
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and biological evaluation of the new ring system benzo[f]pyrimido[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepine and its cycloalkane and cycloalkene condensed analogues†
Mohamed El Haimer a,
Márta Palkó*a,
Matti Haukkab,
Márió Gajdácsc,
István Zupkóc and
Ferenc Fülöpad
a,
Márta Palkó*a,
Matti Haukkab,
Márió Gajdácsc,
István Zupkóc and
Ferenc Fülöpad
aInstitute of Pharmaceutical Chemistry, University of Szeged, Interdisciplinary Excellence Centre, Eötvös utca 6, Szeged H-6720, Hungary. E-mail: palko.marta@szte.hu
bDepartment of Chemistry, University of Jyväskylä, FIN-40014 Turku, Finland
cPharmacodynamics and Biopharmacy, University of Szeged, Interdisciplinary Excellence Centre, Eötvös utca 6, Szeged H-6720, Hungary
dMTA-SZTE Stereochemistry Research Group, Hungarian Academy of Sciences, Eötvös utca 6, Szeged H-6720, Hungary
First published on 10th February 2021
Abstract
Derivatives of the new ring system benzo[f]pyrimido[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepinone and its cycloalkane and cycloalkene condensed analogues have been conveniently synthesized through a three-step reaction sequence. An atom-economical, one-pot, three-step cascade process engaging five reactive centers (amide, amine, carbonyl, azide, and alkyne) has been performed for the synthesis of alicyclic derivatives of quinazolinotriazolobenzodiazepine using cyclohexane, cyclohexene, and norbornene β-amino amides. The stereochemistry and relative configurations of the synthesized compounds were determined by 1D and 2D NMR spectroscopy and X-ray crystallography. The reaction was also performed using enantiomeric starting materials leading to enantiomeric quinazolinotriazolobenzodiazepine with an ee of 95%. The synthesis of 9H-benzo[f]pyrimido[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepinone, a new heterocyclic system, was achieved in a good yield using a retro Diels–Alder (RDA) procedure. Some compounds were tested for antiproliferative activities against five human cancer cell lines of gynecological.
Introduction
The development and synthesis of architecturally diverse and complex molecules in an efficient, environmentally benign, and atom-economic fashion have become extremely important in the last several decades.1 As a solution to this challenging problem, a multistep, one-pot procedure has been developed. Because this type of procedure is based on several transformations with bond forming taking place in a domino reaction manner – which is also referred to as “pot economy”,2 – it minimizes time consumption and chemical waste generation.Therefore, the development of a reaction sequence, that assembles several components or transformations engaging several reactive centers, is ideal for preparing complex structures. In general, the number of possible diastereomers increases along with the number of components. Although several successful examples have been reported,3 because of the difficulty of performing “one-pot” domino reactions with high diastereoselectivity, the task is still challenging.4
Because of their high hit rates and pharmacological profiles, privileged scaffold derivatives are especially suited for the preparation of molecular libraries as leads in medicinal chemistry.5 Indeed, only a few frameworks, described as privileged scaffolds, are the main substructure for nearly 25% of all known drugs.6 Being considered as privileged heterocycles, quinazolinones, benzodiazepines, and triazoles are found in many bioactive compounds.7–9
Furthermore, various biologically active compounds containing a triazole-fused 1,4-benzodiazepine unit, such as protease inhibitors,10 alprazolam,11 estazolam,12 and triazolam,13 continuously attract the attention of organic and medicinal chemists due to their medicinal potential.14–16
Following our studies on diastereoselective, two-component multi-functional domino ring-closure reactions and our project for the synthesis of novel N-heterocycles with the focus on the biological potential of fused quinazolinones and triazoles,17–22 herein we report our study with respect to the diastereoselectivity of a one-pot, two-step cascade synthesis procedure of quinazolinotriazolobenzodiazepines described by Guggenheim et al.23 and the synthesis of a new series of alicyclic derivatives.
Our aim was (i) the synthesis of alicyclic derivatives of quinazolinotriazolobenzodiazepine in an atom-economical, one-pot, three-step cascade process engaging five reactive centers, (ii) to examine the diastereoselectivity of the domino ring-closure reaction of N-propargyl-substituted amino acids with substituted and unsubstituted 2-azidobenzaldehydes, (iii) elaboration of a retro Diels–Alder reaction (RDA) on norbornene derivatives to prepare a new heterocyclic ring system and, finally, (iv) to determine the antiproliferative properties of the synthesized derivatives against a panel of human adherent cancer cell lines by means of MTT assay.
Results and discussion
Alicyclic N-Boc-protected propargyl amides (±)-3, (±)-4, (±)-9, and (±)-10 were prepared according to a previous procedure, starting from the corresponding Boc-protected amino acids (±)-1, (±)-2, (±)-7, and (±)-8, using a mixture of N,N′-diisopropylcarbodiimide (DIC) and hydroxybenzotriazole (HOBt) in tetrahydrofuran.22,24 The other starting materials, 2-azidobenzaldehyde derivatives, were obtained by the addition of sodium azide to 2-nitrobenzaldehydes in hexamethylphosphoramide (HMPA) following the procedure already described in the literature.25Afterwards, the free amide bases, prepared by an acidic deprotection of amides (±)-3, (±)-4, (±)-9, and (±)-10 were reacted further without purification. Namely, a one-pot, two-step cascade process was carried out by reacting the corresponding 2-azidobenzaldehyde derivatives with the alicyclic propargyl amides under reflux in EtOH in the presence of iodine or p-toluenesulfonic acid as catalysts for 2 h. The main products (±)-5a–c, (±)-6a–c, (±)-11a–c, and (±)-12a–c were obtained after crystallization from Et2O followed by recrystallization from diisopropyl ether–EtOH (Schemes 2 and 3).
In the cascade process of alicylic 2-aminocarboxamides with 2-azidobenzaldehydes involving five reactive centers, first a Schiff base is produced, which undergoes a ring-closure reaction to give quinazoline epimers through a ring–chain tautomerism. The next step is an intramolecular azide–alkyne 1,3-dipolar cycloaddition delivering the penta- and hexacyclic ring systems (Scheme 1).23
 | ||
| Scheme 1 The domino reaction pathway. | ||
 | ||
| Scheme 2 Synthesis of octahydrobenzo[5,6][1,2,3]triazolo-[5′,1′:3,4][1,4]diazepino[7,1-b]quinazolin-11(9H)-one (±)-5a–c and (±)-6a–c. Reagents and conditions: (i) DIC, HOBt, propargylamine, THF, r.t., 24 h, 84% (ii) 1. HCl/H2O 10%, r.t., 4 h 2. NaOH, H2O/CHCl3, 3. I2 or p-TSA, 2-azidobenzaldehyde derivative, EtOH, reflux, 2 h, 69–76%. | ||
 | ||
| Scheme 3 Synthesis of hexahydrobenzo[5,6][1,2,3]triazolo-[5′,1′:3,4][1,4]diazepino[7,1-b]quinazolin-11(9H)-one (±)-11a–c and (±)-12a–c. Reagents and conditions: (i) DIC, HOBt, propargylamine, THF, r.t., 24 h, 72–74% (ii) 1. HCl/H2O 10%, r.t., 4 h 2. NaOH, H2O/CHCl3, 3. I2 or p-TSA, 2-azidobenzaldehyde derivative, EtOH, reflux, 2 h, 66–77%. | ||
This designed cascade process is atom economic as described by Trost,26,27 as the process maximizes the incorporation of both reagents into the final compound without any by-product, with a given consideration to the use of stoichiometric quantities for both reagents.28 Moreover, it is also employing environmentally benign iodine as a catalyst. Furthermore, the process is step economic, minimizing the number of reactions steps to the bare minimum, as described by Wender,29,30 with the possibility to form two epimers of (±)-5a–c, (±)-6a–c, (±)-11a–c, and (±)-12a–c.
In all cases, the 1H NMR spectra revealed the formation of the single epimers of quinazolino[1,2,3]triazolo-[1,4]benzodiazepines (±)-5a–c, (±)-6a–c, (±)-11a–c, and (±)-12a–c. The total NMR signal assignment was carried out for these compounds. Characteristic NOE crosspeaks were found between the protons C11aH, C15aH, and C16aH for cyclohexane cis-condensed (±)-5a–c and cyclohexene cis-condensed (±)-11a–c derivatives.
This allowed the deduction of the relative configuration of the new asymmetric center, which is in cis arrangement with the C11aH and C15aH hydrogens. For cyclohexane trans-condensed (±)-6a–c and cyclohexene trans-condensed (±)-12a–c, the relative configuration of the new asymmetric center C16aH is in cis arrangement with the C15aH proton as shown by their NOE crosspeaks. Furthermore, the stereochemistry of (±)-5a and (±)-6a was also confirmed by X-ray diffraction analysis (Fig. 1).
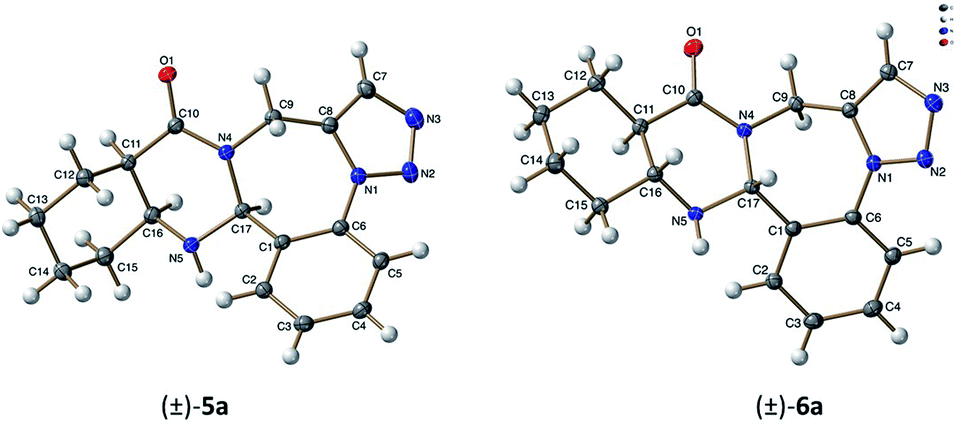 | ||
| Fig. 1 ORTEP plot of the X-ray structure of (±)-5a, (±)-6a. | ||
Our study was extended to racemic norbornene derivatives (Scheme 4), and for further examination, the reaction was performed with enantiomerically pure norbornene starting materials to obtain enantiomerically pure products, followed by the investigation of a potential RDA reaction. The racemic N-Boc-protected amino acids were prepared from the corresponding amino acids according to an earlier method.31 Following the same procedure, the synthetic route was fully diastereoselective giving the single epimers (±)-17a–c and (±)-18a–c.
 | ||
| Scheme 4 Synthesis of hexahydro-methanobenzo[5,6][1,2,3]triazolo-[5′,1′:3,4][1,4]diazepino[7,1-b]quinazolin-11(9H)-one (±)-17a–c and (±)-18a–c. Reagents and conditions: (i) DIC, HOBt, propargylamine, THF, r.t., 24 h, 72–75% (ii) 1. HCl/H2O 10%, r.t., 4 h 2. NaOH, H2O/CHCl3, 3. I2 or p-TSA, 2-azidobenzaldehyde derivative, EtOH, reflux, 2 h, 62–70%. | ||
Full NMR signal assignment followed by stereochemistry investigation by NOE crosspeaks revealed the relative configuration of the C16aH protons to be in a cis arrangement with the annelated hydrogen atoms C11aH and C15aH for diendo (±)-17a–c and diexo (±)-18a–c derivatives. The stereochemistry was further confirmed by X-ray diffraction analysis of the single crystal of (±)-17c (Fig. 2).
 | ||
| Fig. 2 ORTEP plot of the X-ray structure of (±)-17c. | ||
In further studies, enantiomerically pure diendo N-Boc-protected amino acids, prepared following the procedure described in a previous work, were used as chiral sources.31 The reaction was performed with both (−)-13 and (+)-13. Note, however, that only a single enantiomer is shown in Scheme 5 to represent the process. The reaction was carried out under conditions applied previously, starting from enantiomeric N-Boc-protected amino-acid (−)-13 (ee > 90%). Product (+)-17a was obtained with a relatively good enantiomeric excess (ee > 84%). On the other hand, (−)-17a was isolated with higher enantiomeric excess (ee > 95%) starting from (+)-13 (ee > 95%).
 | ||
| Scheme 5 Transformation of enantiomeric diendo N-Boc protected amino acid (−)-13. Reagents and conditions: (i) DIC, HOBt, propargylamine, THF, r.t., 24 h, 75% (ii) 1. HCl/H2O 10%, r.t., 4 h 2. NaOH, H2O/CHCl3, 3. I2 or p-TSA, 2-azidobenzaldehyde derivative, EtOH, reflux, 2 h, 70%. | ||
The next goal of our present work was the application of the RDA reaction of these specific structures to obtain the new ring system benzo[f]pyrimido[1,2-d][1,2,3]triazolo[1,5-a][1,4]diazepinone.
The investigation of the RDA reaction conditions was performed on both diendo (±)-17a and diexo (±)-18a derivatives. Unfortunately, the diexo (±)-18a product led only to degradation or no reaction under all tested conditions. Consequently, only the transformation of diendo (±)-17a derivative is shown in Scheme 6 and discussed below.
 | ||
| Scheme 6 RDA reaction of diendo (±)-17a. Reagents and conditions: see in Table 1. | ||
Table 1 lists the results of our efforts to find appropriate reaction conditions for the transformation of (±)-17a including both classic and modern organic synthesis techniques for the synthesis of compound 19. The flow reactor showed temperature limitations not reaching high enough temperatures for our procedure. Likewise, the use of classic batch reactor gave unsatisfactory results even at high temperature.
| Methods | Conditions | Compound (yield) |
|---|---|---|
| Batch reactor | Toluene, reflux, 30 min | No reaction |
| Batch reactor | 1,2-Dichlorobenzene, reflux, 30 min | 19 (traces) |
| Batch reactor | Solvent free, 220 °C, 30 min | 19 (traces) |
| Flow reactor | Toluene, 180 °C, 30 min | No reaction |
| Flow reactor | Toluene/Methanol (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), 180 °C, 30 min :1), 180 °C, 30 min |
No reaction |
| Microwave reactor | Toluene, 180 °C, 30 min | No reaction |
| Microwave reactor | 1,2-Dichlorobenzene, 220 °C, 30 min | 19 (66%) |
The only successful attempt was the use microwave irradiation in a microwave vial in 1,2-dichlorobenzene as solvent at 220 °C for 30 min, generating pyrimidotriazolobenzodiazepine 19, a novel N-heterocyclic ring system. The NMR signal assignment of the obtained product shows the loss of both the cyclopentadiene moiety and the hydrogen of the asymmetric center. These results can be explained by the instability of quinazolinotriazolo-benzodiazepine (±)-17a under high temperature leading to an oxidation of the starting compound of the domino process before the RDA reaction can take place.
This statement can also be confirmed by the result of a literature study, where a similar oxidation side product was isolated even at a temperature much lower than that used in our RDA reaction.23
In vitro antiproliferative activity
The effects of the newly synthesized and structurally related compounds on the tested cancer cell lines are presented in Table 2. The synthesis of fused N-heterocyclic quinazoline derivatives 20 was described by Guggenheim et al.,23 21 was reported by Madhubabu et al.,32 while the synthesis of derivates (±)-22, (±)-23, (±)-24, and (±)-25 containing a 1,2,3-triazole ring was published in our previous work (Fig. 3).22| Compound | Concentration (μM) | Growth inhibition (%) ± SEM | ||||
|---|---|---|---|---|---|---|
| A2780 | HeLa | MCF-7 | SiHa | MDA-MB-231 | ||
| a Cell growth inhibition values lower than 10% were considered negligible and were not given numerically. | ||||||
| (±)-5a | 10 | <10a | <10 | <10 | <10 | <10 |
| 30 | <10 | <10 | <10 | <10 | <10 | |
| (±)-6a | 10 | <10 | <10 | <10 | <10 | <10 |
| 30 | <10 | <10 | <10 | <10 | <10 | |
| (±)-11a | 10 | <10 | <10 | <10 | <10 | <10 |
| 30 | <10 | <10 | <10 | 11.17 ± 1.84 | <10 | |
| (±)-12a | 10 | <10 | <10 | <10 | <10 | <10 |
| 30 | <10 | <10 | <10 | <10 | <10 | |
| (±)-17a | 10 | <10 | <10 | <10 | <10 | 11.08 ± 1.06 |
| 30 | <10 | 15.04 ± 2.16 | 36.74 ± 3.51 | 17.81 ± 1.19 | 14.06 ± 0.92 | |
| (±)-18a | 10 | <10 | <10 | <10 | 16.32 ± 1.85 | <10 |
| 30 | <10 | <10 | <10 | 18.93 ± 2.29 | <10 | |
| 20 | 10 | <10 | <10 | <10 | 15.56 ± 1.82 | <10 |
| 30 | <10 | 25.52 ± 1.32 | 55.34 ± 2.15 | 19.07 ± 1.34 | <10 | |
| 21 | 10 | <10 | 17.59 ± 0.88 | <10 | <10 | <10 |
| 30 | <10 | 70.86 ± 2.29 | 67.39 ± 2.37 | 26.03 ± 0.98 | <10 | |
| (±)-22 | 10 | <10 | <10 | <10 | <10 | <10 |
| 30 | <10 | <10 | <10 | <10 | <10 | |
| (±)-23 | 10 | <10 | <10 | <10 | <10 | <10 |
| 30 | <10 | <10 | <10 | <10 | <10 | |
| (±)-24 | 10 | <10 | <10 | <10 | <10 | <10 |
| 30 | <10 | <10 | 19.38 ± 1.17 | <10 | 11.45 ± 0.88 | |
| (±)-25 | 10 | <10 | <10 | <10 | <10 | <10 |
| 30 | <10 | 12.07 ± 1.12 | 39.19 ± 1.98 | 12.44 ± 2.10 | <10 | |
| Cisplatin | 10 | 83.57 ± 1.21 | 42.61 ± 2.53 | 53.02 ± 1.78 | 88.64 ± 0.61 | 20.84 ± 0.81 |
| 30 | 95.02 ± 0.36 | 99.94 ± 0.26 | 89.92 ± 2.13 | 90.15 ± 1.58 | 74.47 ± 1.20 | |
 | ||
| Fig. 3 Structure of literature compounds22,23,27 20–(±)-25 subjected to biological studies. | ||
Most of the compounds elicited negligible growth inhibiting action against the utilized cancer cells. None of the substances elicited >10% inhibition on ovarian (A2780) cancer cells. MCF-7 (breast) and HeLa (cervical) cells are more sensitive than SiHa (cervical) or MDA-MB-231 (breast) cell lines. Concerning the diazepine analogues, the condensed aromatic ring may confer some limited activity and the configuration of the annelation seems irrelevant.
The importance of the aromatic ring has been confirmed in the case of quinazoline derivatives with 21 being the most effective compound in the current set.
Conclusions
In summary, the preparation of alicyclic derivatives of quinazolinotriazolobenzodiazepine was achieved, using iodine or p-TSA as catalyst under green conditions in good yields. The diastereoselectivity of the three-step cascade process engaging five reactive centers (amide, amine, carbonyl, azide, and alkyne) was proved by NMR and X-ray methods. Moreover, this was shown to be consistent throughout the studied scope. The investigation of the RDA process led to a novel heterocyclic ring system, pyrimidotriazolobenzodiazepine, in a relatively high yield using microwave irradiation. The simplicity of this process with the use of accessible starting materials and the wide scope are the major features to make the current protocol valuable. Most of the presented analogues have no substantial antiproliferative activity, while some compounds exerted a modest inhibition against some cancer cell lines at higher concentration.Conflicts of interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data, in the writing of the manuscript, or in the decision to publish the results.Acknowledgements
We are grateful to the Hungarian Research Foundation (OTKA No. K 115731). The financial support of the GINOP-2.3.2-15-2016-00038 project is acknowledged. The Ministry of Human Capacities, Hungary grant, TKP-2020 is acknowledged. M. G. was supported by the János Bolyai Research Scholarship (BO/00144/20/5) of the Hungarian Academy of Sciences, the New National Excellence Programme (ÚNKP-20-5-SZTE-330) of the Ministry of Human Resources, and ESCMID's “30 under 30” Award.Notes and references
- (a) A. Domling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef CAS; (b) A. DiguezVzquez, C. C. Tzschucke, W. Y. Lam and S. V. Le, Angew. Chem., Int. Ed., 2008, 47, 209–212 CrossRef; (c) F. L. Muller, T. Constantieux and J. Rodriguez, J. Am. Chem. Soc., 2005, 127, 17176–17177 CrossRef.
- Y. Hayashi, Chem. Sci., 2016, 7, 866–880 RSC.
- (a) A. Endo, A. Yanagisawa, M. Abe, S. Tohma, T. Kan and T. Fukuyama, J. Am. Chem. Soc., 2002, 124, 6552–6554 CrossRef CAS; (b) G. Sklute, D. Amsallem, A. Shabli, J. P. Varghese and I. Marek, J. Am. Chem. Soc., 2003, 125, 11776–11777 CrossRef CAS; (c) A. B. Smith III, D.-S. Kim and M. Xian, Org. Lett., 2007, 9, 3307–3309 CrossRef.
- (a) D. B. Ramachary and C. F. III Barbas, Chem.–Eur. J., 2004, 10, 5323–5331 CrossRef CAS; (b) N. Selander, A. Kipke, S. Sebelius and K. J. Szabo, J. Am. Chem. Soc., 2007, 129, 13723–13731 CrossRef CAS; (c) X. Xu, J. Zhou, L. Yang and W. Hu, Chem. Commun., 2008, 48, 6564–6566 RSC; (d) F. L. Zhang, A. W. Xu, Y. F. Gong, M. H. Wei and X. L. Yang, Chem.–Eur. J., 2009, 15, 6815–6818 CrossRef CAS; (e) C. G. Evans and J. E. Gestwicki, Org. Lett., 2009, 11, 2957–2959 CrossRef CAS.
- (a) D. A. Horton, G. T. Bourne and M. L. Smythe, Chem. Rev., 2003, 103, 893–930 CrossRef CAS; (b) R. W. DeSimone, K. S. Currie, S. A. Mitchell, J. W. Darrow and D. A. Pippin, Comb. Chem. High Throughput Screening, 2004, 7, 473–493 CrossRef CAS; (c) L. Costantino and D. Barlocco, Curr. Med. Chem., 2006, 13, 65–85 CrossRef CAS; (d) C. D. Duarte, E. J. Barreiro and C. A. M. Fraga, Mini-Rev. Med. Chem., 2007, 7, 1108–1119 CrossRef CAS; (e) M. E. Welsch, S. A. Snyder and B. R. Stockwell, Curr. Opin. Chem. Biol., 2010, 14, 347–361 CrossRef CAS.
- G. W. Bemis and M. A. Murcko, J. Med. Chem., 1996, 39, 2887–2893 CrossRef CAS.
- (a) S. B. Mhaske and N. P. Argade, Tetrahedron, 2006, 62, 9787–9826 CrossRef CAS; (b) J. Zhou and J. Fang, J. Org. Chem., 2011, 76, 7730–7736 CrossRef CAS; (c) B. A. Granger, K. Kaneda and S. F. Martin, Org. Lett., 2011, 13, 4542–4545 CrossRef CAS; (d) S. Rostamizadeh, A. M. Amani, R. Aryan, H. R. Ghaieni and N. Shadjou, Synth. Commun., 2008, 38, 3567–3576 CrossRef CAS.
- E. Sigel, Med. Chem. Rev.--Online, 2005, 2, 251–256 CrossRef CAS.
- (a) K. B. Sharpless and R. Manetsch, Expert Opin. Drug Discovery, 2006, 1, 525–538 CrossRef CAS; (b) D. K. Mohapatra, P. K. Maity, M. Shabab and M. I. Khan, Bioorg. Med. Chem. Lett., 2009, 19, 5241–5245 CrossRef CAS.
- D. K. Mohapatra, P. K. Maity, M. Shabab and M. I. Khan, Bioorg. Med. Chem. Lett., 2009, 19, 5241–5245 CrossRef CAS.
- Z. Ye, M. Ding, Y. Wu, Y. Li, W. Hua and F. Zhang, Green Chem., 2018, 20, 1732–1737 RSC.
- M. R. Ahmad, G. Sajjad, L. H. Ardeshiri and A. Nahad, Med. Chem. Res., 2016, 25, 1538–1550 CrossRef.
- F. Santos, G. Javier and P. Carlos, Molecules, 2006, 11, 583–588 CrossRef.
- A. Dömling and I. Ugi, Angew. Chem., Int. Ed., 2000, 39, 3168–3210 CrossRef.
- A. Dömling, Chem. Rev., 2006, 106, 17–89 CrossRef.
- A. Dömling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef.
- F. Fülöp, F. Miklós and E. Forró, Synlett, 2008, 11, 1687–1689 CrossRef.
- F. Miklós, Z. Tóth, M. M. Hänninen, R. Sillanpää, E. Forró and F. Fülöp, Eur. J. Org. Chem., 2013, 22, 4887–4894 CrossRef.
- F. Miklós, K. Bozó, Z. Galla, M. Haukka and F. Fülöp, Tetrahedron: Asymmetry, 2017, 28, 1401–1406 CrossRef.
- I. Nekkaa, M. Palkó, I. Mándity, F. Miklós and F. Fülöp, Eur. J. Org. Chem., 2018, 32, 4456–4464 CrossRef.
- F. Miklós and F. Fülöp, Eur. J. Org. Chem., 2010, 5, 959–965 CrossRef.
- M. Palkó, M. El Haimer, Z. Kormányos and F. Fülöp, Molecules, 2019, 24, 772 CrossRef.
- K. G. Guggenheim, H. Toru and M. J. Kurth, Org. Lett., 2012, 14, 3732–3735 CrossRef CAS.
- F. Fülöp, M. Palkó, E. Forró, M. Dervarics, T. A. Martinek and R. Sillanpää, Eur. J. Org. Chem., 2005, 15, 3214–3220 Search PubMed.
- B. J. Stokes, C. V. Vogel, L. K. Urnezis, M. Pan and T. G. Driver, Org. Lett., 2010, 12, 2884–2887 CrossRef CAS.
- B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS.
- B. M. Trost, Acc. Chem. Res., 2002, 35, 695–705 CrossRef CAS.
- C. J. Li and B. M. Trost, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 13197–13202 CrossRef CAS.
- P. A. Wender, M. P. Croatt and B. Witulski, Tetrahedron, 2006, 62, 7505–7511 CrossRef CAS.
- P. A. Wender, Nat. Prod. Rep., 2014, 31, 433–440 RSC.
- M. Palkó, E. Sándor, P. Sohár and F. Fülöp, Monatsh. Chem., 2005, 136, 2051–2058 CrossRef.
- M. V. Madhubabu, R. Shankar, G. R. Reddy, T. S. Rao, M. V. B. Rao and R. Akula, Tetrahedron Lett., 2016, 46, 5033–5037 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2048904–2048906. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ra10553h |
| This journal is © The Royal Society of Chemistry 2021 |