
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDecoration of silver nanoparticles on nitrogen-doped nanoporous carbon derived from zeolitic imidazole framework-8 (ZIF-8) via in situ auto-reduction†
Vediyappan Veeramania,
Nguyen Van Chi *b,
Yi-Lin Yanga,
Nguyen Thi Hong Huongc,
Thuan Van Trand,
Tansir Ahamade,
Saad M. Alshehrie and
Kevin C.-W. Wu*a
*b,
Yi-Lin Yanga,
Nguyen Thi Hong Huongc,
Thuan Van Trand,
Tansir Ahamade,
Saad M. Alshehrie and
Kevin C.-W. Wu*a
aDepartment of Chemical Engineering, National Taiwan University, No. 1, Sec. 4, Roosevelt Road, Taipei 10617, Taiwan. E-mail: kevinwu@ntu.edu.tw
bInstitute of Applied Science and Technology, Van Lang University, Ho Chi Minh 700000, Vietnam. E-mail: chi.nv@vlu.edu.vn
cFaculty of Medicine and Pharmacy, Van Lang University, Ho Chi Minh 700000, Vietnam
dNTT Hi-Tech Institute, Nguyen Tat Thanh University, Ho Chi Minh 755414, Vietnam
eDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia
First published on 9th February 2021
Abstract
We discovered an in situ auto-reduction method to embed silver nanoparticles onto a nanoporous carbon (NC) derived from the zeolitic imidazole framework-8 (ZIF-8), without any requirement of the reducing agents. The detailed analysis demonstrated the formation of Ag NPs by the replacement of the metallic Zn residue in the NC with Ag ions. The synthesized Ag@NC exhibited a superior catalytic activity toward the reduction reaction of 4-nitrophenol into 4-aminophenol.
Introduction
Noble metal nanoparticles (NPs), such as Pd, Pt, Ag and Au, play an important role in diverse applications because of their high reactivities and selectivities.1 Among these noble metal NPs, Ag NPs are extensively used for the catalytic reactions under mild conditions owing to their intrinsic physical and chemical properties.2 However, it is well known that the naked metal NPs are unstable because of their ease in aggregation, which leads to diminishing catalytic activities and catalyst recycling. This drawback is unappealing for industrial applications.3 Therefore, metal NPs have been immobilized on templates (i.e. zeolite,4 metal oxide,5 and carbon materials6) or polymers used, which serve as a protecting agent to prevent the metal NPs from agglomeration.7 In addition, to embed noble metal NPs on supports, the researchers have been using various types of reducing agents, such as hydrogen gas at high temperature, sodium borohydride, and sodium citrate, for reducing the metal ions to metals.8 Moreover, the plant extract, polyol group, chemical vapor deposition (CVD) and plasma synthetic strategy were investigated for the synthesis of the metal NPs.9 However, these synthetic strategies required the addition of reducing agents and higher operating temperature and pressure of the tubular furnace, which should be considered in practical applications. Thus, the green approaches to synthesize metal nanoparticles (NPs), such as an in situ auto-reduction and an ion exchange method, have become an interesting topic of study.Metal–organic frameworks (MOFs), known as porous coordination polymers, are hybrid materials of inorganic metal ions and organic ligands, which are extensively used in diverse applications such as drug delivery, gas storage, sensors and catalysts10 owing to their numerous favourable properties like tunable chemical properties, larger internal surface area and the different types of cavities.11 Compared with other MOF materials, the synthesis of a high surface area zeolitic imidazole framework-8 (ZIF-8) is relatively easy, and its preparation can be performed in either aqueous or organic solvent systems.12 After thermal treatment, the framework of a ZIF-8 would be broken and converted into a nitrogen-enriched nanoporous carbon (NC), which was employed as an efficient metal-free catalyst for a chemical reaction or an electrochemical reaction.13 Moreover, the NC could be considered as a green template for loading metal/metal oxide on the framework.6 However, to obtain a pure NC material or metal@NC, the residual zinc atoms within the NC material were removed using concentrated acids (i.e., HCl or H2SO4), which raised a concern about the corrosion and environmental issues.13a Therefore, to take advantage of the zinc residual in the NC material and to decrease the production cost by ignoring the reducing agents and post-treatment, herein we proposed an in situ auto-reduction method to synthesize Ag NPs in the NC at room temperature.
Fig. 1A shows the simple process of preparing the Ag NPs on the ZIF-8 derived nitrogen-enriched nanoporous carbon (Ag@NC) composite. It was the first time that the synthesis of the Ag NCs does not require any acid treatment or addition of any reducing agents. Briefly, the Zn@NC sample was obtained by the pyrolysis of the ZIF-8 nanocrystal. Without the removal of the residual metallic Zn from the nitrogen-enriched nanoporous carbon, the Ag(NO3)2 solution was then added into Zn@NC and stirred at RT for 3 h. The ionic Ag+ could automatically exchange with the metallic Zn to form the Ag NPs in NC (named Ag@NC). The presence of Ag@NC material was confirmed using various physicochemical studies and its catalytic behaviour was tested. Owing to its catalytic activity, the synthesized Ag@NC was used as the effective catalyst for the reduction reaction of 4-nitrophenol (4-NP) into 4-aminophenol (4-AP) using NaBH4 as the hydrogen donor. Here, NaBH4 was used as the additive reagent for the reduction reaction, but no reducing agent was used for the synthetic process.
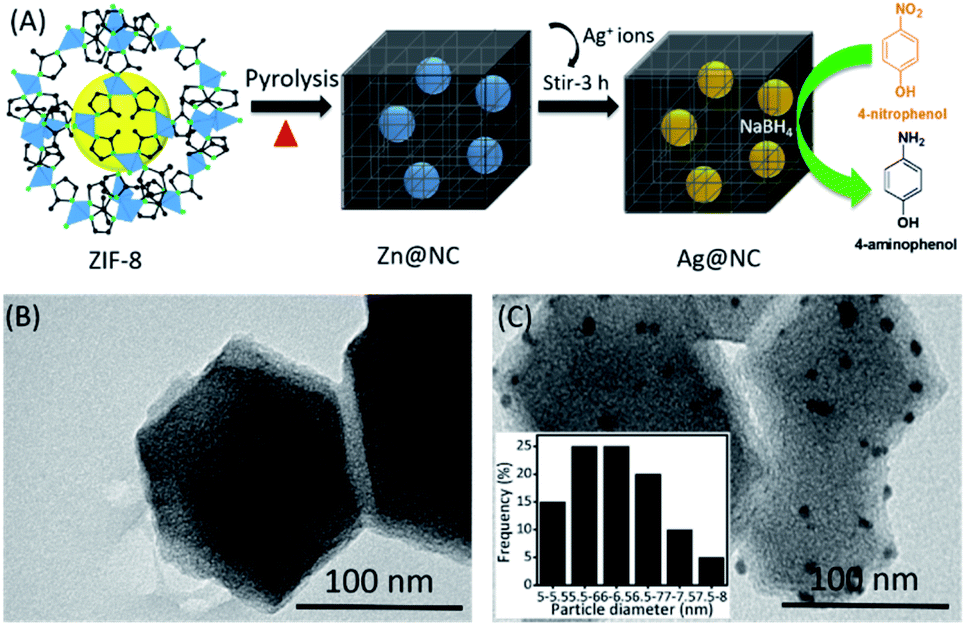 | ||
| Fig. 1 (A) Schematic representation of the synthesis of the Ag@NC by an ion exchange method. (B) and (C) TEM images of the Zn@NC and Ag@NC samples, respectively. | ||
Results and discussion
Scanning Electron Microscopy (SEM) was performed to observe the shape of the synthesized material (Fig. S1†). After the carbonation process, Zn@NC still retained its polyhedron morphology similar to that of the ZIF-8. However, we could observe roughness on the surface of the carbon sample (Fig. S1B and C†). The transmission electron microscopic (TEM) images of the Zn@NC and the Ag@NC are shown in Fig. 1B and C. The Zn particles were not observed, whereas the images clearly exhibited that the Ag NPs were uniformly embedded on the NC (Fig. 1C). The average particle size of the Ag NP was evaluated to be approximately 6 nm. The X-ray diffraction patterns of the as-synthesized samples (ZIF-8, Zn@NC and Ag@NC) are shown in Fig. 2A. The Zn@NC sample displayed two broad peaks at 24° and 44°, which corresponded to the graphitic carbon and the low degree of disordered carbon, respectively, which were confirmed by the Raman spectrum (Fig. S2†), thereby indicating that the ZIF-8 was carbonated to carbon. There were no observations of the diffraction peaks of Zn in both the Zn@NC and the Ag@NC samples (Fig. 2A). However, the XPS survey spectra (Fig. 2C) and the EDS mapping (Fig. S3†) of the Zn@NC sample indicated the existence of the Zn residue. It was supposed that the particle size of Zn was too small and located inside the NC framework. Ag@NC revealed the diffraction peaks at 38.2°, 44.3°, 64.3°, 75.5° and 81.4°, which corresponded to the (111), (200), (220), (311), and (222) planes of the metallic Ag.14 According to the ICP-OES measurements, the Ag content in the Ag@NC sample was 8.46 wt% (Table S1†), which suggested the presence of the Ag NPs in the NC. | ||
| Fig. 2 (A) XRD pattern and (B) N2 adsorption/desorption analysis of the ZIF-8, the Zn@NC and the Ag@NC samples. (C) XPS survey spectra of the Ag@NC sample and (D) the chemical state of Ag. | ||
Furthermore, the presence of the pore characteristics of the prepared samples is evaluated by using the N2 adsorption–desorption isotherms, and the data are shown in Fig. 2B, S4 and S5.† The obtained specific surface area, total pore volume, and pore size value are provided in Table S1.† The surface area of the Ag@NC decreased to 534.9 m2 g−1, compared to the NC and the Zn@NC that were 807.9 m2 g−1 and 792.5 m2 g−1, respectively. Moreover, the pore size of the Ag@NC still remained similar to that of the Zn@NC, as shown in Table S2.† We supposed that the Ag NPs substituted for the metal Zn and occupied the pores of the NC. Based on the Raman analysis (Fig. S2†), the ratios of the D band over the G band (ID/IG) were calculated to be 0.99 and 1.06 for the Zn@NC and the Ag@NC samples, respectively, indicating that the carbon structure had higher defects after the reduction process. The high-resolution TEM image further confirmed that the Ag NPs were confined within the NC (Fig. S6†). To detect the chemical composition of the Ag@NC nanocomposites, the XPS analysis was performed. Fig. 2C shows the presence of various material compositions such as Ag, C, N, O and Zn. The existence of C, N, and Zn in the Zn@NC was due to the decomposition of the ZIF-8 during the carbonation process.15 Predictably, the presence of Ag in the Ag@NC sample was detected. Moreover, Fig. 2D reveals the peaks of Ag 3d5/2 at 368.3 eV and Ag 3d3/2 at 374.4 eV, which indicate the presence of the metallic Ag on the nanoporous carbon.3 The results demonstrated the successful preparation of Ag@NC.
It was a matter of concern as what reducing agent could be used to reduce Ag ions into Ag metal? To clarify this point, we analysed the materials in detail. The survey XPS spectrum of the Zn@NC revealed the presence of Zn species (Fig. 2C) but Ag species were not observed. After the reduction process, the Ag peak was detected by the XPS (Fig. 2C) with 8.46 wt% (Table S1†). In addition, the Zn residue in the Zn@NC was 33.6 wt%, while the value in the Ag@NC sample was 10.5 wt% (Table S1†). The high-resolution XPS of Zn demonstrated that the binding energy of the Zn species in the Ag@NC (i.e. Zn 2p3/2 = 1022.2 eV, 2p1/2 = 1045.1 eV) was higher than that of the Zn@NC (i.e. Zn 2p3/2 = 1020.8 eV, 2p1/2 = 1044.7 eV) (Fig. S7†), which suggested that the Ag NPs were formed by the ion exchange of Ag+ ions with the metallic Zn residual present in the NC. It was well known that the standard reduction potential of Zn/Ag species was −0.76/+0.80 V. We presumed that Ag+ was automatically reduced by the reaction between Zn and ionic Ag+. Furthermore, we used NCW (NCW = NC was washed by HCl to remove Zn) for the synthesis of the Ag NPs using a similar procedure (see Experimental section). The residual Zn component (8.2 wt%) was still present in the NCW sample; however, the Ag NPs do not form in the nanoporous carbon (Table S1†). In addition, the XRD pattern showed no diffraction peaks of the Ag NPs (Fig. S4A†). We supposed that the Zn content in NCW was not metallic Zn due to the influence of the HCl treatment; thus, it was unable to reduce the Ag ions into Ag metal. These results demonstrated that the Ag NPs were produced by the auto-reduction reaction between the metallic Zn and the ionic Ag+.
4-Nitrophenol (4-NP) is an aromatic nitro compound, and it is highly harmful to humans and causes many sicknesses such as headaches, drowsiness, nausea, and cyanosis.16 In contrast, 4-aminophenol (4-AP) is an important and a valuable product obtained from the reduction of 4-NP. It is a useful intermediate in the pharmaceutical industry and in the manufacture of the drug paracetamol.2a,3 Recently, the reduction of 4-NP to 4-AP in an aqueous media with the presence of NaBH4 is reported to be an important catalytic reaction by the researchers because of its industrial application.
Thus, the conversion of 4-nitrophenol into 4-aminophenol was selected to investigate the catalytic ability of Ag@NC nanocomposites.17 The reduction reaction of 4-NP into 4-AP was carried out in a water medium at room temperature using NaBH4 as the reducing agent. As shown in Fig. 3A and B, the absorption peaks at 303 nm correspond to 4-NP. NaBH4 mixed with a 4-NP solution to form 4-nitrophenolate ions, which could be confirmed by the shifting of the absorption wavelength from 303 nm to 400 nm. In the presence of a catalyst, the absorption peak (400 nm represented 4-nitrophenolate) intensity gradually decreased. Simultaneously, a new absorption peak appeared at 300 nm and decolouration occurred in the reaction mixture (as shown in Fig. 3B), which suggested the formation of 4-AP. As shown in Fig. 3C and S4,† the NC (1 mg) and the Zn@NC (1 mg) are not efficient catalysts for this reaction, with the occurrence of low 4-NP conversion after 20 min of reaction. However, the Ag@NC demonstrated an excellent performance in reducing 4-NP to 4-AP, with 100% conversion in 20 min of reaction. Furthermore, the conversion efficiency was calculated by testing with the various amounts of the Ag@NC catalysts (amount: 0.5 and 1.5 mg) (Fig. S5†). The conversion accelerated rapidly in accordance with the increasing amounts of the Ag@NC catalyst. This indicated that the Ag NP was an active site for the reduction of 4-NP to 4-AP.
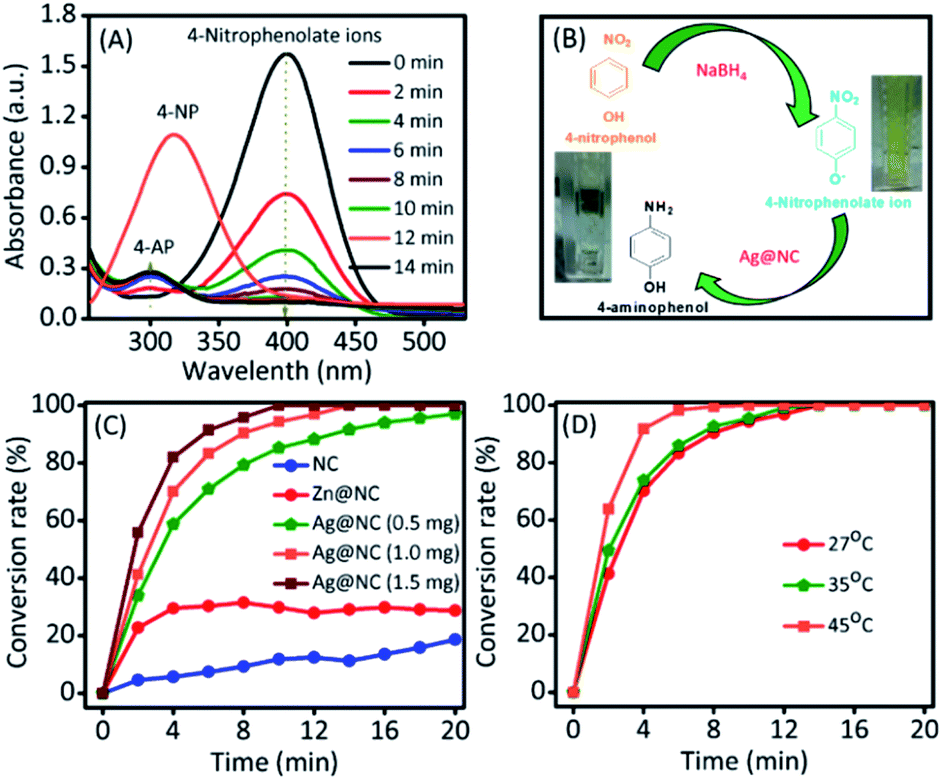 | ||
| Fig. 3 (A) UV-vis spectra of 4-nitrophenol reduction recorded every 2 min using the Ag@NC (1.0 mg) as the catalyst. (B) The reaction scheme and its corresponding photograph images of the color change from a bright yellow to a colorless solution. (C) The catalytic conversion to 4-AP from 4-NP over different catalyst materials. (D) Catalytic conversion to 4-AP from 4-NP at different temperatures using the Ag@NC (1.0 mg) as the catalyst. | ||
To further investigate, the kinetic reaction was studied. Fig. 3D and S6† represent the reaction conversion and the UV-vis spectra for 4-NP reduction at various temperatures, respectively. The kinetic rate constant (k) value could be calculated from the following equation: kt = −ln(Ct/Co), where ‘t’ was the reaction time and Ct/Co were the initial (Co) and final (Ct) concentrations of 4-NP. From Fig. 4A, the linear correlation is obtained between the plot of −ln(Ct/Co) vs. reaction time; hence, it suggests that the ensuing reaction is a pseudo-first order kinetic reaction.17 Accordingly, the calculated kinetic rate constant (k) values were 4.2 × 10−3, 6.5 × 10−3, and 11.4 × 10−3 s−1 at various temperatures of 27, 35 and 45 °C, respectively. The high reaction rate constant indicated the high reductive activity of the Ag@NC catalyst (Table S3†). The obtained rate constant value was higher than that of the Au@Ag core–shell nanoparticles stabilized on a metal–organic framework.18 In addition, the rate constant value was compared with some earlier published Ag NP-based nanocomposite materials.2a,3,19
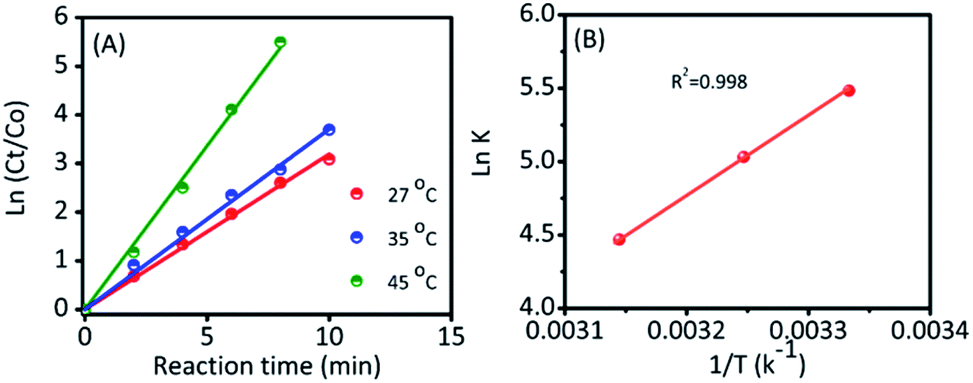 | ||
Fig. 4 (A) The relationship of ln(Ct/Co) versus reaction time for the reduction of 4-NP to 4-AP using the Ag@NC (1.0 mg) catalyst at different temperatures. (B) Arrhenius plot ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) k vs. 1/T (K−1) for the estimation of the activation energy. k vs. 1/T (K−1) for the estimation of the activation energy. | ||
Furthermore, the activation energy was an important parameter to evaluate their catalytic behavior, and it could be calculated from a classic Arrhenius theory. The equation is shown in below:
| lnk = lnA − (Ea/RT) |
To compare the catalytic activity of the Ag@NC catalyst, we synthesized a Ag/NC-SB catalyst using sodium borohydride as the reducing agent (SB: sodium borohydride, see details in synthetic experiment in ESI†). As shown in Fig. S7,† the 100% 4-NP conversion can be afforded in 10 min at RT in the presence of the Ag/NC-SB as the catalyst, which is similar to that of the Ag@NC catalyst. However, the synthesis of the Ag/NC-SB catalyst required an additional amount of sodium borohydride as the reducing agent to reduce Ag ions to Ag nanoparticles. This indicated the novel synthesis of the Ag@NC material using the auto-reduction approach. Moreover, we extended our study to test with other metal catalysts, such as Pt/NC, Au/NC, and Ru/NC, and their corresponding data are shown in Fig. S8.† The results indicated that the catalytic activity of the Ag@NC was almost similar to that of the Pt/NC, Au/NC, and Ru/NC catalysts.
The recyclability and stability of the Ag@NC catalyst were examined. The catalyst was collected and washed several times with deionized water after the reduction reaction. Then, the reused catalyst was activated under vacuum condition before performing the next run. As shown in Fig. 5, the activity of the Ag@NC catalyst remained high after five runs of the reaction.
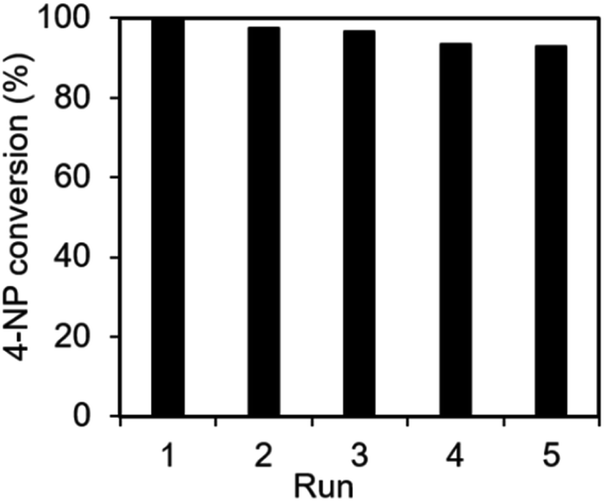 | ||
| Fig. 5 The recycle test of the Ag@NC catalyst for the 4-NP reduction. | ||
Conclusions
We reported a simple route for the synthesis of the Ag@NC nanocomposites without using any reducing agents. The metallic Zn residue played an important role in reducing the Ag ions into metallic Ag NPs, which were formed by the pyrolysis of the ZIF-8 precursor. The Ag NPs embedded into the nitrogen-enriched nanoporous framework and exhibited a high catalytic activity for the 4-NP reduction. The concept of our work might inspire further studies to utilize the metal residue in the carbon framework after the treatment of the high temperature of the metal–organic frameworks.Experimental section
Chemicals
2-Methylimidazole (2-MIM), zinc nitrate hexahydrate (Zn(NO3)2·6H2O), silver nitrate (AgNO3), sodium borohydride (NaBH4) and methanol anhydrous were purchased from Sigma-Aldrich and were used without further purification. Polyvinylpyrrolidone (PVP) was purchased from TCI Chemicals. 4-Nitrophenol (4-NP) was purchased from Acros. Deionized water was purified with a Milli-Q system (Millipore, Bedford, MA, USA).Synthetic procedure of nitrogen-enriched nanoporous carbon (NC)
The nitrogen-enriched nanoporous carbon (NC) was synthesized as described in the previous literatures.1 Briefly, polyvinylpyrrolidone (12 g) and 2-methylimidazole (4.2 g) were dissolved in 150 mL methanol solution, and Zn(NO3)2·6H2O (4.4 g) was dissolved in an another 150 mL methanol solution. The two solutions were stirred separately using a magnetic stirrer for 10 min. Subsequently, the zinc containing solution was added into the 2-methylimidazole solution and stirred for another 1 h. After 24 h of aging, the white sediment was collected by centrifugation and washed with methanol solution. Finally, the white powder was dried in vacuum at RT for 24 h. Furthermore, the ZIF-8 powder was carbonized by using a tubular furnace at 900 °C for 8 h under N2 atmosphere at a heating rate of 5 °C min−1. The as-synthesized carbon samples were labelled as Zn@NC.Synthesis of Ag NPs on the nanoporous carbon by auto-reduction method (Ag@NC)
For the synthesis of Ag@NC nanocomposites, 0.1 g of Zn@NC was dissolved in 30 mL of water. (Notes: Zn@NC sample was used immediately after removing from the tubular furnace to prevent the oxidation of Zn into ZnO.) Further, the aqueous solution of silver nitrate (1 mL; 0.1 M) was directly added to the above solution and stirred for 3 h at room temperature. After that, the black powder was collected by centrifugation and washed using DI water and methanol solution. Finally, the sample was dried under vacuum for 1 day and labelled as Ag@NC.Similarly, we tried synthesizing Ag@NCW (NCW is a nitrogen-doped carbon after washing with HCl). About 0.1 g of NCW was dissolved in 30 mL of water. Further, the aqueous solution of silver nitrate (1 mL; 0.1 M) was directly added to the above solution and stirred for 3 h at room temperature. After that, the black powder was collected by centrifugation and washed using DI water and methanol solution. Finally, the sample was dried under vacuum for 1 day and labelled as Ag@NCW. However, the Ag NPs could not form on the nanoporous carbon (Fig. S2A†).
Synthesis of Ag/NC catalyst by impregnation method
The aqueous solution of silver nitrate (1 mL; 0.1 M) was directly added to 30 mL solution containing NC sample (0.1 g) and stirred for 0.5 h at room temperature. After that, NaBH4 (5 mL, 1 mmol) was dropped into the above solution and was kept stirring for 1 h. The resulting solid was collected by centrifugation and washed with DI water and methanol solution. The obtained solid was dried under vacuum for 1 day and labelled as Ag/NC-SB where SB was sodium borohydride.Characterizations
The crystal structure of the samples was analyzed by using the powder X-ray diffraction (XRD – Rigaku-Ultima IV instrument) patterns. The morphology of the samples was monitored by using scanning/transmission (SEM/TEM) electron microscopy (SEM, NovaTM NanoSEM 230; TEM, JEOL JEM-1200EX II). The internal surface area and the pore size of the as-synthesized samples were determined using the N2-adsorption/desorption measurements (Micromeritics ASAP 2020). The material compositions were recorded by using X-ray photoelectron spectroscopy (XPS, Thermo Scientific, Theta Probe). The present weight percentage of the material composition was analyzed by using an inductively coupled plasma-optical emission spectrometry (ICP-OES).Catalytic reaction of 4-NP
To investigate the catalytic ability and reusability of the Ag@NC nanocomposite used for the hydrogenation reaction of 4-NP, typically, 4-NP (1 × 10−4 M, 20 mL) and NaBH4 (0.5 M, 5 mL) solutions (i.e. NaBH4 as hydrogen donor for the reaction) were mixed together and then stirred for 20 min to form the 4-nitrophenolate ion. After that, Ag@NC catalyst (1 mg) was added to the above mixture, which was designated at a time (to) and stirred at room temperature. During these time periods, 1.5 mL of the reaction mixture was withdrawn every 2 min and monitored using a UV-vis spectroscopy (Jasco V-670 at a wavelength of 400 nm).Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This work was financially supported by the Ministry of Science and Technology (MOST), Taiwan (108-2638-E-002-003-MY2; Shackleton Program Award). The authors also thank to the Researchers Supporting Project Number (RSP-2021/6), King Saud University, Riyadh, Saudi Arabia.Notes and references
- (a) Y.-Z. Chen, G. Cai, Y. Wang, Q. Xu, S.-H. Yu and H.-L. Jiang, Green Chem., 2016, 18, 1212–1217 RSC; (b) P. Zhang, C. Shao, Z. Zhang, M. Zhang, J. Mu, Z. Guo and Y. Liu, Nanoscale, 2011, 3, 3357–3363 RSC.
- (a) J. Dai and M. L. Bruening, Nano Lett., 2002, 2, 497–501 CrossRef CAS; (b) J. Han, P. Fang, W. Jiang, L. Li and R. Guo, Langmuir, 2012, 28, 4768–4775 CrossRef CAS; (c) G. Yang, G. Gao, C. Wang, C. Xu and H. Li, Carbon, 2008, 46, 747–752 CrossRef CAS.
- M. Liang, R. Su, R. Huang, W. Qi, Y. Yu, L. Wang and Z. He, ACS Appl. Mater. Interfaces, 2014, 6, 4638–4649 CrossRef CAS.
- Y. Wu, C. Li, J. Bai and J. Wang, Results Phys., 2017, 7, 1616–1622 CrossRef.
- S. Shuang, R. Lv, Z. Xie and Z. Zhang, Sci. Rep., 2016, 6, 26670 CrossRef CAS.
- Y.-T. Liao, J. E. Chen, Y. Isida, T. Yonezawa, W.-C. Chang, S. M. Alshehri, Y. Yamauchi and K. C.-W. Wu, ChemCatChem, 2016, 8, 502–509 CrossRef CAS.
- J. Safari, A. E. Najafabadi, Z. Zarnegar and S. F. Masoul, Green Chem. Lett. Rev., 2016, 9, 20–26 CrossRef CAS.
- (a) H.-J. Zhang, S.-D. Qi, X.-Y. Niu, J. Hu, C.-L. Ren, H.-L. Chen and X.-G. Chen, Catal. Sci. Technol., 2014, 4, 3013–3024 RSC; (b) Z. Jiang, J. Xie, D. Jiang, J. Jing and H. Qin, CrystEngComm, 2012, 14, 4601–4611 RSC; (c) O. J. Isaac, F. M. Romero, N. G. Bastus and V. Puntes, J. Phys. Chem. C, 2010, 114, 1800–1804 CrossRef.
- (a) M. P. Patil, J. Palma, N. C. Simeon, X. Jin, X. Liu, D. Ngabire, N.-H. Kim, N. H. Tarte and G.-D. Kim, New J. Chem., 2017, 41, 1363–1371 RSC; (b) K. Takahashi, S. Yokoyama, T. Matsumoto, J. L. C. Huaman, H. Kaneko, J.-Y. Piquemal, H. Miyamura and J. Balachandran, New J. Chem., 2016, 40, 8632–8642 RSC; (c) B. Jeyadevan, J. L. Cuya, Y. Inoue, K. Shinoda, T. Ito, D. Mott, K. Higashimine, S. Maenosono, T. Matsumoto and H. Miyamura, RSC Adv., 2014, 4, 26667–26672 RSC; (d) J. L. C. Huaman, S. Fukao, K. Shinoda and B. Jeyadevan, CrystEngComm, 2011, 13, 3364–3369 RSC; (e) C. Dhand, N. Dwivedi, X. J. Loh, A. N. J. Ying, N. K. Verma, R. W. Beuerman, R. Lakshminarayanan and S. Ramakrishna, RSC Adv., 2015, 5, 105003–105037 RSC.
- (a) H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef; (b) O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS; (c) J. He, R. C. C. Yap, S. Y. Wong, Y. Zhang, Y. Hu, C. Chen, X. Zhang, J. Wang and X. Li, CrystEngComm, 2016, 18, 5262–5266 RSC; (d) B. Li, H.-M. Wen, Y. Cui, W. Zhou, G. Qian and B. Chen, Adv. Mater., 2016, 28, 8819–8860 CrossRef CAS; (e) X.-Q. Wu, J.-G. Ma, H. Li, D.-M. Chen, W. Gu, G.-M. Yang and P. Cheng, Chem. Commun., 2015, 51, 9161–9164 RSC.
- Y. V. Kaneti, J. Tang, R. R. Salunkhe, X. Jiang, A. Yu, K. C.-W. Wu and Y. Yamauchi, Adv. Mater., 2017, 1604898 CrossRef.
- (a) J. Cravillon, R. Nayuk, S. Springer, A. Feldhoff, K. Huber and M. Wiebcke, Chem. Mater., 2011, 23, 2130–2141 CrossRef CAS; (b) M. Jian, B. Liu, R. Liu, J. Qu, H. Wang and X. Zhang, RSC Adv., 2015, 5, 48433–48441 RSC.
- (a) C. V. Nguyen, Y.-T. Liao, T.-C. Kang, J. E. Chen, T. Yoshikawa, Y. Nakasaka, T. Masuda and K. C.-W. Wu, Green Chem., 2016, 18, 5957–5961 RSC; (b) X. Ma, Y.-X. Zhou, H. Liu, Y. Li and H.-L. Jiang, Chem. Commun., 2016, 52, 7719–7722 RSC; (c) B. Y. Xia, Y. Yan, N. Li, H. B. Wu, X. W. Lou and X. Wang, Nat. Energy, 2016, 1, 1–8 Search PubMed.
- S. Xiao, W. Xu, H. Ma and X. Fang, RSC Adv., 2012, 2, 319–327 RSC.
- B. Liu, H. Shioyama, H. Jiang, X. Zhang and Q. Xu, Carbon, 2010, 48, 456–463 CrossRef CAS.
- (a) M. Koizumi, Y. Yamamoto, Y. Ito, M. Takano, T. Enami, E. Kamata and R. Hasegawa, J. Toxicol. Sci., 2001, 26, 299–311 CrossRef CAS; (b) P. Veerakumar, R. Madhu, S.-M. Chen, V. Veeramani, C.-T. Hung, P.-H. Tang, C.-B. Wang and S.-B. Liu, J. Mater. Chem. A, 2014, 2, 16015–16022 RSC.
- (a) H. Yang, S. J. Bradley, A. Chan, G. I. N. Waterhouse, T. Nann, P. E. Kruger and S. G. Telfer, J. Am. Chem. Soc., 2016, 138, 11872–11881 CrossRef CAS; (b) Y. Yang, Y. Ren, C. Sun and S. Hao, Green Chem., 2014, 16, 2273–2280 RSC; (c) Y. Yusran, D. Xu, Q. Fang, D. Zhang and S. Qiu, Microporous Mesoporous Mater., 2017, 241, 346–354 CrossRef CAS.
- H.-L. Jiang, T. Akita, T. Ishida, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2011, 133, 1304–1306 CrossRef CAS.
- (a) Y. Chi, L. Zhao, Q. Yuan, X. Yan, Y. Li, N. Li and X. Li, J. Mater. Chem., 2012, 22, 13571–13577 RSC; (b) T. V. Thu, P. J. Ko, N. H. H. Phuc and A. Sandhu, J. Nanopart. Res., 2013, 15, 1975 CrossRef.
- J. Liang, A. Yue, Q. Wang, S. Song and L. Li, RSC Adv., 2016, 6, 29497–29503 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10546e |
| This journal is © The Royal Society of Chemistry 2021 |