray photoelectron spectrometer (Thermo, USA). A Bruker A300 spectrometer (Bruker, Germany) was used to identify the main radicals during the photocatalytic process.2.3. Photocatalytic performances studies
The degradations of 20 mmol L−1 RhB solution under visible light illumination were evaluated to investigate the photocatalytic performances of Bi4V2O11 and Pt–Bi4V2O11 series samples. Moreover, the degradation of 40 mg L−1 ENR solution using the optimal sample was also conducted to study the potential of the as-prepared photocatalyst for antibiotics removal. A 400 W Xe lamp with a UV-cut-off filter (λ > 420 nm) was fixed at 10 cm from the samples to simulate the light source. The photocatalytic experiments were carried out by adding 40 mg of the studied photocatalysts into 40 mL RhB or ENR solutions. The RhB or ENR solutions with the photocatalysts samples were stirred in dark for 2 h to reach the adsorption–desorption equilibrium before light illumination. For RhB, the aqueous samples were taken at 0.5 h intervals, while for ENR, the samples were taken at 1 h intervals. The absorbance spectrum of RhB at 553 nm and the UV-Vis spectra of ENR were determined by a UV-Vis spectrophotometer. The degradation efficiencies were evaluated by C/C0, where C0 (mmol L−1) was the initial concentration of RhB or ENR, and C (mmol L−1) is the concentration of RhB or ENR at a certain time.3. Result and discussion
3.1. XRD analysis
The XRD patterns of Bi4V2O11 and Pt–Bi4V2O11 series samples were presented in Fig. 1. For pure Bi4V2O11, the diffraction peaks were consistent with the standard data for orthorhombic crystal structure of Bi4V2O11 (JCPDS No. 42-135),27 and no impurity peaks were detected, which indicated the high purity of Bi4V2O11. As for Pt–Bi4V2O11 series samples, the XRD patterns of both the Bi4V2O11 and elemental Pt phases were detected, and the characteristic peaks have been labeled respectively. The peaks at 10.9° and 28.3° were attributed to the orthorhombic Bi4V2O11, while the distinctive peak at 18.8° was attributed to the elemental Pt (JCPDS No. 04-802).41 Notably, with the increase of Pt contents, the intensity of peak of Pt was gradually enhanced, which confirmed the formation of Pt on the crystal surface of Bi4V2O11. However, other Pt peaks was hard to observe, which might be caused by the high dispersion and low content of Pt in Pt–Bi4V2O11 series samples. Besides, the intensities of the peaks stand for Bi4V2O11 decreased, which indicated the coupling of the Pt on the surface of Bi4V2O11.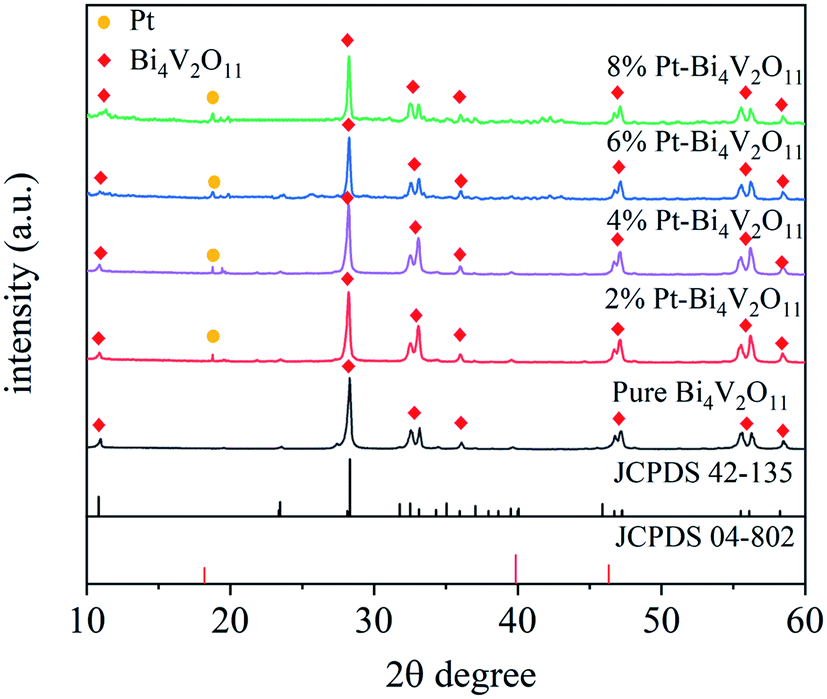 | ||
| Fig. 1 XRD patterns of the pure Bi4V2O11, and Pt loaded Bi4V2O11 samples. | ||
3.2. Morphology characterization
SEM images of Bi4V2O11 and Pt–Bi4V2O11 series samples were depicted in Fig. 2. As shown in Fig. 2A, pure Bi4V2O11 exhibited irregular polygon grains agglomerated in a disordered manner. As shown in Fig. 2B–E, the morphologies of Pt–Bi4V2O11 series samples showed Pt particles dispersed on Bi4V2O11 surface uniformly. Notably, with the increase of the Pt contents, more Pt nanoparticles were observed on Bi4V2O11 surface. Furthermore, in order to observe the distribution of Pt nanoparticles and compare the different Pt loading contents, EDS mapping of Pt–Bi4V2O11 series samples were determined and shown in Fig. S1.† As observed in Fig. S1,† Pt nanoparticles were uniformly dispersed on Bi4V2O11 surface. Besides, with the increase of Pt contents, more Pt nanoparticles were observed, and the sizes of Pt particle were enlarged clearly. Moreover, the loading contents were consistent with the theoretical values. The results of EDS mapping also revealed that Pt were successfully loaded on the surface of Bi4V2O11. | ||
| Fig. 2 SEM images of Bi4V2O11 (A) and Pt–Bi4V2O11 series samples with Pt contents from 2% to 8% (B–E). | ||
Moreover, the TEM and HRTEM of pristine Bi4V2O11 and 6% Pt–Bi4V2O11 composite samples were determined and illustrated in Fig. 3. As shown in Fig. 3A, it can be clearly observed that pristine Bi4V2O11 possess the irregular polygon grain morphology. Moreover, the 0.306 lattice fringe observed in Fig. 3B was attributed to the (0 2 0) plane of pristine Bi4V2O11. As shown in Fig. 3C, TEM image of the 6% Pt–Bi4V2O11 sample also showed irregular polygon morphology, which was consistent with the results of SEM studies. The HRTEM image showed in Fig. 3D could further investigate the growth of Pt on the surface of the Bi4V2O11 bulk, and the lattice fringe of (1 3 1) plane of Bi4V2O11 bulk was 0.176 nm, while the lattice fringe of Pt (2 0 0) was 0.195 nm. The results of TEM and HRTEM strongly proved the formation of Pt–Bi4V2O11 systems.
 | ||
| Fig. 3 TEM image pristine Bi4V2O11 (A), HRTEM image of pristine Bi4V2O11 (B), TEM image of 6% Pt–Bi4V2O11 sample (C), and HRTEM image of 6% Pt–Bi4V2O11 sample (D). | ||
In order to gain further insights into the surface properties of the as-prepared photocatalysts, the surface areas of Bi4V2O11 and Pt–Bi4V2O11 were analyzed. The nitrogen adsorption–desorption isotherms of the as-prepared samples were shown in Fig. S2,† and the BET surface areas and N2 sorption capacities were shown in Table S1.† The N2 adsorption–desorption isotherms of all the as-prepared photocatalysts were categorized as type IV. Besides, with the increase of Pt loading content, the BET surface areas of Pt–Bi4V2O11 increased until the Pt content reached 4%. When the Pt loading content higher than 4%, the surface areas showed a dramatically decrease. This phenomenon might be due to that when the loading content was low, the size of loaded Pt was small, which led to the increase of surface areas. However, with the increase of loading content, the size of loaded Pt would increase, which might wrap the original surface and cause the decrease of surface area.
3.3. Chemical state analysis
Moreover, in order to investigate the elemental compositions and surface chemical states of Pt loaded Bi4V2O11 and pristine Bi4V2O11 samples, the XPS spectra of 6% Pt–Bi4V2O11 and pristine Bi4V2O11 were determined, and the deconvolution spectra of Bi 4f, O 1s, V 2p and Pt 4f were shown in Fig. 4A–D, respectively. Before analyzing, all peaks were calibrated according to the standard C 1s signal at 284.8 eV. The synthesis of Pt–Bi4V2O11 involved nitrogen atoms, however, there is no peak detected corresponding to nitrogen at approximately 400 eV,42 which indicated that there is no nitrogen doping on the Pt–Bi4V2O11 sample. As depicted in Fig. 4A–C, the peaks of Bi 4f, O 1s and V 2p of 6% Pt–Bi4V2O11 showed slightly shift in comparison of pristine Bi4V2O11, which might be due to the interaction between Bi4V2O11 and metallic Pt.43 Moreover, there was a characteristic peak in the deconvolution spectrum of Pt 4f (71.4 eV) at 6% Pt–Bi4V2O11 in Fig. 4D, which was not observed in pristine Bi4V2O11. This result indicated Pt was successfully attached on the surface of Bi4V2O11.44 Overall, the results of XPS analysis confirmed the successful synthesis of Pt loaded Bi4V2O11 samples.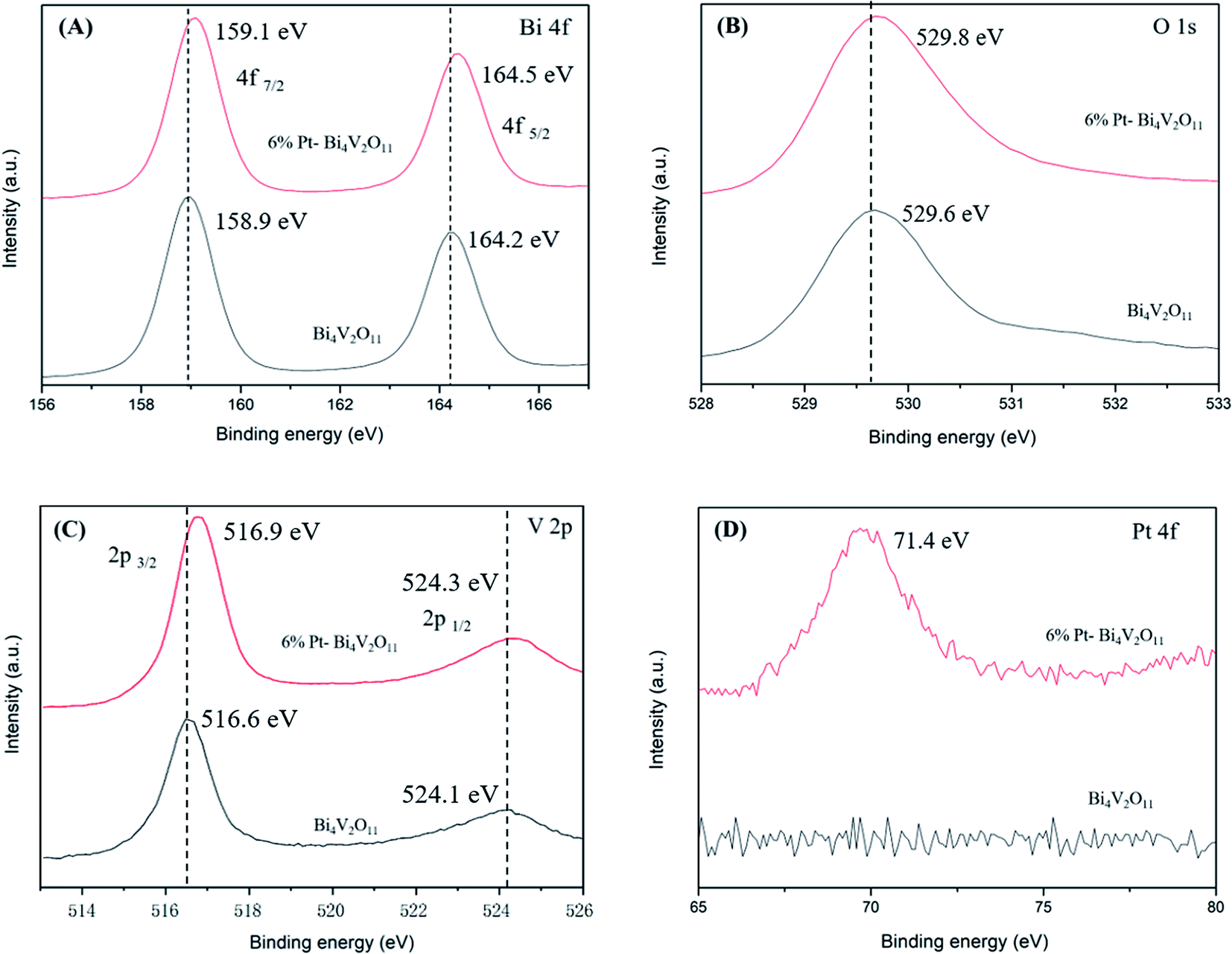 | ||
| Fig. 4 XPS spectra of 6% Pt–Bi4V2O11 and pristine Bi4V2O11: Bi 4f (A), O 1s (B), V 2p (C) and Pt 4f (D). | ||
3.4. Optical properties
The optical properties of the Bi4V2O11 and Pt–Bi4V2O11 series samples at 500–700 nm were studied by UV-Vis DRS, and the results were shown in Fig. 5. It can be clearly observed that compared with pure Bi4V2O11, the light absorption range of Pt–Bi4V2O11 were enlarged after Pt loading, which could react to visible light illumination. Among the as-prepared samples, 8% Pt–Bi4V2O11 had the largest absorption range, while pure Bi4V2O11 had a relatively narrow absorption range. The red shift of the composite sample is obvious, which indicates that the addition of noble metal platinum can improve the optometric absorption of the material, and for 8% Pt–Bi4V2O11, the absorption range is the largest in the composite sample.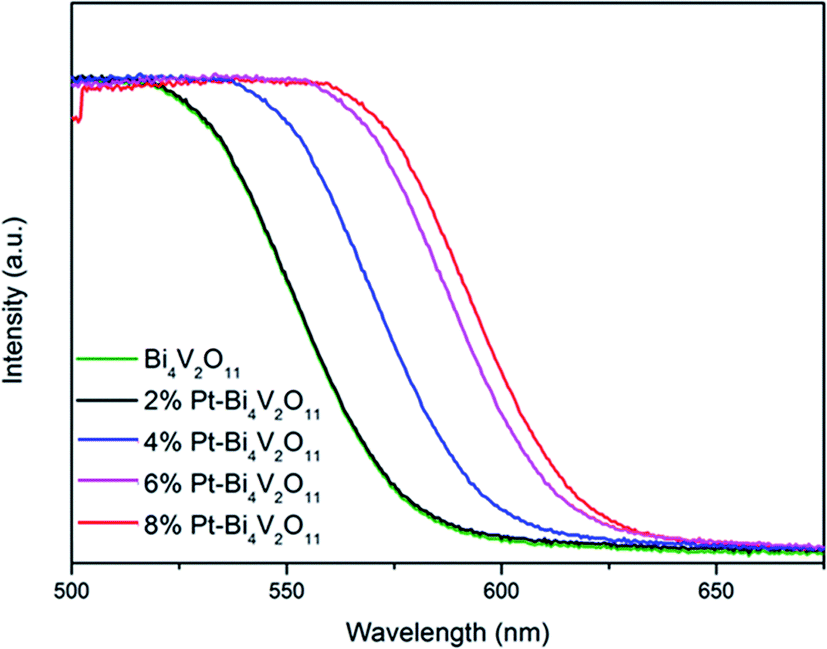 | ||
| Fig. 5 UV-Vis DRS spectra of the Bi4V2O11 and Pt–Bi4V2O11 series samples. | ||
Furthermore, the band gap energies (Eg) of semiconductor photocatalysts were calculated according to the following equation:45
| Ahν = α(hν − Eg)n/2 |
Accordingly, the band gap energy (Eg) of the pure Bi4V2O11 was calculated to be 2.10 eV. As for Pt–Bi4V2O11 series samples, Eg were calculated to be 2.08, 2.02, 1.95 and 1.91 eV respectively for the Pt contents from 2% to 8%. The results demonstrated that loading Pt narrowed the band gap of Bi4V2O11, and with the increase of Pt content, Eg of Pt–Bi4V2O11 showed a tendency of decrease.47
3.5. Photocatalytic properties
The photodegradation performances of the as-prepared samples for RhB under visible light illumination were evaluated. In order to avoid the effect of adsorption,48 the samples were dispersed in RhB solution and continuously stirred in dark for 2 h to ensure the samples reached the adsorption–desorption equilibrium. Before analysis, the adsorption capacities were deducted.As presented in Fig. 6A, the photocatalytic activity of pristine Bi4V2O11 is relatively low, and after 150 min of visible light irradiation, RhB concentration showed no significant change. The poor photocatalytic activity may be due to the rapid recombination of carriers. The photocatalytic activity of Pt–Bi4V2O11 composite samples was enhanced, the decomposition rates enhanced with the increase of Pt content until it reached 6%. As Fig. 6A shown, for 6% Pt–Bi4V2O11 samples, the degradation rate of RhB after irradiation for 150 min reached about 98%. However, when Pt content was further increased to 8%, photocatalytic activity declined, possibly due to excessive Pt coverage that further narrowed the band gap and inhibited carrier separation.49,50
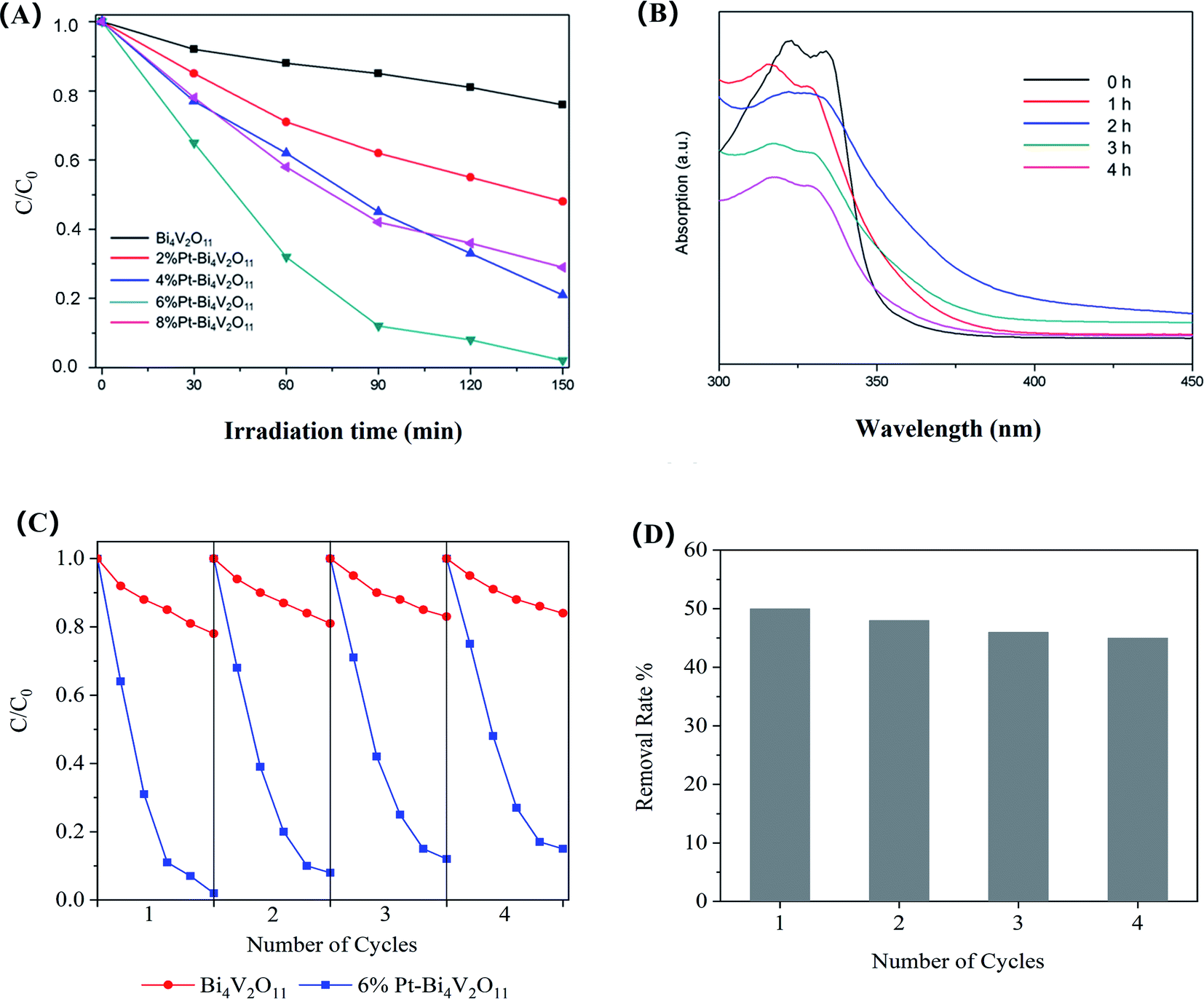 | ||
| Fig. 6 Comparison of the degradation ration of RhB using pure Bi4V2O11 and Pt–Bi4V2O11 series samples (A), the degradation of ENR using 6% Pt–Bi4V2O11 sample (B), cyclic photodegradation of RhB by 6% Pt–Bi4V2O11 and Bi4V2O11 sample (C), and the cyclic photodegradation rate of ENR by 6% Pt–Bi4V2O11 (D). | ||
Moreover, other than organic dye RhB, pristine Bi4V2O11 and the superior catalyst (6% Pt–Bi4V2O11) was used to degrade colorless enrofloxacin (ENR). We found that pristine Bi4V2O11 sample showed no obvious degradation efficiency for ENR, while as shown in Fig. 6B, it can be observed that after 4 h visible light illumination, almost 50% enrofloxacin has been efficiently degraded by 6% Pt–Bi4V2O11. The above results suggested that Pt–Bi4V2O11 could be a promising strategy for ENR degradation. Furthermore, the degradation rates under pH values ranging from 4.0 to 9.0 were also evaluated. However, the results showed that the effect of solution pH was mainly reflected in the adsorption process, which was deducted in the discussion of photocatalytic properties. Therefore, the photocatalytic performances showed no obvious change under the studied pH conditions.51
The stability of the prepared photocatalysts strongly affects its practical application potential.52 The photocatalytic stability of 6% Pt–Bi4V2O11 was confirmed by four cyclic photodegradation of RhB. For comparing, the stability of pristine sample has been tested as well. As revealed in Fig. 6C, no significant inactivation of pristine Bi4V2O11 and 6% Pt–Bi4V2O11 photocatalyst were found, and the degradation rate of 6% Pt–Bi4V2O11 was still superior over pristine Bi4V2O11 after 4 cyclic experiments. Moreover, the cyclic photodegradation of ENR by 6% Pt–Bi4V2O11 was also investigated, and the results were shown in Fig. 6D. After 4 cyclic experiments, the degradation rate of ENR remained at 45%, which also confirmed the stability of 6% Pt–Bi4V2O11. Therefore, the 6% Pt–Bi4V2O11 photocatalyst shows excellent stability to become a promising photocatalysts for regulating organic pollutants.
3.6. Photocatalytic mechanism
To summarize, compared with pristine Bi4V2O11, the Pt–Bi4V2O11 series samples showed highly enhanced photocatalytic properties. Photoluminescence (PL) spectra were utilized to verify the effective inhibition of charge carriers recombination by loading elemental Pt on the substrate surface of Bi4V2O11.53As shown in Fig. 7A, the photoluminescence signal at about 367 nm was contributed to the electron–hole pairs formed on the Bi4V2O11 substrate.54 In general, a lower PL intensity suggests a better electron transfer efficiency, which enhances photocatalytic activity.55 We also observed that compared with pure Bi4V2O11, the photoluminescence intensity of Pt–Bi4V2O11 series samples decreased significantly, indicating that the separation rate of photogenic charge carriers was tremendously enhanced. In particular, for 6% Pt–Bi4V2O11 samples, the loaded Pt effectively transmitted electrons, improving the photocatalytic performance. However, the carrier separation of the 8% Pt–Bi4V2O11 sample was inhibited due to the excessively narrow band gap which greatly promoted the electron–hole pair recombination. In addition, because of the low loading content of Pt, the PL peak has no obvious deviation.
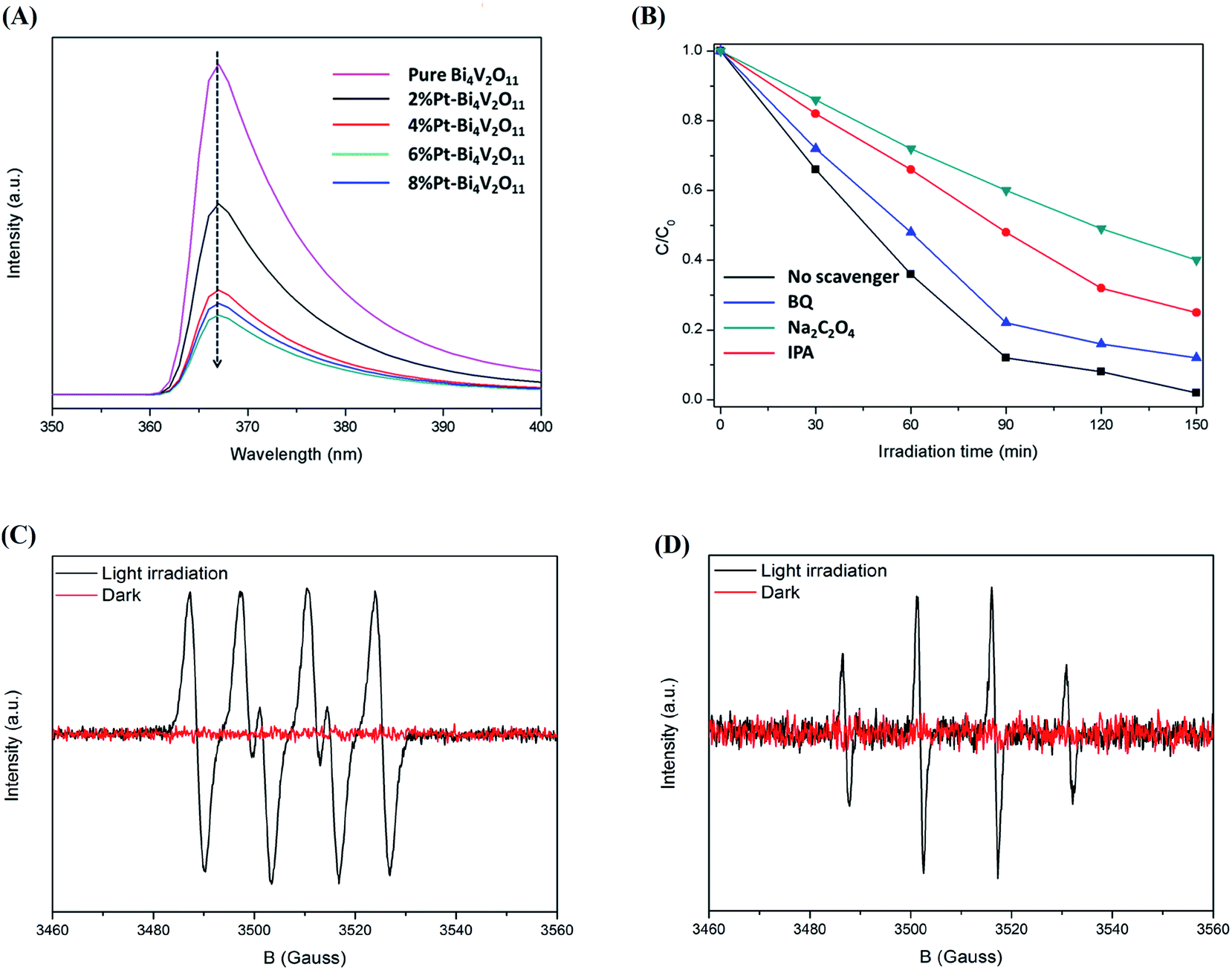 | ||
| Fig. 7 Photoluminescence (PL) spectra of the pure Bi4V2O11 and the Pt–Bi4V2O11 series samples (A), the quenching experiment results of RhB degradation by 6% Pt–Bi4V2O11 photocatalysts (B), ESR spectra of ˙O2− (C) and ˙OH (D) in the system of 6% Pt–Bi4V2O11 under visible light irradiation and dark conditions. | ||
The main active component of 6% Pt–Bi4V2O11 was studied by radical and hole trapping experiment. Three different quenching agents, isopropanol (IPA, 10 mM),56 sodium oxalate (Na2C2O4, 10 mM),57 and benzoquinone (BQ, 1 mM),58 were used as scavengers of hydroxyl radical (˙OH), hole (h+) and superoxide radical (˙O2−), respectively. It could be observed in Fig. 7B that the catalytic properties of 6% Pt–Bi4V2O11 were partially affected by IPA, indicating that hydroxyl radical (˙OH) is one of the main radical during the photocatalytic procedure. After adding Na2C2O4, the photocatalytic activity decreased obviously, this indicates that during the photodegradation of RhB, h+ not only directly oxidizes degrade pollutants, but also generates ˙OH. The addition of BQ affected the photocatalytic activity, suggesting that the superoxide radical also take a big part in the photocatalytic procedure. The conduction band (CB) can be calculated according to the following equation:59
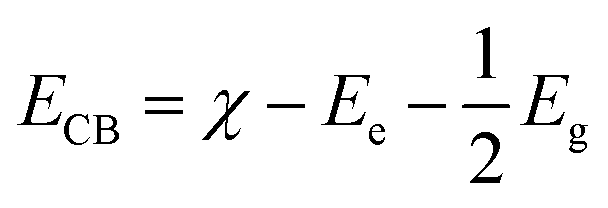
In order to directly determine the main radicals of 6% Pt–Bi4V2O11 in the photocatalytic process, the electron spin resonance (ESR) technique was utilized to capture the ˙O2− (A) and ˙OH (B) signals under light irradiation and dark conditions. As shown in Fig. 7C and D, the 6% Pt–Bi4V2O11 sample showed the obvious signals of both ˙O2− and ˙OH radicals after light irradiation. This result confirmed that Pt loaded Bi4V2O11 could efficiently produce superoxide radicals and hydroxyl radicals during the photocatalytic process.
On the basis of the experimental and computational consequences, the possible photocatalytic mechanism of 6% Pt–Bi4V2O11 under visible light illumination was put forward and the Fig. 8 shows the deduced photocatalytic procedure.
 | ||
| Fig. 8 Schematic illustration of the mechanism of the 6% Pt–Bi4V2O11 photocatalyst activity under visible light irradiation. | ||
The detailed photocatalytic reaction process of 6% Pt–Bi4V2O11 could be described as follows: photoelectrons (e−) were transferred from VB to CB, and holes (h+) were left in VB. For route (I), the adsorbed OH− or H2O were oxidized by the h+ in VB and generated ˙OH, and the ˙OH degraded organic pollutant. For route (II), part of the h+ in VB could also oxidation the organic pollutants directly. Remarkably, the CB potential of pristine Bi4V2O11 is much positive than E(O2/˙O2−), and therefore, it is hard to produce ˙O2− in the Bi4V2O11 sample. However, the loading of Pt could not only efficiently facilitate the transfer of photogenerated charge carriers (Fig. 7A), but also directly utilize the electrons on the CB of Bi4V2O11. Therefore, for route (III), the loaded Pt could react with the absorbed O2 to produce ˙O2−, and degrade the organic pollutants. The following reactions clearly illustrate the degradation process:
| 6% Pt–Bi4V2O11 + hv → electrons (CB) + holes (VB) |
| Electrons (CB) → Pt + O2 → ˙O2− |
| Holes (VB) + H2O → ˙OH + H+ |
| ˙OH + organic waste → carbon dioxide + H2O | (I) |
| hVB+ + organic waste → carbon dioxide + H2O | (II) |
| ˙O2− + organic waste → carbon dioxide + H2O | (III) |
4. Conclusion
In summary, this study successfully synthesized Bi4V2O11 and a series of efficient Pt–Bi4V2O11 visible-light-driven photocatalysts. The photocatalytic performances of Pt–Bi4V2O11 were obviously enhanced compared with pristine Bi4V2O11, and 6% Pt–Bi4V2O11 exhibited the highest photocatalytic activity. The results of UV-DRS and the PL spectra illustrated that the improvement of photocatalytic performance was because of the enhancement of light absorption range and the facilitation of transmission of charge carriers at the same time, which confirmed the hypothesis of loading appropriate noble metal could improve these two contradictory factors. Notably, the quenching experiments and ESR spectra revealed that other than the h+ and ˙OH, abundant ˙O2− was produced in the Pt–Bi4V2O11 system, which were not observed in previous studies of Bi4V2O11 based photocatalysts. Moreover, the mechanism of producing additional ˙O2− was creatively proposed, which might be resulted from the loading of Pt could directly reduce the absorbed O2, and generate ˙O2− to enhance the photocatalytic performance. This study not only provided a novel visible-light-driven photocatalytic system for both organic dyes and antibiotics, but also put forward a promising strategy for producing additional radicals, which could enhance the photocatalytic performance in other applications such as CO2 reduction and hydrogen production.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was jointly supported by the National Key R&D Program of China (No. 2016YFC0700600 and 2016YFC0700901), the National Natural Science Foundation of China (No. 21878018 and 21906006), and the Fundamental Research Funds for the Central Universities (No. FRF-IDRY-19-026, FRF-MP-19-012 and FRF-TP-20-018A2). The authors would like to thank Shiyanjia lab (http://www.shiyanjia.com) for the BET and XPS analysis.References
- W. Wang, J. Fang, S. Shao, M. Lai and C. Lu, Appl. Catal., B, 2017, 217, 57–64 CrossRef CAS.
- Y. Zhao, L. Zhu, W. Li, J. Liu, X. Liu and K. Huang, J. Mol. Liq., 2019, 293, 111516 CrossRef CAS.
- Z. L. Cheng, Y. X. Li and Z. Liu, Ecotoxicol. Environ. Saf., 2018, 148, 585–592 CrossRef CAS PubMed.
- M. Danish, W. A. Khanday, R. Hashim, N. S. B. Sulaiman, M. N. Akhtar and M. Nizami, Ecotoxicol. Environ. Saf., 2017, 139, 280–290 CrossRef CAS PubMed.
- H. Zeng, M. Gao, T. Shen and F. Ding, Colloids Surf., A, 2018, 555, 746–753 CrossRef CAS.
- Y. Zhao, W. Li, Z. Liu, J. Liu, L. Zhu, X. Liu and K. Huang, ACS Sustainable Chem. Eng., 2018, 6, 15264–15272 CrossRef CAS.
- X. J. Wen, C. G. Niu, L. Zhang, C. Liang and G. M. Zeng, Appl. Catal., B, 2018, 221, 701–714 CrossRef CAS.
- H. Yu, L. Jiang, H. Wang, B. Huang, X. Yuan, J. Huang, J. Zhang and G. Zeng, Small, 2019, 15, e1901008 CrossRef PubMed.
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862–4871 CrossRef CAS.
- S. Chandrasekaran, C. Bowen, P. Zhang, Z. Li, Q. Yuan, X. Ren and L. Deng, J. Mater. Chem. A, 2018, 6, 11078–11104 RSC.
- S. Kundu and A. Patra, Chem. Rev., 2017, 117, 712–757 CrossRef CAS PubMed.
- X. Y. Kong, W. Q. Lee, A. R. Mohamed and S.-P. Chai, Chem. Eng. J., 2019, 372, 1183–1193 CrossRef CAS.
- S. Ijaz, M. F. Ehsan, M. N. Ashiq, N. Karamat and T. He, Appl. Surf. Sci., 2016, 390, 550–559 CrossRef CAS.
- X. An, C. Hu, H. Liu and J. Qu, J. Mater. Chem. A, 2017, 5, 24989–24994 RSC.
- Y. Hu, X. Hao, Z. Cui, J. Zhou, S. Chu, Y. Wang and Z. Zou, Appl. Catal., B, 2020, 260, 118131 CrossRef CAS.
- J. Liu, Y. Yu, R. Qi, C. Cao, X. Liu, Y. Zheng and W. Song, Appl. Catal., B, 2019, 244, 459–464 CrossRef CAS.
- C. Lv, G. Chen, J. Sun, Y. Zhou, S. Fan and C. Zhang, Appl. Catal., B, 2015, 179, 54–60 CrossRef CAS.
- K. Deng, Z. Hou, X. Deng, P. Yang, C. Li and J. Lin, Adv. Funct. Mater., 2015, 25, 7280–7290 CrossRef CAS.
- H. Gu, C. Guo, S. Zhang, L. Bi, T. Li, T. Sun and S. Liu, ACS Nano, 2018, 12, 559–567 CrossRef CAS PubMed.
- S. Ning, L. Ding, Z. Lin, Q. Lin, H. Zhang, H. Lin, J. Long and X. Wang, Appl. Catal., B, 2016, 185, 203–212 CrossRef CAS.
- W. Xie, W. Hu, L. L. Zou and D. H. Bao, Ceram. Int., 2015, 41, S265–S273 CrossRef CAS.
- Y. Bai, L. Ye, T. Chen, L. Wang, X. Shi, X. Zhang and D. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 27661–27668 CrossRef CAS PubMed.
- J. Song, L. Zhang, J. Yang, J. S. Hu and X. H. Huang, J. Alloys Compd., 2018, 735, 660–667 CrossRef CAS.
- X. Jin, L. Ye, H. Wang, Y. Su, H. Xie, Z. Zhong and H. Zhang, Appl. Catal., B, 2015, 165, 668–675 CrossRef CAS.
- C. Y. Wang, X. Zhang, H. B. Qiu, G. X. Huang and H. Q. Yu, Appl. Catal., B, 2017, 205, 615–623 CrossRef CAS.
- D. Liu, W. Yao, J. Wang, Y. Liu, M. Zhang and Y. Zhu, Appl. Catal., B, 2015, 172, 100–107 CrossRef.
- C. Lv, G. Chen, X. Zhou, C. Zhang, Z. Wang, B. Zhao and D. Li, ACS Appl. Mater. Interfaces, 2017, 9, 23748–23755 CrossRef CAS PubMed.
- C. Lv, G. Chen, J. Sun and Y. Zhou, Inorg. Chem., 2016, 55, 4782–4789 CrossRef CAS PubMed.
- X. J. Wen, L. Qian, X. X. Lv, J. Sun, J. Guo, Z. H. Fei and C. G. Niu, J. Hazard. Mater., 2020, 385, 121508 CrossRef CAS PubMed.
- X. Chen, J. Liu, H. Wang, Y. Ding, Y. Sun and H. Yan, J. Mater. Chem. A, 2013, 1, 877–883 RSC.
- X. Liu, Y. Zhu, W. Li, F. Wang, H. Li, C. Ren and Y. Zhao, Appl. Surf. Sci., 2018, 458, 1–9 CrossRef CAS.
- X.-l. Li, X.-j. Wang, J.-y. Zhu, Y.-p. Li, J. Zhao and F.-t. Li, Chem. Eng. J., 2018, 353, 15–24 CrossRef CAS.
- A. P. Devi, D. K. Padhi, P. M. Mishra and A. K. Behera, J. Environ. Chem. Eng., 2020, 104778, DOI:10.1016/j.jece.2020.104778.
- P. He, T. Mao, A. Wang, Y. Yin, J. Shen, H. Chen and P. Zhang, RSC Adv., 2020, 10, 26067–26077 RSC.
- B. Quan, W. Liu, Y. Liu, Y. Zheng, G. Yang and G. Ji, J. Colloid Interface Sci., 2016, 481, 13–19 CrossRef CAS PubMed.
- S. Ma, Y. Deng, J. Xie, K. He, W. Liu, X. Chen and X. Li, Appl. Catal., B, 2018, 227, 218–228 CrossRef CAS.
- X. Rong, F. Qiu, J. Yan, H. Zhao, X. Zhu and D. Yang, RSC Adv., 2015, 5, 24944–24952 RSC.
- X. Liu, Y. Su, J. Lang, Z. Chai and X. Wang, J. Alloys Compd., 2017, 695, 60–69 CrossRef CAS.
- Y. J. Yuan, H. W. Lu, Z. T. Yu and Z. G. Zou, ChemSusChem, 2015, 8, 4113–4127 CrossRef CAS.
- X. Zhao, Z. Duan and L. Chen, Ind. Eng. Chem. Res., 2019, 58, 10402–10409 CrossRef CAS.
- Z. Yan, Z. Xu, L. Yue, L. Shi and L. Huang, Chin. J. Catal., 2018, 39, 1919–1928 CrossRef CAS.
- L. Zeng, Z. Lu, M. Li, J. Yang, W. Song, D. Zeng and C. Xie, Appl. Catal., B, 2016, 183, 308–316 CrossRef CAS.
- J. Di, J. Xia, M. Ji, H. Li, H. Xu, H. Li and R. Chen, Nanoscale, 2015, 7, 11433–11443 RSC.
- L. Li, Y. Li, H. Wang, S. Liu and J. J. Bao, Colloids Surf., A, 2019, 570, 322–330 CrossRef CAS.
- Z. Guo, J. Zhou, L. Zhu and Z. Sun, J. Mater. Chem. A, 2016, 4, 11446–11452 RSC.
- V. S. Bystrov, C. Piccirillo, D. M. Tobaldi, P. M. L. Castro, J. Coutinho, S. Kopyl and R. C. Pullar, Appl. Catal., B, 2016, 196, 100–107 CrossRef CAS.
- F. Sun, S. Tan, H. Zhang, Z. Xing, R. Yang, B. Mei and Z. Jiang, J. Colloid Interface Sci., 2018, 531, 119–125 CrossRef CAS.
- A. Younis, D. Chu, Y. V. Kaneti and S. Li, Nanoscale, 2016, 8, 378–387 RSC.
- M. Xiao, Z. Wang, B. Luo, S. Wang and L. Wang, Appl. Catal., B, 2019, 246, 195–201 CrossRef CAS.
- F. K. Naqvi, M. Faraz, S. Beg and N. Khare, ACS Omega, 2018, 3, 11300–11306 CrossRef CAS PubMed.
- H. Wang, Y. Wu, M. Feng, W. Tu, T. Xiao, T. Xiong, H. Ang, X. Yuan and J. W. Chew, Water Res., 2018, 144, 215–225 CrossRef CAS.
- Y. Hong, Y. Jiang, C. Li, W. Fan, X. Yan, M. Yan and W. Shi, Appl. Catal., B, 2016, 180, 663–673 CrossRef CAS.
- M. Ji, R. Chen, J. Di, Y. Liu, K. Li, Z. Chen, J. Xia and H. Li, J. Colloid Interface Sci., 2019, 533, 612–620 CrossRef CAS PubMed.
- N. Alarcos, B. Cohen, M. Ziolek and A. Douhal, Chem. Rev., 2017, 117, 13639–13720 CrossRef CAS.
- B. Chai and X. Wang, RSC Adv., 2015, 5, 7589–7596 RSC.
- J. Tan, X. Wang, W. Hou, X. Zhang, L. Liu, J. Ye and D. Wang, J. Alloys Compd., 2019, 792, 918–927 CrossRef CAS.
- D. Wu, B. Wang, W. Wang, T. An, G. Li, T. W. Ng, H. Y. Yip, C. Xiong, H. K. Lee and P. K. Wong, J. Mater. Chem. A, 2015, 3, 15148–15155 RSC.
- A. G. Mojarrad and S. Zakavi, RSC Adv., 2016, 6, 100931–100938 RSC.
- F. Chen, Q. Yang, X. Li, D. Wang, Y. Zhong, J. Zhao, Y. Deng, C. Niu and G. Zeng, Catal. Commun., 2016, 84, 137–141 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00055a |
| This journal is © The Royal Society of Chemistry 2021 |