
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synergistic influence of mesoporous spinel nickel ferrite on the electrocatalytic activity of nano-structured palladium†
Fariba Kaedia,
Zahra Yavari *ab,
Ahmad Reza Abbasianc,
Milad Asmaeic,
Kagan Kermand and
Meissam Noroozifar*d
*ab,
Ahmad Reza Abbasianc,
Milad Asmaeic,
Kagan Kermand and
Meissam Noroozifar*d
aDepartment of Chemistry, University of Sistan and Baluchestan, Zahedan, P.O. Box 98135-674, Iran. Fax: +98-54-3341-6888; Tel: +98-54-3341-6464E-mail: z_yavari@chem.usb.ac.ir; zahrayavari5@gmail.com
bRenewable Energies Research Institute, University of Sistan and Baluchestan, Zahedan, Iran
cDepartment of Materials Engineering, Faculty of Engineering, University of Sistan and Baluchestan, Zahedan, Iran
dDepartment of Physical and Environmental Sciences, University of Toronto, Scarborough 1265 Military Trail, Toronto, Ontario M1C 1A4, Canada
First published on 23rd March 2021
Abstract
Structure and surface area are critical factors for catalysts in fuel cells. Hence, a spinel nickel ferrite mesoporous (SNFM) is prepared via the solution combustion technique, an efficient and one-step synthesis. Dynamic X-ray analysis has clarified the structural properties of SNFM. The grain size of SNFM is determined to be ∼11.6 nm. The specific surface area (87.69 m2. g−1) of SNFM is obtained via the Brunauer–Emmett–Teller method. The Barrett–Joyner–Halenda pore size distributions revealed that the size of the mesopores in as-synthesized SNFM mainly falls in the size range of 2–16 nm. Scanning electron microscopy studies showed the regularities involved during porous-structure formation. SNFM is employed as the support for nano-structured palladium (PdNS). Field emission scanning electron microscope studies of PdNS-SNFM showed the deposition of PdNS in cavities and on/in the pores of SNFM. The electrochemical surface area obtained for PdNS-SNFM is about 27 times larger than that of PdNS via cyclic voltammetry. The electrochemical studies are utilized to study the features and catalytic performance of PdNS-SNFM in the electro-oxidation of diverse small organic fuels, whereas the electrooxidation of diethylene glycol is reported for first-time.
Introduction
One of the significant factors in fuel cells is modifying the catalytic utility of the supported noble metallic nanoparticles, which relies on the stabilization, distribution, dispersion, and control of particle-size, and on the support options to the necessity of goal reactions.1 A practical support has some characteristics, including large surface area, low cost, stability, and high electrical conductivity in the catalysis process.2 Hence, for the optimal usage of noble metals, it is essential to fabricate components that have more active sites on the outer surface of the nanocatalyst.3 Carbonic supports have a low resistance towards chemical or electrochemical oxidation. Regardless of good conduction (because of carbon structures), these electrocatalysts still are not appropriate because of low usage of noble metal, imperfect mass transportation, and the impermanent stability of carbon-based supports. Accordingly, ceramic materials with durability and high oxidation-resistance are necessary.4 Metallic oxides have been employed for the fine dispersion of nanocatalysts.5,6 Spinel oxides with the general formula AB2O4 are a meaningful type of oxides with A3+/A2+ and B3+/B2+ redox couples that would allow for good electrocatalytic performance.7,8 One of the principal magnetic oxides is nickel ferrite (NiFe2O4). The Fe and Ni elements of NiFe2O4 are numerous, environmentally benign, and relatively cheap.9 NiFe2O4, with its superparamagnetic nature and high thermal stability, is often used as a support for magnetic catalysts.10 For instance, Gao et al. prepared a Pd–NiFe2O4 catalyst, which significantly increased the catalytic efficiency.11,12 Numerous articles have reported various methods of fabricating NiFe2O4 such as spray pyrolysis,13 coprecipitation,14 mechanical activation,15 hydrothermal methods,16 and sol–gel synthesis.17 The solution combustion process is a useful and one-step synthesis for the fabrication of metal-oxide catalysts. Porous structure and high crystallization are obtained due to a lot of gases released during the reaction and the exothermicity of the combustion process; in addition, the precursors can improve the chemical structure of the product.18In the last decade, Pd has been considered in the oxidation processes19 because it exhibits an impressive electrocatalytic ability, and in contrast to Pt electrocatalysts, Pd catalysts has superb resistance to poisoning from the oxidation of alcohol.20 Moreover, the abundance of Pd is about 50 times more than Pt. For this reason, Pd is cheaper than Pt. Consequently, the fabrication of noble metal and mixed oxide on the nanoscale can be the essential factor affecting the efficiency of noble-metal electrocatalysts.21 Also, supported nano-particles have been explored in oxidizing conditions, and it has been demonstrated that an excessively thin surface oxide forms before the onset of bulk oxidation. This thin surface oxide is more active in oxidation than corresponding metallic surfaces.22
Here, the spinel nickel ferrite mesoporous (SNFM) was synthesized via a salt-assisted solution combustion method and characterized via a scanning electron microscopy, X-ray diffraction, and Brunauer–Emmett–Teller analysis. The nano-structured palladium (PdNS) in the presence of deacetylchitin as an adhesive agent on the electrode's surface23 was placed onto the pores of SNFM. Electrochemical studies were performed to investigate the features and catalytic performance of PdNS-SNFM during the oxidation of diverse small organic fuels (SOFs), including methanol (SOF1), ethanol (SOF2), ethylene glycol (SOF3), diethylene glycol (SOF4), formaldehyde (SOF5), and formic acid (SOF6); whereas diethylene glycol electrooxidation is reported for the first time.
Experimental
Chemicals
Nickel(II) nitrate (Ni(NO3)2, Carlo Erba), iron(III) nitrate nonahydrate (Fe(NO3)2·9H2O, May & Baker), deacetylchitin (low molecular weight Fluka), palladium chloride (PdCl2 Sigma-Aldrich), and other reagents (Merck) used were of analytical grade.Preparation of SNFM and PdNS-SNFM
SNFM was prepared via an efficient salt-assisted solution combustion method. The process was as follows: 3.333 g of glycine, 2.908 g of Ni (NO3)2, 8.080 g Fe (NO3)2·9H2O and, 1.49 g potassium chloride were mixed in a known amount of acidic water (pH = 4), respectively. The resulting mixture was continually kept on a heater to facilitate the combustion reaction. The as-prepared sediments were heated in deionized water to eliminate solvable by-products. The sediments were separated via an external magnetic field, washed with deionized water, and eventually dried at 85 °C for 5 h. To fabricate PdNS-SNFM, 3 ml deacetylchitin solution (0.018 g deacetylchitin + 100 ml 1% volumetric acetic acid aqueous solution) was added to 5 mg PdCl2 and 20 μl HCl 37%. In a second container, 3 mg SNFM was added to 0.5 ml deacetylchitin solution. After ultrasound, two as-prepared mixtures were added together and stirred on a rotary apparatus at 200 rpm for a full day. Then, 100 μl of NaBH4 solution (100 mg ml−1) was added and again stirred at the same speed for 4 h.Instrumentation and electrochemical tests
For the physical description of samples, X-ray diffraction (XRD) was applied using Bruker, Advanced D8. The infrared spectra of the as-synthesized samples were recorded in the range of 400–4000 cm−1 on a Fourier transform infrared (FTIR) spectrometer (Bruker, TENSOR II). Microstructure evaluations were carried out via the scanning electron microscopy (SEM, KYKY EM3900) technique. The field emission scanning electron microscopy (FESEM) analyses were done on an SAMX electron microscope (MIRA3 TESCAN). The adsorption/desorption isotherms of SNFM were obtained via the N2 adsorption technique using a BELSORP-mini II instrument. The specific surface areas and the pore size distribution were assessed via the Brunauer–Emmett–Teller (BET) technique and the Barrett–Joyner–Halenda (BJH) concept, respectively. Standard t-curve calculated from eqn (1) by converting the adsorption isotherm of the vertical axis to the t value.
 | (1) |
The electrochemical tests were performed by an SAMA 500 electroanalyzer (SAMA Center, Iran) in a cell containing: a Pt (as the auxiliary electrode) electrode, Hg/HgO (as the reference electrodes) electrode, and glassy carbon (GC, as the working electrode) electrode with 3.14 × 10−6 m2 surface area. The working electrode was prepared as follows: (1) mechanical polishing, (2) electrochemical activation in an acidic medium by an anodic and cathodic sweep, (3) spraying 10 μl of catalytic composite on the surface electrode, and (4) solvent evaporation of the composite and the creation of a catalytic layer on the surface electrode, which were denoted as GC/PdNS-SNFM and GC/PdNS electrodes.
Results and discussions
Evaluation of SNFM and PdNS-SNFM
The XRD pattern of the obtained powder is shown in Fig. 1A. The observed peaks are in good agreement with the pure cubic spinel NiFe2O4 phase (JCPDS No. 044-1485). The peaks appeared at 2θ degrees of 18.43, 30.23, 35.59, 37.14, 43.32, 53.80, 57.22, 62.89 and 74.68 can be well-indexed to the (111), (220), (311), (222), (400), (422), (511), (440) and (533) planes of NiFe2O4 phase, respectively. The grain size of NiFe2O4 obtained via the Debye–Scherrer's relation was ∼11.6 nm.24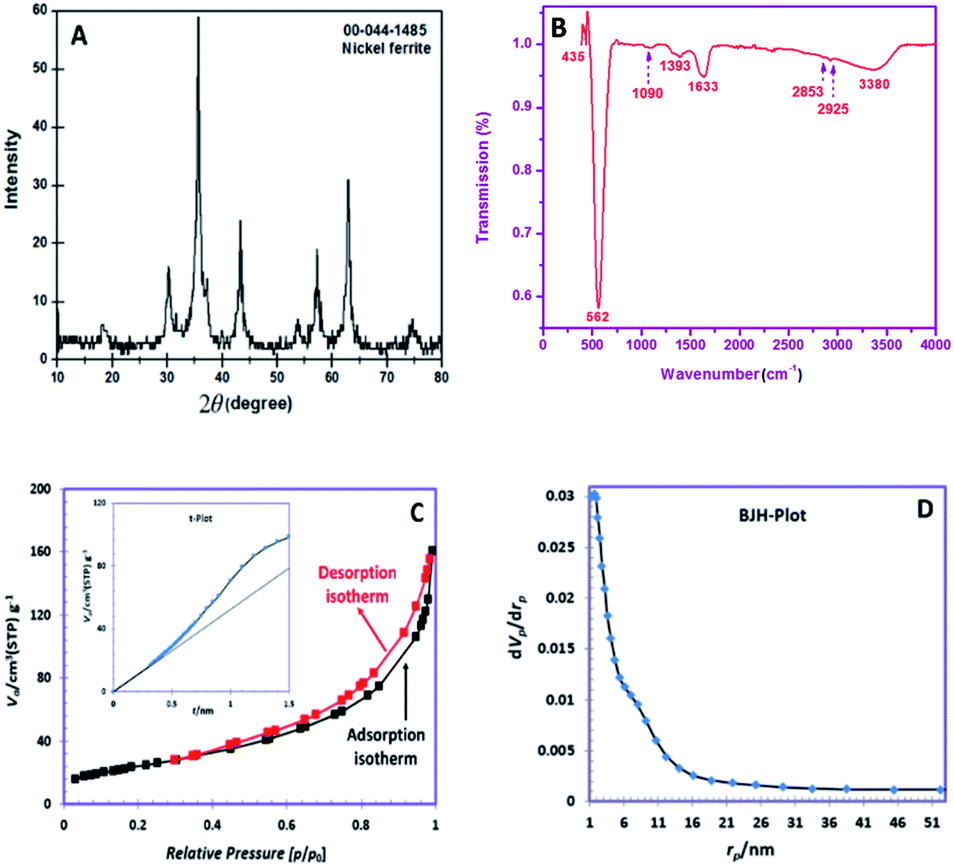 | ||
| Fig. 1 (A). XRD pattern, (B) FTIR spectrum, (C) nitrogen adsorption and desorption isotherms (inset t-plot), (D) BJH pore size distributions. | ||
Fig. 1B depicts the FTIR spectrum of SNFM powders. The broad band at around 3380 cm−1 is responsible for the presence of the O–H group.25 The weak bands at 2926 and 2853 cm−1 can be assigned to C–H asymmetric and C–H symmetric stretching frequencies, respectively.26 The band around 1633 cm−1 correspond to the vibration of the N–H bond.27 The band at 1393 cm−1 corresponds to NO3− ions.28 The observed weak band at 1090 cm−1 is due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching with ring stretching.29 The bands at 562 and 435 cm−1 indicate the cubic spinel structure formation of NiFe2O4. The band at 435 cm−1 proves metal–oxygen stretching in the octahedral position. The intense peak at 562 cm−1 is correlated to the metal-oxygen stretching tetrahedral band.30
O stretching with ring stretching.29 The bands at 562 and 435 cm−1 indicate the cubic spinel structure formation of NiFe2O4. The band at 435 cm−1 proves metal–oxygen stretching in the octahedral position. The intense peak at 562 cm−1 is correlated to the metal-oxygen stretching tetrahedral band.30
The N2 adsorption/desorption isotherms of the obtained SNFM powder is presented in Fig. 1C. The hysteresis profile is detected to form IV class with H3 hysteresis, which suggests the presence of mesopores.31 Also, the t-plot of SNFM powders is shown in the inset of Fig. 1C. The t-plot depicts a sharp straight line that begins from the origin point but becomes smoother after some point. Therefore, SNFM powders belong to mesoporous materials. Likewise, SNFM powder exhibits a specific surface area and total pore volume of 87.69 m2 g−1 and 0.2377 cm3 g−1, respectively. The BJH pore dimension distributions are also depicted in Fig. 1D, which shows that the size of mesopores in the as-synthesized SNFM mainly falls in the range of 2–16 nm. The presence of slit-shaped pores and panel-shaped particles generates the H3 hysteresis. Due to non-rigid particle aggregates, the isotherms revealing type H3 do not limit adsorption at high P/P0.31 Consequently, it seems that the brittle agglomerations of porous particles result in IV isotherms with H3 hysteresis.
In the SEM micrograph with a lower magnification (Fig. 2A), a porous and spongy structure of SNFM is observed. In another image with higher magnification (Fig. 2B), the effect of combustion is more apparent. The combustion created tiny cavities in SNFM powders. Also, it can be seen that agglomerates are formed by assembling nanoparticles. Therefore, Pd can be inserted on the surface and in the cavities of SNFM.
 | ||
| Fig. 2 The SEM images with (A) 10, and (B) 1 μm scales for SNFM; (C) FESEM image of PdNS-SNFM, with 100 μm scale, and (D) TEM micrograph of PdNS-SNFM with 50 μm scale. | ||
To consider the morphology of PdNS-SNFM, FESEM images are shown in Fig. 2C. It is clear that the holes were occupied with PdNS (brighter points) in the SNFM structure (darker places). Therefore, the formation of PdNS into holes and onto the surface of SNFM was demonstrated. Nevertheless, there are unfilled cavities in PdNS-SNFM.
Such cavities improve the fuel storage on the surface catalyst for electrooxidation. The good dispersion of PdNS on SNFM is marked.
Fig. 2D displays the TEM micrograph for PdNS-SNFM in 50 nm scale. It confirmed that the palladium particles formed in nano-size with diameter range of 10–20 nm. As in the FESEM image, the unfilled holes in the TEM image are visible.
Electrocatalytic performance
The electrochemical performance of PdNS and PdNS-SNFM was considered via cyclic voltammetry. Fig. 3A shows CVs for PdNS and PdNS-SNFM after 1 run (inset: 80 run) with a Pd loading of 0.31 mg cm−2. The voltammetric features of the modified electrodes reveal the typical behaviour of palladium. The observed peaks for both electrodes in the different regions are attributed to several electrochemical processes. The forward sweep directed an electrooxidation process on the electrode surface, resulting in the creation of Pd oxides (+0.3 to +0.7 V vs. Hg/HgO) and hydrogen desorption (−0.9 to −0.4 V vs. Hg/HgO), while the reverse sweep reflected the reduction of the Pd oxides (−0.2 to −0.5 V vs. Hg/HgO) and hydrogen adsorption (−0.6 to −0.9 V vs. Hg/HgO).32 In 2005, Ketteler et al. found that the Pd surface oxidation proceeds by the formation of stable and metastable structures,33 such as PdO and PdOH in positive voltages.34 The accumulation of oxygenated compounds affects the adsorption of hydrocarbons on the surface of the working electrode by changing the electrocatalyst surface potential. Respectively, the potential region of −0.2 to +0.4 V vs. Hg/HgO was related to the double layer on the working electrode.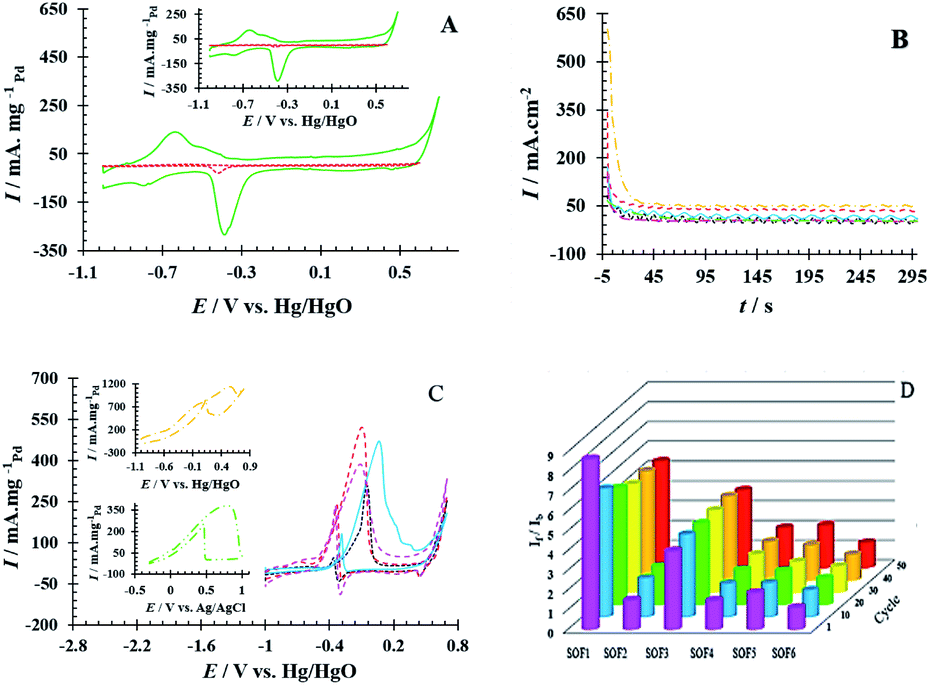 | ||
Fig. 3 (A) The CV curves for GCE/PdNS ( ) and GCE/PdNS-SNFM ( ) and GCE/PdNS-SNFM ( ) electrodes with the Pd loading of 0.31 mg cm−2 in 1 M NaOH (inset: after 80 cycles), (B) The CA curves at E = 0.1 V, (C) CV curves ( ) electrodes with the Pd loading of 0.31 mg cm−2 in 1 M NaOH (inset: after 80 cycles), (B) The CA curves at E = 0.1 V, (C) CV curves ( ) 0.80 M SOF1, ( ) 0.80 M SOF1, ( ) 0.55 M SOF2, ( ) 0.55 M SOF2, ( ) 0.55 M SOF3, ( ) 0.55 M SOF3, ( ) 0.34 M SOF4, ( ) 0.34 M SOF4, ( ) 0.43 M SOF5 and ( ) 0.43 M SOF5 and ( ) 0.85 M SOF6 electroxidation on the GCE/PdNS-SNFM electrode, and (D) bar chart of If/Ib stability. ) 0.85 M SOF6 electroxidation on the GCE/PdNS-SNFM electrode, and (D) bar chart of If/Ib stability. | ||
The electrochemical surface area (EASH/EASPd) is important to define electrocatalyst efficiency.35 EASH and EASPd are respectively estimated over the surface below the peak adsorption/desorption hydrogen (QH) and the surface below the curve of Pd–O reduction (QPd).36,37 The durability of the as-synthesized electrocatalysts was examined by applying potential cycling in an alkaline medium. Comparisons of EASH and EASPd at the beginning and end of the test were used to analyze the loss EAS rate. QH (1th cycle = 104.18 and 80th cycle = 103.28 mC cm−2) and QPd (1th cycle = 127.39 and 80th cycle = 122.36 mC cm−2) were obtained on GCE/PdNS-SNFM, while QH (1th cycle = 4.00 and 80th cycle = 2.66 mC cm−2) and QPd (1th cycle = 7.04 and 80th cycle = 4.01 mC cm−2) were on GCE/PdNS. This data exhibited that EASH and EASPd for PdNS-SNFM were considerably improved relative to PdNS. The dispersion of PdNS was dropped after durability for modified electrodes, representing decrease in EASH and EASPd due to sintering and dissolution of PdNS. The loss percentages were 33.55 and 9.75 for PdNS and 1.26 and 3.94 for PdNS-SNFM on EASH and EASPd, respectively. These data are presented in Table S1.† A comparison showed that using a mesoporous support enhanced the dispersion and electrochemical surface area of PdNS. PdNS-SNFM had superior distribution and durability compared to PdNS. SNFM with three-dimensional porous construction can be sufficient for inhibiting sintering, agglomeration and, dissolution of PdNS. Therefore, the stability of the electrocatalyst can be improved through SNFM. The size and dispersion of PdNS were better due to the creation of PdNS on and into the pores of SNFM as the support.
Also, the stability of PdNS-SNFM and PdNS were studied via chronoamperometry (CA). Fig. 3B shows the chronoamperograms of SOF1–SOF6 oxidation on the PdNS-SNFM catalysts at 0.1 V potential for 300 s. In the beginning, I vs. t curves meaningfully dropped (about 10 first seconds). Next, the current density was approximately stable owing to the falling number of active sites on the electrocatalyst to substitute the SOF molecules. Such enhanced stability is explained via the improved creation of OH groups on the SNFM surface.38 The reaction among such groups and middle species may increase the durability of PdNS-SNFM. As shown in Fig. 3B, SOF5 has a greater current density (637.01 mA cm2) compared to the other SOF.
The number of fuel molecules per electrocatalyst surface per second is equal to the turnover number (TON). This factor determines the current in steady-state for a given electrooxidation process. The number of fuel monolayers that are electrooxidized per unit time is known as the turnover frequency (TOF). These two factors can be estimated according to ref. 39. The electrochemical results from Fig. 3B were presented in Table S2.†
The electrooxidation reaction of SOFs on PdNS-SNFM was evaluated utilizing the CV technique. Fig. 3C shows the CV curves related to SOF1–SOF6 electrooxidation on GCE/PdNS-SNFM with 0.05 V s−1. The peak that appeared in the forward sweep (If) is ascribed to the oxidation of the adsorbed species including SOF. In contrast, the second peak in the backward sweep (Ib) is responsible for the oxidation of created intermediate components that were not completely oxidized in the forward scan.39 A likely pathway for the SOF oxidation reaction is expressed in Table 1.40–44
| SOF1 (ref. 40) | SOF2 (ref. 41) | SOF3 (ref. 42) | SOF5 (ref. 43) | SOF6 (ref. 44) |
|---|---|---|---|---|
| Pd + CH3OH → Pd−CH3OHads | Pd + CH3CH2OH → Pd−CH3CH2OHads | Pd + (CH2OH)2 → Pd-(CH2OH)2 ads | Pd + H2O → Pd–OHads + H+ + e− | The general reactions |
| HCOOH + Pd → HCOO−Pd + H+ + e− | ||||
| HCOOH + Pd → Pd−CO + H2O | ||||
| Pd−CH3OHads + OH− → Pd−CH3Oads + H2O + e− | Pd−CH3CH2OHads + 3OH− → 3H2O + Pd−CH3 ads + 3e− | Pd–(CH2OH)2 ads+ 4OH− → Pd–(HCO)2 ads+ 4H2O + 4e− | Pd + HCHOsol → Pd−HCHOads | Formic acid electrooxidation reaction in the direct pathway |
| HCOO−Pd → Pd−H + CO2 | ||||
| Pd−H → Pd + H+ + e− | ||||
| Pd−CH3Oads + OH− → H2O + Pd−CH2Oads + e− | Pd−CH3COads + Pd−OHads → Pd + Pd−CH3COOH | Pd−HCO2 ads + 4OH− → Pd−HCOO2 ads + 2H2O + 4e− | Pd−HCHOads → Pd–CHOads + H+ + e− | Formic acid electrooxidation reaction in the indirect pathway |
| Pd + H2O → Pd−OH + H+ + e− | ||||
| Pd−CO + Pd−OH → Pd + CO2 + H+ + e− | ||||
| Pd−CH2Oads + OH− → Pd−CHOads + H2O + e− | Pd−CH3COOH + OH− → Pd + CH3COO− + H2O | Pd–HCOOads + e− → Pd–COads + OH− | Pd–CHOads → Pd–COads + Pd–OHads + H+ + e− | Net reaction |
| Pd−CHOads + OH− → Pd−COads + 4H2O + e− | Pd–HCOads + e− → Pd–COads + OH− | Pd–COads + Pd–OHads → Pd + Pd–COOHads | HCOOH → CO2 + 2H+ + 2e− | |
| Pd−COads +2OH− → Pd + CO2 + H2O + 2e− | Pd−OH− → Pd + OHads + e− | Pd + Pd–COOHads → 2Pd + CO2 + H+ + e− | ||
| Pd–COads + Pd + OHads → 2Pd + CO2 + H+ + e− |
The stability of the If/Ib ratio during 50 cycles is shown in the bar chart in Fig. 3D. It is responsible for the earlier elimination of intermediates on the PdNS surface due to the higher surface area. It indicates that the synergistic properties of SNFM on PdNS performance enhance the electrocatalytic ability of PdNS-SNFM as a multi-functional electrocatalyst in SOF electrooxidation. Structural oxygens in SNFM are the active species to eliminate the intermediates of SOF electrooxidation. Support containing interface metal like NiFe2O4 is a promoter agent for the dehydrogenation as the initial step of oxidation of small organic fuels due to their multi-oxidative state via the orienting C–H bond; because it can be desirable to create the cycling redox among the high and low valences.
The Ni3+/Ni2+ and Fe3+/Fe2+ redox couples exhibit good electrocatalytic performance. As stated before, the thin surface oxide formed is more active in oxidation than corresponding metallic surfaces.22 Presumably, the surface oxygen of ferrite activated the oxidation process. On the other hand, the porous network of NiFe2O4 prevents PdNS aggregation. The synergistic influence can be because of the earlier removal of intermediates due to increased surface area and weakened agglomeration of PdNS, thus decreasing the resistance of mass transfer to store small organic fuels into the holes of NiFe2O4 ceramic support.
Adsorption isotherms on the PdNS-SNFM surface
Simultaneously, with the SOF adsorption process on the electrode surface, a competition happens among the adsorbed hydrogen on the modified electrode surface and SOF in the bulk solution. The adsorption is assessed via isotherms45,46 (See Fig. 4 and Table 2). The initial stage in SOF oxidation is the adsorption of SOF molecules as obtained by Temkin isotherm,47 which assumes that the interaction among the adsorbed species and that the as mentioned exists a competition among adsorbed species (SOFs and H2).46,48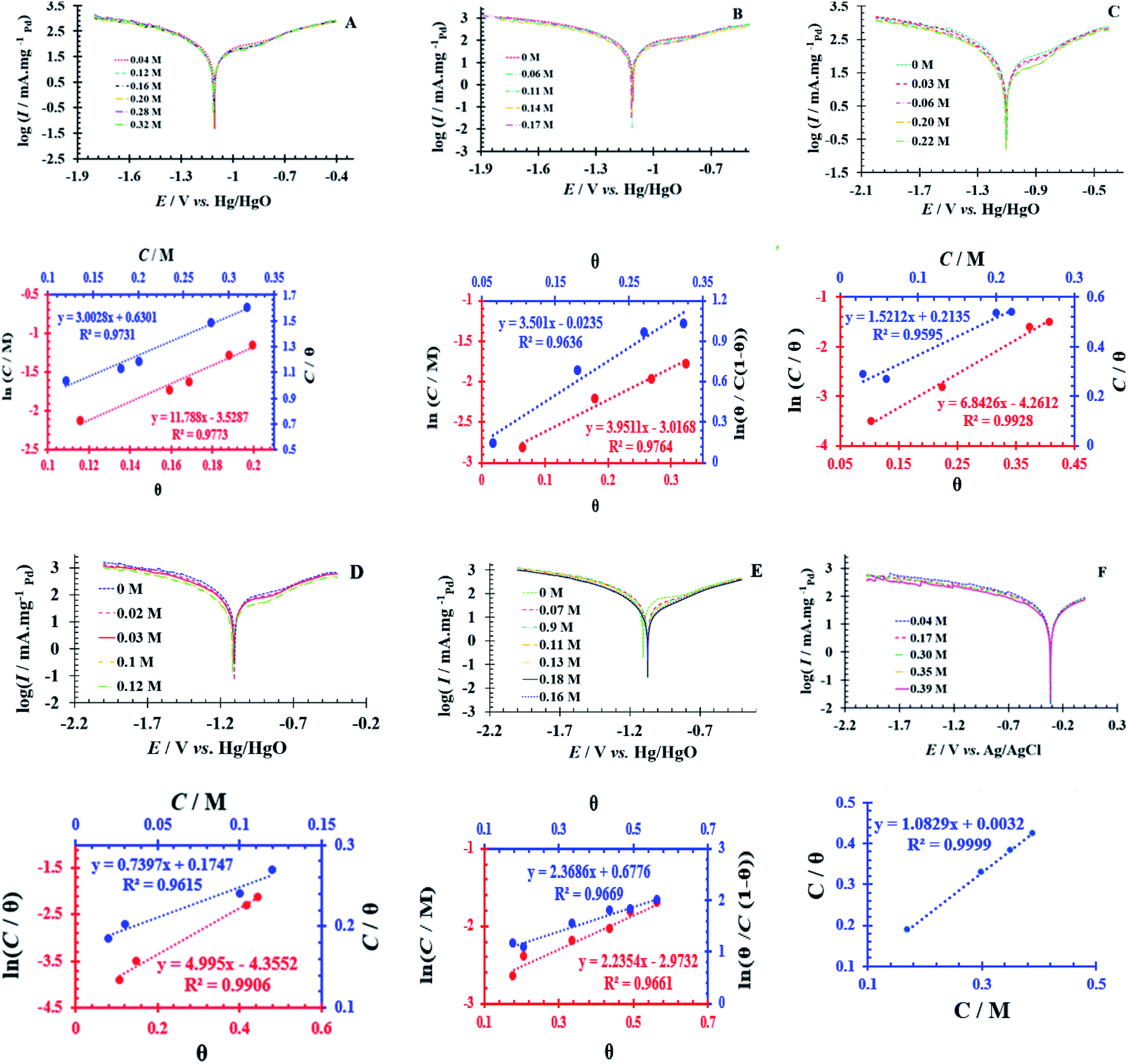 | ||
Fig. 4 Tafel curves at different concentrations of SOF1–SOF6 and the resulting charts: the linear outcomes fitted according Temkin (ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) C vs. θ), Langmuir (C/θ vs. C) and Frumkin (lnθ/C (1 − θ) vs. θ) adsorption isotherms on the surface of the PdNS-SNFM catalyst. C vs. θ), Langmuir (C/θ vs. C) and Frumkin (lnθ/C (1 − θ) vs. θ) adsorption isotherms on the surface of the PdNS-SNFM catalyst. | ||
| SOF1 | SOF2 | SOF3 | SOF4 | SOF5 | SOF6 | |
|---|---|---|---|---|---|---|
| Temkin: lnC = −lnK + aθ |
0.977 | 0.976 | 0.992 | 0.990 | 0.966 | 0.638 |
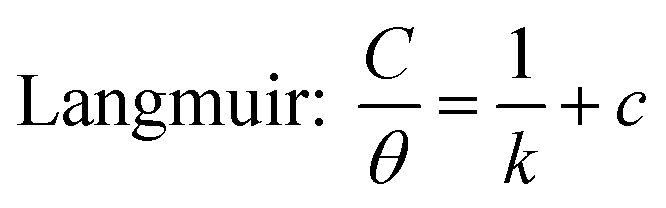 |
0.973 | 0.864 | 0.959 | 0.961 | 0.495 | 0.999 |
 |
0.758 | 0.963 | 0.466 | 0.810 | 0.966 | 0.549 |
C = SOF concentration; Kads = equilibrium constant; θ = surface coverage;  |
||||||
The accessible sites on PdNS were particular for H2 adsorption during enhancing SOF concentration. Consequently, the exchange current was decreased. The degree of surface coverage (θ) determines the outcome of species competition in the adsorption. Hence, the range of potential for linear polarization the gradient of Langmuir isotherm displays the formation of the SOF mono-layer on the electrode surface. The SOF was adsorbed on the active sites of PdNS-SNFM without any interaction among them.46 The slope amounts of Langmuir isotherm for SOF1–SOF6 were not equal to the unit. Hence, it is clear that there are the significant interactions among absorbed species.
Conclusion
SNFM was prepared by the solution combustion technique. SNFM exhibited a specific surface area of 87.69 m2. g−1. Therefore, it was employed for the formation of PdNS into cavities and onto the surface of SNFM. The value of EASH for PdNS-SNFM was about 27 times more than PdNS. The catalytic performance was investigated for SOF electrooxidation on the PdNS-SNFM catalyst by the electrochemical studies. In an overview, the PdNS-SNFM activity was decreased relative to the electro-oxidation of different fuels as follows: SOF5 > SOF2 > SOF3 > SOF4 > SOF6 > SOF1. It is concluded that the synergistic influence of SNFM on the catalytic activity of PdNS can be due to the improvement of the surface area, distribution, and stability of PdNS. In addition, it enlarged the ability of removing the compounds by blocking the active sites of the catalyst for the electro-oxidation of SOFs with the help of surface oxygens, and the creation of the cycling redox between the high and low chemical valents of the intermediate metals such as nickel and iron.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The current work was supported by the University of Sistan and Baluchestan.References
- Q. Wang, X. Cai, Y. Liu, J. Xie, Y. Zhou and J. Wang, Appl. Catal., B, 2016, 189, 242–251 CrossRef CAS.
- J. C. Ferreira, R. V. Cavallari, V. S. Bergamaschi, R. M. Antoniassi, A. A. Teixeira-Neto, M. Linardi and J. C. M. Silva, J. Renew. Sustain. Energy, 2018, 23, 7–23 Search PubMed.
- L. X. Ding, A. L. Wang, G. R. Li, Z. Q. Liu, W. X. Zhao, C. Y. Su and Y. X. Tong, J. Am. Chem. Soc., 2012, 134, 5730–5733 CrossRef CAS PubMed.
- H. Lv and Sh. Mu, Nanoscale, 2014, 6, 4945–5556 RSC.
- K. Jukk, N. Kongi, A. Tarre, A. Rosental, A. B. Treshchalov, J. Kozlova, P. Ritslaid, L. Matisen, V. Sammelselg and K. Tammeveski, J. Electroanal. Chem., 2014, 735, 68–76 CrossRef CAS.
- R. X. Wang, Y. J. Fan, L. Wang, L. N. Wu, S. N. Sun and S. G. Sun, J. Power Sources, 2015, 287, 341–348 CrossRef CAS.
- F. Cheng, J. Shen, B. Peng, Y. Pan, Z. Tao and J. Chen, Nat. Chem., 2011, 3, 79–84 CrossRef CAS PubMed.
- L. Luo, Q. Li, Y. Xu, Y. Ding, X. Wang, D. Deng and Y. Xu, Sens. Actuators, B, 2010, 145, 293–298 CrossRef CAS.
- K. M. Naik and S. Sampath, Electrochim. Acta, 2018, 292, 268–275 CrossRef CAS.
- Zh. Liang, Q. Li, F. Li, Sh. Zhao and X. Xia, Int. J. Hydrogen Energy, 2017, 42, 3971–3980 CrossRef CAS.
- Z. Gao, Y. Feng, F. Cui, Z. Hua, J. Zhou, Y. Zhu and J. Shi, J. Mol. Catal. A: Chem., 2011, 336, 51–57 CrossRef CAS.
- A. Ren, Ch. Liu, Y. Hong, W. Shi, Sh. Lin and P. Li, Chem. Eng. J., 2014, 258, 301–308 CrossRef CAS.
- D. S. Jung and Y. C. Kang, J. Magn. Magn. Mater., 2009, 321, 619–623 CrossRef CAS.
- P. Sivakumar, R. Ramesh, A. Ramanand, S. Ponnusamy and C. Muthamizhchelvan, J. Mater. Sci. Lett., 2011, 65, 483–485 CrossRef CAS.
- Z. H. Zhou, J. M. Xue, J. Wang, H. S. O. Chan, T. Yu and Z. X. Shen, J. Appl. Phys., 2002, 91, 6015–6020 CrossRef CAS.
- A. Indra, P. W. Menezes, N. Ranjbar Sahraie, A. Bergmann, C. Das, M. Tallarida, D. Schmeißer, P. Strasser and M. Driess, J. Am. Chem. Soc., 2014, 136, 17530–17536 CrossRef CAS PubMed.
- P. Lavela and J. L. Tirado, J. Power Sources, 2007, 172, 379–387 CrossRef CAS.
- M. Piumetti, S. Bensaid, T. Andana, N. Russo, R. Pirone and D. Fino, Appl. Catal., B, 2017, 205, 455–468 CrossRef CAS.
- R. Kumar, J. H. Oh, H. J. Kim, J. H. Jung, C. H. Jung, W. G. Hong, H. J. Kim, J. Y. Park and I. K. Oh, ACS Nano, 2015, 9, 7343–7351 CrossRef CAS PubMed.
- A. L. Wang, H. Xu, J. X. Feng, L. X. Ding, Y. X. Tong and G. R. Li, J. Am. Chem. Soc., 2013, 135, 10703–10709 CrossRef CAS PubMed.
- M. H. Dehghan, Z. Yavari and M. Noroozifar, ACS Omega, 2018, 3, 16795–16804 CrossRef CAS PubMed.
- K. Qadir, S. H. Joo, B. S. Mun, D. R. Butcher, J. R. Renzas, F. Aksoy, Zh. Liu, G. A. Somorjai and J. Y. Park, Nano Lett., 2012, 12, 5761–5768 CrossRef CAS PubMed.
- Z. Yavari, M. Noroozifar and M. Khorasani-Motlagh, J. Appl. Electrochem., 2015, 45, 439–451 CrossRef CAS.
- S. A. Hosseini, V. Majidi and A. R. Abbasian, J. Sulfur Chem., 2018, 39, 119–129 CrossRef CAS.
- S. A. Hosseini, M. Davodian and A. R. Abbasian, J. Taiwan Inst. Chem. Eng., 2017, 75, 97–104 CrossRef CAS.
- H. Khosravi and R. Eslami-Farsani, Polym. Test., 2016, 55, 135–142 CrossRef CAS.
- A. Abdi, R. Eslami-Farsani and H. Khosravi, Fibers Polym., 2018, 19, 635–640 CrossRef CAS.
- Q. Zhou, Y. Mou, X. Ma, L. Xue and Y. Yan, J. Eur. Ceram. Soc., 2014, 34, 801–807 CrossRef CAS.
- V. Parameswaran, N. Nallamuthu, P. Devendran, A. Manikandan and E. R. Nagarajan, J. Nanosci. Nanotechnol., 2018, 18, 3944–3953 CrossRef CAS PubMed.
- S. A. Hosseini, A. R. Abbasian, O. Gholipoor, S. Ranjan and N. Dasgupta, Int. J. Environ. Sci. Technol., 2018, 16, 7455–7466 CrossRef.
- Z. A. ALOthman, Mater, 2012, 5, 2874–2902 CrossRef CAS.
- F. Alcaide, G. Álvarez, P. L. Cabot, H. J. Grande, O. Miguel and A. Querejeta, Int. J. Hydrogen Energy, 2011, 36, 4432–4439 CrossRef CAS.
- G. Ketteler, D. F. Ogletree, H. Bluhm, H. Liu, E. L. D. Hebenstreit and M. Salmeron, J. Am. Chem. Soc., 2005, 127, 18269–18273 CrossRef CAS PubMed.
- B. Habibi, Int. J. Hydrogen Energy, 2013, 38, 5464–5472 CrossRef CAS.
- G. F. Alvarez, M. Mamlouk, S. M. Senthil Kumar and K. Scott, J. Appl. Electrochem., 2011, 41, 925–937 CrossRef CAS.
- R. C. Venkateswara, S. K. Singh and B. Viswanathan, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem2E, 2008, 47, 1619–1625 Search PubMed.
- A. L. Wang, X. J. He, X. F Lu, H. Xu, Y. X. Tong and G. R. Li, Angew. Chem., Int. Ed., 2015, 54, 3669–3673 CrossRef CAS PubMed.
- N. Alfi, A. R. Rezvani, M. Khorasani-Motlagh and M. Noroozifar, New J. Chem., 2017, 41, 10652–10658 RSC.
- F. Kaedi, Z. Yavari, M. Noroozifar and H. Saravani, J. Electroanal. Chem., 2018, 827, 204–212 CrossRef CAS.
- E. D. Wang, J. B. Xu and T. S. Zhao, J. Phys. Chem. C, 2010, 114, 10489–10497 CrossRef CAS.
- L. P. A. Guerrero-Ortega, E. Ramírez-Meneses, R. Cabrera-Sierra, L. M. Palacios-Romero, K. Philippot, C. R. Santiago-Ramírez, L. Lartundo-Rojas and A. Manzo-Robledo, J. Mater. Sci., 2019, 54, 13694–13714 CrossRef CAS.
- Z. X. Liang, T. S. Zhao, J. B. Xu and L. D. Zhu, Electrochim. Acta, 2009, 54, 2203–2208 CrossRef CAS.
- H. Gao, Ch. Zhai, Ch. Yuan, Zh. Q. Liu and M. Zhu, Electrochim. Acta, 2020, 330, 135214 CrossRef CAS.
- A. Ejaz, M. Sh. Ahmed and S. Jeon, J. Electrochem. Soc., 2016, 163, B163–B168 CrossRef CAS.
- S. Yang, J. Yang, Y. Chung and Y. Kwon, Int. J. Hydrogen Energy, 2017, 42, 17211–17220 CrossRef CAS.
- M. Dehdab, Z. Yavari, M. Darijani and A. Bargahi, Desalination, 2016, 400, 7–17 CrossRef CAS.
- O. A. Khazova, A. A. Mikhailova, A. M. Skundin, E. K. Tuseeva, A. Havránek and K. Wippermann, Fuel Cells, 2002, 2, 99–108 CrossRef CAS.
- V. S. Bagotzicy, Y. B. Vassiliev, O. A. Khazova and S. S. Sedova, Electrochim. Acta, 1971, 16, 913–938 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10944d |
| This journal is © The Royal Society of Chemistry 2021 |