
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBottom-up fabrication of triazine-based frameworks as metal-free materials for supercapacitors and oxygen reduction reaction†
Ronghan Caoa,
Fangyuan Hu *b,
Tianpeng Zhangb,
Wenlong Shaob,
Siyang Liub and
Xigao Jian*ab
*b,
Tianpeng Zhangb,
Wenlong Shaob,
Siyang Liub and
Xigao Jian*ab
aState Key Laboratory of Fine Chemicals, Department of Polymer Materials & Engineering, Liaoning Province Engineering Research Centre of High Performance Resins, Dalian University of Technology, Dalian, 116024, China. E-mail: jian4616@dlut.edu.cn
bSchool of Materials Science and Engineering, Key Laboratory of Energy Materials and Devices (Liaoning Province), State Key Laboratory of Fine Chemicals, Liaoning Province Engineering Centre of High Performance Resins, Dalian University of Technology, Dalian 116024, China. E-mail: hufangyuan@dlut.edu.cn
First published on 22nd February 2021
Abstract
Doping porous carbon materials with heteroatoms is an effective approach to enhance the performance in the areas of supercapacitors and the oxygen reduction reaction (ORR). However, most traditional heteroatom-doped metal-free porous carbon materials have random structures and pore distributions with high uncertainty, which is harmful for a deep understanding of supercapacitors and the ORR mechanism. Basing on the molecular design, a series of N, O co-doped porous carbon frameworks (p-PYPZs) has been prepared through the template-free trimerization of cyano groups from our designed and synthesized 2,8-bis(4-isocyanophenyl)-2,3,7,8-tetrahydropyridazino[4,5-g]phthalazine-1,4,6,9-tetraone (PYPZ) monomer and subsequent ionothermal synthesis, which has the advantage that the type, position, content of the heteroatom and the pore structure in the porous carbon material can be regulated. Nitrogen and oxygen atoms introduced via covalent bond and the hierarchically porous structure endow the material with excellent cycling stability, and 110% capacitance retention after 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles in 1 M H2SO4. A symmetric supercapacitor was assembled with the material and shows an energy density of 32 W h kg−1. The material can be applied to the area of oxygen reduction reaction as a metal-free catalyst with an onset potential of 0.85 V versus RHE, indicating the good catalytic ability. The material exhibits excellent methanol crossover resistance and a four-electron pathway mechanism. Results also indicate a positive correlation between the N-Q content and the selectivity of the four-electron pathway. In this paper, the electrochemical properties of materials are regulated at the molecular level, which provides a new idea for further understanding the electrochemical mechanism of energy storage devices.
000 cycles in 1 M H2SO4. A symmetric supercapacitor was assembled with the material and shows an energy density of 32 W h kg−1. The material can be applied to the area of oxygen reduction reaction as a metal-free catalyst with an onset potential of 0.85 V versus RHE, indicating the good catalytic ability. The material exhibits excellent methanol crossover resistance and a four-electron pathway mechanism. Results also indicate a positive correlation between the N-Q content and the selectivity of the four-electron pathway. In this paper, the electrochemical properties of materials are regulated at the molecular level, which provides a new idea for further understanding the electrochemical mechanism of energy storage devices.
1. Introduction
Human beings are facing a series of environment-related issues, including global warming and the depletion of traditional fossil fuels. Therefore, sustainable energy storage devices, such as supercapacitors and efficient energy conversion devices, including fuel cells and metal-air batteries, are urgently needed.1–5 Supercapacitors with high energy densities and specific capacitance are foremost required.6–9 As for fuel cells and metal-air batteries, developing stable and economical oxygen reduction reaction (ORR) electrocatalysts to replace the state-of-art expensive commercial Pt/C is crucial.10–13 These challenging issues have drawn considerable attention.Porous carbon materials are widely used due to their high surface areas, chemical stability, low costs, excellent electrical conductivity, and nontoxic nature.14–16 To enhance the performance of porous carbon materials, several strategies have been implemented. Doping materials with heteroatoms (N, O, S, P, and B) is one of the effective approaches.17–20 Moreover, owing to the synergetic effects of heteroatoms, multi-heteroatom doping is a more effective and versatile approach than using a single dopant.21,22 The use of different elements in doping has various effects on material properties in supercapacitors and ORR. Nitrogen plays a crucial role in these heteroatom-doping materials. Nitrogen groups affect the inert nature of carbon materials on the atomic level, and enhance capacitance through the pseudocapacitance effect.23,24 As for ORR, nitrogen has a close relationship with the final performance, and different nitrogen species have different effects on the catalytic behavior.25 Guo et al.26 suggested that the carbon atom next to pyridinic N is responsible for ORR catalysis instead of the pyridinic N itself. Zhang et al.27 elaborated the point that the existence of quaternary N in a graphene layer can provide electrons to the π-system, and enhance the nucleophile characteristic of the adjacent carbon atom. Such effect enhances the O2 adsorption and catalytic performance. Aryloxy radicals are responsible for the increase in the paramagnetism, which accompanies the chemical and electrochemical oxidation of carbon materials.28,29 Oxygen doping alone may not have much effect on the performance, but a significant increase is observed when another heteroatom, such as nitrogen, is added to the carbon material.30 In aqueous electrolytes, nitrogen and oxygen-containing functional groups boost the efficiency of ion transfer by enhancing the property of hydrophily and provide extra pseudocapacitance through the reversible redox reaction.31–33 Meanwhile, the fabrication of the hierarchically porous distribution is of paramount importance. Micropores increase the number of exposed active sites, and mesopores facilitate the mass transport in supercapacitors and fuel cells.34–38 However, most traditional heteroatom-doped metal-free porous carbon materials have random structures and pore distributions with high uncertainty, which render the understanding of ORR and supercapacitor mechanisms difficult.39 Therefore, materials with precise heteroatom and pore distribution structures should be explored.
The development of porous organic frameworks (POFs) enables a controllable fabrication that meets the abovementioned requirements. Among all kinds of POFs, covalent triazine frameworks (CTFs), which were first developed by Antonietti and Thomas et al.,40 possess robust thermal and chemical stability, large specific surface areas, and high N contents. The thermal and chemical stabilities are beneficial for the structural maintenance after high-temperature carbonization.41–44 Large specific surface areas have large advantages in terms of the properties related to the electrochemical energy storage and electrocatalysis for the reasons mentioned above. More importantly, the heteroatoms introduced to the materials can be adjusted by using different monomers, and the defined pore sizes can be acquired by selecting monomers with different sizes.
Based on a molecular design, we designed a monomer containing nitrogen and oxygen heteroatoms, namely, 2,8-bis(4-isocyanophenyl)-2,3,7,8-tetrahydropyridazino[4,5-g]phthalazine-1,4,6,9-tetraone (PYPZ). The high N content of the monomer enables the production of N-rich final products. The polymerization product CTFs (p-PYPZs) were synthesized through the high-temperature ionothermal polymerization of the nitril precursor by using ZnCl2 as a solvent and catalyst. The following carbonization procedures afford materials with significantly enhanced charge transfer and mass transport properties. The holes formed through the ionothermal method are more homogeneously distributed, and have more regular pore sizes than other porous carbons synthesized through KOH activation or template-assistance methods. In summary, the strategy used for polymerization determines the structure of products, and structure determines the final performance. The as-synthesized nitrogen, oxygen co-doped p-PYPZ products have several merits as follows: uniform distribution of oxygen and nitrogen heteroatoms, high nitrogen content, oxygen content, hierarchical porous structures, high specific surface area, and easy to acquire without using templates. p-PYPZ@600 exhibits satisfactory energy storage performance (256 F g−1 at a current density of 0.1 A g−1) and high cycling stability (110% capacitance retention after 35000 cycles) in aqueous electrolyte (1 M H2SO4). As for a two-electrode system, p-PYPZ@600 exhibits a capacitance of 121 F g−1 at 0.1 A g−1, energy density of 22 W h kg−1 in TEABF4/AN, 74.23 F g−1 at the current density of 0.1 A g−1, and energy density of 32 W h kg−1 in [BMIM][BF4]. With regard to ORR, p-PYPZ@700 shows a four-electron dominant pathway and excellent methanol crossover resistance in an alkaline electrolyte. Moreover, the bottom-up design is an ingenious approach for the facile synthesis of defined heteroatom-doped carbon materials with hierarchical pore structures for energy storage and electrocatalysts.
2. Experimental
2.1 Synthesis of PYPZ and p-PYPZs
All of the starting materials were obtained from commercial suppliers and used without further purification unless otherwise indicated.The synthesis of a monomer containing nitrogen and oxygen heteroatoms, 2,8-bis(4-isocyanophenyl)-2,3,7,8-tetrahydropyridazino[4,5-g]phthalazine-1,4,6,9-tetraone (PYPZ), has the following procedures: (1) 2.25 g of 1H,3H-benzo[1,2-c:4,5-c′]difuran-1,3,5,7-tetraone and 3.355 g of (4-isocyanophenyl)hydrazine were added to a three-neck flask with 175 ml glacial acetic acid. (2) The mixture was stirred for 5 h under 125 °C, and then cooled down to room temperature. (3) The final product was obtained after filtration, washed with absolute ethyl alcohol, and dried under vacuum. Finally, 4.165 g of a faint yellow product was obtained (yield: 74.3%).
The final products were obtained by mixing the precursor monomer PYPZ and anhydrous ZnCl2 in a molar ratio of 1:5, and transferring the mixture to a quartz tube ampoule in a glovebox (argon with <0.1 ppm water and <0.1 ppm oxygen). ZnCl2 acted as the catalyst and solvent simultaneously. The ampoule was then evacuated and sealed rapidly, and heated at a rate of 5 °C min−1 to 550 °C and maintained for 30 h. Eventually, the resulting black powder was dispersed in a 5% HCl solution, and deionized water for the removal of impurities. Then, the final p-PYPZ@550 product was obtained by drying the solution under vacuum at 100 °C for 24 h. The samples were denoted as p-PYPZ@x, where x represents the treatment temperature. Other products, p-PYPZ@600, p-PYPZ@650, and p-PYPZ@700, were synthesized under similar conditions, but higher temperatures (600 °C, 650 °C, and 700 °C, respectively).
2.2 Material characterization
All of the materials synthesized above were characterized by various methods, as follows. The 1H-NMR spectrum was tested to detect the purity of the monomer. Mass spectrometry (MS) spectra were measured on a LTQ Orbitrap XL mass spectrometer. Thermogravimetric analysis (TGA) was conducted on a Mettler TGA/SDTA851 TGA thermal analysis meter in a flowing nitrogen atmosphere at a heating rate of 20 °C min−1 from 30 °C to 800 °C. Thermal parameters of the products were measured using a NETZSCH DSC 204 differential scanning calorimetry (DSC) instrument in flowing nitrogen at a different heating rate from 25 °C to 400 °C. The Fourier transform infrared (FT-IR) spectra were acquired by a Bruker Equinox 55 spectrometer. Samples were analyzed using pellets prepared by compressing a dispersed mixture of the sample and KBr powder. For the PYPZ monomer and p-PYPZs, elemental analysis (EA, a Flash EA1112 analyzer) was carried out to determine the elemental content in the materials. As for p-PYPZs, nitrogen sorption/desorption isotherms of the samples were measured at 77 K on an Autosorb iQ (Quantachrome) analyzer. Before sorption measurements, the samples were degassed at 120 °C under vacuum for 30 h. The Brunauer–Emmett–Teller (BET) method and density functional theory (DFT) pore model were utilized to calculate the specific surface area and pore size distribution respectively. The X-ray diffraction (XRD) spectrum was obtained by a SmartLab 9KW Rigaku at the scanning rate of 20° min−1. Raman spectra were taken by DXR Microscope. X-Ray photoelectron spectroscopy (XPS) was conducted to observe the surface functional groups and bonding characterization of the composites via an ESCALAB 250 device with a base pressure of 1 × 10−9 mbar and Al Kα X-ray radiation. The morphologies and the structure of the as-obtained products were observed by scanning electron microscopy (SEM) (Quanta-450, FEI), transmission electron microscopy (TEM) (Eindhoven, The Netherlands, 120 kV), and high-resolution transmission electron microscopy (HR-TEM). The elemental composition of the materials was studied via SEM in cooperation with energy dispersive spectroscopy (EDS).2.3 Electrochemical measurements
To check the energy storage properties of the products, three-electrode system and two-electrode system measurements were performed. The working electrodes were fabricated as follows: (1) for the three-electrode system, a p-PYPZ film pressed on a titanium mesh was used as the working electrode, a platinum sheet (10 mm × 20 mm) was used as the counter electrode, and an aqueous Ag/AgCl was used as the reference electrode. The electrolyte was 1 M H2SO4 solution, and these three electrodes were immersed into the electrolyte to form the three-electrode testing system; (2) for the two-electrode system, the materials were prepared by mixing p-PYPZ (80 wt%, 32 mg), carbon black (10 wt%, 4 mg), and polytetrafluoroethylene (10 wt%, 4 mg) evenly. Hereafter, the as-prepared thin film was dried at the temperature of 80 °C under vacuum for 12 h, and then cut into a circular shape with a diameter of 10 mm. Two p-PYPZ electrodes with the same weight were selected as the symmetrical working electrodes, and assembled in a coin cell with tetraethylammonium tetrafluoroborate in an acetonitrile solution (TEABF4/AN) or 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]) with glassy fiber as the separator.Cyclic voltammograms (CV), galvanostatic charge/discharge (GCD) curves, and electrochemical impedance spectroscopy (EIS) were recorded via a BioLogic Potentiostat (Model A VMP-3) at room temperature. The potential window ranged from −0.2 to 0.8 V in 1 M H2SO4 for the three-electrode system, and 0 to 2.3 V in TEABF4/AN, 0 to 3.5 V in [BMIM][BF4] for the two-electrode system. The specific capacitance was calculated using the following equations:
| C = It/mV | (1) |
| C = 2It/mV | (2) |
| E = CV2/(8 × 3.6) | (3) |
| P = 3600E/t | (4) |
2.4 Electrocatalytic measurements
For the establishment of a catalyst ink, 4 mg of the catalyst material, 700 μl of ethyl alcohol, 250 μl of H2O, and 50 μl of Nafion117 solution were mixed evenly through 1 h of ultrasonic treatment. The slurry of the active material was then coated and dried on a glassy carbon electrode with a geometrical surface area of 0.196 cm2 (all of the current densities in the curves of the electrochemical measurements were normalized to the glassy carbon geometric area). An ink suspension (15 μl) was loaded on a freshly polished glassy carbon electrode with a catalyst loading of 300 μg cm−2. For comparison, Pt/C (Alfa Aesar, 20 wt% Pt on carbon black) was also fabricated on a glassy carbon electrode with a loading density of 150 μg cm−2 with the same procedure. A three-electrode cell was constructed using Ag/AgCl, Pt, and Teflon-coated glassy carbon as the reference, counter, and working electrodes, respectively. All electrochemical measurements in this work were performed using a BioLogic Potentiostat (Model A VMP-3) at room temperature (∼24 °C). All of the potentials reported in this work were referred to the RHE, based on the Nernst equation.| E(RHE) = E(Ag/AgCl) + 0.0592 × pH + 0.1957V | (5) |
The electrolyte (0.1 M KOH) was saturated by bubbling oxygen and nitrogen for at least 30 min before the test. Electrochemical catalytic activities of the materials were evaluated in oxygen and nitrogen saturated 0.1 M KOH electrolytic solution by rotating disk electrode (RDE), and then rotating ring disk electrode (RRDE) via a rotating electrode system (Pine) to monitor the formation of the intermediate peroxide species. CV measurements were performed in an N2 or O2 saturated 0.1 M KOH solution under a potential range from 0 to 1 V at a scan rate of 10 mV s−1. Linear sweep voltammogram (LSV) curves were obtained in an O2 saturated 0.1 M KOH solution. The test was scanned under the potential from 0.1 to 1 V at a scan rate of 5 mV s−1, and the N2 background current was subtracted to eliminate the contributions of the capacitive currents. While using RRDE to detect the H2O2 yield, the ring potential was set to 1.2 V vs. RHE to oxidize the H2O2 transferred from the glassy carbon disk electrode. The H2O2 yield rate and the electron transfer number (n) were calculated by the following equation:
| H2O2 (%) = 200 × (Ir/N)/(Id + Ir/N) | (6) |
| n = 4 × Id/(Id + Ir/N) | (7) |
3. Results and discussion
3.1 Structure characterization
Fig. 1a illustrates the ionothermal strategy for the polymerization of p-PYPZs. The monomer PYPZ was first synthesized by using 1H,3H-benzo[1,2-c:4,5-c′]difuran-1,3,5,7-tetraone and (4-isocyanophenyl)hydrazine. Then, PYPZ was polymerized for the preparation of p-PYPZs with a facile ionothermal strategy. PYPZ was characterized through Fourier transform infrared (FT-IR) spectroscopy, 1H NMR spectroscopy, elemental analysis (EA), TGA, and DSC (Fig. 1f, S1 and Table S1†). MS calculated for C24H12N6O4, 448.26; found, 448.23; anal. calcd for C24H12N6O4: C, 64.29; H, 2.7; N, 18.74; found: C, 64.13; H, 2.73; N, 18.78. According to FT-IR spectroscopy for the monomer in Fig. 1f, the signals at 2221 and 1680 cm−1 indicate the existence of the cyano and carbonyl groups in the PYPZ monomer. The strong and intense characteristic cyano stretching band at 2221 cm−1 disappeared, and the peaks of the characteristic triazine appeared at 1250 and 1550 cm−1. This finding indicates that p-PYPZs were obtained successfully via cyano polymerization. The TGA curves in Fig. S2† and DSC curves in Fig. S3† indicate that the monomer is stable below 350 °C. Fig. S4† shows the good thermal stability of p-PYPZs with a high carbon yield at 800 °C. Such good thermal characteristics can be attributed to the triazine-based framework of the materials.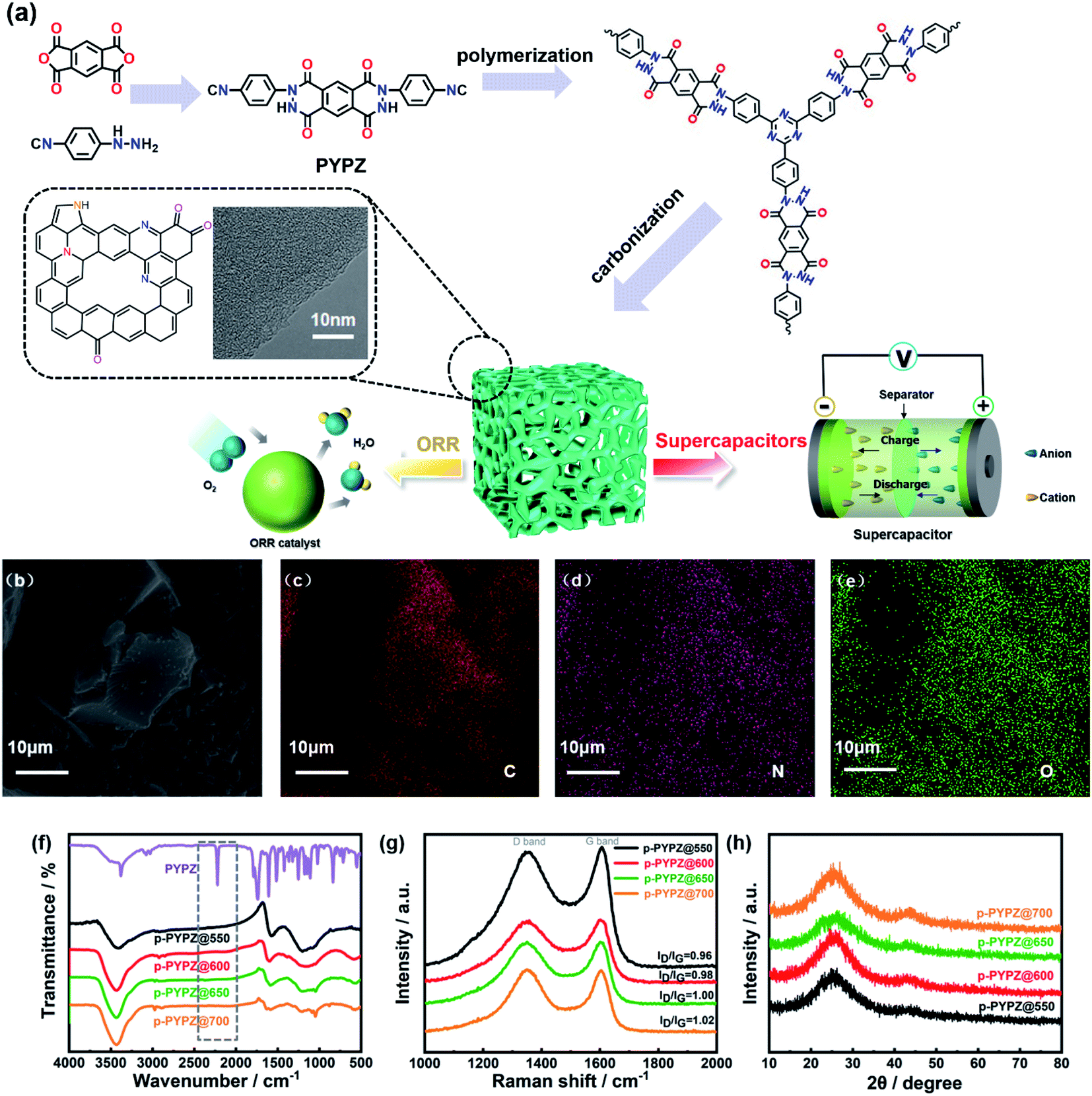 | ||
| Fig. 1 (a) Schematic of the synthesis of p-PYPZs; (b) SEM image of p-PYPZ@600; (c–e) SEM-EDS mapping images of p-PYPZ@600; (f) FT-IR spectra of PYPZ and p-PYPZs; (g) Raman spectra of p-PYPZs; and (h) XRD patterns of p-PYPZs. | ||
Raman spectroscopy is a conventional and effective method for characterizing the degree of disorder of carbonaceous materials.45 The D-band (defect) located at 1347 cm−1 and G-band (graphite) located at 1602 cm−1 are the two main peaks in Fig. 1g. The intensity ratios of the D and G bands reflect the degree of material defect. The intensity ratios (ID/IG) of the p-PYPZs were 0.96, 0.98, 1.00, and 1.02 for p-PYPZ@550, p-PYPZ@600, p-PYPZ@650, and p-PYPZ@700, respectively. The defects increased when the reaction temperature increased from 550 °C to 700 °C. The degree of crystallization in all samples was determined on the basis of the XRD patterns. Fig. 1h shows no intense peak and two broad peaks at 24° and 44°, corresponding to the (002) and (101) lattice planes of amorphous carbon.46 The result indicates that the products exhibited the dominant features of the amorphous carbon structures. SEM was used in intuitively observing the morphology of p-PYPZs. The images shown in Fig. S5† are consistent with the conclusion above; that is, the number of fragmental products appearing at high polymerization temperatures increases.
According to the TEM and HR-TEM images (Fig. 1a, S6 and S7†), the p-PYPZs possess vermicular pore structures. The pore structures were further determined using N2 adsorption/desorption isotherm curves, and the BET method was used. Their specific surfaces were determined at 77 K. The density functional theory (DFT) method was used in calculating the pore size distributions. The results of the specific surface area, total pore volume, and average pore size are summarized in Table S4.† The porosity of the resultant product was influenced by the reaction temperature because the surface area of p-PYPZs increased from 981 m2 g−1 to 1464 m2 g−1 when the polymerization temperature increased from 550 °C to 700 °C. As illustrated in Fig. 2a, under increasing reaction temperature, the isotherm curves gradually changed from type I (p-PYPZ@550 and p-PYPZ@600) to type IV (p-PYPZ@650 and p-PYPZ@700), and the total pore volume increased from 0.65 cm3 g−1 to 0.93 cm3 g−1. The N2 adsorption/desorption isotherms of p-PYPZs showed high absorption at a low relative pressure (P/P0), indicating the existence of numerous micropores (Fig. 2b). The formation of hysteresis loops is related to the capillary condensation and evaporation in mesopores, and therefore confirmed the coexistence of mesoporous structures.47 Fig. 2b shows the pore size distribution plots obtained through DFT. The samples had a similar pore size distribution with a micropore distribution peak between 0.5 and 1.5 nm, and a mesopore peak between 2 and 3 nm. The proportion of micropores increased with polymerization temperature, and micropores contribute more to the specific surface area. Therefore, a higher temperature will lead to a larger specific surface area, which is consistent with the result in Table S4.†
 | ||
| Fig. 2 (a) Nitrogen adsorption/desorption isotherm and (b) pore size distributions for p-PYPZs; (c) Overall XPS analysis scanned spectrum of p-PYPZs; (d) high resolution N 1s XPS spectra of p-PYPZs. | ||
The EDS mapping images show the even distribution of nitrogen (pink) and oxygen (green) atoms in the p-PYPZ@600 skeleton (Fig. 1b–e). The elemental chemical composition and bonding configurations of p-PYPZs were explored through XPS and EA. As shown in Fig. S8,† three element species existed in p-PYPZs, and their contents are listed in Table S1 and S2.† The EA test shows that the nitrogen content decreased from 8.85 wt% to 3.5 wt%, and XPS shows the nitrogen content decreases from 8.1 to 3.46 at% when the polymerization temperatures were changed from 550 °C to 700 °C, consistent with that from EA. The complex N 1s spectra of the p-PYPZ could be further deconvoluted into three typical peaks as follows: 398.3 eV (N-6), 399.6 eV (N-5), and 400.7 eV (N-Q), which correspond to pyridinic N (or triazine N), pyrrolic N (or carbazole N), and graphitic N, respectively.48 The contents of different nitrogen species were influenced by the reaction temperature. The results are shown in Fig. 2d. The content of N-Q increased with the reaction temperature. This phenomenon was attributed to the conversion of the pyridinic N and pyrrolic N to graphitic N as the pyrolysis temperature increases.49 The hydrophilicity properties of p-PYPZs were measured by the contact angles test. The result showed that the wettability of the material is improved with the increment total content of oxygen and nitrogen atoms (Fig. S9†).
3.2 Electrochemical performance of the supercapacitor
Owing to their high specific surface areas and unique hierarchical pore structures, the as-synthesized materials are promising candidates as metal-free porous carbon materials for supercapacitors. The prepared materials were tested using a three-electrode cell in 1 M H2SO4 acidic aqueous electrolyte for the evaluation of their electrochemical performance through CV, galvanostatic charge/discharge (GCD), electrochemical impedance spectroscopy (EIS), and long cycle life test. The CV curves of p-PYPZs in Fig. 3a were tested at a scan rate of 10 mV s−1. The curves had irregular rectangle shapes and broad redox bumps at approximately 0.1–0.6 V, which are the results of the redox reactions between the electrolytes and nitrogen- and oxygen-containing functional groups. Another conclusion that can be drawn from the CV curves is that p-PYPZ@600 exhibited the largest integrated area under a potential window of −0.2–0.8 V. This conclusion indicates that the specific capacity of p-PYPZ@600 was higher than that of the other p-PYPZs materials under the same measurement conditions. | ||
| Fig. 3 Electrochemical performance of p-PYPZs in 1.0 M H2SO4 aqueous solution: (a) CV curves at 10 mV s−1; (b) GCD curves at 1 A g−1; (c) specific capacitance of p-PYPZs at different current densities; (d) Nyquist plots of p-PYPZs with frequency ranging from 100 kHz to 10 mHz; (e) cycling performance of the p-PYPZ@600 electrode at 10 A g−1 for 35000 cycles (inset: GCD curves for the first and last 10 cycles; contact angles before and after 35000 cycles). | ||
The GCD curves of p-PYPZs (Fig. 3b) were performed at 1 A g−1, and reflect the same trend of capacitance as the results from CV. p-PYPZ@600 shows the longest charge–discharge time at 1 A g−1, consistent with the largest integrated area of CV curves, and had the best capacitance characteristics. The specific capacitance of different materials was calculated by the discharge slope in the GCD curves, which were obtained at different current densities (Fig. S10†). p-PYPZ@600 also exhibited superior capacitance of 256 F g−1 at 0.1 A g−1, which is higher than p-PYPZ@550 (241 F g−1), p-PYPZ@650 (212 F g−1), and p-PYPZ@700 (142 F g−1). The good supercapacitor performance can be attributed to the high content of heteroatoms and proper hierarchical pore structure. On the one hand, the pyrrolic nitrogen, pyridinic nitrogen, and quinine-type O can provide pseudocapacitance through faradaic redox reactions in acidic electrolytes. On the other hand, the phenol-type O can enhance the carbon wettability and improve the infiltration of the electrolyte.50 Thus, the appropriate presence of different types of nitrogen and oxygen is important for enhancing the supercapacitor properties. Fig. S10† displays the GCD curves of different materials at increasing current densities (0.1, 0.2, 0.5, 1, 2, 5, and 10 A g−1). The small mesopores facilitate the fast ion transport through the porous structures, especially when a large current density is employed.51 The as-synthesized p-PYPZs possessed a good rate capacitance property due to the existence of the small mesopores.
The conductive and diffusion behavior of the materials were measured using the EIS technique (Fig. 3d) in 1 M H2SO4 solution at a frequency ranging from 100 kHz to 10 mHz. The equivalent circuit for the impedance spectra and the fitting data is shown in Fig. S10.† The Nyquist plots of p-PYPZs clearly exhibited vertical lines in the low-frequency area, indicating a low ionic diffusion resistance. In the high-frequency region, the intercept of the Nyquist plots with the Z′ axis represented the equivalent series resistance (Rs). Furthermore, the Rs of p-PYPZ@550, p-PYPZ@600, p-PYPZ@650, and p-PYPZ@700 were 0.25, 0.21, 0.38 and 0.21 Ω, respectively. The small Rs of p-PYPZ@600 and p-PYPZ@700 can be attributed to the combined effect of both graphitization degree and the N content. The charge transfer resistance (Rct) can be obtained from the diameter of the semicircle in the high-frequency region. Rct decreased from 2.67, 1.25, 0.81 to 0.62 Ω for p-PYPZ@550, p-PYPZ@600, p-PYPZ@650, and p-PYPZ@700, respectively. The small Rct indicated that it was conducive to transfer electrons on the surface of the electrode material. The electrochemical stability is another crucial parameter determining the final long cycle life of a material. Hence, the materials were tested at a current density of 10 A g−1 for 35000 cycles, and the result shows that the specific capacitance has an increasing tendency rather than a declining tendency after 35000 cycles. This result can be attributed to the covalent-based heteroatom-doped structure, which effectively prevented the loss of heteroatoms from the electrode, and ensured an efficient charge transfer. Such a structure ensured the stability of the capacitance, and the increasing tendency was mainly due to the active process occurring after the infiltration of the electrolyte. The contact angles figure in the inset of Fig. 3e showed the improved hydrophilicity property of p-PYPZ@600 before and after 35000 cycles. The GC curves in the inset in Fig. 3e were symmetrically triangular in shape after 35000 cycles, which also indicates the outstanding cycling performance of p-PYPZ@600. The cycling stability of p-PYPZ@600 was better than that of other reported heteroatom-doped carbon electrodes (Table S5†). In addition, the preparation method is much easier than that of other reported materials.
All of the above results illustrated that p-PYPZ@600 exhibited excellent long-term durability as the supercapacitor electrode material, and the good performance of p-PYPZ@600 was mainly due to the combined advantages of the defined hierarchical pore structure, high specific surface area, good conductivity and large amount of stable surface functional groups.
Given the good three-electrode performance of p-PYPZ@600, we further evaluated the electrochemical properties of p-PYPZ@600 by assembling symmetric two-electrode supercapacitors. The energy density of a supercapacitor requires urgent improvement, and thus symmetric supercapacitors use the organic electrolyte TEABF4/AN with a potential window of 2.3 V. A wide potential window can greatly enhance the energy density properties of the supercapacitors.
Fig. 4 shows the performance of p-PYPZ@600 in TEABF4/AN. As shown in Fig. 4a, p-PYPZ@600 exhibited rectangular CV curves and triangular-shaped GCD curves in TEABF4/AN, indicating enhanced capacitive performance with facilitated electron and ion propagation process. The specific capacitance was calculated using the discharge slope in GCD curves at different current densities in TEABF4/AN. In Fig. 4d (inset), the specific capacitance of p-PYPZ@600 is 121 F g−1 at the current density of 0.1A g−1. The Ragone plot is a link between the power and energy densities, and is a useful indicator for the performance of the supercapacitor devices. The two-electrode device showed a high energy density of up to 22 W h kg−1 at 0.1 A g−1, and 10 W h kg−1 at 10 A g−1 with a power density of 5737 W kg−1. Fig. 4e shows the high stability of p-PYPZ@600 (only 10% capacitance decay after 15000 cycles at 10 A g−1) in TEABF4/AN, indicating a promising application prospect.
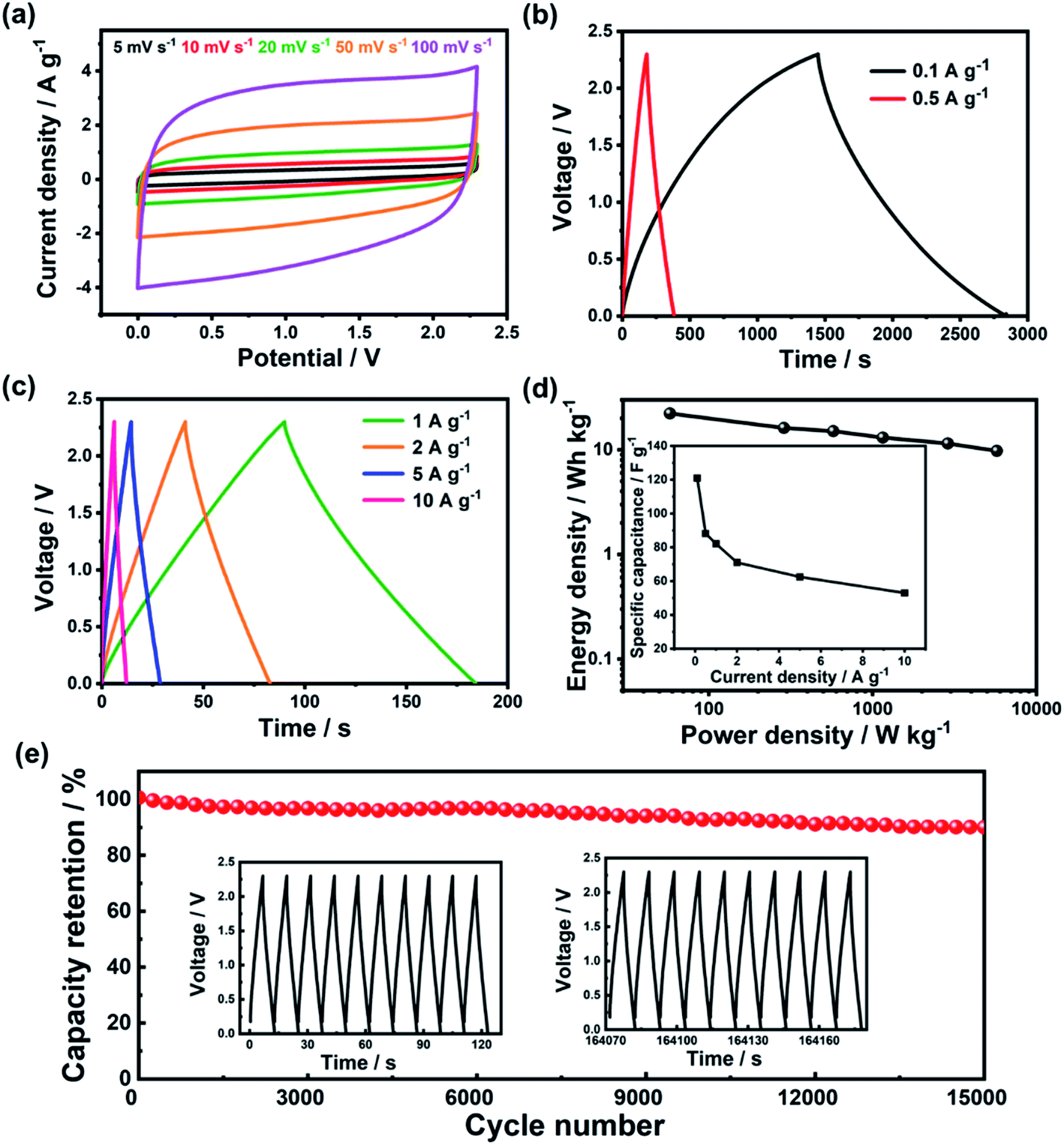 | ||
| Fig. 4 Electrochemical performance of p-PYPZ@600 in the two-electrode system with TEABF4/AN: (a) CV curves at different scan rates; (b) GCD curves at 0.1 A g−1 and 0.5 A g−1; (c) GCD curves at 1, 2, 5, and 10 A g−1; (d) Ragone plots (inset: specific capacitance at different current densities); (e) long-term cycling tests of the p-PYPZ@600 electrode with the electrolyte of TEABF4/AN (inset: GCD curves for the first and last 10 cycles). | ||
The ionic liquid as 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM][BF4]) was used as the electrolyte in the two-electrode system. We aimed to broaden the voltage window to 3.5 V. Fig. S12† shows the performance of p-PYPZ@600 in [BMIM][BF4]. The shapes of the CV curves for p-PYPZ@600 were not rectangular, indicating inferior electrochemical performance compared with the performance of the organic electrolyte. The specific capacitance in Fig. S12(d)† was calculated using the GCD technique at different current densities in [BMIM][BF4]. The device had a specific capacitance of 74 F g−1 at 0.1 A g−1. The Ragone plots shown in Fig. S12† indicate that the p-PYPZ@600-based supercapacitor can deliver a high-energy density of up to 32 W h kg−1 at 0.1 A g−1. As the power density increased to 8845 W kg−1 at 10 A g−1, the energy density was maintained at about 11 W h kg−1.
In summary, the enhanced electrochemical performance of p-PYPZ@600 can be ascribed to the synergistic effects. First, abundant heteroatoms in the materials not only act as functional groups for pseudocapacitive performance, but also tune the hydrophilicity and polarity of the carbon frameworks. More importantly, the heteroatoms exist in the materials via covalent bonds, which contribute to the stability. Second, the hierarchical pore structure provided the rapid electrochemical kinetics process, as well as the exposure of the surface area for adsorbing the charged ions. All of these good properties are attributed to the precise structure by bottom-up method and molecular design.
3.3 Electrocatalytic properties in ORR
The sluggish kinetic process of the cathodic oxygen reduction reaction in fuel cells greatly hinders the commercial application of the process. Therefore, a four-electron pathway-dominant efficient ORR catalyst is of vital importance. The presence of N and O greatly affects the electroneutrality of carbon atoms, and promotes catalytic reaction. The large difference in the electronegativity among N, O, and C promotes the adsorption of the oxygen reactant.52 Therefore, in this study, the ORR performance of p-PYPZs was evaluated using a rotating disk electrode (RDE) and rotating ring disk electrode (RRDE) method in 0.1 M KOH. Fig. S14† shows the linear sweep voltammetry (LSV) curves collected at a rotation speed of 1600 rpm by RDE. The synthesized p-PYPZ@700 has an onset potential of 0.86 V vs. RHE and a diffusion limiting current density of 4.8 mA cm−2, which is better than other p-PYPZs, indicating a better mass transport ability and better catalytic activity of p-PYPZ@700 on ORR. The CV curves in Fig. S13† show a reduction peak at 0.8 V in O2-saturated 0.1 M KOH, whereas no such peak was found under N2-saturated solutions, suggesting an efficient conversion of oxygen. The low overpotential can be comparable to that of Pt/C for about 0.87 V.The electron transfer number and peroxide yield rate are the two crucial parameters reflecting the selectivity property of a catalyst. The two-electron pathway produces peroxide as a catalytic product in contrast to the four-electron pathway, which results in water products. Peroxide generated from a two-electron pathway limits the practical application of the materials in fuel cells, impairing the ESI† or membrane and poisoning the cells. Therefore, RRDE was used in analyzing these properties. By calculating the data of the RRDE test in Fig. 5a, we obtained the final results in Fig. 5b. The average electron transfer number value of p-PYPZ@700 was calculated to be in the range of 3.5–3.7 under the potential of 0.2–0.6 V, and was the highest among the values of p-PYPZs. Additionally, the peroxide yield rate decreased from 60% for p-PYPZ@550 to 20% for p-PYPZ@700. To summarize, p-PYPZ@700 showed an efficient four-electron reaction process similar to commercial Pt/C. The polymerization temperature can greatly affect the ORR activities and the selectivity of the reaction pathway.
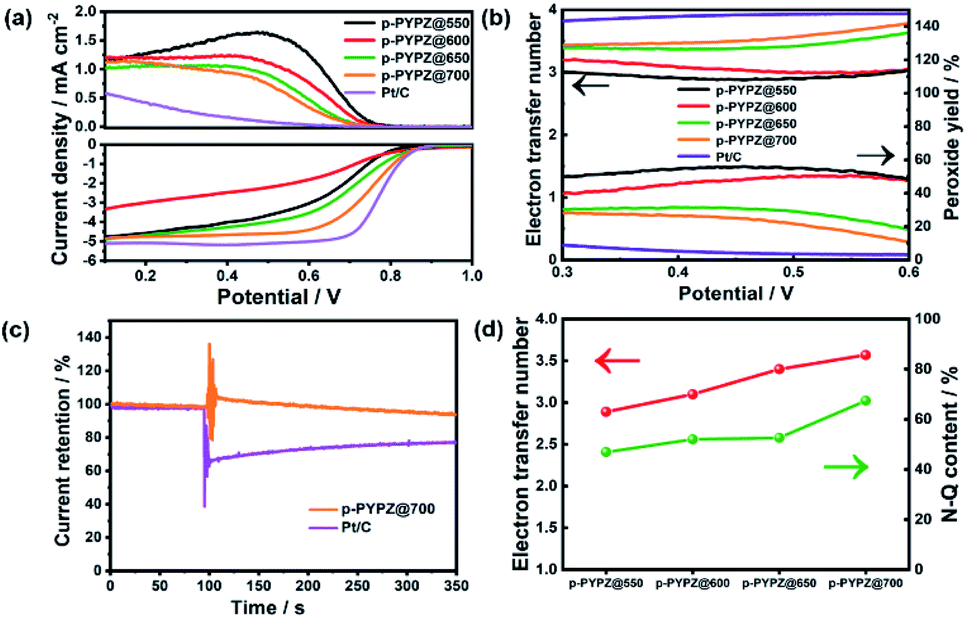 | ||
| Fig. 5 (a) RRDE voltammogram curves of p-PYPZs samples and Pt/C recorded in 0.1 M KOH at 1600 rpm and 5 mV s−1; (b) electron transfer number and peroxide yield rate calculated from the RRDE tests; (c) methanol-tolerance test for p-PYPZ@700 and commercial Pt/C; (d) electron transfer number n and N-Q content. | ||
Table S3† lists the contents of different nitrogen species in p-PYPZs. The content of N-Q increased with temperature. The increase was caused by the conversion of N-6 and N-5 to N-Q. Fig. 5d shows the positive correlation between the N-Q content and the four-electron pathway selectivity. The result can be attributed to the contribution of N-Q atoms to improving the conductivity, and providing strong physicochemical interactions and adsorption of the oxygen reactant. Thus, the high content of N-Q leads to the best ORR performance of p-PYPZ@700.
The methanol-tolerance capacity of p-PYPZ@700 was tested through the chronoamperometric measurement and Pt/C as a reference (Fig. 5c). Methanol (10 ml) was added into 200 ml 0.1 M KOH electrolyte at 100 s. The current density of Pt/C declined sharply due to the electrochemical oxidization of methanol. However, such phenomenon did not occur on p-PYPZ@700, suggesting its excellent tolerance to methanol crossover and high selectivity for electrocatalytic oxygen reduction. This phenomenon indicates a promising prospect of the application in the methanol fuel cell.
All of these properties showed p-PYPZ@700 to be an excellent candidate as the metal-free ORR catalyst, and implied that N-Q plays an important role in the catalytic ORR reaction process.
4. Conclusion
A series of multi-heteroatom porous carbon frameworks (p-PYPZs) was designed and synthesized with a bottom-up method through the trimerization of the molecular designed monomer, PYPZ. The as-synthesized p-PYPZs show high ORR activities and energy storage capacitance owing to the abundant N, O heteroatoms, large surface areas, and microporous and mesoporous structures. The large surface area and hierarchical porous structures can shorten the length of ion diffusion and enlarge the effective surface area. The electrochemical performance of p-PYPZs was evaluated in an aqueous electrolyte with a three-electrode system. The as-made p-PYPZ@600 exhibits a specific capacitance of 256 F g−1 at 0.1 A g−1, and a superb cycling stability with a capacity retention of 110% after 35000 cycles in 1 M H2SO4. The material shows high stability as a symmetric two-electrode supercapacitor (only 10% capacitance decay after 15000 cycles at 10 A g−1) in TEABF4/AN and a high energy density of 32 W h kg−1 at 0.1 A g−1 in [BMIM][BF4]. Such good electrochemical stability can be attributed to the homogeneous distribution of N, O heteroatoms at the atomic level. In addition, p-PYPZ@700 possesses good ORR catalysis ability, excellent methanol tolerance, and the four-electron transfer dominant pathway, which is relative to the content of N-Q. The effect of different heteroatoms and different types of nitrogen should be clarified for the further enhancement of the electrochemical and electrocatalytic performance, which highly depends on the precise control of the structure of the materials. Therefore, the facile synthesis route and the conception of the bottom-up strategy show the superiority of the prepared materials, and such approach can be extended to other cyano-containing monomers to tune the structure of the carbonaceous materials by changing the element composition of the monomer.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support from the National Key Research and Development Program (No. 2018YFB1107500), Liao Ning Revitalization Talents Program (XLYC1907144), the National Natural Science Foundation of China (No. 51503024), Dalian Youth Science and Technology Star Project Support Program (No. 2017RQ104).References
- C. Wei, R. R. Rao, J. Peng, B. Huang, I. E. L. Stephens, M. Risch, Z. J. Xu and Y. Shao-Horn, Adv. Mater., 2019, 31, e1806296 CrossRef
.
- B. C. Kim, J.-Y. Hong, G. G. Wallace and H. S. Park, Adv. Energy Mater., 2015, 5, 1500959 CrossRef
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797–828 RSC
- B. Liang, K. Li, Y. Liu and X. Kang, Chem. Eng. J., 2019, 358, 1002–1011 CrossRef CAS
- Y. Liang, Y. Li, H. Wang and H. Dai, J. Am. Chem. Soc., 2013, 135, 2013–2036 CrossRef CAS
- C. Guan, X. Li, Z. Wang, X. Cao, C. Soci, H. Zhang and H. J. Fan, Adv. Mater., 2012, 24, 4186–4190 CrossRef CAS
- Q. Wu, L. Yang, X. Wang and Z. Hu, Adv. Mater., 2020, 32, e1904177 Search PubMed
- M. Nawwar, R. Poon, R. Chen, R. P. Sahu, I. K. Puri and I. Zhitomirsky, Carbon Energy, 2019, 1, 124–133 CrossRef CAS
- Y. Yuan and J. Lu, Carbon Energy, 2019, 1, 8–12 CrossRef
- Y. Huang, Y. Wang, C. Tang, J. Wang, Q. Zhang, Y. Wang and J. Zhang, Adv. Mater., 2019, 31, e1803800 CrossRef
- M. Wang, Y. Li, J. Fang, C. J. Villa, Y. Xu, S. Hao, J. Li, Y. Liu, C. Wolverton, X. Chen, V. P. Dravid and Y. Lai, Adv. Energy Mater., 2019, 10, 1902736 CrossRef
- C. X. Zhao, B. Q. Li, J. N. Liu and Q. Zhang, Angew. Chem., Int. Ed., 2020, 60(9), 4448–4463 CrossRef
- S. Liu, Z. Li, C. Wang, W. Tao, M. Huang, M. Zuo, Y. Yang, K. Yang, L. Zhang, S. Chen, P. Xu and Q. Chen, Nat. Commun., 2020, 11, 938 CrossRef CAS
- G. He, G. Yan, Y. Song and L. Wang, Front. Chem., 2020, 8, 226 CrossRef CAS
- H. Lu, L. Zhuang, R. R. Gaddam, X. Sun, C. Xiao, T. Duignan, Z. Zhu and X. S. Zhao, J. Mater. Chem. A, 2019, 7, 22579–22587 RSC
- B. Liu, M. Yang, D. Yang, H. Chen and H. Li, J. Power Sources, 2020, 456 CAS
- C. C. Jianhua Hou, F. Idrees and X. Ma, ACS Nano, 2015, 9, 2556–2564 CrossRef
- L. L. Z. Jongwoo Han, S. Lee, J. Oh, Kyoung-Seok Lee, J. R. Potts, J. Ji, X. Zhao, R. S. Ruoff and S. Park, ACS Nano, 2013, 7, 19–26 CrossRef
- A. M. P. Denisa Hulicova-Jurcakova, O. I. Poddubnaya, F. Suárez-García, J. M. D. Tascón and G. Qing Lu, J. Am. Chem. Soc., 2009, 131, 5026–5027 CrossRef
- X. Zhao, Q. Zhang, C.-M. Chen, B. Zhang, S. Reiche, A. Wang, T. Zhang, R. Schlögl and D. Sheng Su, Nano Energy, 2012, 1, 624–630 CrossRef CAS
- J. P. Paraknowitsch, J. Zhang, D. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87–92 CrossRef CAS
- T. Li, W. Zhang, L. Zhi, H. Yu, L. Dang, F. Shi, H. Xu, F. Hu, Z. Liu, Z. Lei and J. Qiu, Nano Energy, 2016, 30, 9–17 CrossRef CAS
- F. o. B. Elzbieta Frackowiak, Carbon, 2002, 40, 1775–1787 CrossRef
- T. Lin, I.-W. Chen, F. Liu, C. Yang, H. Bi, F. Xu and F. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS
- J. Quílez-Bermejo, E. Morallón and D. Cazorla-Amorós, Carbon, 2020, 165, 434–454 CrossRef
- R. S. Donghui Guo, C. Akiba, S. Saji, T. Kondo and J. Nakamura, Science, 2018, 351, 361–365 Search PubMed
- L. Zhang, C. Y. Lin, D. Zhang, L. Gong, Y. Zhu, Z. Zhao, Q. Xu, H. Li and Z. Xia, Adv. Mater., 2019, 31, e1805252 CrossRef
- W. H. Jun Xu and R. L. McCreery, J. Electroanal. Chem., 1996, 235–242 Search PubMed
- C. Paliteiro, J. Electroanal. Chem., 1987, 147–159 CrossRef CAS
- M. J. Mostazo-Lopez, D. Salinas-Torres, R. Ruiz-Rosas, E. Morallon and D. Cazorla-Amoros, Materials, 2019, 12, 1346 CrossRef CAS
- T. Lin, I.-W. Chen, F. Liu, C. Yang, H. Bi, F. Xu and F. Huang, Science, 2015, 350, 1508–1513 CrossRef CAS
- S. Zhang, S. Tsuzuki, K. Ueno, K. Dokko and M. Watanabe, Angew. Chem., Int. Ed., 2015, 54, 1302–1306 CrossRef CAS
- D. Zhu, J. Jiang, D. Sun, X. Qian, Y. Wang, L. Li, Z. Wang, X. Chai, L. Gan and M. Liu, J. Mater. Chem. A, 2018, 6, 12334–12343 RSC
- G. Y. J. Chmiola, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2018, 313, 1760–1763 CrossRef
- G. A. Ferrero, K. Preuss, A. B. Fuertes, M. Sevilla and M. M. Titirici, J. Mater. Chem. A, 2016, 4, 2581–2589 RSC
- B. Guo, R. Ma, Z. Li, S. Guo, J. Luo, M. Yang, Q. Liu, T. Thomas and J. Wang, Nano-Micro Lett., 2020, 12, 20 CrossRef CAS
- Z. Chen, S. Ye, S. D. Evans, Y. Ge, Z. Zhu, Y. Tu and X. Yang, Small, 2018, 14, e1704015 CrossRef
- Y. Qiu, M. Hou, J. Gao, H. Zhai, H. Liu, M. Jin, X. Liu and L. Lai, Small, 2019, 15, e1903836 CrossRef
- Z. L. Yuxi Xu, X. Huang, Y. Wang, Yu Huang and X. Duan, Adv. Mater., 2013, 25, 1–6 CrossRef
- P. Kuhn, M. Antonietti and A. Thomas, Angew. Chem., Int. Ed., 2008, 47, 3450–3453 CrossRef CAS
- L. Hao, S. Zhang, R. Liu, J. Ning, G. Zhang and L. Zhi, Adv. Mater., 2015, 27, 3190–3195 CrossRef CAS
- Y. Cao, Y. Zhu, X. Chen, B. S. Abraha, W. Peng, Y. Li, G. Zhang, F. Zhang and X. Fan, Catal. Sci. Technol., 2019, 9, 6606–6612 RSC
- S. Cao, B. Li, R. Zhu and H. Pang, Chem. Eng. J., 2019, 355, 602–623 CrossRef CAS
- Y. Zheng, S. Chen, H. Song, H. Guo, K. A. I. Zhang, C. Zhang and T. Liu, Nanoscale, 2020, 12, 14441–14447 RSC
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li, J. Lin and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 RSC
- H. Yu, L. Shang, T. Bian, R. Shi, G. I. Waterhouse, Y. Zhao, C. Zhou, L. Z. Wu, C. H. Tung and T. Zhang, Adv. Mater., 2016, 28, 5080–5086 CrossRef CAS
- L. Jiao, G. Wan, R. Zhang, H. Zhou, S. H. Yu and H. L. Jiang, Angew. Chem., Int. Ed., 2018, 57, 8525–8529 CrossRef CAS
- W. Yu, S. Gu, Y. Fu, S. Xiong, C. Pan, Y. Liu and G. Yu, J. Catal., 2018, 362, 1–9 CrossRef CAS
- G. H. Tiva Sharifi, X. Jia and T. Wagberg, ACS Nano, 2012, 6, 8904–8912 CrossRef
- F. Hu, J. Wang, S. Hu, L. Li, G. Wang, J. Qiu and X. Jian, Nanoscale, 2016, 8, 16323–16331 RSC
- M. Kim, I. Oh and J. Kim, J. Power Sources, 2015, 282, 277–285 CrossRef CAS
- T. Oh, K. Kim and J. Kim, J. Energy Chem., 2019, 38, 60–67 CrossRef
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra00043h |
| This journal is © The Royal Society of Chemistry 2021 |