DOI:
10.1039/D0RA09476E
(Paper)
RSC Adv., 2021,
11, 6620-6627
Preparation of conductive self-healing hydrogels via an interpenetrating polymer network method†
Received
7th November 2020
, Accepted 29th January 2021
First published on 9th February 2021
Abstract
Conductive self-healing hydrogels and related soft sensor devices are gaining extensive attention from academia to industry because of their impacts on the lifetime and ergonomic design of artificial skins and soft robotics, as well as health monitoring systems. However, so far the development of such a material has been limited considering performance and availability. In this work, we developed composite hydrogels of acrylamide, polyacrylamide, dialdehyde-functionalized poly(ethylene glycol) and conductive carbon black through an interpenetrating polymer network strategy. After optimizing the composition ratio, the resultant hydrogel exhibited self-healing reversibility mechanically and electrically when cut and self-healed. We used 1H NMR and FT-IR spectroscopy to determine the self-healing mechanism of the system, thus demonstrating that the cooperative effect of the dynamic covalent and noncovalent interactions contributes to the self-healing capability of the gel. Rheology, scanning electron microscopy and light-emitting diode circuits were carried out to examine its macroscopic and microscopic properties, making it possible to apply in soft and conformable electronics.
1. Introduction
Hydrogels are a kind of soft material that are composed of water and hydrophilic polymer chains arranged in a three-dimensional network structure. Due to their high water content, porous structure, biocompatibility, tunable mechanical strength and biodegradability, they have been investigated in various fields such as biomedical, electrical engineering, environmental fields, food industry etc.1–5 For long-term durability requirements of engineering applications, self-healing hydrogels are being intensively studied to mimic natural systems to have the ability to self-repair damage inflicted on them.6–11 Based on the healing mechanisms, self-healing hydrogels divided into two different classes, depending on the dynamic equilibrium of physical interactions or chemical bonds in damaged regions.12,13 The process and outcome of self-healing can be characterized by the restoration of micro- and macrostructure14–16 as well as the recovery of mechanical and rheological properties.17–19
In recent decades, conductive hydrogels are gaining increasing attention as an important class of functional polymer materials because of their combined mechanical flexibility and electrical conductivity.20–25 Conductive hydrogels exhibit a healing capability which is helpful to extend the applications in health monitoring, artificial skins and implantable bioelectronics.26–32 Han and coworkers designed multifunctional hybrid conducting polymer hydrogels based on polyvinyl alcohol-borax and cellulose nanofiber–polypyrrole complexes.29 Hu and Wang synthesized polypyrrole or Zn-functionalized chitosan materials, which are cross-linked with poly(vinyl alcohol) to form a conductive and self-healing hydrogel (PCPZ hydrogel) for chronic wound treatment.30 Wan and Zhang prepared functionalized single-wall carbon nanotube, polyvinyl alcohol and polydopamine hydrogel, which is self-adhesive, self-healable and wearable, and can be applied in the field of bioelectronics.31 Zhai et al. developed a one-pot synthesis method to prepare a stretchable, conductive and self-healing hydrogel (GO3SPNB), that has great potential in smart electronic device adhesives.32
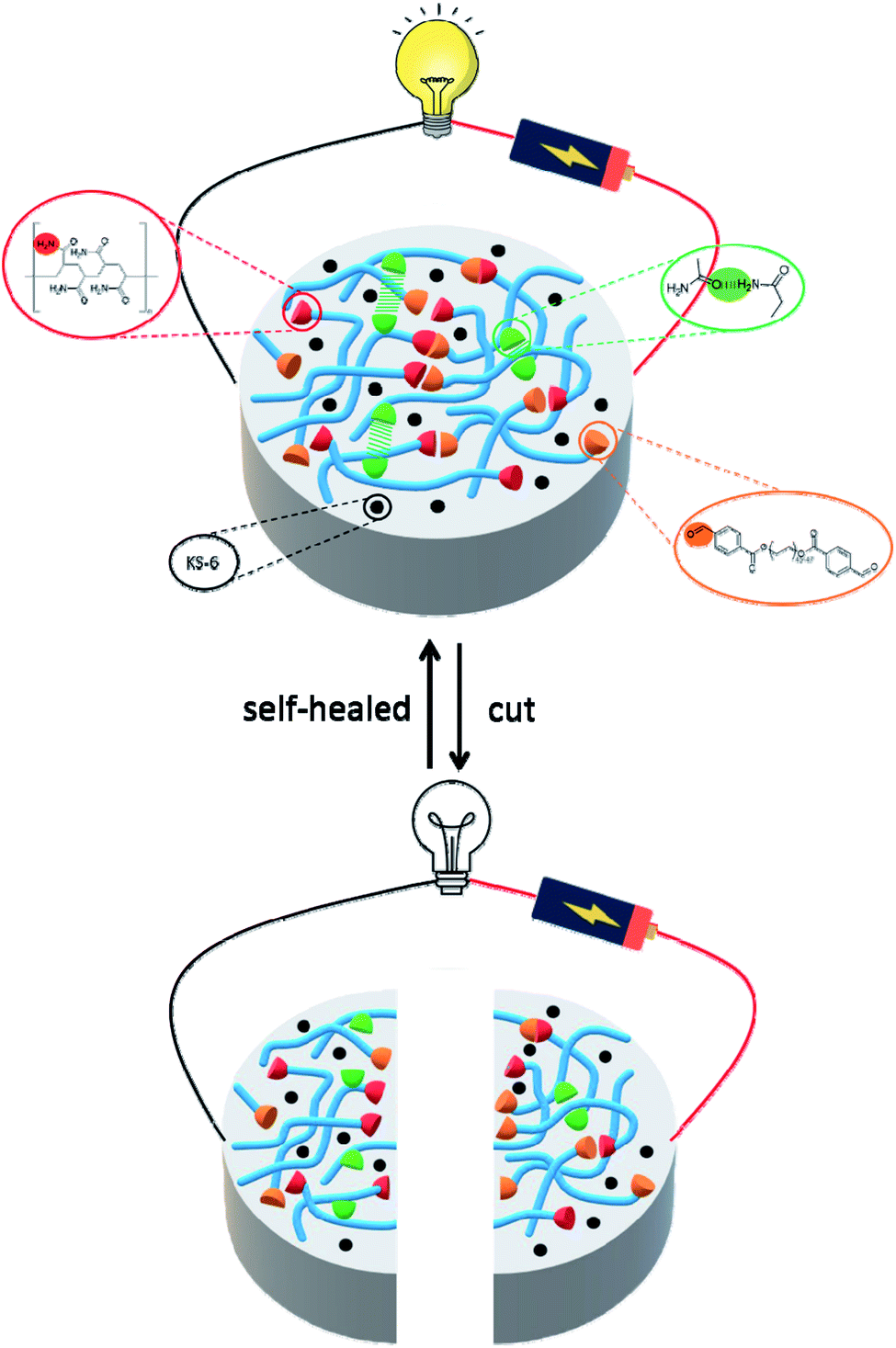
Additionally, hydrogels prepared by the interpenetrating polymer network (IPN) strategy could effectively expand their functionality and applicability.33–36 Therefore, in this work new self-healing and conductive IPN hydrogels are presented. We developed and fabricated the composite hydrogels of acrylamide, polyacrylamide, dialdehyde-functionalized poly(ethylene glycol) and conductive carbon black. Here carbon black is chosen as the conductive material, because of its high chemical and thermal stability, permanent electrical conductivity, light weight and low cost.37,38 After optimizing the composition ratio, the resultant hydrogel exhibited self-healing reversibility mechanically and electrically when cut and self-healed. Fig. 1 shows the molecular design strategy of the conductive hydrogel, the cooperative effect of the dynamic covalent (Schiff base reaction) and noncovalent interactions (hydrogen bond) contributes to the self-healing capability of the gel. 1H NMR and FT-IR spectroscopy were used to determine the self-healing mechanism of the system. The rheological measurement was conducted to understand the performance of the mechanical properties. An electron microscopic study was carried out to characterize the morphology of the hydrogels. The light-emitting diode circuit was utilized to demonstrate the good stability of the self-healed gel. We found that the combination of chemical and physical crosslinking can be beneficial for the mechanical performance of the conductive hydrogels, making it possible to apply in soft and conformable electronics.
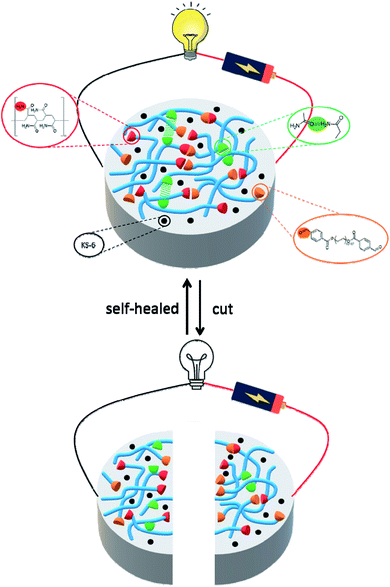 |
| | Fig. 1 Schematic illustration the molecular design strategy of conductive self-healing hydrogels. The synergistic interaction of the covalent interaction (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N) and noncovalent interaction (hydrogen bond) contributes to the self-healing capability of the gel. N) and noncovalent interaction (hydrogen bond) contributes to the self-healing capability of the gel. | |
2. Experimental section
2.1 Materials and method
Acrylamide, polyacrylamide average MW 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and poly(ethylene glycol) (PEG) average MW 2000 were purchased from Acros Organics. Potassium persulfate, 4-formylbenzoic acid, 4-(dimethylamino)pyridine (DMAP) and N,N′-dicyclohexylcarbodiimide (DCC) were obtained from Sigma-Aldrich. Food dyes and conductive carbon black graphite KS-6 were from LorAnn Oils and TIMCAL TIMREX®, respectively. The particle size of KS-6 is 5.8–7.1 μm (d90, laser Malvern), crystallinity hight is 60 nm, specific surface area (BET) is 26 m2 g−1 and bulk density is 0.00253 lb/in3. Dialdehyde-functionalized PEG (DF-PEG) was synthesized according to literature protocol.39 PEG (2.0 g), 4-formylbenzoic acid (0.6 g), DMAP (0.03 g) and DCC (1.13 g) were reaction at room temperature for overnight. The precipitate was filtered and re-dissolved in THF, followed by precipitation with diethyl ether to obtain the DF-PEG polymer.
000 and poly(ethylene glycol) (PEG) average MW 2000 were purchased from Acros Organics. Potassium persulfate, 4-formylbenzoic acid, 4-(dimethylamino)pyridine (DMAP) and N,N′-dicyclohexylcarbodiimide (DCC) were obtained from Sigma-Aldrich. Food dyes and conductive carbon black graphite KS-6 were from LorAnn Oils and TIMCAL TIMREX®, respectively. The particle size of KS-6 is 5.8–7.1 μm (d90, laser Malvern), crystallinity hight is 60 nm, specific surface area (BET) is 26 m2 g−1 and bulk density is 0.00253 lb/in3. Dialdehyde-functionalized PEG (DF-PEG) was synthesized according to literature protocol.39 PEG (2.0 g), 4-formylbenzoic acid (0.6 g), DMAP (0.03 g) and DCC (1.13 g) were reaction at room temperature for overnight. The precipitate was filtered and re-dissolved in THF, followed by precipitation with diethyl ether to obtain the DF-PEG polymer.
2.2 Preparation of P1–P8 hydrogels
2.2.1 P1–P6 hydrogels. Acrylamide (0.375 g, 15% w/v) and potassium persulfate (0.0035 g, 0.15% w/v) were dissolved in 2.5 mL deionized water and then ultrasonic treated for 5 min. The resultant homogeneous solution was heated to 70 °C until the formation of P1 hydrogel (ca. 7 h). In a manner similar to that described above, acrylamide (0.375 g, 15% w/v) and potassium persulfate (0.0035 g, 0.15% w/v) were treated with ultrasonic for 5 min. Subsequently, the DF-PEG (0.025 g, 1% w/v) and DF-PEG (0.050 g, 2% w/v) were added to the mixture to obtain P2 and P3, respectively. In a manner similar to that described above, acrylamide (0.375 g, 15% w/v) and potassium persulfate (0.0035 g, 0.15% w/v) were treated with ultrasonic for 5 min. Subsequently, the poly(acrylamide) (0.0125 g, 0.5% w/v) was added to achieve P4. In a manner similar to that described above, acrylamide (0.375 g, 15% w/v) and potassium persulfate (0.0035 g, 0.15% w/v) were treated with ultrasonic for 5 min. Subsequently, the DF-PEG (0.025 g, 1% w/v), poly(acrylamide) (0.0125 g, 0.5% w/v) and DF-PEG (0.050 g, 2% w/v), poly(acrylamide) (0.0125 g, 0.5% w/v) were added to the mixture to get P5 and P6, respectively.
2.2.2 P7 and P8 hydrogels. We prepared the solution of P6 and then conductive carbon black graphite KS-6 (0.0773 g, 3% w/v) was added. After stirring for 1.5 h, the solution was heated to 70 °C for 7 h to obtain P7. In a manner similar to that described above, a mixture of P6 solution and KS-6 (0.1315 g, 5% w/v) to convert P8.
2.3 Characterizations
The hydrogels were prepared according to the method of the Section 2.2 and freeze-dried to analyze the quality and functional characteristics. 1H NMR spectra were measured on a Bruker AVANCEII 400 NMR spectrometer using D2O as solvent. The morphology of the materials was observed by scanning electron microscopy (SEM, JEOL JSM-7600F). Fourier-transform infrared spectroscopy (FT-IR) spectra were obtained on Jasco 4600 FT/IR infrared spectrophotometer with the wavelength ranged from 4000 to 750 cm−1. For FT-IR, the samples were dissolved in deionized water, dropped on ZnSe plate (size: 20 mm × 2 mm) and left to dry to form a thin membrane.
2.4 Self-healing of the hydrogels
The hydrogels were prepared according to the method of the Section 2.2 and then cut into two pieces from the middle. The damaged hydrogels were contacted with a drop of 0.1 M NaOH for 1 h at room temperature to obtain the healed hydrogels. The macroscopic and microscopic images were recorded to demonstrate the self-healing capability.
2.5 Rheological measurements
The rheological measurements of the P1–P8 hydrogels were performed using TA rheometer (DHR-1, USA) with a parallel plate setup (diameter of 20 mm and a gap of 1 mm). A dynamic oscillatory frequency sweep experiments, including shear storage modulus (G′) and loss modulus (G′′) as functions of angular frequency (ω), were measured over the ω range of 0.1–100 rad s−1 at strain (γ) = 1% at 25 °C. The complex modulus (G*) and complex viscosity (η*) were calculated based on the values of G′ and G′′.24 The strain amplitude sweep test (γ = 0.1–100000%, ω = 1 rad s−1) at 25 °C were performed to study the viscoelastic properties of the hydrogels. Amplitude oscillatory strains were switched from small strain (γ = 10%) to subsequent large strain (γ = 2900%, 5000%, and 7000% for hybrid hydrogels of P6 as well as 1900%, 3000%, and 5000% for conductive hydrogels of P7) with 200 s for every strain interval.
3. Results and discussion
3.1 Preparation of self-healing hydrogels
We firstly prepared and tested the hydrogelation properties of 5, 10, 15 and 20% w/v of acrylamide, respectively. As depicted in Fig. S1,† stable hydrogels were obtained when the concentration higher than 15% w/v, thus we use 15% w/v of acrylamide for further investigation (P1). Since the acrylamide gel don't have the self-healing feature, we incorporated the DF-PEG into 15% w/v acrylamide gel. We speculate that the DF-PEG may interact with acrylamide to gain Schiff base (i.e. CN bond), thus making gels with self-healing characteristic. As can be seen from Fig. 2 and Table 1, we found that P2 is a transparent and sticky gel, while P3 is semi-transparent gel with some elasticity. However, P2 and P3 still don't have the self-healing though, we then decided to add poly(acrylamide) and expected the polymer can provide the hydrogen bond interaction to improve the molecular recognition between acrylamide and DF-PEG. We compared the properties of hydrogels of P4, P5 and P6, and found that P5 and P6 have self-healing abilities. Additionally, P6 has better elasticity and tensility than that of P5, therefore we take P6 for self-healing testing (Fig. 2).
Table 1 Physical properties of hydrogels of P1–P8a
| No |
Acrylamideb |
DF-PEGb |
Poly(acrylamide)b |
KS-6b |
Appr.c |
G′, G′′ (Pa) |
| Potassium persulfate 0.15% w/v, 2.5 mL deionized water. Unit: % w/v. TG: transparent gel; STG: semi-transparent gel; OG: opaque gel. |
| P1 |
15 |
— |
— |
— |
TG |
6.85 × 101, 2.65 × 101 |
| P2 |
15 |
1 |
— |
— |
TG |
9.83 × 101, 3.43 × 101 |
| P3 |
15 |
2 |
— |
— |
STG |
1.84 × 102, 6.39 × 101 |
| P4 |
15 |
— |
0.5 |
— |
TG |
3.45 × 102, 1.36 × 102 |
| P5 |
15 |
1 |
0.5 |
— |
TG |
5.57 × 101, 2.13 × 101 |
| P6 |
15 |
2 |
0.5 |
— |
STG |
1.40 × 102, 5.17 × 101 |
| P7 |
15 |
2 |
0.5 |
3 |
OG |
8.97 × 101, 3.66 × 101 |
| P8 |
15 |
2 |
0.5 |
5 |
OG |
7.14 × 101, 2.14 × 101 |
 |
| | Fig. 2 Optical images of hydrogels of (a) P1, (b) P2, (c) P3, (d) P4, (e) P5 and (f) P6. | |
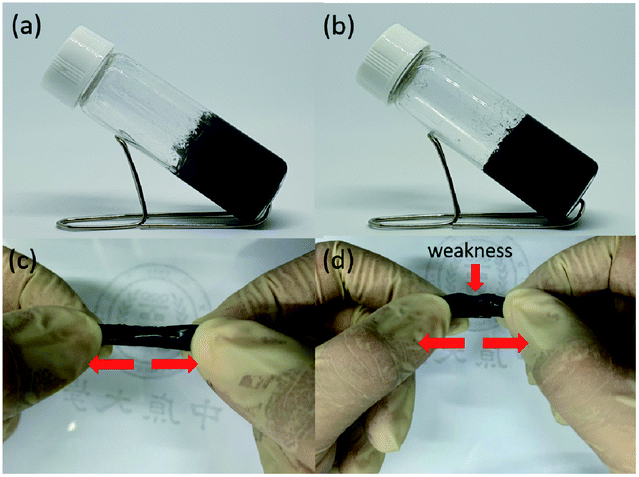
To evaluate the self-healing ability of P6 hydrogel, the two disk-shaped hydrogels with blue and white colors, which stained with food dyes, were cut into equal pieces and then two different colored semicircles were put together to form a united disk (Fig. 3a and b). After 1 h at room temperature, no fracture was observed when the hydrogel was stretched by hands, indicating the self-healing ability of the hydrogel (Fig. 3c). The good self-repair properties of P6 encourage us to further think about the development of conductive hydrogels for flexible devices applications. As displayed in Fig. 4 and Table 1, 3% w/v and 5% w/v conductive carbon black graphite (KS-6) were used to prepare the composite hydrogels of P7 and P8, respectively. Both are shown stable hydrogels, we therefore cut the hydrogels into two pieces and put together for an hour. We can obviously observe a better elastic property for P7 compared to that of P8 (Fig. 4c and d).
 |
| | Fig. 3 Photographs of self-healing testing of damaged and healed P6 hydrogel. | |
 |
| | Fig. 4 Optical images of hydrogels of (a) P7 and (b) P8. Photographs of self-healing testing of (c) P7 and (d) P8 hydrogels. | |
3.2 Mechanism of self-healing hydrogels
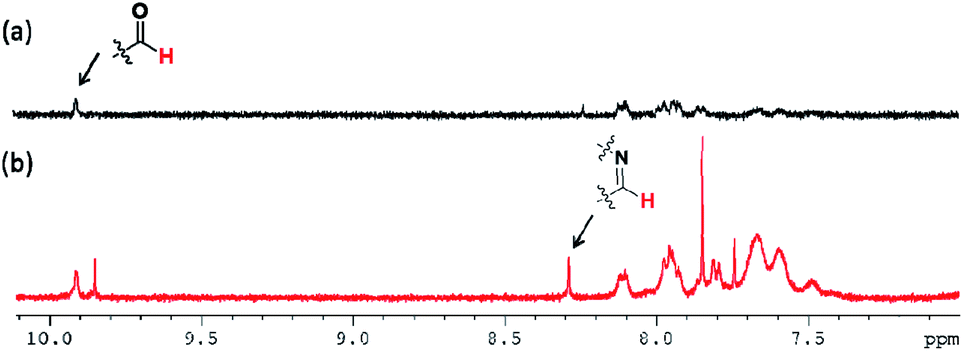
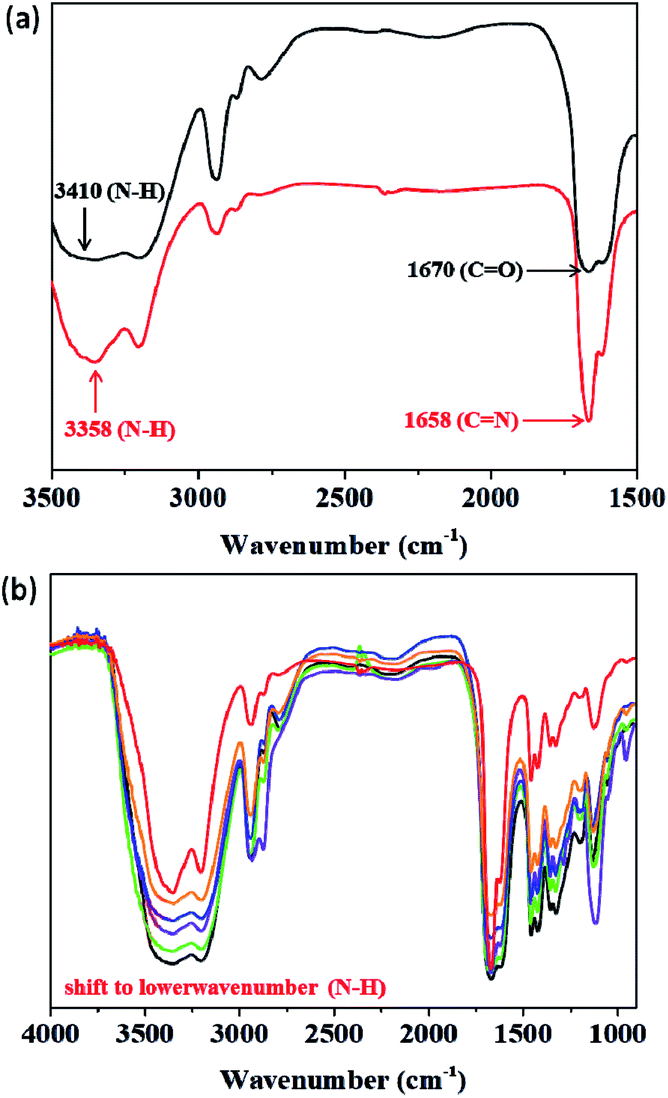
To identify the self-healing mechanism of P5–P8 hydrogels, 1H NMR and FT-IR spectroscopies have been conducted. For 1H NMR study, we take P6 hydrogel for example to examine the molecular interactions between acrylamide, DF-PEG as well as poly(acrylamide). Fig. 5 reveals the partial 1H NMR spectra of DF-PEG polymer and freeze-dried P6 hydrogel in a solution of D2O. From the result of the spectrum, a sharp singlet is observed at 9.9 ppm in DF-PEG, which can be assigned to the benzaldehyde proton (CHO). On the other hand, a new peak is appeared at 8.3 ppm in P6, which is attributed to iminic hydrogen (CHN).40,41 Noted that we also can observe the aldehyde signal in the spectrum, demonstrating the possibility of dynamic imine bond formation in P6 system (Fig. 5b). Furthermore, the FT-IR spectra of P1–P6 are measured and presented in Fig. 6. In Fig. 6a, we compared the P1 and P6, the peak at 1670 cm−1 corresponding to the aldehyde group (CO) and the peak at 1658 cm−1 corresponding to the imine group (CN), respectively, indicating the Schiff base linkage formed that resulted from the condensation reaction between amine and aldehyde in P6.41,42 The peaks in the range of 1200–1000 cm−1 could be identified as C–O, C–O–C and C–C bond.43 The amide band around 1600 cm−1, as well as the amine shown two absorption peaks about 3400 and 3200 cm−1, which represent asymmetrical and symmetrical N–H stretching.44 Interestingly, P5 and P6 hydrogels exhibit lower wavenumber shift by ca. 50 cm−1 in the N–H band as compared with that of P1–P4, illustrating P5 and P6 may have intermolecular hydrogen bonding interaction among acrylamide, DF-PEG and poly(acrylamide) (Fig. 6b).29,45,46 We also used heating of the gel at 70 °C to see the reversibility of the interactions (Fig. S2†). By combining the experimental results of NMR and FT-IR, we can conclude that the cooperative effect of Schiff base and intermolecular hydrogen bonding interactions would the driving force for the formation of the self-healing hydrogels.
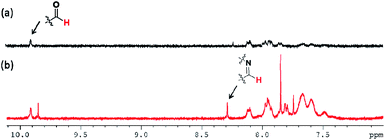 |
| | Fig. 5 NMR spectra of (a) DF-PEG and (b) P6 in D2O. | |
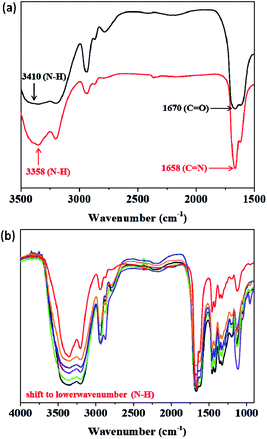 |
| | Fig. 6 FT-IR spectra of (a) P1 (black) and P6 (red) in 3500–1500 cm−1 region. (b) P1–P6 (black for P1; green for P2; violet for P3; indigo for P4; orange for P5; red for P6). | |
3.3 Mechanical properties of hydrogels
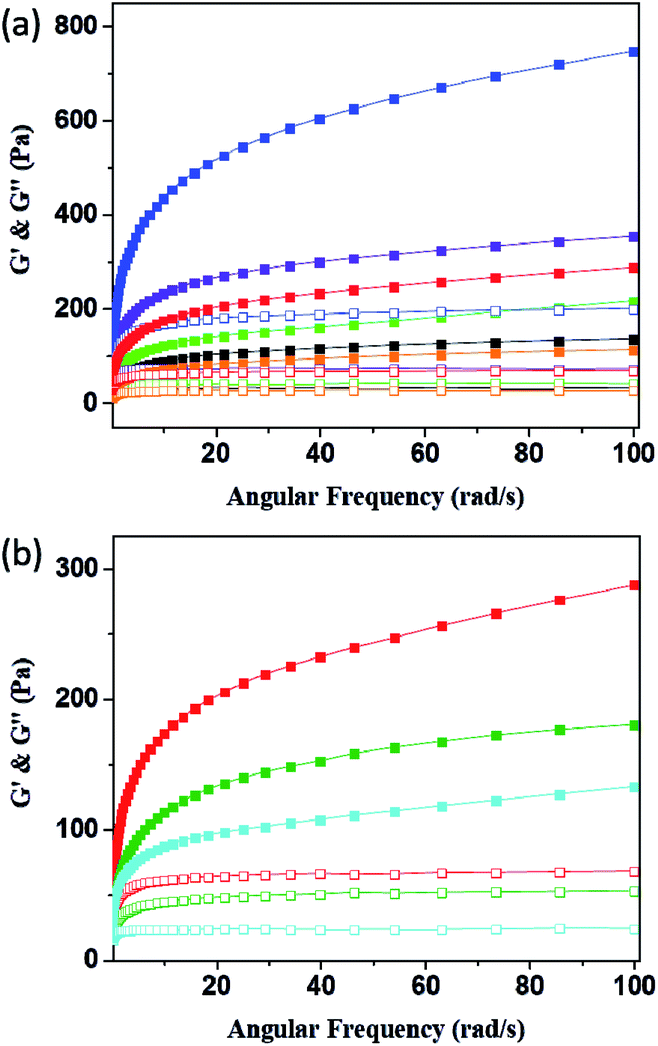
Rheology is the study of deformation and flow of matter, especially liquids and soft matter. The rheology is very important to materials because rheological characterization can give a good indication of the viscoelastic properties that are relevant for final structures of the system.47–49 Fig. 7 and Table 1 revealed the G′ (elasticity) and G′′ (viscosity) of P1–P8 hydrogels as a function of angular frequency within the linear viscoelastic region (ω = 0.1–100 rad s−1). In all samples, the values of G′ increased with increasing sweep frequency as well as the G′′ were lower than G′ in all the frequency range, suggesting they exhibited viscoelastic solid behaviour.50 It is clear from the rheological measurement data that the G′ value can increase when we add DF-PEG and poly(acrylamide) into the 15% w/v acrylamide gel, probably due to the formation of dynamic imine bonds between DF-PEG and acrylamide as well as hydrogen bond interaction between poly(acrylamide) and acrylamide, respectively (P1–P4). We further combine the acrylamide, DF-PEG and poly(acrylamide) in one system to obtain self-healing hydrogels of P5 and P6. Because of the better mechanical property of P6, we prepared the conductive hydrogels of P7 and P8 based on P6. As shown in Fig. 7b, with increasing the amount of KS-6 graphite, the G′ value gradually decreases, probably due to the fact that the doping-induced decrease in the mechanical strength.
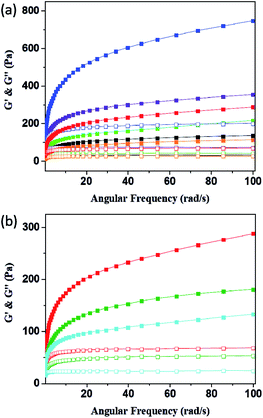 |
| | Fig. 7 Frequency dependent (ω = 0.1–100 rad s−1) of rheology measurement of (a) P1–P6 and (b) P6–P8 hydrogels (solid for G′ and open for G′′; black for P1; green for P2; violet for P3; indigo for P4; orange for P5; red for P6; olive for P7; blue for P8). | |
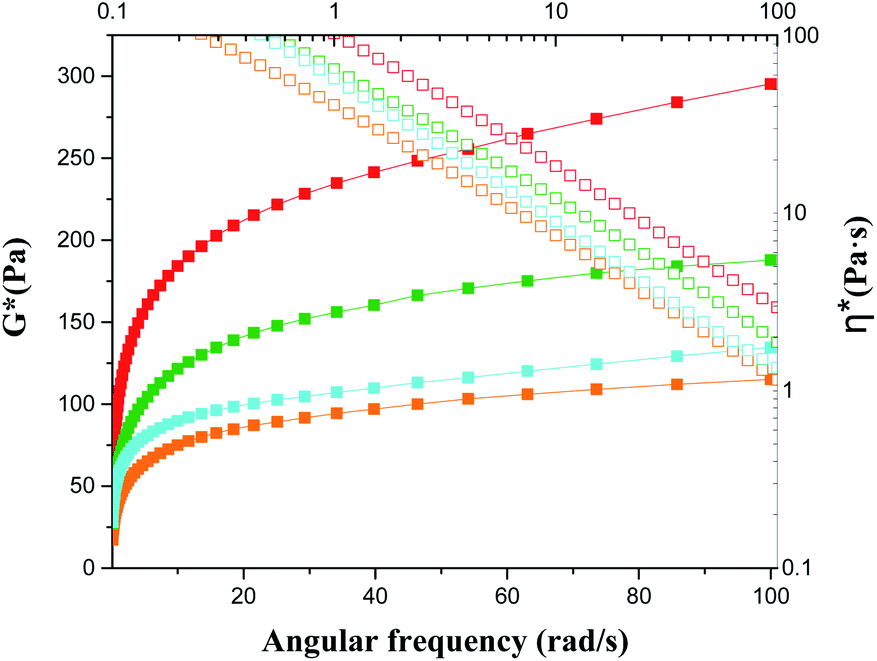
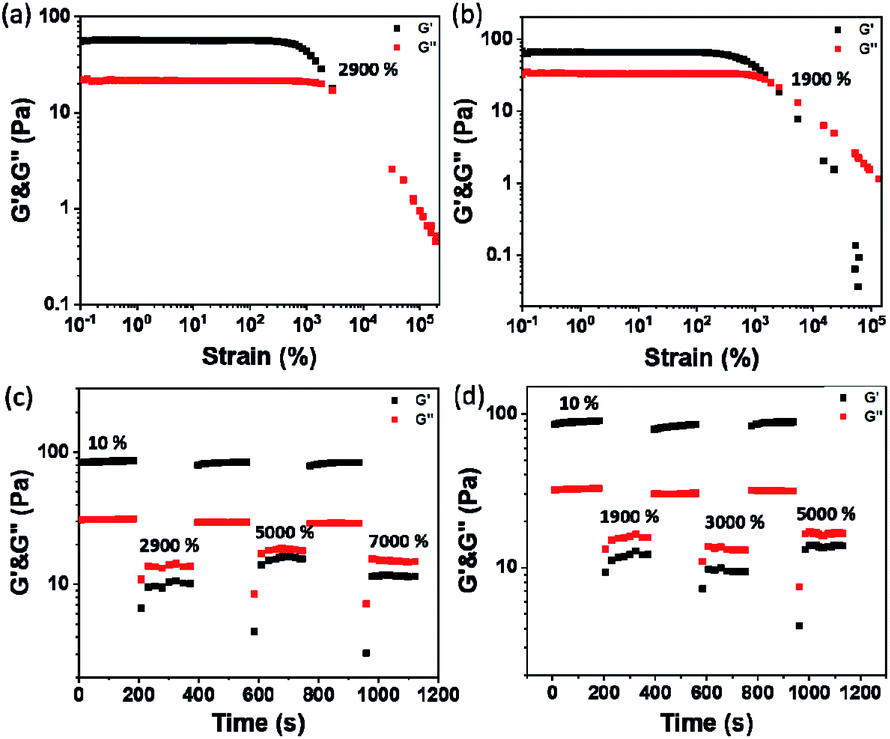
The plots of complex modulus (G*) and complex viscosity (η*) as a function of angular frequency provided a sharper contrast of P5–P8 hydrogels (Fig. 8). We found that P6 and P7 exhibit the higher G* and η* within the whole frequency range, which means better resistance to deformation and better recovery of P6 and P7.51 Moreover, the viscosity of the complex decreases linearly with frequency variation owing to shear-thinning.52 To determine the self-healing efficiency of P6 and P7, we achieved the rheological recovery tests. Fig. 9a and b depicted the strain amplitude sweep of P6 and P7 hydrogels, the G′ and the G′′ curves intersect at the strain of 2900% and 1900%, respectively, indicating that the critical point of transition between solid and fluid state was larger in P6 than in P7 gel. In addition, we assessed the self-recovering of hydrogels by alternate-step strain measurement (Fig. 9c and d). Since a strain deformation of 10% was within the linear viscoelastic range in both gels, we used 10% as a low-magnitude strain to monitor the recovery of the hydrogel structure. The critical point of P6 was 2900%, hence a high-magnitude strain (2900%) was applied to damage the P6 hydrogel network. We observed that the G′ dramatically decreases and becomes smaller than G′′ under large strains (2900%, 5000%, 7000%) and the G′ restores its strength value after strain changes to 10%. Similar results were obtained in P7 hydrogel with the strain cycles (large strain up to 5000%, Fig. 9d). Moreover, the stress–strain mechanical properties of P6 and P7 hydrogels were also investigated. As can be seen from Fig. S3,† P6 and P7 exhibit high elongation-at-break beyond 10000% strain, suggesting they are stretchable materials.53 After being cut into two pieces and self-healed for 1 h, the stress–strain curves of P6 and P7 hydrogels were almost identical to their original counterparts. These results prove that the P6 and P7 hydrogels have excellent self-healing abilities, which are consistent with the observation in Fig. 3c and 4c.
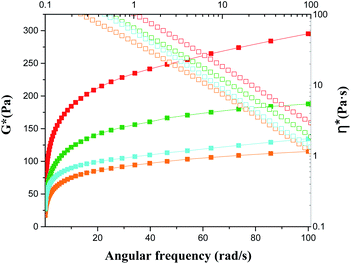 |
| | Fig. 8 Frequency dependent of G* and η* of P5–P8 (solid for G*; open for η*; orange for P5; red for P6; olive for P7; blue for P8). | |
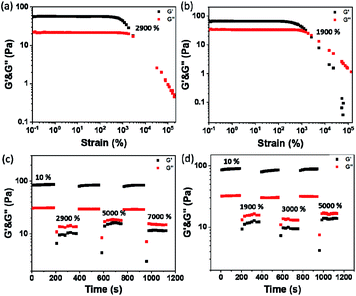 |
| | Fig. 9 Strain dependent of G′ and G′′ of (a) P6 and (b) P7. The cyclic G′ and G′′ values of the (c) P6 and (d) P7 hydrogels at alternate-step strain sweep. | |
3.4 Morphology investigation of hydrogels
From the macroscopic investigation of the hydrogels given in Fig. 3 and 4, it was evident that P6 and P7 exhibited the good self-healing performance. We therefore conducted the microscopic analysis by SEM to better understand the morphology and physical structure of the hydrogels. As can be seen from Fig. 10, the SEM image of P6 showed the layered microstructure, illustrating the elastic properties of P6 hydrogel. While adding the KS-6 graphite into the hybrid hydrogel, the particle-like structures appear in the network, demonstrating the content validity of the carbon black. Notably, P7 still retain the layered morphology, in spite of the adding of 3% w/v carbon black. With increasing the amount of carbon black to 5% w/v, the poor interfacial interaction between the polymer chains (matrix) and the carbon black (filler) was observed and resulted in relatively low G′ compared to that of P7 (Fig. 7b, 10b and c). This result is consistent with the literature report that high filler content may sacrifice the mechanical properties of the composites.38,54,55 Furthermore, the self-healing performance of P6–P8 hydrogels were also investigated by SEM analysis, and shown in insets of Fig. 10. From the SEM images of the scratched area of P6 and P7, the cracks were sealed and healed, which suggest the high performance of self-healing ability. On the contrary, the sample of P8 was partially healed on the scratched region, this result indicate that the conductive hydrogel of P7 has better healing performance than P8. These findings are consistent with the observation of macroscopic features in Fig. 3 and 4.
 |
| | Fig. 10 SEM images of (a) P6, (b) P7 and (c) P8 hydrogels. Inset: SEM images of the scratch test of hydrogels after 1 h (red arrow: scratched region). Scale bar: 10 μm. | |
3.5 Electrical properties of conductive hydrogels
Self-healing materials can improve the durability of many electronic and electrochemical devices, such as electronic skins, sensors, solar cells, energy storage devices and so on.56–58 For the electrical application, we measured the resistances of the P6–P8 hydrogels (before and after self-healed), among them, P7 showed better resistance recovery (Fig. S4†). In addition, the electrical conductivity of original and healed P7 hydrogel is 1.52 × 10−2 and 1.47 × 10−2 S cm−1, respectively, as measured by the four-point probe measurement.59 Since P7 hydrogel exhibited high self-healing capability (Fig. S3, S4,† 4c, 9d and 10b inset), we further examine the P7 hydrogel by connecting it to the light-emitting diode (LED). As displayed in Fig. 11, the LED light up due to the conductive hydrogel of P7 was used. When the P7 gel was cut and the LED was immediately extinguished, and the LED light up again autonomously after healing the gel.
 |
| | Fig. 11 Demonstration of a cutting and healing process for the P7 hydrogel connected in a circuit with a LED lamp. (a) Original sample, (b) cut sample and (c) self-healing sample. | |
4. Conclusions
In summary, we have developed the composite hydrogels of acrylamide, polyacrylamide, DF-PEG and conductive carbon black. After optimizing the composition ratio, the resultant hydrogel of P7 exhibited self-healing reversibility mechanically and electrically when cut and self-healed. We used the 1H NMR and FT-IR spectroscopy to determine the self-healing mechanism of the system, thus demonstrating the cooperative effect of the dynamic covalent (Schiff base reaction) and noncovalent interactions (hydrogen bond) contributes to the self-healing capability of the gel. The rheology, scanning electron microscope and light-emitting diode circuit were carried out to examine its macroscopic and microscopic properties, making it possible to apply in soft and conformable electronics.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported by the Ministry of Science and Technology of the Republic of China, Taiwan (MOST 109–2113-M-033-006-).
Notes and references
- Y. X. Lin, H. Y. Zhang, H. Y. Liao, Y. Zhao and K. Li, Chem. Eng. J., 2019, 367, 139–148 CrossRef CAS.
- T. L. Hu, Y. B. Wu, X. Zhao, L. Wang, L. Y. Bi, P. X. Ma and B. L. Guo, Chem. Eng. J., 2019, 366, 208–222 CrossRef CAS.
- N. Broguiere, L. Isenmann, C. Hirt, T. Ringel, S. Placzek, E. Cavalli, F. Ringnalda, L. Villiger, R. Zullig, R. Lehmann, G. Rogler, M. H. Heim, J. Schuler, M. Zenobi-Wong and G. Schwank, Adv. Mater., 2018, 30, 1801621 CrossRef.
- M. J. Rowland, C. C. Parkins, J. H. McAbee, A. K. Kolb, R. Hein, X. J. Loh, C. Watts and O. A. Scherman, Biomaterials, 2018, 179, 199–208 CrossRef CAS.
- W. J. Zhang, W. G. Xu, C. Ning, M. Q. Li, G. Q. Zhao, W. Q. Jiang, J. X. Ding and X. S. Chen, Biomaterials, 2018, 181, 378–391 CrossRef CAS.
- Y. Yang and M. W. Urban, Chem. Soc. Rev., 2013, 42, 7446–7467 RSC.
- M. D. Hager, P. Greil, C. Leyens, S. van der Zwaag and U. S. Schubert, Adv. Mater., 2010, 22, 5424–5430 CrossRef CAS.
- S. K. Ghosh, Self-Healing Materials: Fundamentals, Design Strategies, and Applications, John Wiley & Sons, Hoboken, NJ, 2009 Search PubMed.
- D. Y. Wu, S. Meure and D. Solomon, Prog. Polym. Sci., 2008, 33, 479–522 CrossRef CAS.
- K. Jud, H. H. Kausch and J. G. Williams, J. Mater. Sci., 1981, 16, 204–210 CrossRef CAS.
- E. Acome, S. K. Mitchell, T. G. Morrissey, M. B. Emmett, C. Benjamin, M. King, M. Radakovitz and C. Keplinger, Science, 2018, 359, 61–65 CrossRef CAS.
- Q. Li, C. Liu, J. Wen, Y. Wu, Y. Shan and J. Liao, Chin. Chem. Lett., 2017, 28, 1857–1874 CrossRef CAS.
- Z. Wei, J. Hai, J. Zhou, F. Xu, M. Zrinyi, P. Dussault, Y. Osada and Y. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- F. Yu, X. Cao, J. Du, G. Wang and X. Chen, ACS Appl. Mater. Interfaces, 2015, 7, 24023–24031 CrossRef CAS.
- W. J. Yang, X. Tao, T. Zhao, L. Weng, E.-T. Kang and L. Wang, Polym. Chem., 2015, 6, 7027–7035 RSC.
- Z. Wei, J. H. Yang, X. J. Du, F. Xu, M. Zrinyi, Y. Osada, F. Li and Y. M. Chen, Macromol. Rapid Commun., 2013, 34, 1464–1470 CrossRef CAS.
- V. Can, Z. Kochovski, V. Reiter, N. Severin, M. Siebenbürger, B. Kent, J. Just, J. P. Rabe, M. Ballauff and O. Okay, Macromolecules, 2016, 49, 2281–2287 CrossRef CAS.
- E. A. Appel, M. W. Tibbitt, M. J. Webber, B. A. Mattix, O. Veiseh and R. Langer, Nat. Commun., 2015, 6, 6295 CrossRef CAS.
- F. Luo, T. L. Sun, T. Nakajima, T. Kurokawa, Y. Zhao, A. B. Ihsan, H. L. Guo, X. F. Li and J. P. Gong, Macromolecules, 2014, 47, 6037–6046 CrossRef CAS.
- Z. Deng, H. Wang, P. X. Ma and B. Guo, Nanoscale, 2020, 12, 1224–1246 RSC.
- B. Zhang, J. He, M. Shi, Y. Liang and B. Guo, Chem. Eng. J., 2020, 400, 125994 CrossRef CAS.
- Y. Liang, X. Zhao, T. Hu, Y. Han and B. Guo, J. Colloid Interface Sci., 2019, 556, 514–528 CrossRef CAS.
- J. He, M. Shi, Y. Liang and B. Guo, Chem. Eng. J., 2020, 394, 124888 CrossRef CAS.
- Z. Deng, T. Hu, Q. Lei, J. He, P. X. Ma and B. Guo, ACS Appl. Mater. Interfaces, 2019, 11, 6796–6808 CrossRef CAS.
- Z. Deng, Y. Guo, X. Zhao, P. X. Ma and B. Guo, Chem. Mater., 2018, 30, 1729–1742 CrossRef CAS.
- J. Kang, J. B.-H. Tok and Z. Bao, Nat. Electron., 2019, 2, 144–150 CrossRef.
- R. N. Dong, X. Zhao, B. L. Guo and P. X. Ma, ACS Appl. Mater. Interfaces, 2016, 8, 17138–17150 CrossRef CAS.
- L. Han, X. Lu, M. H. Wang, D. L. Gan, W. L. Deng, K. F. Wang, L. M. Fang, K. Z. Liu, C. W. Chan, Y. H. Tang, L. T. Weng and H. P. Yuan, Small, 2017, 13, 1601916 CrossRef.
- Q. Ding, X. Xu, Y. Yue, C. Mei, C. Huang, S. Jiang, Q. Wu and J. Han, ACS Appl. Mater. Interfaces, 2018, 10, 27987–28002 CrossRef CAS.
- J. Zhang, C. Wu, Y. Xu, J. Chen, N. Ning, Z. Yang, Y. Guo, X. Hu and Y. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 40990–40999 CrossRef CAS.
- M. H. Liao, P. B. Wan, J. R. Wen, M. Gong, X. X. Wu, Y. G. Wang, R. Shi and L. Q. Zhang, Adv. Funct. Mater., 2017, 27, 1703852 CrossRef.
- Y. M. Wang, F. R. Huang, X. B. Chen, X. W. Wang, W. B. Zhang, J. Peng, J. Q. Li and M. L. Zhai, Chem. Mater., 2018, 30, 4289–4297 CrossRef CAS.
- Y. Liu, Y.-H. Hsu, A. P.-H. Huang and S.-h. Hsu, ACS Appl. Mater. Interfaces, 2020, 12, 40108–40120 CrossRef CAS.
- S.-H. Shin, W. Lee, S.-M. Kim, M. Lee, J. M. Koo, S. Y. Hwang, D. X. Oh and J. Park, Chem. Eng. J., 2019, 371, 452–460 CrossRef CAS.
- F. Ding, Y. Zou, S. Wu and X. Zou, Polymer, 2020, 206, 122907 CrossRef CAS.
- L. J. Macdougall, M. M. Pérez-Madrigal, J. E. Shaw, M. Inam, J. A. Hoyland, R. O'Reilly, S. M. Richardson and A. P. Dove, Biomater. Sci., 2018, 6, 2932–2937 RSC.
- I. Mironi-Harpaz and M. Narkis, J. Appl. Polym. Sci., 2001, 81, 104–115 CrossRef CAS.
- Z. Zhou, S. Wang, Y. Zhang and Y. Zhang, J. Appl. Polym. Sci., 2006, 102, 4823–4830 CrossRef CAS.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS.
- M. E. Bracchi and D. A. Fulton, Chem. Commun., 2015, 51, 11052–11055 RSC.
- S.-H. Lee, S.-R. Shin and D.-S. Lee, Mater. Des., 2019, 172, 107774 CrossRef CAS.
- P.-C. Zhao, W. Li, W. Huang and C.-H. Li, Molecules, 2020, 25, 597 CrossRef CAS.
- Y. Zhang, L. Tao, S. Li and Y. Wei, Biomacromolecules, 2011, 12, 2894–2901 CrossRef CAS.
- Y. Zheng, Y. Liang, D. Zhang, X. Sun, L. Liang, J. Li and Y.-N. Liu, ACS Omega, 2018, 3, 4766–4775 CrossRef CAS.
- C. N. R. Rao, G. C. Chaturvedi and R. K. Gosavi, J. Mol. Spectrosc., 1968, 28, 526–535 CrossRef CAS.
- S. Schäfer and G. Kickelbick, Macromolecules, 2018, 51, 6099–6110 CrossRef.
- H. A. Barnes, Rheol. Rev., 2003, 1–36 Search PubMed.
- D. T. N. Chen, Q. Wen, P. A. Janmey, J. C. Crocker and A. G. Yodh, Annu. Rev. Condens. Matter Phys., 2010, 1, 301–322 CrossRef CAS.
- S. O. Ilyin, V. G. Kulichikhin and A. Y. Malkin, Rheol. Acta, 2016, 55, 223–233 CrossRef CAS.
- M.-Y. Yeh, J.-Y. Zhao, Y.-R. Hsieh, J.-H. Lin, F.-Y. Chen, R. D. Chakravarthy, P.-C. Chung, H.-C. Lin and S.-C. Hung, RSC Adv., 2017, 7, 21252–21257 RSC.
- H. Fazaeli, H. Behbahani, A. A. Amini, J. Rahmani and G. Yadollahi, Adv. Mater. Sci. Eng., 2012, 406791 Search PubMed.
- L. Zhong, M. Oostrom, M. J. Truex, V. R. Vermeul and J. E. Szecsody, J. Hazard. Mater., 2013, 244–245, 160–170 CrossRef CAS.
- Z. Lei and P. Wu, Nat. Commun., 2019, 10, 3429 CrossRef.
- F. Gubbels, R. Jerome, P. Teyssie, E. Vanlathem, R. Deltour, A. Calderone, V. Parente and J. L. Bredas, Macromolecules, 1994, 27, 1972–1974 CrossRef CAS.
- M. Narkis, G. Lidor, A. Vaxman and L. Zuri, J. Electrost., 1999, 47, 201–214 CrossRef CAS.
- Y. J. Tan, J. Wu, H. Li and B. C. K. Tee, ACS Appl. Mater. Interfaces, 2018, 10, 15331–15345 CrossRef CAS.
- M. C. LeMieux and Z. Bao, Nat. Nanotechnol., 2008, 3, 585–586 CrossRef CAS.
- D. H. Kim, J. H. Ahn, W. M. Choi, H. S. Kim, T. H. Kim, J. Song, Y. Y. Huang, Z. Liu, C. Lu and J. A. Rogers, Science, 2008, 320, 507 CrossRef CAS.
- S. Liu, M. Kang, K. Li, F. Yao, O. Oderinde, G. Fu and L. Xu, Chem. Eng. J., 2018, 334, 2222–2230 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09476e |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*