
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAccess and modulation of substituted 1-methyl-1,6-dihydropyrazolo[3,4-c]pyrazoles†
Nicu-Cosmin Ostache ab,
Marie-Aude Hiebela,
Adriana-Luminiţa Fînarub,
Hassan Allouchic,
Gérald Guillaumeta and
Franck Suzenet*a
ab,
Marie-Aude Hiebela,
Adriana-Luminiţa Fînarub,
Hassan Allouchic,
Gérald Guillaumeta and
Franck Suzenet*a
aInstitut de Chimie Organique et Analytique (ICOA), Université d'Orléans, UMR CNRS 7311, BP 6759, 45067 Orléans Cedex 2, France. E-mail: franck.suzenet@univ-orleans.fr
bCenter of Applied Chemistry and Process Engineering (CAIP), University “Vasile Alecsandri” of Bacau, Calea Mărăşesşti, Bacău, 600115, Romania
cSynthèse et Isolement de Molécules BioActives (SIMBA), EA 7502, Laboratoire de Chimie Physique, Université de Tours, 31, Avenue Monge, 37200 Tours, France
First published on 5th March 2021
Abstract
Despite the pharmacological potential of the pyrazolo[3,4-c]pyrazoles, only a few methods of preparation and direct functionalization of this moiety have been described. We report herein a convenient design of new pyrazolo[3,4-c]pyrazoles with a high therapeutic impact. The effective chosen strategy consists of hydrazine condensations and C–N Ullmann-type cross-coupling reactions with microwave activation. Moreover, chemoselective bromination of the newly formed bipyrazoles followed by Suzuki–Miyaura cross-coupling reactions allowed the synthesis of a variety of modulated heterobicycles.
Introduction
Fused bicyclic heterocycles are ubiquitous in nature. The most prominent include biological macromolecules (DNA and RNA with their purine bases), proteins and peptides, as well as physiologically important biomolecules (serotonin, melatonin etc.).1 In contrast, bicyclic heterocycles containing a nitrogen–nitrogen (N–N) bond are relatively rare in nature, but nonetheless prevalent in the pharmaceutical industry.2 Such a class is well represented by the ring-contracted [5:5] bicyclic aromatic rings. Among this group, the pyrazolo[3,4-c]pyrazole nucleus stands out from the little attention it was given, despite previous reports of interesting biologically activities.3–7 Differences in the distribution of ring electron densities and in spatial orientation between [5:5] bicyclic aromatic rings and their [6:5] and [6:6] analogs could explain these interesting therapeutic effects.8The synthesis of this bicyclic scaffold are scarcely found in the literature and are almost exclusively based on an existing pyrazole motif.9–11 The desired bicyclic framework is afterward obtained either according to a mononuclear heterocyclic rearrangement1,8 (known as Boulton–Katritzky rearrangement – Route A), an annulation strategy (Route B)12–17 or other miscellaneous methods (using isothiocyanates,18 nitrilimines,9 aryl-semicarbazides19 or 2-cyano-N-methyl-acrylamide20).
These methods, while apparently abundant, exhibit nonetheless several major limitations such as a narrow functional group tolerance, a lack of further modulation, a non-convergent strategy, multiple steps and complex starting materials (Scheme 1a). In order to overcome these difficulties, we hereby present an efficient one-pot approach and a robust process in the design of novel pyrazolo[3,4-c]pyrazoles, using the 5-bromo-1-methyl-1H-pyrazole-4-carbaldehyde21,22 (1) (Scheme 1b). The first modulation arises from the use of diverse aromatic hydrazines, followed by a C–N Ullmann-type cross-coupling reaction under microwave irradiation, leading to compound 2. Chemoselective bromination of the formed bipyrazole ensures the formation of further functionalized scaffolds by a Suzuki–Miyaura cross-coupling. The main text of the article should appear here with headings as appropriate.
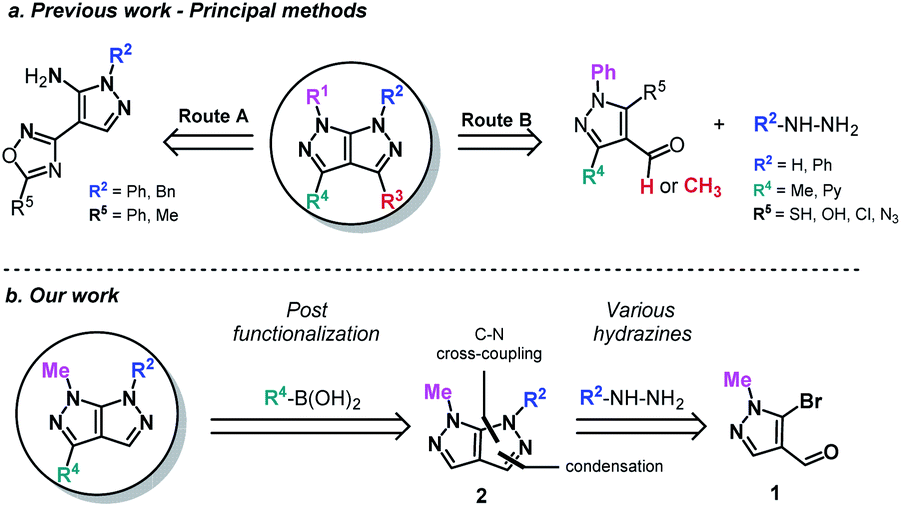 | ||
| Scheme 1 State of the art and retrosynthetic approach. | ||
Results and discussion
The one-pot sequence for the synthesis of the bipyrazole 2a relies on previous studies on similar heterocycles.23–26 The condensation step occurred quantitatively with a slight excess of phenylhydrazine. After a change of solvent and addition of the Ullmann-type catalysts, the expected fused heterocycle was obtained in an encouraging yield (Table 1, entry 1).
|
|||||
|---|---|---|---|---|---|
| Entry | DMEDA (mol%) | CuI (mol%) | t (min) | T (°C) | Yielda (%) |
| a Isolated yield after silica gel chromatography purification.b Yield obtained with conventional heating.c Result obtained with 5-chloro-1-methyl-1H-pyrazole-4-carbaldehyde as starting material.d Traces of starting material present. | |||||
| 1 | 10 | 5 | 30 | 120 | 36 |
| 2 | 10 | 5 | 30 | 150 | 64 (30)b (47)c |
| 3 | 20 | 10 | 30 | 150 | 56 |
| 4 | 5 | 2.5 | 30 | 150 | 64d |
| 5 | 5 | 2.5 | 60 | 150 | 73 |
| 6 | 10 | — | 30 | 150 | 28 (0)c |
| 7 | — | — | 30 | 150 | 13 (0)c |
| 8 | — | 2.5 | 60 | 150 | 76 |
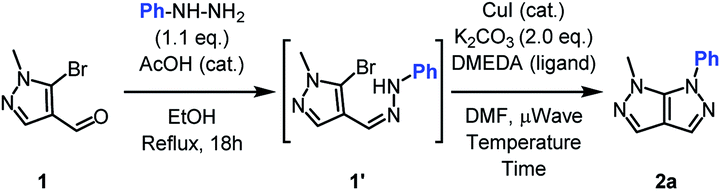
Increasing the reaction temperature to 150 °C allowed a significant yield improvement (Table 1, entries 2). The amount of catalyst and ligand was investigated and reducing to half the initial quantities lead to an identical yield, alongside traces of starting material (Table 1, entries 3–4). The chlorinated analogue was tried but gave lower yield. In order to increase the conversion to its fullest, the reaction time was doubled (Table 1, entry 5). Finally, the sole use of copper catalyst was proven to be essential for the cyclization giving access to the optimized reaction conditions (Table 1, entries 6–8).
Next, the scope and generality of the cyclization step were examined (Table 2). Nonetheless, the condensation of various monosubstituted aromatic hydrazines requires two different reaction conditions, depending on the nature of the arylhydrazines. When salt free arylhydrazines are involved, a catalytic amount of acid is used (1) while under their hydrochloride salt form, a stoichiometric amount of sodium acetate is required (1*).
|
||
|---|---|---|
| Entry | R-NH-NH2 | Product (yield%) |
| a Hydrazine used in a hydrochloride salt form.b Reaction performed on 1.00 g of 1.c Reaction performed on 2.40 g of 1.d Reaction performed on 1.45 g of 1. | ||
| 1 | Ph | 2a (76, 69b) |
| 2 | 4-CH3-C6H4a | 2b (64) |
| 3 | 4-CH3O-C6H4a | 2c (46, 62c) |
| 4 | 4-CN-C6H4a | 2d (76) |
| 5 | 4-CF3-C6H4 | 2e (54, 50d) |
| 6 | 4-F-C6H4a | 2f (62) |
| 7 | 3-F-C6H4a | 2g (85) |
| 8 | 2-F-C6H4a | 2h (46) |
| 9 | 4-Pyridine | 2i (46) |
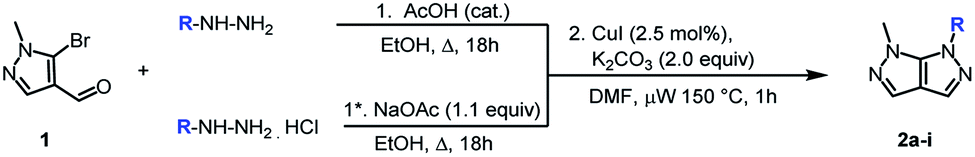
Hydrazines bearing electron-donating (Table 2, entries 1–3) or electron-withdrawing groups (Table 2, entries 4 and 5) were equally well tolerated since the corresponding pyrazolo[3,4-c]pyrazoles (2a–e) were isolated with good to excellent yields over the two steps. Regarding the fluoro substituent position (ortho, meta or para) on the aromatic ring, a slight effect was observed in favor of the meta position (Table 2, entries 6–8). Furthermore, heterocyclic hydrazines were found to be an accessible partner for the annulation step (Table 2, entry 9).
The scope of the reaction is unfortunately limited to aryl hydrazines since hydrazine lead to a bis-condensated compound, and alkylhydrazines to degradation or a lack of conversion.
We afterwards focused our attention on the insertion of a second point of diversity through a bromination step followed by a Suzuki–Miyaura cross-coupling reaction. The bromide was selectively introduced on C3.27 The ratio of the monobrominated (3a) and dibrominated (3a′) compounds was optimized through diverse reaction conditions (Table 3).
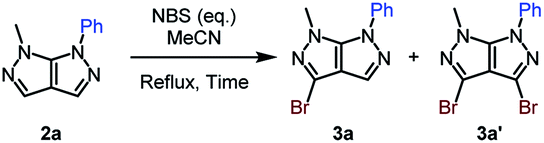
A partial conversion was noticed, with the formation a mixture of a mono- and dibrominated analogue when using a slight excess of halogenation agent (Table 3, entry 1). Determined by this result, the amount of NBS was increased up to 1.5 equivalents, action that lead to a total conversion and an increased yield of the monobrominated bicycle of 69% (Table 3, entries 2 and 3). In order to render the reaction faster, conventional heating was replaced by microwave irradiation. Better results were attained after only 2 hours of reaction time (Table 3, entry 4).
After establishing the best reaction conditions which allow the major formation of the monobrominated bipyrazole, the impact of the electronic nature of a substituent on the aromatic moiety was investigated (Table 4).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R | NBS (eq.) | Heating | t | 3b/ca (%) | SMa (%) |
| a Isolated yield after silica gel chromatography purification. | ||||||
| 1 | 1.0 | μWave | 15 min | 47 | 9 | |
| 2 | 4-CH3O | 1.2 | μWave | 15 min | 49 | 0 |
| 3 | 1.0 | μWave | 30 min | 50 | 8 | |
| 4 | 1.0 | Conventional | 4 h | 55 | 0 | |
| 5 | 1.5 | μWave | 4 h | 92 | 0 | |
| 6 | 4-CF3 | 1.5 | Conventional | 24 h | 58 | 42 |
| 7 | 1.5 | Conventional | 72 h | 82 | 10 | |
| 8 | 2.0 | Conventional | 24 h | 73 | 21 | |
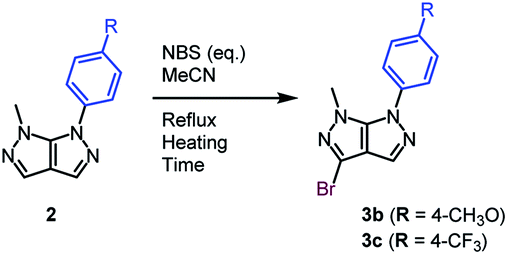
When the substituent present on the aromatic ring is electron-rich in nature, 15 minutes of microwave irradiation and a stoichiometric amount of NBS allowed the synthesis of the halogenated compound with a correct yield, alongside traces of starting material, as well as an inseparable mixture of mono and dibrominated by-products (Table 4, entry 1). Neither a slight bromination agent excess, nor longer reaction times allowed results superior to 50% (Table 4, entries 2 and 3). However, switching to conventional heating lead to the identification of the optimal conditions, as well as obtaining a slightly higher yield of the expected molecule of 55% (Table 4, entry 4). When the aromatic ring from position 6 is substituted by an electron-withdrawing substituent, the bromination reaction performed under microwave irradiation turned out to be highly chemoselective. It required 1.5 equivalents of NBS generating total conversion and an excellent yield of a monobrominated heterocycle after 4 hours of reaction time (Table 4, entry 5). Conventional heating was also studied, but despite increased amounts of NBS and longer reaction times, incomplete conversion and lower yield values were noticed (Table 4, entries 6–8).
Subsequently, two different reaction conditions developed by our group for the Suzuki–Miyaura coupling reaction were tested, both of which proved to be effective (see ESI† for details).22,28
The scope of the Suzuki–Miyaura cross-coupling reaction was next examined (Table 5). To our delight, both electron-rich (Table 5, entries 1, 4 and 5) and electron-poor boronic acid partners (Table 5, entries 6–8) lead towards the expected product in good to excellent yields, without any impact of the position of the substituent on the aromatic ring (Table 5, entries 1–3). Additionally, heterocyclic and even styryl boron derivatives were equally compatible with this transformation (Table 5, entries 9–11).
|
||||
|---|---|---|---|---|
| Entry | SM (R1) | R2 | Yield (%) | |
| a 2 h of μWave irradiation necessary for total conversion. | ||||
| 1 | 3a (R1 = H) | 4-CH3-C6H4 | 83 | 4a |
| 2 | 3-CH3-C6H4 | 89 | 4b | |
| 3 | 2-CH3-C6H4 | 86 | 4c | |
| 4 | Ph | 92 | 4d | |
| 5 | 4-CH3O-C6H4 | 90 | 4e | |
| 6 | 4-CF3-C6H4 | 86 | 4f | |
| 7 | 4-CN-C6H4 | 72 | 4g | |
| 8 | 3-F-C6H4 | 90 | 4h | |
| 9 | 3-Thiophene | 85a | 4i | |
| 10 | 4-Pyridine | 78 | 4j | |
| 11 | Styril | 64 | 4k | |
| 12 | 3b (R1 = OCH3) | 4-CN-C6H4 | 75 | 4l |
| 13 | 4-Pyridine | 76 | 4m | |
| 14 | 4-CH3O-C6H4 | 80 | 4n | |
| 15 | 3c (R1 = CF3) | 4-CN-C6H4 | 68 | 4o |
| 16 | 4-Pyridine | 79 | 4p | |
| 17 | 4-CH3O-C6H4 | 94 | 4q | |
| 18 | 4-CH3-C6H4 | 86 | 4r | |
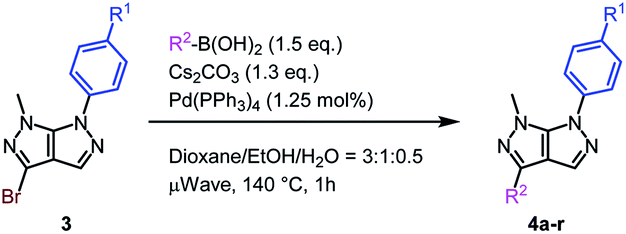
Lastly, the electronic impact of the aryl substituent at the N6 position of the pyrazolo[3,4-c]pyrazoles 3b and 3c was explored. Suitably, independent of the electronic nature of the functional group present on the aromatic moiety, the Suzuki–Miyaura cross-coupling reaction allowed very good to excellent yields (Table 5, entries 12–18).
Compound 4n was found appropriate for X-ray single crystallography, which confirmed that the bromination and subsequent cross-coupling reaction take place at the C-3 position on the pyrazolo[3,4-c]pyrazoles (Fig. 1).29
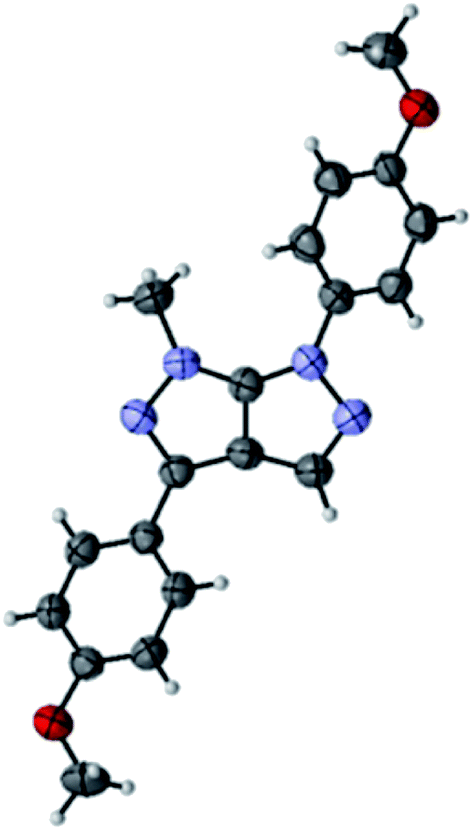 | ||
| Fig. 1 ORTEP representation of pyrazolo[3,4-c]pyrazole 4n. | ||
Conclusions
In summary, the quick access towards variously functionalized pyrazolo[3,4-c]pyrazoles is herein described, starting from 5-bromo-1-methyl-1H-pyrazole-4-carbaldehyde (1). The approach relies on the use of substituted aromatic hydrazines engaged in efficient one-pot sequences. Thus the formed bipyrazoles were subsequently modulated by chemoselective bromination followed by high-yielding Suzuki–Miyaura cross-coupling reactions.Experimental section
Materials and methods
All reagents and organic solvents were purchased from commercial suppliers and were used without further purification. Microwave assisted reactions were carried out in a Biotage Initiator microwave synthesis instrument and temperatures were measured by an IR sensor. Solvents mentioned as dry were purified with a dry station GT S100 immediately prior to use. The reactions were monitored by thin-layer chromatography (TLC) analysis using 0.2 mm precoated Kieselgel 60 F254 (Merck) silica gel plates visualized with a Macherey Nagel UV254 lamp. Column chromatography was performed on silica gel 60 (230–400 mesh, 40–63 μm). Solvent ratios for chromatography and are reported as v/v ratios and are indicated for each compound. Melting points (mp [°C]) were taken on samples in open capillary tubes and are uncorrected on an IA9200 Thermo Scientific Electrothermal Melting Point apparatus/instrument. Infrared analyses were determined on a Thermo Scientific ATR Nicolet iS10 and interpreted using OMNIC software. 1H NMR and 13C NMR spectra were recorded either on a Bruker ULTRASHIELD® PLUS 400 MHz spectrometer (13C, 100 MHz) or on a Bruker AVANCE 250 MHz (13C, 62.9 MHz) spectrometer, as solutions in deuterated solvents. Unless otherwise indicated, chemical shifts (δ) are reported in parts per million (ppm) values, and coupling constants (J) are reported in Hertz. Peak multiplicities are designated by the following abbreviations: s = singlet; d = doublet; t = triplet; q = quartet; m = multiplet; dd = double doublet, br = broadened. High-resolution mass spectrometry analyses (HRMS) were performed on a Maxis Bruker 4G Spectrometer. X-ray diffraction data were collected on Xcalibur CCD area detector diffractometer equipped with monochromatized Mo-Kα radiation (0.71073 Å) at 296 K. The data collection, unit cell refinement, and data reduction were performed using the CrysAlis Pro, Oxford Diffraction Ltd. software package. Analytical numeric absorption correction using a multifaceted crystal model based on expressions derived was carried. The positions of non-H atoms were determined and refinement by SHELXS-2014 program. Non-hydrogen atoms were first refined isotropically followed by anisotropic refinement by full matrix least-squares calculations based on F2 using SHELXL-2014 program.Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 141.6 (CH), 117.7 (C), 113.6 (C), 60.1 (CH2), 37.8 (N–CH3), 14.1 (CH3). IR ν (cm−1): 2995, 2974, 2931, 2871, 1709, 1528, 1470, 1389, 1214, 1173, 1214, 1173, 1040, 770. HRMS (ESI): (m/z) [M + H]+ calculated for [C7H1079BrN2O2]+ 232.9920; found 232.9917, calculated for [C7H1081BrN2O2]+ 234.9900; found 234.9897. A solution of ethyl 5-bromo-1-methyl-1H-pyrazole-4-carboxylate (5.33 g, 0.0228 mol) in anhydrous tetrahydrofuran (75 mL) at 0 °C was treated with diisobutylaluminum hydride (1 M solution in tetrahydrofuran, 58 mL, 0.0571 mol), and the mixture was allowed to warm to room temperature and stir for 2 hours. A cold saturated aqueous sodium potassium tartrate solution was added, and stirring was continued for 4 hours. The aqueous layer was extracted with dichloromethane, and the combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulted crude mixture needed no further purification and was engaged in the next step. Yield: 94% (4.14 g). Colorless solid. Mp: 55–56 °C. 1H NMR (400 MHz, chloroform-d): δ 7.48 (s, 1H), 4.46 (s, 2H), 3.82 (s, 3H), 2.53 (s, 1H). 13C NMR (101 MHz, chloroform-d): δ 139.3 (CH), 120.9 (C), 113.7 (C), 55.6 (CH2), 37.6 (N–CH3). IR ν (cm−1): 3255, 2947, 2896, 2848, 2741, 1554, 1413, 1398, 1322, 1189, 1012, 995, 880. HRMS (ESI): (m/z) [M + H]+ calculated for [C5H879BrN2O]+ 190.9815; found 190.9815, calculated for [C5H881BrN2O]+ 192.9794, found 192.9796. A solution of 5-(bromo-1-methyl-1H-pyrazol-4-yl)methanol(III) (4.14 g, 0.0216 mol) in dichloromethane (25 mL) was treated with activated manganese(IV) oxide (18.8 g, 0.216 mol) and the mixture was stirred at room temperature for 24 hours. Afterwards, the reaction mixture was filtered on Celite® and the filter pad washed with dichloromethane. The recovered filtrates were then concentrated under reduced pressure to yield the crude compound that required no further purification. Yield: 97% (4.01 g). Light yellow solid. Mp: 75–76 °C. 1H NMR (400 MHz, chloroform-d): δ 9.78 (s, 1H), 7.97 (s, 1H), 3.94 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 183.4 (CO), 141.0 (CH), 121.7 (C), 119.7 (C), 37.6 (N–CH3). IR ν (cm−1): 2953, 2920, 2852, 1655, 1524, 1501, 1454, 1419, 1388, 1368, 1190, 1094, 763. HRMS (ESI): (m/z) [M + H]+ calculated for [C5H679BrN2O]+ 188.9658; found 188.9653, calculated for [C5H681BrN2O]+ 190.9638, found 190.9634.
O), 141.6 (CH), 117.7 (C), 113.6 (C), 60.1 (CH2), 37.8 (N–CH3), 14.1 (CH3). IR ν (cm−1): 2995, 2974, 2931, 2871, 1709, 1528, 1470, 1389, 1214, 1173, 1214, 1173, 1040, 770. HRMS (ESI): (m/z) [M + H]+ calculated for [C7H1079BrN2O2]+ 232.9920; found 232.9917, calculated for [C7H1081BrN2O2]+ 234.9900; found 234.9897. A solution of ethyl 5-bromo-1-methyl-1H-pyrazole-4-carboxylate (5.33 g, 0.0228 mol) in anhydrous tetrahydrofuran (75 mL) at 0 °C was treated with diisobutylaluminum hydride (1 M solution in tetrahydrofuran, 58 mL, 0.0571 mol), and the mixture was allowed to warm to room temperature and stir for 2 hours. A cold saturated aqueous sodium potassium tartrate solution was added, and stirring was continued for 4 hours. The aqueous layer was extracted with dichloromethane, and the combined organic layers were dried over magnesium sulfate, filtered, and concentrated under reduced pressure. The resulted crude mixture needed no further purification and was engaged in the next step. Yield: 94% (4.14 g). Colorless solid. Mp: 55–56 °C. 1H NMR (400 MHz, chloroform-d): δ 7.48 (s, 1H), 4.46 (s, 2H), 3.82 (s, 3H), 2.53 (s, 1H). 13C NMR (101 MHz, chloroform-d): δ 139.3 (CH), 120.9 (C), 113.7 (C), 55.6 (CH2), 37.6 (N–CH3). IR ν (cm−1): 3255, 2947, 2896, 2848, 2741, 1554, 1413, 1398, 1322, 1189, 1012, 995, 880. HRMS (ESI): (m/z) [M + H]+ calculated for [C5H879BrN2O]+ 190.9815; found 190.9815, calculated for [C5H881BrN2O]+ 192.9794, found 192.9796. A solution of 5-(bromo-1-methyl-1H-pyrazol-4-yl)methanol(III) (4.14 g, 0.0216 mol) in dichloromethane (25 mL) was treated with activated manganese(IV) oxide (18.8 g, 0.216 mol) and the mixture was stirred at room temperature for 24 hours. Afterwards, the reaction mixture was filtered on Celite® and the filter pad washed with dichloromethane. The recovered filtrates were then concentrated under reduced pressure to yield the crude compound that required no further purification. Yield: 97% (4.01 g). Light yellow solid. Mp: 75–76 °C. 1H NMR (400 MHz, chloroform-d): δ 9.78 (s, 1H), 7.97 (s, 1H), 3.94 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 183.4 (CO), 141.0 (CH), 121.7 (C), 119.7 (C), 37.6 (N–CH3). IR ν (cm−1): 2953, 2920, 2852, 1655, 1524, 1501, 1454, 1419, 1388, 1368, 1190, 1094, 763. HRMS (ESI): (m/z) [M + H]+ calculated for [C5H679BrN2O]+ 188.9658; found 188.9653, calculated for [C5H681BrN2O]+ 190.9638, found 190.9634.General procedures
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:0.5), a solution of cesium carbonate (1.3 eq.) in 0.5 mL of water was added. The tube was argon flushed three times and then Pd(PPh3)4 (1.25 mol%) was added. The tube was again argon flushed, sealed and microwave irradiated at 140 °C for 1 h. The reaction mixture was then filtrated on Celite®, rinsed and extracted with EtOAc (3 × 10 mL). The organic layers collected were washed, dried on MgSO4, filtered and evaporated. The crude obtained was purified by silica gel column chromatography using appropriate solvents, affording the desired compound.
:1:0.5), a solution of cesium carbonate (1.3 eq.) in 0.5 mL of water was added. The tube was argon flushed three times and then Pd(PPh3)4 (1.25 mol%) was added. The tube was again argon flushed, sealed and microwave irradiated at 140 °C for 1 h. The reaction mixture was then filtrated on Celite®, rinsed and extracted with EtOAc (3 × 10 mL). The organic layers collected were washed, dried on MgSO4, filtered and evaporated. The crude obtained was purified by silica gel column chromatography using appropriate solvents, affording the desired compound.
1-Methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole (2a). Prepared as described in the general procedure A (phenylhydrazine: 63 mg, 57 μL, 0.58 mmol, CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 76% (80 mg). Orange oil. Column chromatography eluents: PE/EtOAc = 8/2. 1H NMR (400 MHz, acetone-d6): δ 7.67 (d, J = 7.9 Hz, 2H), 7.62 (s, 1H), 7.58 (t, J = 7.9 Hz, 2H), 7.45 (s, 1H), 7.43 (t, J = 7.9 Hz, 1H), 3.85 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 149.1 (C), 139.8 (C), 130.2 (2 × CH), 130.0 (CH), 128.0 (CH), 127.6 (CH), 124.1 (2 × CH), 121.0 (C), 37.7 (N–CH3). IR ν (cm−1): 3056, 2942, 1682, 1595, 1508, 1457, 1436, 1372, 1189, 1112, 1034, 1004, 990, 969, 844, 759, 715, 696. HRMS (ESI): (m/z) [M + H]+ calculated for [C11H11N4]+ 199.0978; found 199.0978.
1-Methyl-6-(p-tolyl)-1,6-dihydropyrazolo[3,4-c]pyrazole (2b). Prepared as described in the general procedure A (4-methylphenylhydrazine hydrochloride: 92 mg, 0.58 mmol; NaOAc: 48 mg, 0.58 mmol; CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 64% (72 mg). Beige solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 88–89 °C. 1H NMR (400 MHz, acetone-d6): δ 7.59 (s, 1H), 7.52 (d, J = 8.2 Hz, 2H), 7.44 (s, 1H), 7.37 (d, J = 8.2 Hz, 2H), 3.81 (s, 3H), 2.41 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 149.1 (C), 137.9 (C), 137.4 (C), 130.6 (2 × CH), 129.6 (CH), 127.5 (CH), 124.2 (2 × CH), 120.8 (C), 37.5 (N–CH3), 21.0 (CH3). IR ν (cm−1): 3108, 3041, 2918, 2854, 1711, 1604, 1583, 1515, 1436, 1416, 1376, 1365, 1216, 1198, 1108, 1036, 999, 977, 843, 819, 720. HRMS (ESI): (m/z) [M + H]+ calculated for [C12H13N4]+ 213.1134; found 213.1136.
1-(4-Methoxyphenyl)-6-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole (2c). Prepared as described in the general procedure A (4-methoxyphenylhydrazine hydrochloride: 102 mg, 0.58 mmol; NaOAc: 48 mg, 0.58 mmol; CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 46% (56 mg). Light orange oil. Column chromatography eluents: PE/EtOAc = 8/2. 1H NMR (400 MHz, chloroform-d): δ 7.57 (s, 1H), 7.46 (s, 1H), 7.44 (d, J = 8.9 Hz, 2H), 7.01 (d, J = 8.9 Hz, 2H), 3.86 (s, 3H), 3.77 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.3 (C), 148.7 (C), 131.6 (C), 129.1 (CH), 127.6 (CH), 125.7 (2 × CH), 119.4 (C), 114.6 (2 × CH), 55.7 (O–CH3), 36.9 (N–CH3). IR ν (cm−1): 3001, 2935, 2837, 1600, 1585, 1514, 1444, 1421, 1370, 1299, 1246, 1182, 1137, 1109, 1035, 1013, 994, 969, 832, 718, 704. HRMS (ESI): (m/z) [M + H]+ calculated for [C12H13N4O]+ 229.1083; found 229.1085.
4-(6-Methylpyrazolo[3,4-c]pyrazol-1(6H)-yl)benzonitrile (2d). Prepared as described in the general procedure A (4-cyanophenylhydrazine hydrochloride: 99 mg, 0.58 mmol; NaOAc: 48 mg, 0.58 mmol; CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 76% (90 mg). Light brown solid. Column chromatography eluents: PE/EtOAc = 7/3. Mp: 154–155 °C. 1H NMR (250 MHz, chloroform-d): δ 7.82 (d, J = 8.9 Hz, 2H), 7.72–7.67 (m, 3H), 7.52 (s, 1H), 3.94 (s, 3H). 13C NMR (63 MHz, chloroform-d): δ 148.1 (C), 142.1 (C), 133.7 (2 × CH), 131.6 (CH), 128.3 (CH), 122.8 (2 × CH), 121.0 (C), 118.2 (CN), 110.6 (C), 38.1 (N–CH3). IR ν (cm−1): 3133, 3104, 2924, 2853, 2224, 1605, 1591, 1512, 1438, 1411, 1378, 1344, 1287, 1185, 1172, 1110, 1032, 996, 967, 837, 711, 704. HRMS (ESI): (m/z) [M + H]+ calculated for [C12H10N5]+ 224.0930; found 224.0930.
1-Methyl-6-(4-(trifluoromethyl)phenyl)-1,6-dihydropyrazolo[3,4-c]pyrazole (2e). Prepared as described in the general procedure A (4-(trifluoromethyl)-phenylhydrazine: 103 mg, 0.58 mmol; CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 54% (77 mg). Light yellow solid. Column chromatography eluents: DCM/EtOAc = 95/5. Mp: 89–90 °C. 1H NMR (400 MHz, chloroform-d): δ 7.78 (d, J = 8.5 Hz, 2H), 7.68 (d, J = 8.5 Hz, 2H), 7.66 (s, 1H), 7.50 (s, 1H), 3.90 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 148.2 (C), 141.4 (C), 130.9 (CH), 129.2 (q, 2JC–F = 33.0 Hz, C), 128.1 (CH), 126.8 (q, 3JC–F = 3.8 Hz, 2 × CH), 123.8 (q, 1JC–F = 272.1 Hz, CF3), 122.9 (2 × CH), 120.6 (C), 37.8 (N–CH3). 19F NMR (235 MHz, chloroform-d): δ −62.4. IR ν (cm−1): 3106, 2923, 2854, 1616, 1597, 1523, 1507, 1143, 1318, 1193, 1180, 1157, 1118, 1107, 1065, 1031, 999, 970, 872, 839, 823, 714, 690. HRMS (ESI): (m/z) [M + H]+ calculated for [C12H10F3N4]+ 267.0852; found 267.0852.
1-(4-Fluorophenyl)-6-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole (2f). Prepared as described in the general procedure A (4-fluorophenylhydrazine hydrochloride: 95 mg, 0.58 mmol; NaOAc: 48 mg, 0.58 mmol; CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 62% (71 mg). Colorless oil. Column chromatography eluents: PE/EtOAc = 8/2. 1H NMR (400 MHz, chloroform-d): δ 7.60 (s, 1H), 7.51 (dd, J = 8.9, 4.7 Hz, 2H), 7.48 (s, 1H), 7.24–7.16 (dd, J = 8.9, 8.8 Hz, 2H), 3.80 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 161.9 (d, 1JC–F = 248.0 Hz, C), 148.5 (C), 134.7 (d, 4JC–F = 3.1 Hz, C), 129.8 (CH), 127.8 (CH), 125.7 (d, 3JC–F = 8.6 Hz, 2 × CH), 119.8 (C), 116.5 (d, 2JC–F = 23.1 Hz, 2 × CH), 37.1 (N–CH3). IR ν (cm−1): 3111, 3066, 2943, 1701, 607, 1594, 1513, 1443, 1417, 1371, 1218, 1190, 1218, 1190, 1152, 1033, 999, 969, 838, 815, 718, 701. HRMS (ESI): (m/z) [M + H]+ calculated for [C11H10FN4]+ 217.0884; found 217.0884.
1-(3-Fluorophenyl)-6-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole (2g). Prepared as described in the general procedure A (3-fluorophenylhydrazine hydrochloride: 95 mg, 0.58 mmol; NaOAc: 48 mg, 0.58 mmol; CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 85% (98 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 103–104 °C. 1H NMR (400 MHz, chloroform-d): δ 7.62 (s, 1H), 7.51–7.42 (m, 2H), 7.36–7.29 (m, 2H), 7.09 (d, J = 8.8 Hz, 1H), 3.88 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 163.0 (d, 1JC–F = 248.4 Hz, C), 148.2 (C), 140.0 (d, 3JC–F = 10.2 Hz, CH), 130.7 (d, 3JC–F = 9.2 Hz, C), 130.2 (CH), 127.9 (CH), 120.2 (C), 118.6 (d, 4JC–F = 3.2 Hz, CH), 114.4 (d, 2JC–F = 21.1 Hz, CH), 110.9 (d, 2JC–F = 24.7 Hz, CH), 37.5 (N–CH3). IR ν (cm−1): 3091, 2992, 2934, 2853, 1161, 1595, 1509, 1464, 1429, 1414, 1379, 1199, 1171, 1148, 1113, 1081, 1030, 970, 869, 784, 712. HRMS (ESI): (m/z) [M + H]+ calculated for [C11H10FN4]+ 217.0884; found 217.0883.
1-(2-Fluorophenyl)-6-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole (2h). Prepared as described in the general procedure A (2-fluorophenylhydrazine hydrochloride: 95 mg, 0.58 mmol; NaOAc: 48 mg, 0.58 mmol; CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 46% (53 mg). Light brown solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 57–58 °C. 1H NMR (400 MHz, acetone-d6): δ 7.72–7.67 (m, 2H), 7.61–7.53 (ddd, J = 8.2, 8.1, 4.7 Hz, 1H), 7.48–7.40 (m, 3H), 3.71 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 157.1 (d, 1JC–F = 248.7 Hz, C), 150.1 (C), 131.0 (d, 3JC–F = 7.8 Hz, CH), 130.8 (CH), 129.2 (C), 127.6 (CH), 127.5 (CH), 126.1 (d, 3JC–F = 3.9 Hz, CH), 120.1 (C), 117.4 (d, 2JC–F = 19.5 Hz, CH), 36.2 (s, N–CH3). IR ν (cm−1): 3113, 3075, 2924, 2852, 1607, 1515, 1494, 1479, 1463, 1445, 1432, 1417, 1370, 1260, 1216, 1187, 1103, 1025, 1000, 972, 846, 836, 755, 725, 717, 704. HRMS (ESI): (m/z) [M + H]+ calculated for [C11H10FN4]+ 217.0884; found 217.0882.
1-Methyl-6-(pyridin-4-yl)-1,6-dihydropyrazolo[3,4-c]pyrazole (2i). Prepared as described in the general procedure A (4-hydrazinopyridine: 64 mg, 0.58 mmol; CuI: 2.5 mg, 0.013 mmol; K2CO3: 146 mg, 1.06 mmol). Yield: 46% (49 mg). White yellow solid. Column chromatography eluents: EtOAc/MeOH = 95/5. Mp: 105–106 °C. 1H NMR (400 MHz, chloroform-d): δ 8.75 (br s, 2H), 7.68 (s, 1H), 7.53 (br s, 2H), 7.50 (s, 1H), 4.00 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 151.2 (2 × CH), 147.9 (C), 145.3 (C), 131.8 (CH), 128.3 (CH), 121.2 (C), 116.1 (2 × CH), 38.5 (N–CH3). IR ν (cm−1): 3088, 3036, 2943, 1682, 1647, 1587, 1569, 1508, 1440, 1417, 1381, 1191, 1115, 1030, 1009, 993, 696, 842, 820, 715. HRMS (ESI): (m/z) [M + H]+ calculated for [C10H10N5]+ 200.0930; found 200.0930.
3-Bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole (3a). Prepared as described in the general procedure B (1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 2a: 100 mg, 0.5 mmol; NBS: 1.5 eq., 135 mg, 0.75 mmol; MeCN: 2 mL; microwave irradiation at 100 °C for 2 h). Yield: 77% (108 mg). Light orange oil. Column chromatography eluents: PE/EtOAc = 95/5. 1H NMR (400 MHz, chloroform-d): δ 7.60 (s, 1H), 7.55–7.50 (m, 4H), 7.41 (t, J = 8.6 Hz, 1H), 3.79 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 148.8 (C), 138.1 (C), 129.6 (2 × CH), 129.1 (CH), 128.1 (CH), 123.8 (2 × CH), 119.9 (C), 113.1 (C–Br), 37.6 (N–CH3). IR ν (cm−1): 3066, 2928, 1726, 1594, 1506, 1457, 1431, 1367, 1238, 1148, 1079, 1038, 1008, 983, 905, 840, 759, 715, 696. HRMS (ESI): (m/z) [M + H]+ calculated for [C11H1079BrN4]+ 277.0083; found 277.0084, calculated for [C11H1081BrN4]+ 279.0064, found 279.0065.
3-Bromo-6-(4-methoxyphenyl)-1-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole (3b). Prepared as described in the general procedure B (1-(4-methoxyphenyl)-6-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole 2c: 100 mg, 0.43 mmol; NBS: 78 mg, 0.43 mmol, 1.0 eq.; MeCN: 2 mL; conventional heating at reflux for 4 h). Yield: 55% (74 mg). Colorless oil. Column chromatography eluents: PE/EtOAc = 9/1. 1H NMR (400 MHz, chloroform-d): δ 7.54 (s, 1H), 7.41 (d, J = 8.9 Hz, 2H), 7.00 (d, J = 8.9 Hz, 2H), 3.85 (s, 3H), 3.71 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.5 (C), 149.0 (C), 131.0 (C), 128.5 (CH), 125.8 (2 × CH), 119.4 (C), 114.7 (2 × CH), 112.8 (C–Br), 55.7 (O–CH3), 37.2 (N–CH3). IR ν (cm−1): 3107, 2936, 2836, 1600, 1585, 1514, 1462, 1366, 1299, 1246, 1150, 1040, 904, 832, 716. HRMS (ESI): (m/z) [M + H]+ calculated for [C12H1279BrN4O]+ 307.0189; found 307.0189, calculated for [C12H1281BrN4O]+ 309.0169, found 309.0168.
3-Bromo-1-methyl-6-(4-(trifluoromethyl)-1,6-dihydrophenyl)pyrazolo[3,4-c]pyrazole (3c). Prepared as described in the general procedure B (1-methyl-6-(4-(trifluoromethyl)phenyl)-1,6-dihydropyrazolo[3,4-c]pyrazole 2e: 100 mg, 0.37 mmol; NBS: 101 mg, 0.56 mmol, 1.5 eq.; MeCN: 2 mL; microwave irradiation at 100 °C for 4 h). Yield: 92% (120 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 7/3. Mp: 132–133 °C. 1H NMR (400 MHz, chloroform-d): δ 7.79 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 8.3 Hz, 2H), 7.62 (s, 1H), 3.85 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 148.5 (C), 140.9 (C), 130.2 (CH), 129.7 (q, 2JC–F = 33.2 Hz, C), 126.9 (q, 3JC–F = 3.7 Hz, 2 × CH), 123.7 (q, 1JC–F = 272.2 Hz, CF3), 123.1 (2 × CH), 120.6 (C), 113.4 (C–Br), 38.1 (N–CH3). 19F NMR (235 MHz, chloroform-d): −62.5. IR ν (cm−1): 3108, 2924, 2853, 1613, 1596, 1583, 1523, 1500, 1438, 1416, 1320, 1249, 1151, 1107, 1065, 1034, 1009, 904, 838. HRMS (ESI): (m/z) [M + H]+ calculated for [C12H979BrF3N4]+ 344.9957; found 344.9958, calculated for [C12H981BrF3N4]+ 346.9937, found 346.9938.
1-Methyl-6-phenyl-3-(p-tolyl)-1,6-dihydropyrazolo[3,4-c]pyrazole (4a). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 4-methylphenylboronic acid: 74 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 83% (87 mg). Light brown solid. Column chromatography eluents: PE/EtOAc = 9/1. Mp: 126–127 °C. 1H NMR (400 MHz, acetone-d6): δ 7.98 (s, 1H), 7.87 (d, J = 8.0 Hz, 2H), 7.70 (d, J = 7.4 Hz, 2H), 7.60 (t, J = 7.4 Hz, 2H), 7.45 (t, J = 7.4 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 3.88 (s, 3H), 2.37 (s, 3H). 13C NMR (101 MHz, acetone-d6): δ 150.1 (C), 139.6 (C), 139.3 (C), 138.5 (C), 131.0 (C), 130.6 (CH), 130.3 (2 × CH), 130.2 (2 × CH), 128.1 (CH), 126.6 (2 × CH), 124.3 (2 × CH), 118.0 (C), 37.8 (N–CH3), 21.3 (CH3). IR ν (cm−1): 3013, 2919, 1854, 1596, 1510, 1496, 1459, 1416, 1310, 1295, 1278, 1206, 1087, 1040, 1017, 1002, 990, 909, 820, 766, 753, 724, 696. HRMS (ESI): (m/z) [M + H]+ calculated for [C18H17N4]+ 289.1447; found 289.1446.
1-Methyl-6-phenyl-3-(m-tolyl)-1,6-dihydropyrazolo[3,4-c]pyrazole (4b). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 3-methylphenyl-boronic acid: 74 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 89% (93 mg). White yellow solid. Column chromatography eluents: PE/EtOAc = 9/1. Mp: 137–138 °C. 1H NMR (400 MHz, chloroform-d): δ 7.85 (s, 1H), 7.75 (s, 1H), 7.70 (d, J = 7.7 Hz, 1H), 7.59 (d, J = 7.6 Hz, 2H), 7.54 (t, J = 7.6 Hz, 2H), 7.41 (t, J = 7.6 Hz, 1H), 7.36 (t, J = 7.7 Hz, 1H), 7.19 (d, J = 7.7 Hz, 1H), 3.88 (s, 3H), 2.44 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.4 (C), 139.9 (C), 138.6 (C), 138.5 (C), 132.5 (C), 130.3 (CH), 129.6 (2 × CH), 129.2 (CH), 128.8 (CH), 127.8 (CH), 126.6 (CH), 123.8 (2 × CH), 123.5 (CH), 117.4 (C), 37.5 (N–CH3), 21.6 (CH3). IR ν (cm−1): 3059, 2920, 2851, 1596, 1579, 1505, 1456, 1417, 1315, 1196, 1172, 1081, 1039, 1025, 1014, 939, 825, 838, 796, 762. HRMS (ESI): (m/z) [M + H]+ calculated for [C18H17N4]+ 289.1447; found 289.1447.
1-Methyl-6-phenyl-3-(o-tolyl)-1,6-dihydropyrazolo[3,4-c]pyrazole (4c). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 2-methylphenyl-boronic acid: 74 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 86% (90 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 134–135 °C. 1H NMR (400 MHz, chloroform-d): δ 7.71–7.66 (m, 2H), 7.59 (d, J = 7.3 Hz, 2H), 7.53 (t, J = 7.3 Hz, 2H), 7.40 (t, J = 7.3 Hz, 1H), 7.32–7.28 (m, 3H), 3.88 (s, 3H), 2.61 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.1 (C), 140.6 (C), 138.9 (C), 136.7 (C), 132.2 (C), 131.5 (CH), 131.0 (CH), 129.9 (CH), 129.8 (2 × CH), 128.5 (CH), 128.0 (CH), 126.3 (CH), 124.1 (2 × CH), 119.5 (C), 37.8 (N–CH3), 21.7 (CH3). IR ν (cm−1): 3059, 3039, 2953, 2923, 1597, 1526, 1507, 1457, 1414, 1379, 1302, 1270, 1206, 1074, 1041, 1010, 989, 908, 846, 760. HRMS (ESI): (m/z) [M + H]+ calculated for [C18H17N4]+ 289.1447; found 289.1447.
1-Methyl-3,6-diphenyl-1,6-dihydropyrazolo[3,4-c]pyrazole (4d). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; phenylboronic acid: 66 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 92% (91 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 9/1. Mp: 133–135 °C. 1H NMR (400 MHz, chloroform-d): δ 7.92 (d, J = 8.3 Hz, 2H), 7.85 (s, 1H), 7.58 (d, J = 7.3 Hz, 2H), 7.52 (t, J = 8.3 Hz, 2H), 7.46 (t, J = 7.3 Hz, 2H), 7.43–7.33 (m, 2H), 3.86 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.3 (C), 139.6 (C), 138.4 (C), 132.5 (C), 130.1 (CH), 129.5 (2 × CH), 128.8 (2 × CH), 128.3 (CH), 127.7 (CH), 126.1 (2 × CH), 123.7 (2 × CH), 117.3 (C), 37.4 (N–CH3). IR ν (cm−1): 3089, 3056, 2923, 2852, 1595, 1504, 1455, 1418, 1208, 1081, 1016, 988, 907, 758. 694. HRMS (ESI): (m/z) [M + H]+ calculated for [C17H15N4]+ 275.1291; found 275.1289.
3-(4-Methoxyphenyl)-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole (4e). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 4-methoxyphenyl-boronic acid: 83 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 90% (99 mg). Light brown solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 108–109 °C. 1H NMR (400 MHz, chloroform-d): δ 7.84 (d, J = 8.8 Hz, 2H), 7.82 (s, 1H), 7.57 (d, J = 8.0 Hz, 2H), 7.54 (t, J = 8.0 Hz, 2H), 7.40 (t, J = 8.0 Hz, 1H), 6.99 (d, J = 8.8 Hz, 2H), 3.86 (s, 3H), 3.85 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.9 (C), 149.4 (C), 139.6 (C), 138.5 (C), 130.2 (CH), 129.5 (2 × CH), 127.7 (CH), 127.5 (2 × CH), 125.4 (C), 123.7 (2 × CH), 117.1 (C), 114.3 (2 × CH), 55.4 (O–CH3), 37.4 (N–CH3). IR ν (cm−1): 3052, 2999, 2941, 2837, 1596, 1538, 1506, 1458, 1439, 1417, 1245, 1173, 1109, 1087, 1024, 1011, 989, 908, 827, 841, 792. HRMS (ESI): (m/z) [M + H]+ calculated for [C18H17N4O]+ 305.1396; found 305.1396.
1-Methyl-6-phenyl-3-(4-(trifluoromethyl)phenyl)-1,6-dihydropyrazolo[3,4-c]pyrazole (4f). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 4-trifluoro-methylphenylboronic acid: 103 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 86% (107 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 136–137 °C. 1H NMR (400 MHz, chloroform-d): δ 8.01 (d, J = 8.1 Hz, 2H), 7.85 (s, 1H), 7.71 (d, J = 8.1 Hz, 2H), 7.61–7.51 (m, 4H), 7.43 (t, J = 8.1 Hz, 1H), 3.89 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.4 (C), 138.3 (C), 138.0 (C), 136.0 (C), 130.3 (q, 2JC–F = 32.5 Hz, C), 129.8 (CH), 129.6 (2 × CH), 128.0 (CH), 126.2 (2 × CH), 125.9 (q, 3JC–F = 3.8 Hz, 2 × CH), 124.3 (q, 1JC–F = 272.0 Hz, CF3), 123.9 (2 × CH), 117.4 (C), 37.6 (N–CH3). 19F NMR (235 MHz, chloroform-d): δ −62.5. IR ν (cm−1): 3113, 3063, 2950, 1616, 1590, 1590, 1575, 1540, 1495, 1169, 1110, 1082, 1041, 1010, 990, 955, 854, 845. HRMS (ESI): (m/z) [M + H]+ calculated for [C18H14F3N4]+ 343.1165; found 343.1166.
4-(1-Methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazol-3-yl)benzonitrile (4g). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl1,6-dihydro-pyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 4-cyanophenyl- boronic acid: 80 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 72% (78 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 7/3. Mp: 203–204 °C. 1H NMR (400 MHz, chloroform-d): δ 8.00 (d, J = 8.4 Hz, 2H), 7.85 (s, 1H), 7.74 (d, J = 8.4 Hz, 2H), 7.61–7.52 (m, 4H), 7.44 (t, J = 8.0 Hz, 1H), 3.90 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.5 (C), 138.2 (C), 137.4 (C), 137.0 (C), 132.8 (2 × CH), 129.7 (2 × CH), 129.7 (CH), 128.1 (CH), 126.5 (2 × CH), 124.0 (2 × CH), 119.0 (CN), 117.4 (C), 111.5 (C), 37.7 (N–CH3). IR ν (cm−1): 3056, 2943, 2226, 1592, 1581, 1502, 1435, 1417, 1296, 1281, 1206, 1081, 1041, 1019, 990, 908, 844. HRMS (ESI): (m/z) [M + H]+ calculated for [C18H14N5]+ 300.1243; found 300.1244.
3-(3-Fluorophenyl)-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole (4h). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 3-fluorophenyl-boronic acid: 76 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 90% (95 mg). Beige solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 97–98 °C. 1H NMR (400 MHz, chloroform-d): δ 7.83 (s, 1H), 7.67 (d, J = 7.7 Hz, 1H), 7.62 (d, J = 9.8 Hz, 1H), 7.59–7.50 (m, 4H), 7.45–7.39 (m, 2H), 7.04 (dd, J = 8.5, 8.2 Hz, 1H), 3.87 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 163.3 (d, 1JC–F = 245.5 Hz, C), 149.4 (C), 138.4 (C), 138.3 (C), 134.8 (d, 3JC–F = 8.3 Hz, C), 130.4 (d, 3JC–F = 8.4 Hz, CH), 129.9 (CH), 129.6 (2 × CH), 127.9 (CH), 123.8 (2 × CH), 121.9 (d, 4JC–F = 2.8 Hz, CH), 117.3 (C), 115.1 (d, 2JC–F = 21.3 Hz, CH), 112.8 (d, 2JC–F = 22.8 Hz, CH), 37.5 (N–CH3). 19F NMR (235 MHz, chloroform-d): δ −112.9. IR ν (cm−1): 3054, 2931, 2850, 1595, 1580, 1507, 1481, 1449, 1411, 1297, 1183, 1082, 1018, 990, 881, 831. HRMS (ESI): (m/z) [M + H]+ calculated for [C17H14FN4]+ 293.1197; found 293.1197.
1-Methyl-6-phenyl-3-(thiophen-3-yl)-1,6-dihydropyrazolo[3,4-c]pyrazole (4i). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 3-thienylboronic acid: 70 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 85% (86 mg). Light brown solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 100–101 °C. 1H NMR (400 MHz, chloroform-d): δ 7.78 (s, 1H), 7.68 (s, 1H), 7.61–7.50 (m, 5H), 7.43–7.38 (m, 2H), 3.84 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.1 (C), 138.5 (C), 136.0 (C), 134.2 (C), 129.8 (CH), 129.5 (2 × CH), 127.7 (CH), 126.4 (CH), 125.9 (CH), 123.7 (2 × CH), 122.0 (CH), 117.4 (C), 37.3 (N–CH3). IR ν (cm−1): 3095, 3061, 2947, 1596, 1510, 1460, 1413, 1282, 1211, 1188, 1038, 1013, 1001, 951, 870, 793, 779. HRMS (ESI): (m/z) [M + H]+ calculated for [C15H13N4S]+ 281.0855; found 281.0858.
1-Methyl-6-phenyl-3-(pyridin-4-yl)-1,6-dihydropyrazolo[3,4-c]pyrazole (4j). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; 4-pyridinylboronic acid: 67 mg, 0.54 mmol 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 78% (78 mg). Beige solid. Column chromatography eluents: PE/EtOAc = 1/9. Mp: 158–159 °C. 1H NMR (400 MHz, chloroform-d): δ 8.68 (br s, 2H), 7.84 (s, 1H), 7.75 (br s, 2H), 7.58–7.47 (m, 4H), 7.41 (t, J = 6.8 Hz, 1H), 3.86 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 150.3 (2 × CH), 149.3 (C), 139.8 (C), 138.1 (C), 136.7 (C), 129.6 (2 × CH), 129.5 (CH), 128.0 (CH), 123.8 (2 × CH), 120.3 (2 × CH), 117.5 (C), 37.7 (N–CH3). IR ν (cm−1): 3034, 2925, 1609, 1594, 1556, 1506, 1432, 1407, 1388, 1323, 1291, 1204, 1094, 1019, 1002, 992, 917, 826. HRMS (ESI): (m/z) [M + H]+ calculated for [C16H14N5]+ 276.1243; found 276.1244.
(E)-1-Methyl-6-phenyl-3-styryl-1,6-dihydropyrazolo[3,4-c]pyrazole (4k). Prepared as described in the general procedure C (3-bromo-1-methyl-6-phenyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3a: 100 mg, 0.36 mmol; trans-2-phenylvinylboronic acid: 81 mg, 0.54 mmol, 1.5 eq.; Cs2CO3: 153 mg, 0.47 mmol, 1.3 eq.; Pd(PPh3)4: 5.5 mg, 0.004 mmol, 1.25 mol%). Yield: 64% (70 mg). Light brown solid. Column chromatography eluents: PE/EtOAc = 8/2. Mp: 140–141 °C. 1H NMR (400 MHz, chloroform-d): δ 7.84 (s, 1H), 7.60–7.47 (m, 6H), 7.37 (q, J = 7.8 Hz, 3H), 7.27 (t, J = 7.4 Hz, 1H), 7.18 (d, J = 18.5 Hz, 1H), 7.14 (d, J = 18.5 Hz, 1H), 3.79 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.0 (C), 139.9 (C), 138.3 (C), 136.7 (C), 132.8 (CH), 130.0 (CH), 129.5 (2 × CH), 128.8 (2 × CH), 128.1 (CH), 127.7 (CH), 126.6 (2 × CH), 123.7 (2 × CH), 120.2 (CH), 116.6 (C), 37.3 (N–CH3). IR ν (cm−1): 3100, 3061, 3030, 2946, 1591, 1580, 1505, 1494, 1429, 1304, 1261, 1201, 1046, 1013, 1001, 989, 961, 906, 874, 852, 744. HRMS (ESI): (m/z) [M + H]+ calculated for [C19H17N4]+ 301.1447; found 301.1449.
4-(6-(4-Methoxyphenyl)-1-methyl-1,6-dihydropyrazolo[3,4-c]pyrazol-3-yl)benzonitrile (4l). Prepared as described in the general procedure C (3-bromo-6-(4-methoxyphenyl)-1-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3c: 100 mg, 0.32 mmol; 4-cyanophenylboronic acid: 72 mg, 0.48 mmol, 1.5 eq.; Cs2CO3: 138 mg, 0.42 mmol, 1.3 eq.; Pd(PPh3)4: 5 mg, 0.004 mmol, 1.25 mol%). Yield: 75% (81 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 7/3. Mp: 186–187 °C. 1H NMR (400 MHz, chloroform-d): δ 7.99 (d, J = 8.3 Hz, 2H), 7.81 (s, 1H), 7.73 (d, J = 8.3 Hz, 2H), 7.47 (d, J = 8.9 Hz, 2H), 7.04 (d, J = 8.9 Hz, 2H), 3.88 (s, 3H), 3.83 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.6 (C), 149.7 (C), 137.3 (C), 137.1 (C), 132.7 (2 × CH), 131.1 (C), 129.1 (CH), 126.4 (2 × CH), 126.0 (2 × CH), 119.0 (CN), 116.9 (C), 114.7 (2 × CH), 111.3 (C), 55.7 (O–CH3), 37.3 (N–CH3). IR ν (cm−1): 3105, 3082, 3012, 2942, 2842, 2221, 1610, 1593, 1580, 1517, 1439, 1299, 1253, 1203, 1110, 1086, 1045, 1017, 989, 910, 841, 826. HRMS (ESI): (m/z) [M + H]+ calculated for [C19H16N5O]+ 330.1349; found 330.1353.
6-(4-Methoxyphenyl)-1-methyl-3-(pyridin-4-yl)-1,6-dihydropyrazolo[3,4-c]pyrazole (4m). Prepared as described in the general procedure C (3-bromo-6-(4-methoxyphenyl)-1-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3c: 100 mg, 0.32 mmol; 4-pyridinylboronic acid: 61 mg, 0.48 mmol, 1.5 eq.; Cs2CO3: 138 mg, 0.42 mmol, 1.3 eq.; Pd(PPh3)4: 5 mg, 0.004 mmol, 1.25 mol%). Yield: 76% (76 mg). Colorless solid. Column chromatography eluents: DCM/EtOAc = 5/5. Mp: 198–199 °C. 1H NMR (400 MHz, chloroform-d): δ 8.68 (d, J = 6.2 Hz, 2H), 7.83 (s, 1H), 7.76 (d, J = 6.2 Hz, 2H), 7.47 (d, J = 8.9 Hz, 2H), 7.04 (d, J = 8.9 Hz, 2H), 3.88 (s, 3H), 3.83 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 159.6 (C), 150.5 (2 × CH), 149.7 (C), 140.0 (C), 136.7 (C), 131.1 (C), 129.1 (CH), 126.0 (2 × CH), 120.3 (2 × CH), 117.0 (C), 114.7 (2 × CH), 55.7 (O–CH3), 37.3 (N–CH3). IR ν (cm−1): 3067, 2951, 2847, 1593, 1556, 1518, 1455, 1438, 1409, 1298, 1255, 1208, 1166, 1108, 1094, 1017, 991, 917, 821. HRMS (ESI): (m/z) [M + H]+ calculated for [C17H16N5O]+ 306.1349; found 306.1349.
3,6-Bis(4-methoxyphenyl)-1-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole (4n). Prepared as described in the general procedure C (3-bromo-6-(4-methoxyphenyl)-1-methyl-1,6-dihydropyrazolo[3,4-c]pyrazole 3c: 100 mg, 0.32 mmol; 4-methoxyphenylboronic acid: 75 mg, 0.48 mmol, 1.5 eq.; Cs2CO3: 138 mg, 0.42 mmol, 1.3 eq.; Pd(PPh3)4: 5 mg, 0.004 mmol, 1.25 mol%). Yield: 80% (88 mg). Colorless solid. Column chromatography eluent: DCM. Mp: 212–213 °C. 1H NMR (400 MHz, chloroform-d): δ 7.84 (d, J = 8.7 Hz, 2H), 7.78 (s, 1H), 7.47 (d, J = 8.9 Hz, 2H), 7.02 (d, J = 8.9 Hz, 2H), 6.99 (d, J = 8.7 Hz, 2H), 3.87 (s, 3H), 3.85 (s, 3H), 3.78 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 160.1 (C), 159.6 (C), 150.0 (C), 139.8 (C), 131.8 (C), 129.9 (CH), 127.7 (2 × CH), 126.1 (2 × CH), 125.8 (C), 116.9 (C), 114.9 (2 × CH), 114.6 (2 × CH), 56.0 (O–CH3), 55.7 (O–CH3), 37.2 (N–CH3). IR ν (cm−1): 3105, 3002, 2974, 2941, 1840, 1596, 1537, 1513, 1500, 1452, 1438, 1302, 1244, 1168, 1106, 1043, 1024, 1013, 907, 839, 824. HRMS (ESI): (m/z) [M + H]+ calculated for [C19H19N4O2]+ 335.1502; found 335.1505.
4-(1-Methyl-6-(4-(trifluoromethyl)phenyl)-1,6-dihydropyrazolo[3,4-c]pyrazol-3-yl)benzonitrile (4o). Prepared as described in the general procedure C (3-bromo-1-methyl-6-(4-(trifluoromethyl)phenyl)-1,6-dihydro pyrazolo[3,4-c]pyrazole 3e: 100 mg, 0.28 mmol; 4-cyanophenylboronic acid: 64 mg, 0.43 mmol, 1.5 eq.; Cs2CO3: 123 mg, 0.37 mmol, 1.3 eq.; Pd(PPh3)4: 4.5 mg, 0.003 mmol, 1.25 mol%). Yield: 68% (73 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 9/1. Mp: 226–227 °C. 1H NMR (400 MHz, chloroform-d): δ 7.98 (d, J = 8.4 Hz, 2H), 7.88 (s, 1H), 7.82 (d, J = 8.6 Hz, 2H), 7.76–7.69 (m, 4H), 3.96 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.2 (C), 141.0 (C), 137.7 (C), 136.6 (C), 132.7 (2 × CH), 130.7 (CH), 129.7 (q, 2JC–F = 33.1 Hz, C), 126.9 (q, 3JC–F = 3.7 Hz, 2 × CH), 126.5 (2 × CH), 123.8 (d, 1JC–F = 272.2 Hz, CF3), 123.3 (2 × CH), 118.9 (CN), 118.0 (C), 111.7 (C), 38.2 (N–CH3). 19F NMR (235 MHz, chloroform-d): δ −62.5. IR ν (cm−1): 3343, 2923, 1852, 2225, 1616, 1579, 1522, 1541, 1421, 1384, 1315, 1168, 1103, 1038, 1013, 989, 955, 909, 839. HRMS (ESI): (m/z) [M + H]+ calculated for [C19H13F3N5]+ 368.1117; found 368.1116.
1-Methyl-3-(pyridin-4-yl)-6-(4-(trifluoromethyl)phenyl)-1,6-dihydro pyrazolo[3,4-c]pyrazole (4p). Prepared as described in the general procedure C (3-bromo-1-methyl-6-(4-(trifluoromethyl)phenyl)-1,6-dihydro pyrazolo[3,4-c]pyrazole 3e: 100 mg, 0.28 mmol; 4-pyridinylboronic acid: 54 mg, 0.43 mmol, 1.5 eq.; Cs2CO3: 123 mg, 0.37 mmol, 1.3 eq.; Pd(PPh3)4: 4.5 mg, 0.003 mmol, 1.25 mol%). Yield: 79% (79 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 5/5. Mp: 196–197 °C. 1H NMR (400 MHz, chloroform-d): δ 8.69 (d, J = 5.4 Hz, 2H), 7.90 (s, 1H), 7.82 (d, J = 8.4 Hz, 2H), 7.76 (d, J = 5.4 Hz, 2H), 7.71 (d, J = 8.4 Hz, 2H), 3.96 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 150.5 (2 × CH), 149.2 (C), 141.0 (C), 139.5 (C), 137.2 (C), 130.7 (CH), 129.7 (q, 2JC–F = 33.1 Hz, C), 127.0 (q, 3JC–F = 3.7 Hz, 2 × CH), 123.8 (q, 1JC–F = 272.2 Hz, CF3), 123.3 (2 × CH), 120.3 (2 × CH), 118.2 (C), 38.2 (N–CH3). 19F NMR (235 MHz, chloroform-d): δ −62.5. IR ν (cm−1): 3067, 2952, 1614, 1581, 1556, 1526, 1420, 1324, 1219, 1184, 1113, 1094, 1016, 992, 917, 846, 739. HRMS (ESI): (m/z) [M + H]+ calculated for [C17H13F3N5]+ 344.1117; found 344.1118.
3-(4-Methoxyphenyl)-1-methyl-6-(4-(trifluoromethyl)phenyl)-1,6-dihydro pyrazolo[3,4-c]pyrazole (4q). Prepared as described in the general procedure C (3-bromo-1-methyl-6-(4-(trifluoromethyl)phenyl)-1,6-dihydro pyrazolo[3,4-c]pyrazole 3e: 100 mg, 0.28 mmol; 4-methoxyphenylboronic acid: 67 mg, 0.43 mmol, 1.5 eq.; Cs2CO3: 123 mg, 0.37 mmol, 1.3 eq.; Pd(PPh3)4: 4.5 mg, 0.003 mmol, 1.25 mol%). Yield: 94% (102 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 9/1. Mp: 205–206 °C. 1H NMR (400 MHz, chloroform-d): δ 7.88–7.77 (m, 5H), 7.71 (d, J = 8.4 Hz, 2H), 7.00 (d, J = 8.7 Hz, 2H), 3.91 (s, 3H), 3.86 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 160.0 (C), 149.2 (C), 141.4 (C), 140.0 (C), 131.4 (CH), 129.3 (q, 2JC–F = 33.1 Hz, C), 127.5 (2 × CH), 126.8 (q, 3JC-F = 3.7 Hz, 2 × CH), 125.0 (C), 123.0 (2 × CH), 121.2 (d, 1JC-F = 272.1 Hz, CF3), 117.8 (C), 114.4 (2 × CH), 55.4 (O–CH3), 37.8 (N–CH3). 19F NMR (235 MHz, chloroform-d): δ −62.4. IR ν (cm−1): 3338, 3057, 2937, 2845, 1596, 1614, 1540, 1425, 1323, 1251, 1107, 1088, 1031, 1007, 858, 832, 721, 715. HRMS (ESI): (m/z) [M + H]+ calculated for [C19H16F3N4O]+ 373.1270; found 373.1268.
1-Methyl-3-(p-tolyl)-6-(4-(trifluoromethyl)phenyl)-1,6-dihydro pyrazolo[3,4-c]pyrazole (4r). Prepared as described in the general procedure C (3-bromo-1-methyl-6-(4-(trifluoromethyl)phenyl)-1,6-dihydro pyrazolo[3,4-c]pyrazole 3e: 100 mg, 0.28 mmol; 4-methylphenylboronic acid: 60 mg, 0.43 mmol, 1.5 eq.; Cs2CO3: 123 mg, 0.37 mmol, 1.3 eq.; Pd(PPh3)4: 4.5 mg, 0.003 mmol, 1.25 mol%). Yield: 86% (89 mg). Colorless solid. Column chromatography eluents: PE/EtOAc = 9/1. Mp: 204–205 °C. 1H NMR (400 MHz, chloroform-d): δ 7.87 (s, 1H), 7.79 (d, J = 7.8 Hz, 4H), 7.70 (d, J = 8.4 Hz, 2H), 7.27 (d, J = 7.8 Hz, 2H), 3.92 (s, 3H), 2.40 (s, 3H). 13C NMR (101 MHz, chloroform-d): δ 149.2 (C), 141.4 (C), 140.2 (C), 138.6 (C), 131.4 (CH), 129.6 (2 × CH), 129.5 (C), 129.3 (q, 2JC–F = 33.0 Hz, C), 126.9 (q, 3JC–F = 3.7 Hz, 2 × CH), 126.1 (2 × CH), 123.9 (q, 1JC–F = 272.1 Hz, CF3), 123.1 (2 × CH), 118.0 (C), 37.9 (N–CH3), 21.5 (CH3). 19F NMR (235 MHz, chloroform-d): δ −62.4. IR ν (cm−1): 2926, 1614, 1594, 1582, 1522, 1504, 1424, 1317, 1162, 1110, 1085, 1066, 1039, 1014, 990, 965, 952, 909, 842, 825, 737. HRMS (ESI): (m/z) [M + H]+ calculated for [C19H16F3N4]+ 357.1249; found 357.1247.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by “Vasile Alecsandri” University of Bacau (ERASMUS grants and CNFIS-FDI-0151/2017), Francophone University Agency in Central and Eastern Europe (CE_124_FF_2016), Campus France (N 857352 B, N 902192 B) and the Eiffel Scholarship Program of Excellence (N 870738 C) as well as partially supported by Orléans University, CNRS, Labex SynOrg (ANR-11-LABX-0029), Labex IRON (ANR-11-LABX-0018-01), région Centre-Val de Loire.Notes and references
- K. W. Knouse, L. E. Ator, L. E. Beausoleil, Z. J. Hauseman, R. L. Casaubon and G. R. Ott, Tetrahedron Lett., 2017, 58, 202 CrossRef CAS.
- L. M. Blair and J. Sperry, J. Nat. Prod., 2013, 76, 794 CrossRef CAS PubMed.
- S. A. Bahashwan, A. A. Fayed, M. A. Ramadan, A. E.-G. E. Amr and N. O. Al-Harbi, Int. J. Mol. Sci., 2014, 15, 21587 CrossRef PubMed.
- G. H. Sayed, M. E. Azab, N. A. Negm and K. E. Anwer, J. Heterocycl. Chem., 2018, 55, 1615 CrossRef CAS.
- S. M. Gomha, H. M. Abdel-aziz and A. A. M. El-Reedy, J. Heterocycl. Chem., 2018, 55, 1960 CrossRef CAS.
- S. Ojha, A. Bapna and G. L. Talesara, Arkivoc, 2008, 112 Search PubMed.
- R. Mallikarjuna Rao, J. Sreeramulu, L. K. Ravindranath, G. Nagaraja Reddy, K. Hanumanthurayudu, G. Nageswara Reddy, A. Jayaraju and P. Madhusudhan, J. Chem. Pharm. Res., 2012, 4, 272 Search PubMed.
- D. A. Berry, T.-C. Chien and L. B. Townsend, Heterocycles, 2004, 63, 2475 CrossRef CAS.
- A. S. Shawali, Chem. Rev., 1993, 93, 2731 CrossRef CAS.
- T. K. Shkineva, I. L. Dalinger and S. A. Shevelev, Chem. Heterocycl. Compd., 1995, 31, 509 CrossRef.
- R. E. Khidre, H. A. Mohamed and B. F. Abdel-Wahab, Turk. J. Chem., 2013, 37, 1 CAS.
- I. M. A. Awad, Monatsh. Chem., 1990, 121, 1023 CrossRef CAS.
- S. Paul, M. Gupta, R. Gupta and A. Loupy, Tetrahedron Lett., 2001, 42, 3827 CrossRef CAS.
- E.-S. A. Aly, M. A. Abdo and A. A. El-Gharably, J. Chin. Chem. Soc., 2004, 51, 983 CrossRef CAS.
- F. M. Abd El Latif, M. A. Barsy, E. A. Elrady and M. Hassan, J. Chem. Res., Synop., 1999, 696 CrossRef CAS.
- R. A. Pawar and A. A. Patil, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1994, 33, 156 Search PubMed.
- F. M. Abd El Latif, J. Indian Chem. Soc., 1994, 71, 631 Search PubMed.
- R. M. Mohareb, A. Habashi, N. S. Ibrahim and S. M. Sherif, Synthesis, 1987, 228 CrossRef CAS.
- G. H. Elgemeie, H. A. Ali, A. H. Elghandour and A. M. Hussein, Heterocycl. Commun., 2002, 8, 443 CAS.
- S. Asadi, F. Alizadeh-Bami and H. Mehrabi, Arkivoc, 2020, 238 Search PubMed.
- P. Gillespie, R. A. Goodnow and Q. Zhang, US Pat., 20060223852A1, 2006Chem. Abstr., 2006, 145, 397512 Search PubMed.
- C. J. Helal, T. A. Chappie and J. M. Humphrey, WO2012168817A1, 2012Chem. Abstr., 2012, 158, 56278 Search PubMed.
- N.-C. Ostache, M.-A. Hiebel, A.-L. Fînaru, G. Guillaumet and F. Suzenet, ChemCatChem, 2019, 11, 3530 Search PubMed.
- K. Liubchak, A. Tolmachev and K. Nazarenko, J. Org. Chem., 2012, 77, 3365 CrossRef CAS PubMed.
- X. Xiong, Y. Jiang and D. Ma, Org. Lett., 2012, 14, 2552 CrossRef CAS PubMed.
- L. Xu, Y. Peng, Q. Pan, Y. Jiang and D. Ma, J. Org. Chem., 2013, 78, 3400 CrossRef CAS PubMed.
- M. Naas, PhD dissertation, Université d'Orléans, Orléans, 2016.
- S. Grosse, C. Pillard, F. Himbert, S. Massip, J. M. Léger, C. Jarry, P. Bernard and G. Guillaumet, Eur. J. Org. Chem., 2013, 4146 CrossRef CAS.
- Crystallographic data for the structure 4n have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1994906.†.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1994906. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra00314c |
| This journal is © The Royal Society of Chemistry 2021 |