
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceVisible-light enabled room-temperature dealkylative imidation of secondary and tertiary amines promoted by aerobic ruthenium catalysis†
Dong Yang,
Jingqi Shi,
Jiaming Chen,
Xiaoqi Jia,
Cuiying Shi,
Lifang Ma and
Ziyuan Li *
*
Department of Pharmaceutical and Biological Engineering, School of Chemical Engineering, Sichuan University, Chengdu 610065, China. E-mail: liziyuan@scu.edu.cn
First published on 25th May 2021
Abstract
Employing sulfonyl azide as a nitrogen donor, a visible-light-enabled aerobic dealkylative imidation of tertiary and secondary amines involving C(sp3)–C(sp3) bond cleavage with moderate to excellent yields at room temperature is described. It has been demonstrated that this imidation could take place spontaneously upon visible-light irradiation, and could be facilitated considerably by a ruthenium photocatalyst and oxygen. An alternative mechanism to the previous aerobic photoredox pathway has also been proposed.
Introduction
Imine1 and, particularly, amidine2 are among the most ubiquitous and pivotal moieties in miscellaneous ligands, organocatalysts, functional materials, synthetic building blocks, bioactive molecules and pharmaceutics. Beside the most traditional approach through condensation between carbonyl and amine/amide to gain access to such significant scaffolds, various methodologies through transition-metal catalysed direct C–N cross-coupling of amine via C–H cleavage have been reported in the past decades.3 Alternatively, it has also been widely reported that imine and imide could be acquired from azide instead of amine or amide.4Among these methods with azide, imidation of the aliphatic chain of tertiary amine with sulfonyl azide through C(sp3)–C(sp3) bond cleavage (Scheme 1a)5 has drawn our attention for C(sp3)–C(sp3) cleavage under mild conditions because it is generally challenging due to its high bond-dissociation energy and low polarity. In 2008, Li et al. disclosed a pioneering azo-mediated imidation of tertiary amines with sulfonyl azide at room temperature,5a followed by a couple of reports on the same transformation under mild conditions with other oxidants beside the azo compound, such as FeCl3 (ref. 5b) and peroxides,5c or at high temperatures,5d while sulfamide could serve as a surrogate of sulfonyl azide in the presence of NBS5e or peroxide.5f Apart from these imidations through stoichiometric employment of oxidant, homogenous or heterogeneous copper-catalysed imidation of tertiary amine with sulfonyl azide has also been reported.5g–j Meanwhile, such imidation could not only be achieved with tertiary amine via C–C cleavage as illustrated above, it can also be realized on secondary amine involving C(sp3)–N cleavage by electrochemical anodic oxidation (Scheme 1b),5k in which single electron transfer (SET) oxidation of tertiary or secondary amine to form nitrogen-centred radical cation has been proposed to initiate this imidation. Tertiary enamine 1 has been suggested to be the common intermediate for both tertiary and secondary amine, dividing the mechanism of these reaction into two processes: a dehydrogenation process to give this enamine intermediate, and a 1,3-dipolar cycloaddition-decomposition process to afford the final amidine product.5
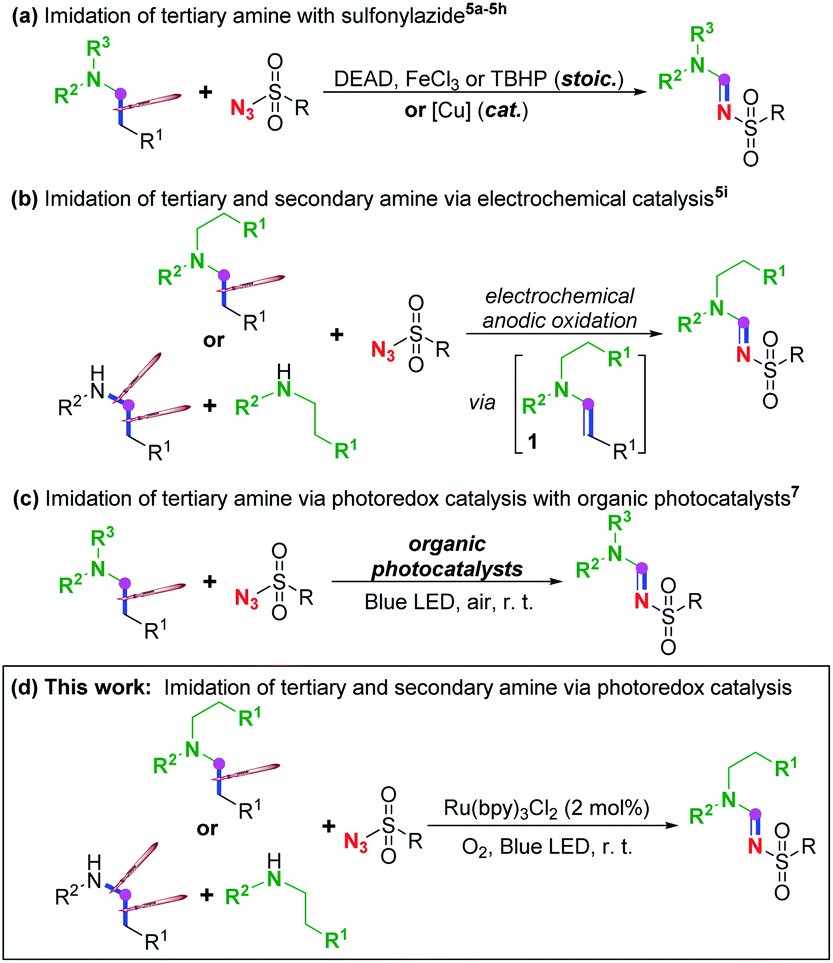 | ||
| Scheme 1 Imidation of amine with sulfonylazide. | ||
Our attention was then emphasized on other means beside electrochemical oxidation for the SET oxidation to generate nitrogen-centred radical cation that initiates such dealkylative imidation, and photoredox catalysis6 which provides a potent synthetic strategy at room temperature and ambient pressure is considered to be the most efficient and green solution. Recently, Zeng7a and Pan7b disclosed two separate studies on visible-light-induced sulfonylimidation of tertiary amine using Eosin Y and acridinium salt, respectively, as organic photocatalysts in air (Scheme 1c),7 based on an earlier report in which it had been proved that enamine 1 can be engendered from tertiary amine through photocatalysis.8 However, employing secondary amine, such as diethylamine, instead of tertiary amine afforded sulfonylated diethylamine7a rather than the amidine product.5k Such sulfonyl azide participated imidation of secondary amine via photoredox catalysis is still unrealized.
Herein, as a continuation of our previous efforts on imidation9 and other methodologies on the synthesis of nitrogen-containing compounds,10 we would like to report our recent study on photoredox dealkylative imidation of tertiary and secondary amine with sulfonyl azide facilitated by aerobic ruthenium-catalysis to afford sulfonyl amidine at room temperature (Scheme 1d).
Results and discussions
Our investigation commenced with the optimization of the reaction conditions employing triethylamine (TEA, 2a) as the tertiary amine substrate, as summarized in Table 1.
|
||
|---|---|---|
| Entry | Changes to standard conditionsa | Yieldb (%) |
| a Reaction conditions: TEA 2a (0.5 mmol), TsN3 (1.5 mmol), Ru(bpy)3Cl2 (0.01 mmol) in 1,4-dioxane (2 mL) at room temperature in O2 (1 atm) under 12 W blue LED irradiation for 6 hours.b Isolated yields of 4a based on 2a. | ||
| 1 | None | 91 |
| 2 | In EtOAc (2 mL) | 25 |
| 3 | In DCE (2 mL) | 73 |
| 4 | In DME (2 mL) | 76 |
| 5 | In THF (2 mL) | 70 |
| 6 | In toluene (2 mL) | 32 |
| 7 | In MeNO2 (2 mL) | 25 |
| 8 | In H2O (2 mL) | 18 |
| 9 | With Ir(ppy)3 (2 mol%) | 48 |
| 10 | In open air | 76 |
| 11 | In Ar (1 atm) | 49 |
| 12 | No photocatalyst | 29 |
| 13 | No photocatalyst in Ar (1 atm) | 27 |
| 14 | No light | 0 |
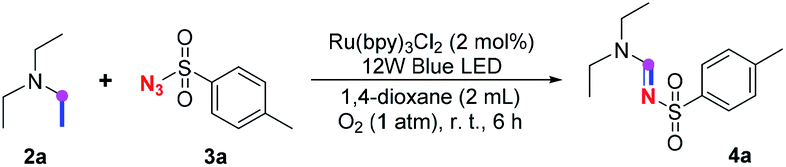
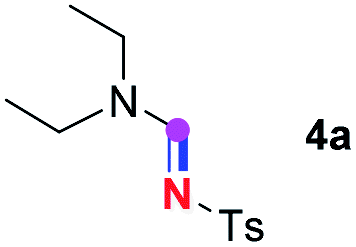
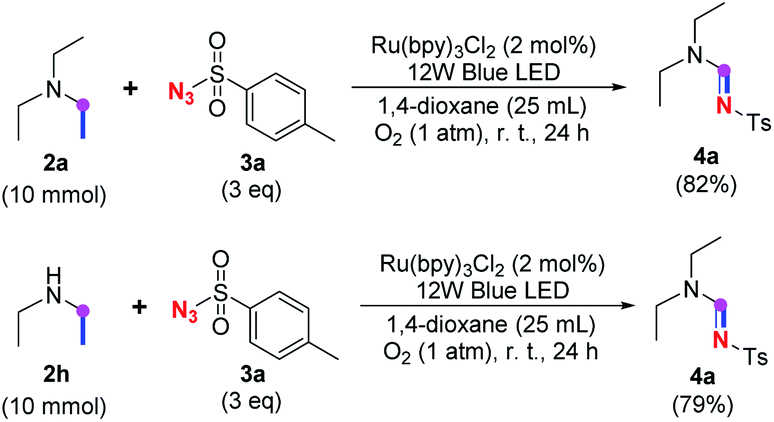
When 2a and tosyl azide (TsN3, 3a) was subjected to the photocatalyst Ru(bpy)3Cl2 in 1,4-dioxane under O2 at room temperature, amidine 4a was obtained in an excellent yield of 91% after 6 hours (entry 1). A series of widely used solvents were tested instead of 1,4-dioxane (entries 2–8), but none of them was found to provide 4a in competent yields comparing to 1,4-dioxane. The yield of 4a declined dramatically if the photocatalyst was replaced with Ir(ppy)3 (entry 9). When the reaction was conducted in open air (entry 10), 4a was acquired in a decreased yield of 76%, while the yield of 4a further declined to 49% under argon (entry 11), demonstrating that the molecular oxygen could considerably promoted this imidation, although it is not prerequisite. Interestingly, the desired amidine product could still been generated, albeit in relatively low yields, in the absence of the photocatalyst either in O2 (entry 12) or under argon (entry 13), whereas no 4a was engendered in the absence of LED irradiation even with the photocatalyst and O2 (entry 14), suggesting that this imidation with azide might spontaneously take place upon visible-light irradiation and neither photocatalyst nor molecular oxygen is prerequisite.
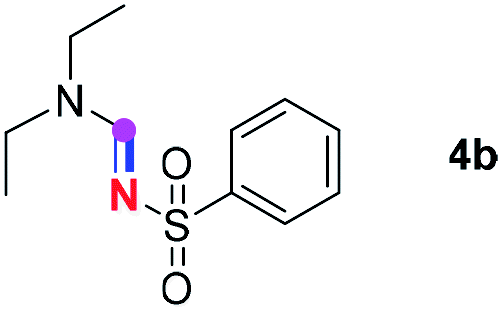
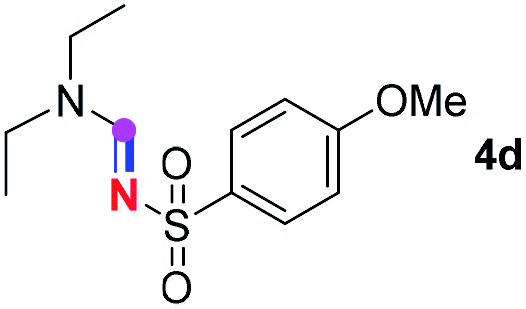
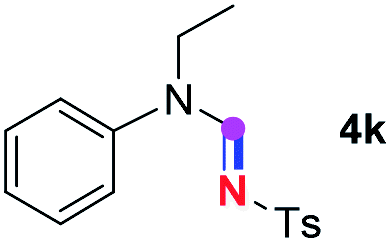
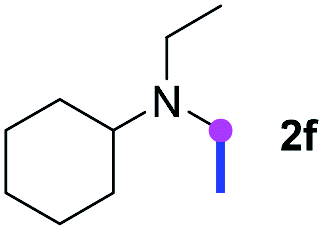
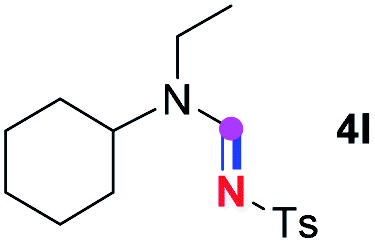
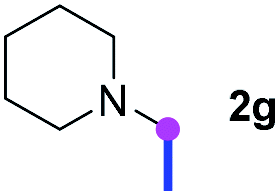
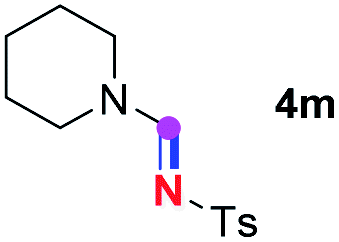
Subsequently, the scope of amine 2 and sulfonyl azide 3 was explored, as shown in Table 2 and 3. With regard to the photoredox imidation of tertiary amine (Table 2), a series of benzenesulfonyl azides 3a–3g with diverse para-substitutions on the benzene ring were tested under the optimized conditions (entries 1–7), and the excellent yields of corresponding amidines were generally maintained, except for amidine 4d obtained from the electron-rich sulfonyl azide 3d with strongly electron-donating methoxyl substituent. Reactions between other acyclic aliphatic tertiary amines and tosyl azide 3a also provided amidines 4h–4j in excellent yields, whereas dialkyl aniline 2e afforded amidine 4k in a moderate yield. In addition, two cyclic tertiary amines 2f and 2g could generate the desired product 4l and 4m, respectively, in synthetically acceptable yields.
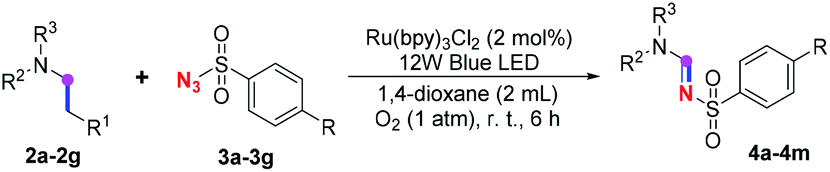
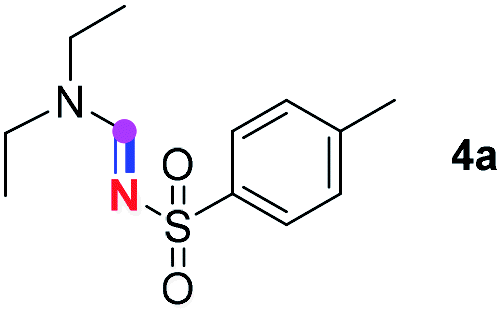
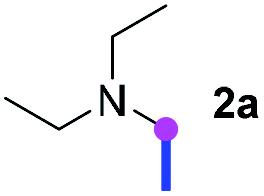
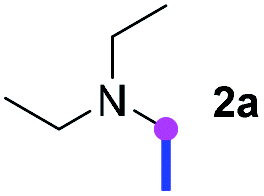
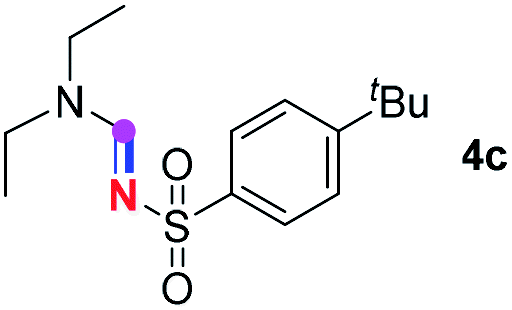
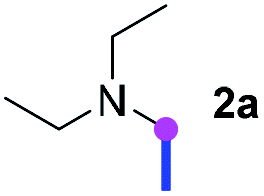
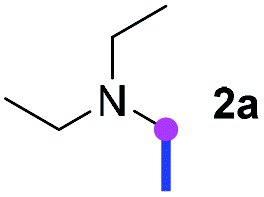
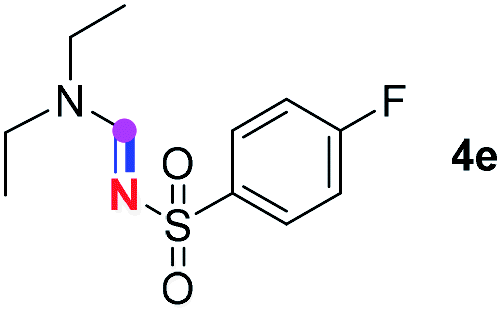
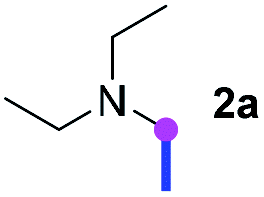
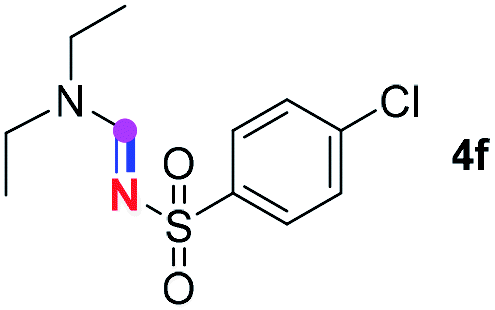
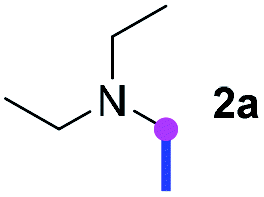
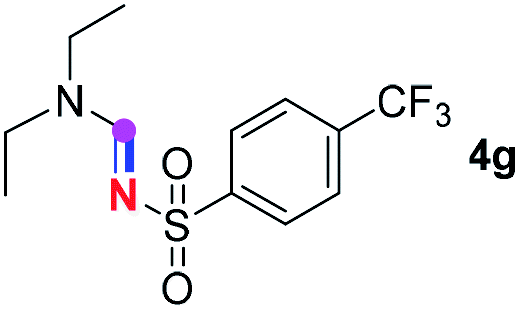
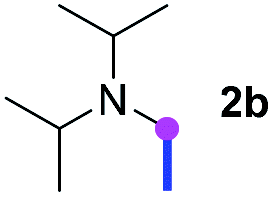
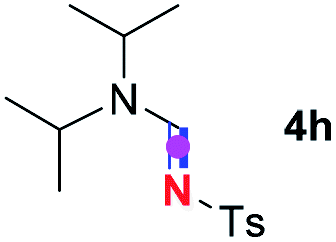
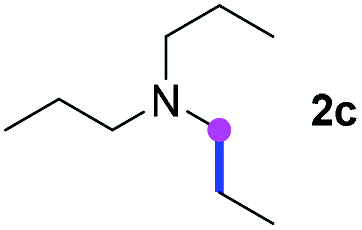
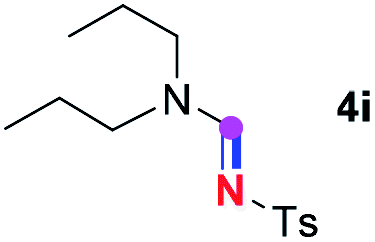
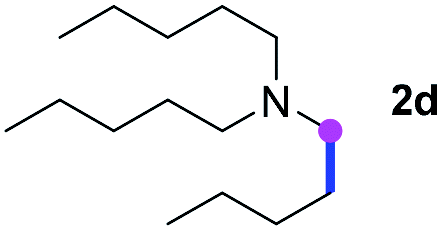
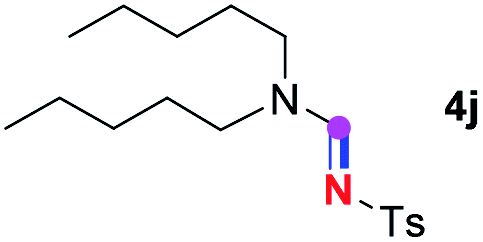
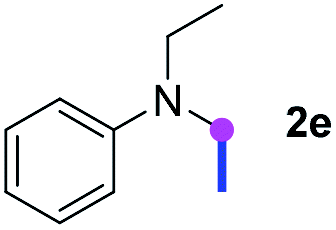
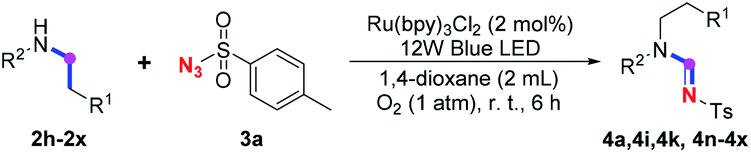
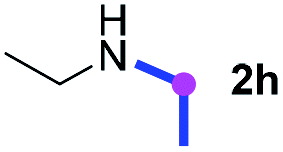
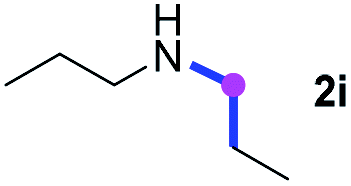
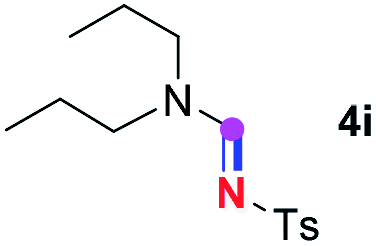
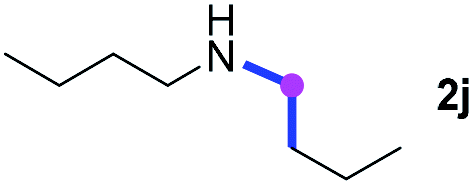
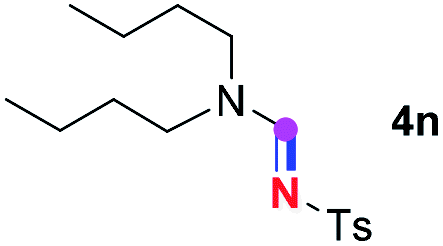
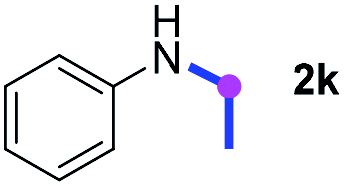
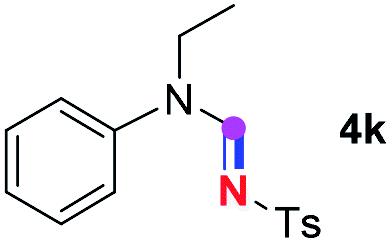
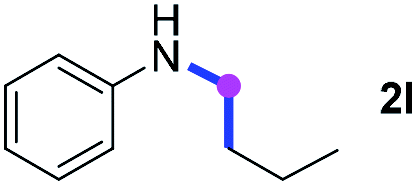
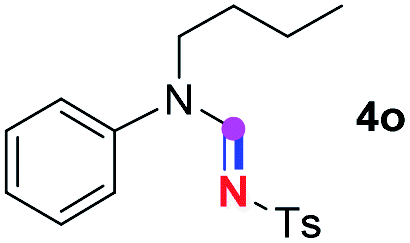
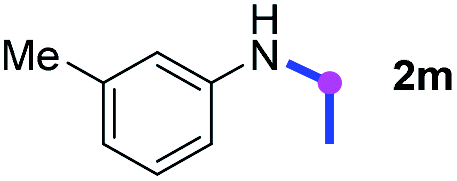
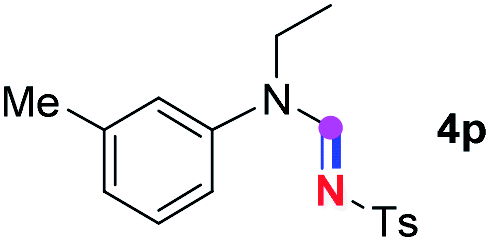
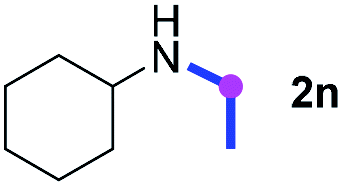
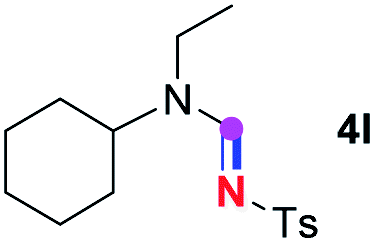
Meanwhile, several secondary amines were investigated, as illustrated in Table 3. Since two molecules of secondary amine could provide only one molecule of the amidine product according to the mechanism proposed in previous work on the electrochemical imidation,5k the photoredox imidation of 0.5 mmol of the secondary amines could theoretically afford 0.25 mmol of corresponding amidines at most, and the yields in Table 3 were calculated accordingly. Results shown in Table 3 suggested that a series of secondary amines 2h–2n were well tolerated with the optimized conditions established on tertiary amines, providing amidines 4a, 4i, 4k, 4l and 4n–4p in moderate to excellent yields which are comparable to their tertiary amine siblings, although some of them required prolonged reaction time to guarantee the good yields. In addition, two gram-scale reactions with triethylamine (2a) and diethylamine (2h), respectively, under the optimized conditions have been conducted as shown in Scheme 2, and the good yields could be generally maintained.
 | ||
| Scheme 2 Gram scale reactions. | ||
Furthermore, the spontaneity of this visible-light enabled imidation in the absence of oxygen and photocatalyst (entry 13, Table 1) attracted our attentions, for such observation suggested that an anaerobic pathways to realize this imidation probably coexist with the aerobic photoredox pathway proposed by Zeng and Pan,7 where oxygen was proposed to serve a vital role in dehydrogenation. To gain more insights into the mechanistic picture, some control experiments were conducted, especially focusing on identification of byproducts generated along with the amidine product (Table 4).
|
||||||
|---|---|---|---|---|---|---|
| Entry | O2/Ar (1 atm) | [Ru] (mol%) | Yieldb (%) | |||
| 4a | 5a | 5b | 5c | |||
| a Reaction conditions: TEA 2a (0.5 mmol), TsN3 (1.5 mmol), Ru(bpy)3Cl2 (0.01 or 0 mmol) in 1,4-dioxane (2 mL) at room temperature in O2 or argon (1 atm) under 12 W blue LED irradiation for 6 hours.b Isolated yields of 4a and 5a–c based on 2a.c Without LED irradiation.d With TEMPO (2.5 mmol).e Without 2a. | ||||||
| 1 | O2 | 2 | 86 | 89 | 7 | 6 |
| 2 | Ar | 2 | 52 | 90 | 5 | 8 |
| 3 | O2 | 0 | 25 | 45 | 6 | 0 |
| 4 | Ar | 0 | 26 | 49 | 5 | 0 |
| 5c | O2 | 2 | 0 | 0 | 0 | 0 |
| 6d | O2 | 2 | 0 | 0 | 0 | 0 |
| 7e | O2 | 2 | 0 | 0 | 0 | 0 |
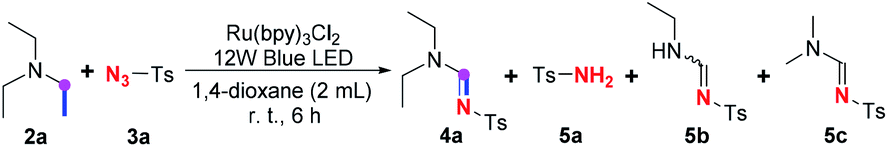
It is discovered that sulfonamide 5a, dealkylated11 amidine 5b, and amidine 5c with an exchanged amino group12 could be obtained under standard conditions (entry 1). However, when the imidation was conducted without oxygen, the yield of amidine 4a dramatically declined, whereas the yields of all of the three byproducts 5a–c generally maintained (entry 2). On the contrary, in the absence of the photocatalyst, 4a and 5a could only be acquired in very low yields regardless if the reactions were conducted in oxygen or argon, while no 5c was obtained, suggesting that the ruthenium catalyst is crucial to the generation of these compounds. Furthermore, no amidine 4a was engendered without visible-light irradiation (entry 5) or with TEMPO (entry 6), and neither were all of the three byproducts. A blank control experiment in the absence of amine 2a also suggested that none of these byproducts were converted from the solvent, 1,4-dioxane (entry 7).
Previous studies on the imidation of amine with sulfonyl azide have almost reached a consensus that essential measures or reagents, such as heating, external oxidant, transition-metal catalyst, electrochemical anodic oxidation and/or photoredox process with photocatalyst, are necessary to the dehydrogenation of amine to form the transient enamine intermediate 1.5,7 Nevertheless, Zhu13 has discovered that perfluoroalkanesulfonyl azide could readily decompose even at 0 °C in the presence of either tertiary or secondary amine via an intermolecular single electron transfer (SET) process between the azide and the amine, giving corresponding sulfonamide and N-substituted enamine equivalently. The formation of byproduct 5a, therefore, suggested that, merely induced by visible-light irradiation at room temperature (entry 4, Table 4), a similar intermolecular SET process between sulfonylazide and amine could take place spontaneously without any photocatalyst and oxidant. It has also been demonstrated that such SET process can be facilitated by the ruthenium catalyst without oxygen, due to the amount of 5a acquired in entry 2 that indicated most of 2a could be converted to the enamine intermediate 1a with the assistance of the ruthenium catalyst in the absence of oxygen. On the contrary, it seems that oxygen cannot promote the spontaneous formation of enamine 1a (entry 3), and the previously proposed aerobic photocatalysis pathway7 seems to contribute little in this ruthenium-promoted visible-light induced imidation, since the yield of 5a indicated that most of 1a was engendered through the above anaerobic SET process even in aerobic environment (entry 1). According to the above observations, it could be concluded that oxygen might be unnecessary to the conversion from amine 2a to enamine 1a, while it might enhance the following cycloaddition and ring-opening decomposition process, comparing the yields of 4a in entries 1 and 2.
To better understand the roles of visible-light, oxygen and the photocatalyst in the subsequent 1,3-dipolar cycloaddition and instantaneous ring-opening process to give the final product, a series of control experiments using a less active but stable and commercially available N-substituted enamine 1b as the substrate were performed (Table 5). Enamine 1b was converted to corresponding amidine product 4q in 31% yield under the standard conditions (entry 1), indicating that this visible-light enabled imidation is probably achieved through enamine intermediate as previous studies proposed.5,7 The yield of 4q significantly declined when this transformation took place in the absence of oxygen (entry 2), confirming the crucial role of oxygen in the cycloaddition and decomposition process which is suggested by the results of control experiments shown in Table 4. However, only trace amount of amidine 4q could be observed on TLC when the reaction was conducted in the absence of the ruthenium photocatalyst or without visible-light irradiation (entries 3–6), demonstrating that the cycloaddition and decomposition process, although it might occur spontaneously with a extremely low rate (entry 6),14 could be considerably facilitated by aerobic ruthenium photocatalysis (entry 1).
|
||||
|---|---|---|---|---|
| Entry | O2/Ar (1 atm) | [Ru] (mol%) | LED irradiation | Yieldb (%) |
| a Reaction conditions: 1b (0.5 mmol), 3a (1.5 mmol), Ru(bpy)3Cl2 (0.01 or 0 mmol) in 1,4-dioxane (2 mL) at room temperature in O2 or argon (1 atm) under 12 W blue LED irradiation or in dark for 24 hours.b Isolated yields of 4q based on 1b. | ||||
| 1 | O2 | 2 | Yes | 31 |
| 2 | Ar | 2 | Yes | 11 |
| 3 | O2 | 0 | Yes | Trace |
| 4 | Ar | 0 | Yes | Trace |
| 5 | O2 | 2 | No | Trace |
| 6 | Ar | 0 | No | Trace |
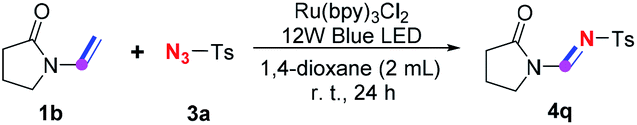
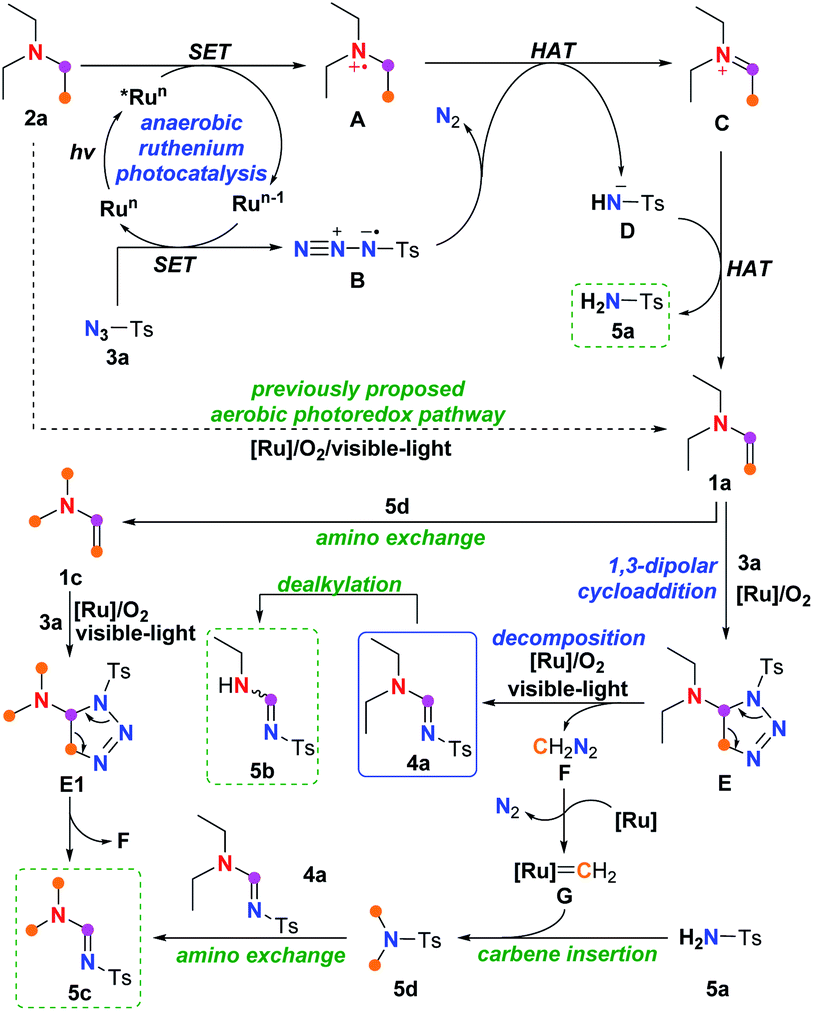
Beside the proposed aerobic photoredox mechanism,7 a plausible alternative mechanism was therefore proposed according to the experimental observations discussed above and previously reported literatures.5,7,13,14 This mechanism comprises two processes: the anaerobic ruthenium-promoted photoredox SET dehydrogenation process to form enamine intermediate; and the cycloaddition and ring-opening decomposition process facilitated by aerobic ruthenium photocatalysis to afford the final amidine product (Scheme 3).
 | ||
| Scheme 3 Plausible alternative mechanism for imidation of tertiary amine beside the aerobic photoredox pathway. | ||
Initially, SET oxidation of amine 2a by the excited state *Run produced via photoexcitation of the higher valent ruthenium catalyst forms tertiary amine radical cation A,7 along with the reduced ruthenium species Run−1, which undergoes another SET process with the azide TsN3, giving azide radical anion B5c,13 and closing the ruthenium catalytic cycle. In the absence of the ruthenium catalyst, the above SET process from amine 2a to sulfonyl azide could take place spontaneously upon visible-light irradiation (entry 4, Table 4), albeit with lower efficiency, and such automatic SET process could also be realized at high reaction temperature5c or with perfluoroalkanesulfonyl azide.13 Then, hydrogen atom transfer (HAT) from the amine radical cation A to the azide radical anion B,13 along with the loss of N2 from radical anion B, generates iminium C7 and sulfonimide anion D5c,13. The iminium C would be deprotonated again by the sulfonimide anion D to provide the vital enamine intermediate 1a, along with equivalent sulfonamide byproduct 5a. Considering that approximately 90% of the starting amine 2a was converted to 1a and 5a no matter if the reaction was conducted under aerobic or anaerobic conditions (entries 1 and 2, Table 4), the previously proposed aerobic photoredox pathway in which oxygen was required both in deprotonation of amine 2a and in reoxidation of the organic photocatalysts,7 might contribute little to this ruthenium-promoted visible-light enabled imidation.
Eventually, 1,3-dipolar addition of enamine 1a with tosyl azide 3a engenders a triazoline intermediate E, followed by decomposition of triazoline E to release diazomethane F,14 along with the final amidine product 4a, which might undergo oxidative dealkylation11 to generated the de-ethylated byproduct 5b. Results shown in Table 5 indicated that such cycloaddition-decomposition process, which might take place spontaneously albeit in very low efficiency (entry 6), could be promoted by aerobic ruthenium photocatalysis (entry 1). Additionally, in the presence of the ruthenium catalyst (entries 1 and 2, Table 4), diazomethane F could be converted to ruthenium carbene species G, which might be trapped by sulfonamide 5a to form a N,N-dimethyl sulfonamide 5d via N–H carbene insertion.15 Amino group exchange12d–f between 5d and the amidine product 4a could directly afford the dimethyl amidine byproduct 5c. Alternatively, such amino exchange might also take place on enamine 1a,12a–c giving dimethyl enamine 1c that also leads to 5c through triazoline E1 by cycloaddition–decomposition. The observation of byproduct 5c, thus, could be considered as an indicator for the generation of diazomethane F.
With regard to secondary amine, control experiments on amine 2h (Scheme 4) suggested that almost all of the secondary amine could be efficiently dehydrogenated through the anaerobic ruthenium promoted photoredox SET process either in the presence (Scheme 4a) or in the absence (Scheme 4b) of oxygen, indicating that the previously proposed aerobic photoredox pathway7 also contributes little to the conversion of secondary amine to give corresponding enamine. Meanwhile, the yield of 4a is higher in the presence of oxygen (Scheme 4a), demonstrating the significance of oxygen to the following cycloaddition-decomposition process as observed in the imidation of tertiary amine.
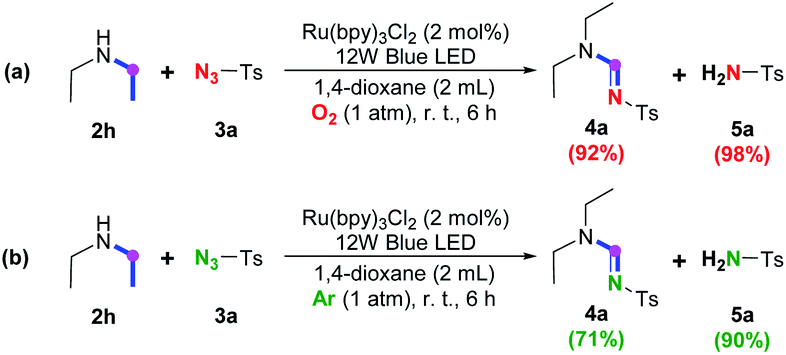 | ||
| Scheme 4 Control experiments on secondary amine 2h. | ||
The mechanism of imidation on secondary amine was therefore proposed as shown in Scheme 5. First, the secondary amine 2h is converted to an enamine intermediate H through the same route proposed in Scheme 3. Unlike previous literature5d,5k in which the amino exchange between enamine H and amine 2h was proposed to lead to the formation of enamine 1a, it might be the amino exchange between two molecules of enamine H that is responsible for the generation of 1a, since most of amine 2h has been converted to enamine H either in aerobic or anaerobic environment according to the control experiments in Scheme 4, leaving little 2h available for the following amino exchange. Finally, enamine 1a undergoes the same cycloaddition-decomposition pathway shown in Scheme 3, leading to the amidine product 4a though triazoline E, along with release of diazomethane F.
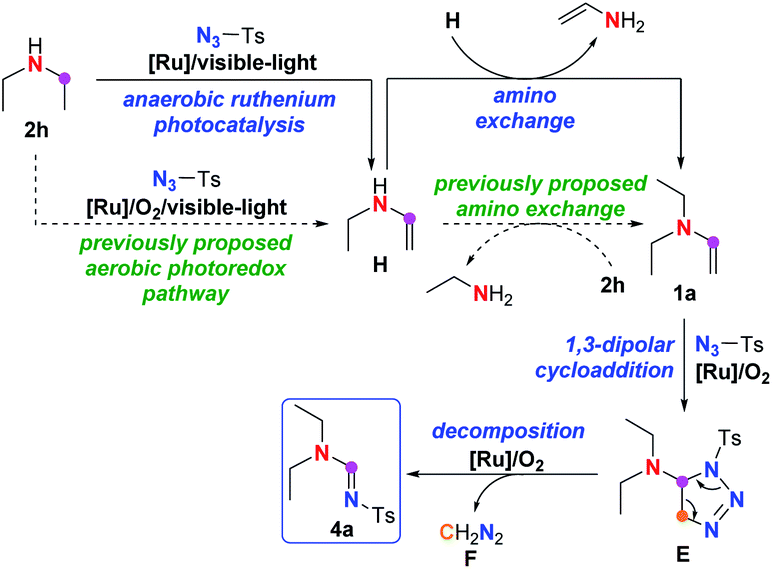 | ||
| Scheme 5 Plausible mechanism for imidation of secondary amine. | ||
Conclusions
To summarize, a photo-induced and aerobic ruthenium-promoted dealkylative imidation of tertiary and secondary amine with sulfonyl azide through C(sp3)–C(sp3) bond cleavage at room temperature has been developed, providing a concise and efficient route toward the significant amidine scaffold under mild conditions. This protocol avoids the employment of metal complexes as the catalyst described previously,5i though both reactions could be realized at room temperature. To our knowledge, this is the first example for photocatalysed imidation of secondary amine with azide. Furthermore, unlike previously proposed mechanism for visible-light-induced imidation with nonmetal organic photocatalysts, a plausible mechanism for this ruthenium photocatalyst participated imidation, featuring the anaerobic ruthenium-promoted photoredox SET dehydrogenation of amine to form the key enamine intermediate and subsequent aerobic ruthenium photocatalysis facilitated cycloaddition-decomposition of this intermediate, have been proposed. Further studies to fully uncover the precise roles of visible-light, oxygen and photocatalyst, as well as application of this mild imidation to the preparation of amidine-based functional molecules, are undergoing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from the National Natural Science Foundation of China (No. 21801177), as well as the NMR technical assistance from the Engineering Experimental Teaching Centre, School of Chemical Engineering, Sichuan University, are highly appreciated. We also thank Zhong Shao and Fang Wang in this group for reproducing the results of 4b, 4h, 4i–l and 4n–p.Notes and references
- (a) J. Brawley, E. Etter, D. Heredia, A. Intasiri, K. Nennecker, J. Smith, B. M. Welcome, R. K. Brizendine, T. W. Gould, T. W. Bell and C. Cremo, Synthesis and evaluation of 4-hydroxycoumarin imines as inhibitors of class II myosins, J. Med. Chem., 2020, 63, 11131 CrossRef CAS PubMed; (b) Z. Dong, Z. Wang, Z.-Q. Guo, S. Gong, T. Zhang, J. Liu, C. Luo, H. Jiang and C.-G. Yang, Structure–activity relationship of SPOP inhibitors against kidney cancer, J. Med. Chem., 2020, 63, 4849 CrossRef CAS PubMed; (c) K. E. Archer, E. E. Reid, M. Shizuka, J. Woods, L. Harris, E. K. Maloney, L. M. Bartle, O. Ab, A. Wilhelm, Y. Setiady, J. F. Ponte, R. Singh, T. A. Keating, R. V. J. Chari and M. L. Miller, Synthesis of highly potent N-10 amino-linked DNA-alkylating indolinobenzodiazepine antibody–drug conjugates (ADCs), ACS Med. Chem. Lett., 2019, 10, 1211 CrossRef CAS PubMed; (d) R. G. Almeida, W. O. Valenca, L. G. Rosa, C. A. de Simone, S. L. de Castro, J. M. C. Barbosa, D. P. Pinheiro, C. R. K. Paier, G. G. C. de Carvalho, C. Pessoa, M. O. F. Goulart, A. Kharma and E. N. da Silva Junior, Synthesis of quinone imine and sulphur-containing compounds with antitumor and trypanocidal activities: redox and biological implications, RSC Med. Chem., 2020, 11, 1145 RSC.
- (a) R. G. Doveston, R. Steendam, S. Jones and R. J. K. Taylor, Total synthesis of an oxepine natural product, (±)-janoxepin, Org. Lett., 2012, 14, 1122 CrossRef CAS PubMed; (b) M. Y. Lee, M. H. Kim, J. Kim, S. H. Kim, B. T. Kim, I. H. Jeong, S. Chang, S. H. Kim and S.-Y. Chang, Synthesis and SAR of sulfonyl- and phosphoryl amidine compounds as anti-resorptive agents, Bioorg. Med. Chem. Lett., 2010, 20, 541 CrossRef CAS PubMed; (c) G. Brasche and S. L. Buchwald, C–H functionalization/C–N bond formation: copper-catalyzed synthesis of benzimidazoles from amidines, Angew. Chem., Int. Ed., 2008, 47, 1932 CrossRef CAS PubMed; (d) Y.-F. Wang, X. Zhu and S. Chiba, Copper-catalyzed aerobic [3 + 2]-annulation of N-alkenyl amidines, J. Am. Chem. Soc., 2012, 134, 3679 CrossRef CAS PubMed; (e) S. Li, Z. Li, Y. Yuan, Y. Li, L. Zhang and Y. Wu, Gold(I)-catalyzed aminohalogenation of fluorinated N′-aryl-N-propargyl amidines for the synthesis of imidazole derivatives under mild conditions, Chem.–Eur. J., 2013, 19, 1496 CrossRef CAS PubMed; (f) S. H. Oakley, D. B. Soria, M. P. Coles and P. B. Hitchcock, Structural diversity in the coordination of amidines and guanidines to monovalent metal halides, Dalton Trans., 2004, 537 RSC.
- (a) J. Kim and S. S. Stahl, Cu-Catalyzed aerobic oxidative three-component coupling route to N-sulfonyl amidines via an ynamine intermediate, J. Org. Chem., 2015, 80, 2448 CrossRef CAS PubMed; (b) I. Bae, H. Han and S. Chang, Highly efficient one-pot synthesis of N-sulfonylamidines by Cu-catalyzed three-component coupling of sulfonyl azide, alkyne, and amine, J. Am. Chem. Soc., 2005, 127, 2038 CrossRef CAS PubMed; (c) J. Kim, S. Y. Lee, J. Lee, Y. Do and S. Chang, Synthetic utility of ammonium salts in a Cu-catalyzed three-component reaction as a facile coupling partner, J. Org. Chem., 2008, 73, 9454 CrossRef CAS PubMed; (d) S. Chang, M. Lee, D. Y. Jung, E. J. Yoo, S. H. Cho and S. K. Han, Catalytic one-pot synthesis of cyclic amidines by virtue of tandem reactions involving intramolecular hydroamination under mild conditions, J. Am. Chem. Soc., 2006, 128, 12366 CrossRef CAS PubMed; (e) Q. Gou, Z. Liu, T. Cao, X. Tan, W. Shi, M. Ran, F. Cheng and J. Qin, Copper-catalyzed coupling of sulfonamides with alkylamines: synthesis of (E)-N-sulfonylformamidines, J. Org. Chem., 2020, 85, 2092 CrossRef CAS PubMed; (f) B. Huang, C. Yang, J. Zhou and W. Xia, Electrochemically generated N-iodoaminium species as key intermediates for selective methyl sulphonylimination of tertiary amines, Chem. Commun., 2020, 56, 5010 RSC.
- (a) S. Y. Chow and L. R. Odell, Synthesis of N-sulfonyl amidines and acyl sulfonyl ureas from sulfonyl azides, carbon monoxide, and amides, J. Org. Chem., 2017, 82, 2515 CrossRef CAS PubMed; (b) Z. Y. Gu, Y. Liu, F. Wang, X. Bao, S. Y. Wang and S. J. Ji, Cobalt(II)-catalyzed synthesis of sulfonyl guanidines via nitrene radical coupling with isonitriles: a combined experimental and computational study, ACS Catal., 2017, 7, 3893 CrossRef CAS; (c) J. Y. Kim, S. H. Kim and S. Chang, Highly efficient synthesis of α-amino amidines from ynamides by the Cu-catalyzed three-component coupling reactions, Tetrahedron Lett., 2008, 49, 1745 CrossRef CAS.
-
(a) X. Xu, X. Li, L. Ma, N. Ye and B. Weng, An unexpected diethyl azodicarboxylate-promoted dehydrogenation of tertiaryamine and tandem reaction with sulfonyl azide, J. Am. Chem. Soc., 2008, 130, 14048 CrossRef CAS PubMed;
(b) S. Wang, Z. Wang and X. Zheng, Facile synthesis of sulfonylamidines via carbon-nitrogen bond formation mediated by FeCl3, Chem. Commun., 2009, 7372 RSC;
(c) A. Rouzi, R. Hudabaierdi and A. Wusiman, Synthesis of N-sulfonylformamidines by tert-butyl hydroperoxide-promoted, metal-free, direct oxidative dehydrogenation of aliphatic amines, Tetrahedron, 2018, 74, 2475 CrossRef CAS;
(d) B. Kaboudin, S. Torabi, F. Kazemi and H. Aoyama, Transition metal- and catalyst-free one-pot green method for the synthesis of N-sulfonyl amidines via direct reaction of sulfonyl azides with amines, RSC Adv., 2020, 10, 26701 RSC;
(e) J. Chen, Y.-P. Guo, M.-H. Sun, G.-T. Fan and L. Zhou, Bromoform reaction of tertiary amines with N,N-dibromosulfonamides or NBS/sulfonamides, Chem. Commun., 2014, 50, 12367 RSC;
(f) Y. Zheng, J. Mao, J. Chen, G. Rong, D. Liu, H. Yan, Y. Chi and X. Xu, Unexpected C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond formation via NaI-catalyzed oxidative de-tetra-hydrogenative cross-couplings between N,N-dimethyl aniline and sulfamides, RSC Adv., 2015, 5, 50113 RSC;
(g) X. Xu, Z. Ge, D. Cheng, L. Ma, C. Lu, Q. Zhang, N. Yao and X. Li, CuCl/CCl4-Promoted convenient synthesis of sulfonyl amidines from tertiary amines and sulfonyl azides, Org. Lett., 2010, 12, 897 CrossRef CAS PubMed;
(h) N. Liu, B.-Y. Tang, Y. Chen and L. He, Catalyzed imidation of tertiary amines by simple copper salts, Eur. J. Org. Chem., 2009, 2059 CrossRef;
(i) S. Shojaei, Z. Ghasemi and A. Shahrisa, Three-component synthesis of N-sulfonylformamidines in the presence of magnetic cellulose supported N-heterocyclic carbene–copper complex, as an efficient heterogeneous nanocatalyst, Tetrahedron Lett., 2017, 58, 3957 CrossRef CAS;
(j) W.-Z. Bi, W.-J. Zhang, Z.-J. Li, X.-Y. Xia, X.-L. Chen, L.-B. Qu and Y.-F. Zhao, Air-induced one-pot synthesis of N-sulfonylformamidines from sulfonyl chlorides, NaN3, and tertiary/secondary amines, Eur. J. Org. Chem., 2019, 6071 CrossRef CAS;
(k) L. Zhang, J.-H. Su, S. Wang, C. Wan, Z. Zha, J. Du and Z. Wang, Direct electrochemical imidation of aliphatic amines via anodic oxidation, Chem. Commun., 2011, 47, 5488 RSC.
N bond formation via NaI-catalyzed oxidative de-tetra-hydrogenative cross-couplings between N,N-dimethyl aniline and sulfamides, RSC Adv., 2015, 5, 50113 RSC;
(g) X. Xu, Z. Ge, D. Cheng, L. Ma, C. Lu, Q. Zhang, N. Yao and X. Li, CuCl/CCl4-Promoted convenient synthesis of sulfonyl amidines from tertiary amines and sulfonyl azides, Org. Lett., 2010, 12, 897 CrossRef CAS PubMed;
(h) N. Liu, B.-Y. Tang, Y. Chen and L. He, Catalyzed imidation of tertiary amines by simple copper salts, Eur. J. Org. Chem., 2009, 2059 CrossRef;
(i) S. Shojaei, Z. Ghasemi and A. Shahrisa, Three-component synthesis of N-sulfonylformamidines in the presence of magnetic cellulose supported N-heterocyclic carbene–copper complex, as an efficient heterogeneous nanocatalyst, Tetrahedron Lett., 2017, 58, 3957 CrossRef CAS;
(j) W.-Z. Bi, W.-J. Zhang, Z.-J. Li, X.-Y. Xia, X.-L. Chen, L.-B. Qu and Y.-F. Zhao, Air-induced one-pot synthesis of N-sulfonylformamidines from sulfonyl chlorides, NaN3, and tertiary/secondary amines, Eur. J. Org. Chem., 2019, 6071 CrossRef CAS;
(k) L. Zhang, J.-H. Su, S. Wang, C. Wan, Z. Zha, J. Du and Z. Wang, Direct electrochemical imidation of aliphatic amines via anodic oxidation, Chem. Commun., 2011, 47, 5488 RSC. - (a) D. M. Yan, J. R. Chen and W. J. Xiao, New roles for photoexcited Eosin Y in photochemical reactions, Angew. Chem., Int. Ed., 2019, 58, 378 CrossRef CAS PubMed; (b) Q. Q. Zhou, Y. Q. Zou, L. Q. Lu and W. J. Xiao, Visible-light-induced organic photochemical reactions through energy-transfer pathways, Angew. Chem., Int. Ed., 2019, 58, 1586 CrossRef CAS PubMed; (c) H. Jiang and A. Studer, α-Aminoxy-acid-auxiliary-enabled intermolecular radical γ-C(sp3)–H functionalization of ketones, Angew. Chem., Int. Ed., 2018, 57, 1692 CrossRef CAS PubMed; (d) X. Zhang and D. W. C. MacMillan, Direct aldehyde C–H arylation and alkylation via the combination of nickel, hydrogen atom transfer, and photoredox catalysis, J. Am. Chem. Soc., 2017, 139, 11353 CrossRef CAS PubMed; (e) S. Mukherjee, R. A. Garza-Sanchez, A. Tlahuext-Aca and F. Glorius, Alkynylation of Cusp2(O)–H bonds enabled by photoredox-mediated hydrogen-atom transfer, Angew. Chem., Int. Ed., 2017, 56, 14723 CrossRef CAS PubMed; (f) T. Niwa, H. Yorimitsu and K. Oshima, Palladium-catalyzed 2-pyridylmethyl transfer from 2-(2-pyridyl)ethanol derivatives to organic halides by chelation-assisted cleavage of unstrained Csp3–Csp3 bonds, Angew. Chem., Int. Ed., 2007, 46, 2643 CrossRef CAS PubMed.
- (a) J. Gui, H. Xie, H. Jiang and W. Zeng, Visible-light-mediated sulfonylimination of tertiary amines with sulfonylazides involving Csp3–Csp3 bond cleavage, Org. Lett., 2019, 21, 2804 CrossRef CAS PubMed; (b) R. Ding, H. Chen, Y.-L. Xu, H.-T. Tang, Y.-Y. Chen and Y.-M. Pan, Photoinduced cascade reaction of tertiary amines with sulfonyl azides: synthesis of amidine derivatives, Adv. Synth. Catal., 2019, 361, 3656 CrossRef.
- G. Q. Xu, J. T. Xu, Z. T. Feng, H. Liang, Z. Y. Wang, Y. Qin and P. F. Xu, Dual C(sp3)–H bond functionalization of N-heterocycles through sequential visible-light photocatalyzed dehydrogenation/[2 + 2] cycloaddition reactions, Angew. Chem., Int. Ed., 2018, 57, 5110 CrossRef CAS PubMed.
- (a) Q. Miao, Z. Shao, C. Shi, L. Ma, F. Wang, R. Fu, H. Gao and Z. Li, Metal-free C–H amination of arene with N-fluorobenzenesulfonimide catalysed by nitroxyl radicals at room temperature, Chem. Commun., 2019, 55, 7331 RSC; (b) X. Wang, B. Lei, L. Ma, H. Jiao, W. Xing, J. Chen and Z. Li, Iron-catalyzed C(5)–H imidation of azole with N-fluorobenzenesulfonimide, Adv. Synth. Catal., 2017, 359, 4284 CrossRef CAS; (c) B. Lei, X. Wang, L. Ma, Y. Li and Z. Li, NFSI-participated intermolecular aminoazidation of alkene through iron catalysis, Org. Biomol. Chem., 2018, 16, 3109 RSC; (d) B. Lei, Q. Miao, L. Ma, R. Fu, F. Hu, N. Ni and Z. Li, Efficient metal-free aminoiodination of alkenes with N-fluorobenzenesulfonimide under mild conditions, Org. Biomol. Chem., 2019, 17, 2126 RSC.
- (a) Z. Li, X. Huang, F. Chen, C. Zhang, X. Wang and N. Jiao, Cu-Catalyzed concise synthesis of pyridines and 2-(1H)-pyridones from acetaldehydes and simple nitrogen donors, Org. Lett., 2015, 17, 584 CrossRef CAS PubMed; (b) Z. Li, X. Wang, L. Ma and N. Jiao, Copper-catalyzed aerobic oxidation and oxygenation of anilines and acetaldehydes with dioxygen for the concise synthesis of 2-aroylquinolines, Synlett, 2017, 28, 1581 CrossRef CAS; (c) C. Shi, Q. Miao, L. Ma, T. Lu, D. Yang, J. Chen and Z. Li, Room-temperature C–H bromination and iodination with sodium bromide and sodium iodide using N-fluorobenzenesulfonimide as an oxidant, ChemistrySelect, 2019, 4, 6043 CrossRef CAS; (d) Z. Li, L. Ma, J. Xu, L. Kong, X. Wu and H. Yao, Pd(II)-catalyzed direct C5-arylation of azole-4-carboxylates through double C–H bond cleavage, Chem. Commun., 2012, 48, 3763 RSC.
- For dealkylation of tertiary amine, see: (a) S. Kim, J. W. Ginsbach, J. Y. Lee, R. L. Peterson, J. J. Liu, M. A. Siegler, A. A. Sarjeant, E. I. Solomon and K. D. Karlin, Amine oxidative N-dealkylation via cupric hydroperoxide Cu–OOH homolytic cleavage followed by site-specific Fenton chemistry, J. Am. Chem. Soc., 2015, 137, 2867 CrossRef CAS PubMed; (b) C. X. Zhang, H.-C. Liang, E.-I. Kim, Q.-F. Gan, Z. Tyeklar, K.-C. Lam, A. L. Rheingold, S. Kaderli, A. D. Zuberbuhler and K. D. Karlin, Dioxygen mediated oxo-transfer to an amine and oxidative N-dealkylation chemistry with a dinuclear copper complex, Chem. Commun., 2001, 631 RSC; (c) J. P. Ferris, R. D. Gerwe and G. R. Gapski, Detoxication mechanisms. III. Scope and mechanism of the iron-catalyzed dealkylation of tertiary amine oxides, J. Org. Chem., 1968, 33, 3493 CrossRef CAS PubMed; (d) S. B. Karki, J. P. Dinnocenzo, J. P. Jones and K. R. Korzekwa, Mechanism of oxidative amine dealkylation of substituted N,N-dimethylanilines by cytochrome P-450: application of isotope effect profiles, J. Am. Chem. Soc., 1995, 117, 3657 CrossRef CAS; (e) J. R. L. Smith and D. N. Mortimer, Model systems for cytochrome P450-dependent mono-oxygenases. Part 5. Amine oxidation. Part 17. Oxidative N-dealkylation of tertiary amines by metalloporphyrin-catalysed model systems for cytochrome P450 mono-oxygenases, J. Chem. Soc., Perkin Trans. 2, 1986, 1743 RSC; for dealkylation of imine, see: (f) D. E. Fogg and B. R. James, Net amine dealkylation at a diruthenium center: dehydrogenation of a secondary amine and hydrolysis of a coordinated imine, Inorg. Chem., 1995, 34, 2557 CrossRef CAS.
- For amino exchange occurring on enamine, see: (a) R. F. Abdulla, K. H. Fuhr and J. C. Williams Jr, New synthetic approaches to 4(1H)-pyridinone derivatives. 3. 2-Bromoacetylated enamines as pyridine ring synthons, J. Org. Chem., 1979, 44, 1349 CrossRef CAS; (b) Y. Zhang, S. Xie, M. Yan and O. Ramstrom, Dynamic covalent chemistry of aldehyde enamines: BiIII- and ScIII-catalysis of amine–enamine exchange, Chem.–Eur. J., 2017, 23, 11908 CrossRef CAS PubMed; (c) J. T. Gupton, N. Telang, X. Jia, B. C. Giglio, J. E. Eaton, P. J. Barelli, M. Hovaizi, K. E. Hall, R. S. Welden, M. J. Keough, E. F. Worrall, K. L. Finzel, E. J. Kluball, R. P. F. Kanters, T. M. Smith, S. Q. Smith, S. R. Nunes, M. T. Wright and J. M. Birnstihl, Further studies on vinamidinium salt amine exchange reactions, borohydride reductions, and subsequent transformations, Tetrahedron, 2010, 66, 8485 CrossRef CAS PubMed; for amino exchange occuring on amidine, see: (d) M. dF. Capela, N. J. Mosey, L. Xing, R. Wang and A. Petitjean, Amine exchange in formamidines: an experimental and theoretical study, Chem.–Eur. J., 2011, 17, 4598 CrossRef CAS PubMed; (e) N. T. Tran and C. S. Cho, An efficient synthesis of benzimidazoles via palladium-catalyzed amine exchange reaction from trialkylamines to o-phenylenediamine in an aqueous medium, Bull. Korean Chem. Soc., 2012, 33, 4188 CrossRef CAS; (f) D. D. Diaz, W. G. Lewis and M. G. Finn, Acid-mediated amine exchange of N,N-dimethylformamidines: preparation of electron-rich formamidines, Synlett, 2015, 2214 Search PubMed.
- Y. Xu and S. Zhu, The reaction of per(poly)fluoroalkanesulfonyl azides with tertiary and secondary amines: generation and trapping of enamines, Tetrahedron, 2001, 57, 4337 CrossRef CAS.
- X. Zheng and J.-P. Wan, The CC bond decomposition initiated by enamine–azide cycloaddition for catalyst- and additive-free synthesis of N-sulfonyl amidines, Adv. Synth. Catal., 2019, 361, 5690 CrossRef CAS.
- For carbene insertion into N–H bond, see: (a) D. Padin, J. A. Varela and C. Saa, Ruthenium-catalyzed tandem carbene/alkyne metathesis/N–H insertion: synthesis of benzofused six-membered azaheterocycles, Org. Lett., 2020, 22, 2621 CrossRef CAS PubMed; (b) W.-S. Huang, Z. Xu, K.-F. Yang, L. Chen, Z.-J. Zheng and L.-W. Xu, Modular construction of multifunctional ligands for the enantioselective ruthenium-catalyzed carbenoid N–H insertion reaction: an enzyme-like and substrate-sensitive catalyst system, RSC Adv., 2015, 5, 46455 RSC; (c) L. Chen, H. Cui, Y. Wang, X. Liang, L. Zhang and C.-Y. Su, Carbene insertion into N–H bonds with size-selectivity induced by a microporous ruthenium–porphyrin metal–organic framework, Dalton Trans., 2018, 47, 3940 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, 1H and 13C NMR data and spectra. See DOI: 10.1039/d0ra10517a |
| This journal is © The Royal Society of Chemistry 2021 |