
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general method for the synthesis of covalent and ionic amine borane complexes containing trinitromethyl fragments†
Jin Wangac,
Ming-Yue Jua,
Xi-Meng Chen*a and
Xuenian Chen *ab
*ab
aSchool of Chemistry and Chemical Engineering, Henan Key Laboratory of Boron Chemistry and Advanced Energy Materials, Henan Normal University, Xinxiang, Henan 453007, China. E-mail: xnchen@htu.edu.cn; chenximeng@htu.edu.cn
bGreen Catalysis Center and College of Chemistry, Zhengzhou University, Zhengzhou, Henan 450001, China
cCollege of Chemistry and Chemical Engineering, Mudanjiang Normal University, Mudanjiang, Heilongjiang 157011, China
First published on 5th March 2021
Abstract
A general approach for the synthesis of covalent and ionic amine borane complexes containing trinitromethyl fragments has been developed through metathesis reactions between amine chloroborane complexes and potassium salt of trinitromethyl (K[C(NO2)3]). Five covalent and ionic trinitromethyl amine borane complexes have been synthesized in good yields with high purity and it is found that the ionic complex, [H2B(NH3)2][C(NO2)3], might be a promising energetic material on the basis of the investigation of its thermal decomposition behaviour.
Introduction
Nitroform (CH(NO2)3) holds a unique position among nitro compounds as it is a valuable starting material for the preparation of propellant and explosive components due to its high oxygen content.1 Ioffe group reported the syntheses and transformations of trinitromethylborane complexes with cyclic ethers and aromatic N-containing heterocycles, and also discussed the principle of the reactions.2 However, they attempted to obtain N,N-dinitroamidoborane complexes using the same method, but failed.2a Klapötke and coworkers studied the reactions of boron oxide (B2O3) with various nitro-substituted ethanols (2-nitroethanol, 2-fluoro-2,2-dinitroethanol, and 2,2,2-trinitroethanol) to furnish the corresponding nitroethyl borates B(OCH2CH2NO2)3, B(OCH2CF(NO2)2)3, and B(OCH2C(NO2)3)3.3 The compound B(OCH2C(NO2)3)3 can be used as green-light-emitting pyrotechnic composition.4Interestingly, C(NO2)3, as an oxidizing group, can be introduced into the design of high-energy molecules to co-exist with powerful reducing borohydride in a single covalently bonded structure.5 In 2013, Christe group used CH(NO2)3 and NaBH4 as starting materials to synthesize [Na(glyme)2][BH3C(NO2)3].5 Subsequently, they employed metathesis reactions to convert this salt into PNP+ and PPh4+ analogs that were stable for several months at room temperature. In 2015, ammonia-dinitro-amidoborane, NH3BH2N(NO2)2 was synthesized by the reaction of dinitroamine (HN(NO2)2) with ammonia borane (NH3BH3). This compound is expected to have a good performance as an explosive being comparable to that of pentaerythritol tetranitrate and significantly greater than that of trinitrotoluene.6 So, highly energetic oxidized analogs have attracted attention recently.
NH3BH3, a potential hydrogen storage material, has received immense interest in the past twenty years owing to its high percentage of hydrogen (19.6 wt%), excellent stability at room temperature, and release of hydrogen under mild conditions.7 It can also provide both a proton and hydride in chemical reactions under mild conditions.8 In NH3BH3, the nitrogen atom can be bonded to proton, hydrocarbon, hydroxyl, oxygen and other groups,9 and the boron atom can be bonded to hydride, hydrocarbon, oxygen, oxynitride, halogen and other electron donors that can interact with the empty orbital on boron.10 Many derivatives could be obtained from NH3BH3, including ammonia monochloroborane (NH3BH2Cl)11–14 which is an important intermediate for the synthesis of a series of more complex boron compounds15 such as R1R2N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) BH2 (ref. 12a) and amorphous boron nitride.11b
BH2 (ref. 12a) and amorphous boron nitride.11b
In this work, ammonia and amine monochloroborane complexes (ABH2Cl, A = NH3, aliphatic primary, secondary, tertiary amine, and diamines, 1) were treated with K[C(NO2)3], a milder oxidizing reagent in comparison with CH(NO2)3,16 to prepare target product 3, with the general formula of ABH2C(NO2)3, containing both reducing and oxidizing fragments in a single covalently bonded structure. Furthermore, an ionic complex, [H2B(NH3)2][C(NO2)3], was synthesized from 3a.
Results and discussion
The reactions of each amine borane complex ABH3 (2a–d) with HCl diethyl ether solution at room temperature resulted in the formation of amine monochloroborane complexes (1a–d) in good yields. Ethylenediaminebisborane (2e) reacted with HCl·Et2O at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio to form ClBH2NH2CH2CH2NH2BH2Cl (1e). Trimethylamine borane (2f) reacted with iodine to afford trimethylamine monoiodoborane complex (1f) (Scheme 1). These amine halogenated borane complexes (1a–f) further reacted with K[C(NO2)3] to produce the products (Table 1, 3a–c) by metathesis reactions. Compound (3e) (Table 1) was also synthesized by the reaction of 1e with K[C(NO2)3] according to Scheme 2. It should be noted that ammonia monochloroborane (1a) can further react with ammonia gas in THF to afford [H2B(NH3)2]Cl (1g) ((a) in Scheme 3). 1a is a covalent complex in which the Cl–B bond is a typical covalent bond, in accordance with amine monochloroborane complexes 1a–d. However, 1g shows a ionic behaviour, similar to the diammoniate of diborane ([H2B(NH3)2][BH4]).17 The reaction of 1g and K[C(NO2)3] leads to the formation of an ionic compound, 3g (Scheme 3). On the other hand, attempts for the syntheses of (CH3)3NBH2C(NO2)3 have failed, ether by chloroborane or iodoborane intermediates, probably due to the steric hindrance of the trimethylamine group. In general, five amine borane containing the trinitromethyl group, four covalent and one ionic complexes (Table 1, 3a–c, 3e, 3g), were successfully synthesized in good yields with high purity.
:2 ratio to form ClBH2NH2CH2CH2NH2BH2Cl (1e). Trimethylamine borane (2f) reacted with iodine to afford trimethylamine monoiodoborane complex (1f) (Scheme 1). These amine halogenated borane complexes (1a–f) further reacted with K[C(NO2)3] to produce the products (Table 1, 3a–c) by metathesis reactions. Compound (3e) (Table 1) was also synthesized by the reaction of 1e with K[C(NO2)3] according to Scheme 2. It should be noted that ammonia monochloroborane (1a) can further react with ammonia gas in THF to afford [H2B(NH3)2]Cl (1g) ((a) in Scheme 3). 1a is a covalent complex in which the Cl–B bond is a typical covalent bond, in accordance with amine monochloroborane complexes 1a–d. However, 1g shows a ionic behaviour, similar to the diammoniate of diborane ([H2B(NH3)2][BH4]).17 The reaction of 1g and K[C(NO2)3] leads to the formation of an ionic compound, 3g (Scheme 3). On the other hand, attempts for the syntheses of (CH3)3NBH2C(NO2)3 have failed, ether by chloroborane or iodoborane intermediates, probably due to the steric hindrance of the trimethylamine group. In general, five amine borane containing the trinitromethyl group, four covalent and one ionic complexes (Table 1, 3a–c, 3e, 3g), were successfully synthesized in good yields with high purity.
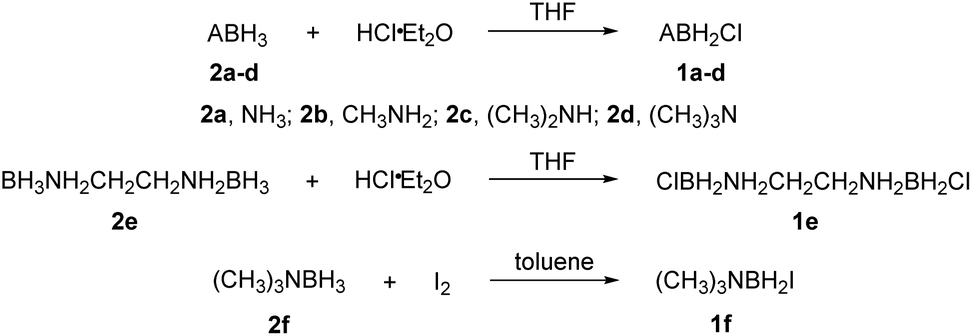 | ||
| Scheme 1 Synthesis of compounds 1a–f. | ||
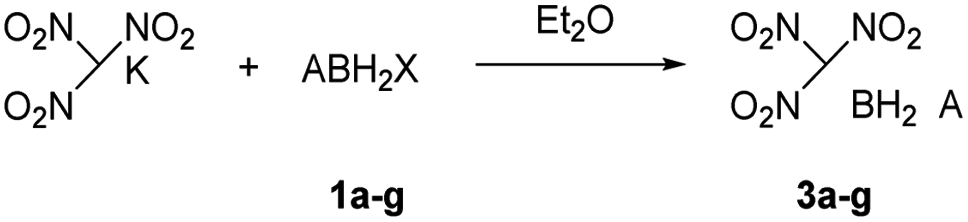
 | ||
| Scheme 2 Synthesis of compound 3e. | ||
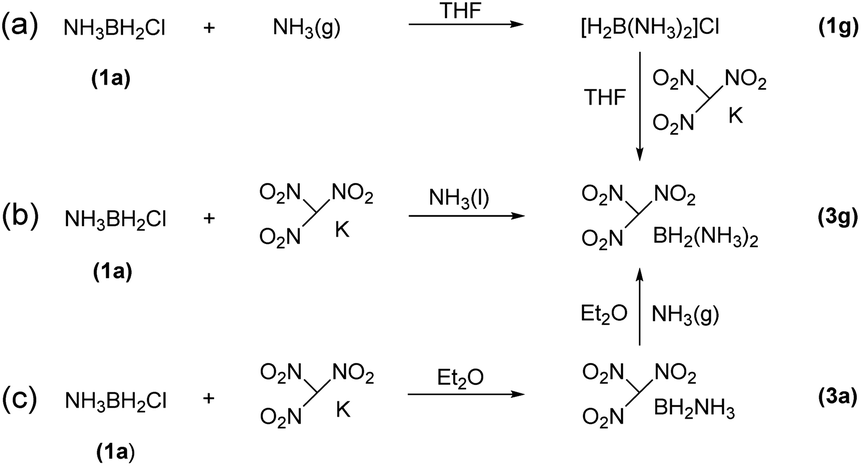 | ||
| Scheme 3 The methods for the synthesis of 3g. | ||
It is worthy to note that 3g was firstly synthesized by Ioffe group in 2014 (3a in Scheme 3).2b We provide two alternative routes in this work. Scheme 3a shows the Ioffe group's method as described above,2b 1a reacted with excess NH3 to convert into 1g and then reacted with K[C(NO2)3] to form a yellow residue 3g in a yield of 65%. In our work, as shown in Scheme 3b, K[C(NO2)3] directly reacted with 1a in liquid NH3 at −78 °C to give yellow product 3g with a yield of 68%; and Scheme 3c, K[C(NO2)3] reacted with 1a in ethyl ether first and then with excess NH3 gas to give 3g, the yield was 59%. In comparison, pathway (b) is a one-step procedure with high efficiency. Further study shows that 3g is an ionic complex, it has good solubility in organic solvents, such as tetrahydrofuran (THF) and acetonitrile (CH3CN), different from (NH3)2BH2Cl.14 3g also showed good stability towards air and moisture even for several months at room temperature. Thus, it has wide potential applications as a high-energy-density material.
The 11B NMR data of ABH2X (X = Cl, I, and C(NO2)3) are summarized in Table 2. The chemical shift of the B atom in ABH2C(NO2)3 shifts downfield about 4 ppm in comparison with that of the B atom in the corresponding ABH2Cl complexes. This can be attributed the strong electron-withdrawing ability of the C(NO2)3 group relative to Cl. With increasing each one methyl group bonded in the N atom, on the other hand, the chemical shift of the B signal shifts downfield about 2 ppm (Table 2) in either ABH2Cl or ABH2C(NO2)3 complexes. These change trends are consistent with those of the chemical shift of the B atom in amine boranes. For the amine chloroborane complexes or trinitromethylamidoborane complexes, whether the compound is mono-substituted (1b and 3b) or bis-substituted (1e and 3e), little effect was observed on the chemical shift of ABH2X (Table 2, entries 2 and 5). The B signal of [H2B(NH3)2]Cl and [H2B(NH3)2][C(NO2)3] are almost identical (Table 2, entry 7) because they are ionic compounds so that the effect of the different counter-anion on the chemical shift of the B atom in the [H2B(NH3)2]+ cation is weak. In contrast, the effect is more pronounced in covalent complexes (1a–f and 3a–c, e) as described above because of the direct N–B and B–C bonding. In addition, the chemical shift of the proton of the BH2 group in ABH2C(NO2)3 in 1H NMR are also summarized in Table S1,† all proton signals appeared at about δ 2 ppm, similar to those of ABH2Cl.11–14 This indicated that the change of substituents may not influence the chemical shift of the BH2 group in ABH2C(NO2)3.
| Entry | Comps. | A | X (1) | 11B NMR (ppm) | X (3) | 11B NMR (ppm) |
|---|---|---|---|---|---|---|
| a A is the lewis base, X is the substituent group.b Molecular formula (NH2CH2BH2Cl)2.c Molecular formula [NH2CH2BH2C(NO2)3]2. | ||||||
| 1 | a | NH3 | Cl | −8.73 | C(NO2)3 | −4.8 |
| 2 | b | CH3NH2 | Cl | −6.36 | C(NO2)3 | −2.32 |
| 3 | c | (CH3)2NH | Cl | −3.47 | C(NO2)3 | −0.22 |
| 4 | d | (CH3)3N | Cl | 0.21 | C(NO2)3 | — |
| 5 | e | (NH2CH2)2 | Cl | −6.47b | C(NO2)3 | −2.93c |
| 6 | f | (CH3)3N | I | −10.57 | C(NO2)3 | — |
| 7 | g | (NH3)2 | Cl | −13.54 | C(NO2)3 | −13.06 |
Thermal decomposition of 3a and 3g was studied by DSC and TGA-MS. As shown in Fig. 1 and 2, thermal decomposition resulted in the generation of H2, N2, NH3, CO2, N2O and NO2, hence the decomposition is believed to proceed according to eqn (1) and (2), respectively.
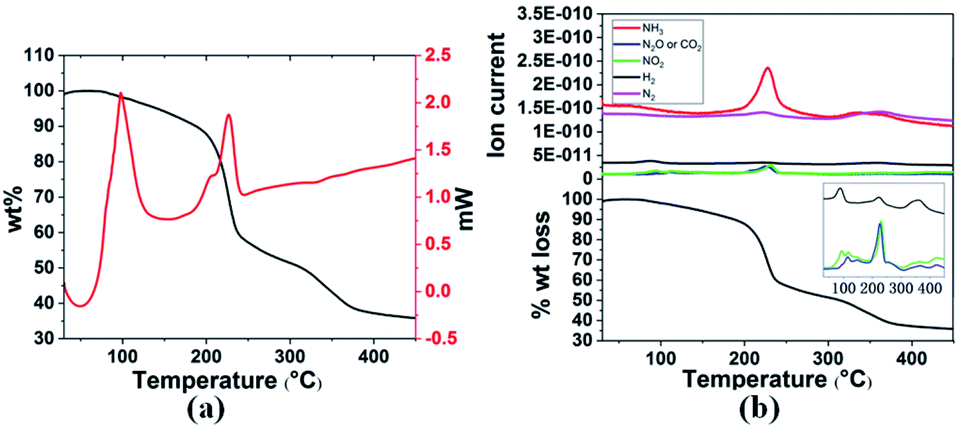 | ||
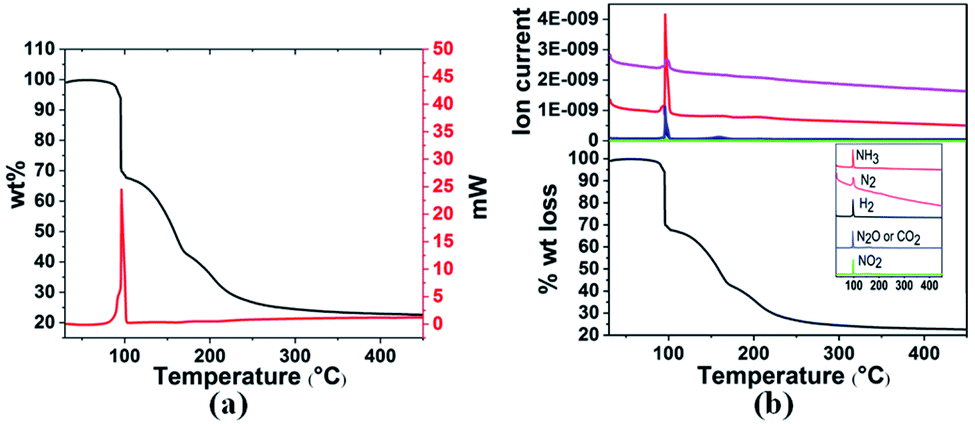
| Fig. 1 (a) TGA and DSC curves and (b) TGA-MS analysis of 3a in the temperature range 30–400 °C with a heating rate of 3 °C min−1. | ||
 | ||
| Fig. 2 (a) TGA and DSC curves (b) TGA-MS analysis of 3g in the temperature range 30–400 °C with a heating rate of 3 °C min−1. | ||
Gaseous products were analysed by mass spectrometry (MS), and solid residues were determined by IR and XRD. It was observed that 3a starts decomposing at 98 °C and the first-stage weight loss is only 1.7 wt%, corresponding to the evolution of hydrogen and nitrogen dioxide, and the m/z 44 signal is assigned to N2O or CO2 evolution. The decomposition behaviour of 3a is similar to that of the energetic oxidizer hydrazinium nitroformate (N2H5C(NO2)3, HNF), as shown in eqn (3).18 The second-stage weight loss is as large as 31.2 wt%, associated with H2, N2, NH3, CO2, N2O and NO2 evolution. Both the first and second steps for 3a are exothermic events, the m/z 2, 28, 17, 44, and 46 signals recorded by the MS during the TGA-MS experiment to 400 °C are overlaid in Fig. 1b. At higher temperatures, it further decomposes and boron oxide was formed (Fig. S1 and S2†).
| 2NH3BH2C(NO2)3 → 2NH3 + 2H2 + 2NO2 + N2O + 2CO2 + B2O3 + N2 | (1) |
| 2[H2B(NH3)2][C(NO2)3] → 4NH3 + 2H2 + 2NO2 + N2O + 2CO2 + B2O3 + N2 | (2) |
| 2N2H5C(NO2)3 → NH4C(NO2)3 + N2O + 2H2O + H2CO | (3) |
The thermal decomposition pattern of 3g is different from 3a. The large weight loss of 30.6 wt%, observed at 95 °C with strongly exothermic, is associated with H2, N2, NH3, CO2, N2O and NO2 evolution. At higher temperature, it further decomposes to release N2O, CO2 and NO2, resulting in boron oxide (Fig. S3 and S4†). The thermal decomposition behaviours of 3a and 3g are different from those of NH3BH2Cl, their parent compound. It was recorded that only H2 was released at the initial stage (eqn (4)), and then the second large weight loss was associated with both H2 and HCl (eqn (5)). At higher temperature, it further decomposes to evolve HCl and H2 to form boron nitride (eqn (6)).11c
| NH3BH2Cl → H2 + (NH2BHCl)x | (4) |
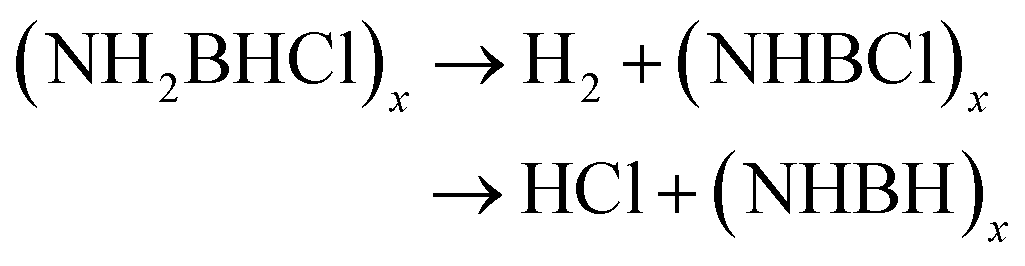 | (5) |
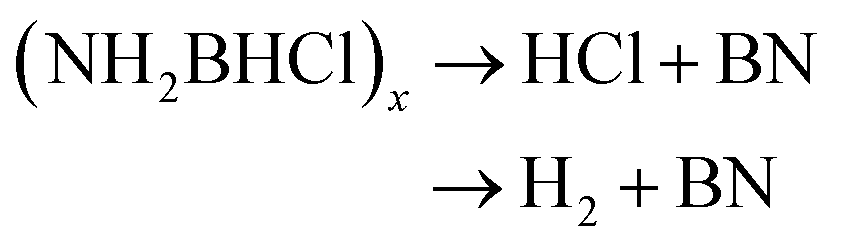 | (6) |
Conclusions
In summary, we have developed general methods for the syntheses of both covalent and ionic complexes containing the amine borane reducing group and the trinitromethyl oxidizing group in one molecule under mild conditions. These complexes were successfully isolated in high yields and characterized by NMR and IR. Thermal decomposition was investigated by TGA-MS and DSC, and results implicated that compound 3g may be a promising explosive material. Further studies on the explosive property of 3g are in progress in our lab.Experimental
CAUTION! All nitrogen- and oxygen-rich compounds are potentially explosive energetic materials which should be handled with great care, although no hazards were observed during preparation and handling of these compounds. In any case, it is necessary to take proper precautions by employing all standard energetic materials safety procedures in experiments involving such substances, such as face shields, a leather apron, gloves, and hearing protection should be employed.General information
All manipulations were carried out under a nitrogen atmosphere using standard Schlenk techniques and glove box. The 11B NMR and 11B{1H} NMR spectra were recorded at 128 or 193 MHz spectrometers and externally referenced to BF3·OEt2 in C6D6 (δ = 0.00 ppm). The 1H NMR and 1H{11B} NMR spectra were obtained at 600 MHz spectrometer. The 13C NMR spectra were recorded at 151 MHz. IR spectra were measured by a Spectrum 400F. X-ray diffraction (XRD) data were obtained with a Rigaku D/max 2500 diffractometer using Cu/Kα radiation, λ = 0.1542 nm, 40 kV, 100 mA. The thermal behaviours of compounds 3a and 3g were determined by synchronous thermal analyses (TGA-DSC, Netzsch 449C Jupiter/QMS 403D). The samples were heated to 500 °C with a heating rate of 3 °C min−1, under a flowing Ar atmosphere.Anhydrous nitric acid, sulphuric acid, acetic anhydride, KOH, anhydrous sodium sulfate, ethanol, and iodine were purchased from Sinopharm Chemical Reagents Co., Ltd. NH3BH3, MeNH2BH3, Me2NHBH3, Me3NBH3, BH3NH2CH2CH2NH2BH3 and HCl diethyl ether solution (1 mol L−1) were purchased from United Boron (Zhengzhou) Energy Materials S&T LLC and used as received. Tetrahydrofuran (THF), diethyl ether (Et2O), n-hexane, and toluene were dried over sodium and freshly distilled prior to use. N,N-Dimethylformamide (DMF) was dried by molecular sieves.
Amine chloroborane (1a–d) and K[C(NO2)3] were prepared according to the literature methods.11,19
Synthesis of ClBH2NH2CH2CH2NH2BH2Cl (1e)
To a solution of ethylenediaminebisborane (0.088 g, 1 mmol) in THF (2 mL) was added HCl diethyl ether solution (2.2 mmol, 2.2 mL) via syringe at ambient temperature. The reaction was monitored by 11B NMR and after about 30 min the reaction was finished. Then the mixture was filtered and solvent was removed from the filtrate under a dynamic vacuum to leave a white product (0.144 g, yield 92%). 11B NMR (193 MHz, CD3CN): δ −6.47 (t, JB–H = 107.7 Hz) ppm (Fig. S9a†). 11B{1H} NMR (193 MHz, THF): δ −6.47 (s) ppm (Fig. S9b†).Synthesis of (CH3)3NBH2I (1f)
To a solution of trimethylamine borane (0.073 g, 1 mmol) in toluene (2 mL) was added I2 (0.127 g, 0.5 mmol) in toluene (5 mL) via syringe at ambient temperature. The reaction was monitored by 11B NMR and after about 1 h the reaction was finished. After reaction, the mixture was filtered and solvent was removed from the filtrate under a dynamic vacuum to leave a white product (0.169 g, yield 85%). 11B NMR (128 MHz, toluene): δ −10.57 (t, JB–H = 130.9 Hz) ppm (Fig. S10a†). 11B{1H} NMR (128 MHz, toluene): δ −10.57 (s) ppm (Fig. S10b†).Synthesis of [(NH3)2BH2]Cl (1g)
Ammonia chloroborane (0.073 g, 1 mmol) was placed in a 10 mL flask, and the 2 mL of THF was injected into the flask. Then excess NH3 was bubbled into the flask at room temperature and white precipitate was formed immediately. After filtration, THF was removed from the filtrate under dynamic vacuum to leave a white powder product (0.078 g, yield 94%). 11B NMR (193 MHz, DMF): δ −13.54 (t, JB–H = 104.4 Hz) ppm (Fig. S11a†). 11B{1H} NMR (193 MHz, DMF): δ −13.54 (s) ppm (Fig. S11b†).General procedure for the synthesis of ABH2C(NO2)3 (3a–c)
To a solution of aminoborane (1 mmol) (2a: 0.031 g; 2b: 0.045 g; 2c: 0.059 g) in THF (2 mL) was added HCl diethyl ether (1.1 mmol, 1.1 mL) via syringe at ambient temperature. The reaction was monitored by 11B NMR and after about 30 min the reaction was finished. Then the mixture was filtered and solvent was removed from the filtrate under a dynamic vacuum to leave the products of 1a–c. The prepared 1a–c and K[C(NO2)3] (0.208 g, 1.1 mmol) were added to the flask and then added 5 mL diethyl ether at ambient temperature. The yellow solid product was separated by filtration using a filter cannula and dried in vacuo.Synthesis of [CH2NH2BH2C(NO2)3]2 (3e)
To a solution of ethylenediaminebisborane (0.088 g, 1 mmol) in THF (2 mL) was added HCl diethyl ether solution (2.2 mmol, 2.2 mL) via syringe at ambient temperature. The reaction was monitored by 11B NMR and after about 30 min the reaction was finished. Then the solvent was removed from the filtrate under a dynamic vacuum to leave product. The prepared 1e and K[C(NO2)3] (0.416 g, 2.2 mmol) were added to the flask and then added 5 mL diethyl ether at ambient temperature. The yellow solid product was separated by filtration using a filter cannula and dried in vacuo.Synthesis of [H2B(NH3)2][C(NO2)3] (3g)
To a solution of ammonia borane (0.031 g, 1 mmol) in THF (2 mL) was dropwise added HCl diethyl ether (1.1 mmol, 1.1 mL) via syringe at ambient temperature. The white solid product (1a) was separated by filtration using a filter cannula dried in vacuo for use.(a) Repeated the literature method. The prepared 1a was placed in a flask, and the 2 mL of THF was injected into the flask. Then excess NH3 was bubbled into the flask for 30 min under stirring at room temperature and white precipitate was produced immediately. Then K[C(NO2)3] (0.208 g, 1.1 mmol) in THF (5 mL) was added into the flask, the reaction mixture was stirred for 2 h and the solution turned from colourless and transparent to yellow. After filtration to remove the formed KCl, THF was removed from the filtrate under dynamic vacuum to give a yellow powder product (3g, 0.129 g, yield 65%).
(b) The prepared 1a and K[C(NO2)3] (0.208 g, 1.1 mmol) were added to a flask and then 5 mL of liquid NH3 was condensed into the flask at −78 °C and stirred for 2 hours. Then the reaction was warm up to room temperature and liquid NH3 was volatilized completely to leave white and yellow powder precipitate (KCl and 3g). The yellow precipitate was extracted with 20 mL of THF. Removal of THF from the filtrate under dynamic vacuum gave a yellow powder product (0.133 g, yield 68%).
(c) The prepared 1a and K[C(NO2)3] (0.208 g, 1.1 mmol) were added to a flask and the 5 mL of ethyl ether was injected into the flask. The reaction was stirred for 2 h, then excess NH3 was bubbled into the flask and white precipitate was produced immediately, the solution turned to yellow. Removal of ethyl ether from the filtrate under dynamic vacuum gave a yellow product (3g, 0.117 g, yield 59%).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Numbers: 21771057 to X. C., 22001061 to X.-M. C.) and China Postdoctoral Science Foundation (Grant Numbers: 2020M682309 to X.-M. C.).Notes and references
- (a) A. Hantzsch and A. Rinckenberger, Ber. Dtsch. Chem. Ges., 1899, 32, 628–641 CrossRef CAS; (b) A. A. Gidaspov, V. V. Bakharev and I. K. Kukushkin, Russ. Chem. Bull., 2009, 58, 2154–2163 CrossRef CAS; (c) V. Thottempudi, H. Gao and J. M. Shreeve, J. Am. Chem. Soc., 2011, 133, 6464–6471 CrossRef CAS PubMed; (d) O. G. Jolodar, K. Ghauri, M. Seddighi, F. Shirini and Y. Bayat, J. Mol. Struct., 2019, 1186, 448–457 CrossRef CAS; (e) A. A. Gidaspov, V. A. Zalomlenkov, V. V. Bakharev, V. E. Parfenov, E. V. Yurtaev, M. I. Struchkova, N. V. Palysaeva, K. Y. Suponitsky, D. B. Lempert and A. B. Sheremetev, RSC Adv., 2016, 6, 34921–34934 RSC.
- (a) O. P. Shitov, V. A. Tartakovsky, I. S. Golovanov, A. Y. Sukhorukov and S. L. Ioffe, Chem.–Asian J., 2017, 12, 2237–2244 CrossRef CAS PubMed; (b) O. P. Shitov, V. A. Tartakovskii and S. L. Ioffe, Chem. Heterocycl. Compd., 2015, 12, 1647–1657 CrossRef; (c) S. L. Ioffe, A. S. Shashkov, A. L. Blyumenfel'd, L. M. Leont'eva, L. M. Makarenkova, O. B. Belkina and V. A. Tartakovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1976, 25, 2371–2379 CrossRef; (d) S. L. Ioffe, A. V. Kalinin, B. N. Khasapov, L. M. Leont'eva and V. A. Tartakovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1978, 27, 1019–1021 CrossRef; (e) O. P. Shitov, L. M. Leont'eva, S. L. Ioffe, B. N. Khasanov, V. M. Novikov, A. U. Stepanyants and V. A. Tartakovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1974, 23, 2684–2691 CrossRef; (f) S. L. Ioffe, A. V. Kalinin, T. N. Golovina, B. N. Khasapov and V. A. Tartakovskii, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1978, 27, 816–819 CrossRef.
- T. M. Klapötke and B. K. Richard Moll, Chem.–Eur. J., 2013, 36, 12113–12123 CrossRef PubMed.
- T. M. Klapötke, B. Krumm, M. Rusana and J. J. Sabatini, Chem. Commun., 2014, 50, 9581–9583 RSC.
- G. Bélanger-Chabot, M. Rahm, R. Haiges and K. O. Christe, Angew. Chem., Int. Ed., 2013, 52, 11002–11006 CrossRef PubMed.
- G. Bélanger-Chabot, M. Rahm, R. Haiges and K. O. Christe, Angew. Chem., Int. Ed., 2015, 54, 11730–11734 CrossRef PubMed.
- (a) M. Liu, L. Zhou, X. Luo, C. Wan and L. Xu, Catalysts, 2020, 10, 788–822 CrossRef CAS; (b) U. B. Demirci, Energies, 2020, 13, 3071–3115 CrossRef CAS; (c) U. B. Demirci, Int. J. Hydrogen Energy, 2017, 42, 9978–10013 CrossRef CAS; (d) A. Rossin and M. Peruzzini, Chem. Rev., 2016, 116, 8848–8872 CrossRef CAS PubMed; (e) Y. Lin and W. L. Mao, Chin. Sci. Bull., 2014, 59, 5235–5240 CrossRef CAS; (f) V. Rizzi, D. Polino, E. Sicilia, N. Russo and M. Parrinello, Angew. Chem., Int. Ed., 2019, 58, 3976–3980 CrossRef CAS PubMed.
- (a) H. Li, Qi. Yang, X. Chen and S. G. Shore, J. Organomet. Chem., 2014, 751, 60–66 CrossRef CAS; (b) H. Li, R. Wang, Q. Xia, Q. Yang, P. Wang, C. Wei, N. Ma and X. Chen, Chem. Res., 2018, 29, 118–124 Search PubMed; (c) C. Cui, Y. Ma, J. Zhang and X. Chen, Chem. Res., 2019, 30, 454–462 Search PubMed.
- (a) P. J. Silva and M. J. Ramos, J. Org. Chem., 2009, 74, 6120–6129 CrossRef CAS PubMed; (b) E.-C. Koch and T. M. Klapötke, Propellants Explos. Pyrotech., 2012, 37, 335–344 CrossRef CAS; (c) P. Srivastava and H. J. Singh, J. Energ. Mater., 2010, 28, 202–218 CrossRef CAS; (d) H. A. LeTourneau, R. E. Birsch, G. Korbeck and J. L. Radkiewicz-Poutsma, J. Phys. Chem. A, 2005, 109, 12014–12019 CrossRef CAS PubMed; (e) D. H. Campbell, T. C. Bissot and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 1549–1552 CrossRef CAS.
- (a) S. Toyota, T. Futawaka, M. Asakura, H. Ikeda and M. Oki, Organometallics, 1998, 17, 4155–4163 CrossRef CAS; (b) A. P. M. Robertson, M. F. Haddow and I. Manners, Inorg. Chem., 2012, 51, 8254–8264 CrossRef CAS PubMed; (c) H. K. Lingam, X. Chen, J.-C. Zhao and S. G. Shore, Chem.–Eur. J., 2012, 18, 3490–3492 CrossRef CAS PubMed; (d) R. Kumar and B. R. Jagirdar, Inorg. Chem., 2013, 52, 28–36 CrossRef CAS PubMed; (e) C. T. Hoang, I. Prokes, G. J. Clarkson, M. J. Rowland, J. H. R. Tucker, M. Shipman and T. R. Walsh, Chem. Commun., 2013, 49, 2509–2511 RSC; (f) B. Icli, E. Sheepwash, T. Riis-Johannessen, K. Schenk, Y. Filinchuk, R. Scopellitia and K. Severin, Chem. Sci., 2011, 2, 1719–1721 RSC.
- (a) M. G. Hu and R. A. Geanangel, Inorg. Chem., 1979, 18, 3297–3301 CrossRef CAS; (b) D. R. Ketchum, A. L. DeGraffenreid, P. M. Niedenzu and S. G. Shore, J. Mater. Res., 1999, 14, 1934–1938 CrossRef CAS; (c) H. K. Lingam, C. Wang, J. C. Gallucci, X. Chen and S. G. Shore, Inorg. Chem., 2012, 51, 13430–13436 CrossRef CAS PubMed.
- (a) O. J. Metters, A. M. Chapman, A. P. M. Robertson, C. H. Woodall, P. J. Gates, D. F. Wass and I. Manners, Chem. Commun., 2014, 50, 12146–12149 RSC; (b) C. R. Balulescu and P. C. Keller, Inorg. Chem., 1978, 17, 3707–3708 CrossRef CAS; (c) O. T. Beachley and B. Washburn, Inorg. Chem., 1975, 14, 120–123 CrossRef CAS; (d) W. H. Myers, G. E. Ryschkewitsch, M. A. Mathur and R. W. King, Inorg. Chem., 1975, 14, 2874–2881 CrossRef CAS; (e) W. H. Myers and G. E. Ryschkewitsch, Inorg. Chem., 1978, 17, 1157–1159 CrossRef CAS.
- (a) P. J. Bratt, K. R. Seddon and I. A. Steer, Inorg. Chim. Acta, 1974, 10, 191–195 CrossRef CAS; (b) M. P. Brown, R. W. Heseltine, P. A. Smith and P. J. Walker, J. Chem. Soc. A, 1970, 410–414 RSC; (c) H. C. Kelly, S. C. Yasui and R. B. Twiss-Brooks, Inorg. Chem., 1984, 23, 2220–2223 CrossRef CAS; (d) W. H. Myers, G. E. Ryschkewitsch, M. A. Mathur and R. W. King, Inorg. Chem., 1975, 14, 2874–2881 CrossRef CAS.
- (a) D. R. Schultz and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 4–8 CrossRef CAS; (b) S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 8–12 CrossRef CAS; (c) S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 12–15 CrossRef CAS; (d) R. W. Parry and S. G. Shore, J. Am. Chem. Soc., 1958, 80, 15–20 CrossRef CAS; (e) H. K. Lingam, X. Chen, J.-C. Zhao and S. G. Shore, Chem.–Eur. J., 2012, 18, 3490–3492 CrossRef CAS PubMed.
- (a) U. Vogel, P. Hoemensch, K.-C. Schwan, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2003, 9, 515–519 CrossRef CAS PubMed; (b) C. Marquardt, A. Adolf, A. Stauber, M. Bodensteiner, A. V. Virovets, A. Y. Timoshkin and M. Scheer, Chem.–Eur. J., 2013, 19, 11887–11891 CrossRef CAS PubMed; (c) C. Marquardt, C. Thoms, A. Stauber, G. Balázs, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 3727–3730 CrossRef CAS PubMed.
- (a) M. Göbel, T. M. Klapötke and P. Mayer, Z. Anorg. Allg. Chem., 2006, 632, 1043–1050 CrossRef; (b) A. A. Gakh, J. C. Bryan, M. N. Burnett and P. V. Bonnesen, J. Mol. Struct., 2000, 520, 221–228 CrossRef CAS.
- (a) R. W. Parry and S. G. Shore, J. Am. Chem. Soc., 1958, 80, 15–20 CrossRef CAS; (b) X. Chen, X. Bao, J.-C. Zhao and S. G. Shore, J. Am. Chem. Soc., 2011, 133, 14172–14175 CrossRef CAS PubMed.
- O. E. Dragomir, M. J. Tummers, E. H. van Veen, A. E. D. M. van der Heijden and D. J. E. M. Roekaerts, Combust. Flame, 2009, 156, 1810–1817 CrossRef CAS.
- Y. Huang, H. Gao, B. Twamley and J. M. Shreeve, Eur. J. Inorg. Chem., 2007, 2025–2030 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00440a |
| This journal is © The Royal Society of Chemistry 2021 |