
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA novel Fe/N/C electrocatalyst prepared from a carbon-supported iron(II) complex of macrocyclic ligands for oxygen reduction reaction†
Dawei Xua,
Yuanyuan Fua,
Dejian Xiaoa,
Xuhui Lia,
Yefei Wanga,
Kai Lia,
Zhongfeng Li a,
Lirong Zhengb and
Xia Zuo*a
a,
Lirong Zhengb and
Xia Zuo*a
aDepartment of Chemistry, Capital Normal University, Beijing 100048, China. E-mail: zuoxia@mail.cnu.edu.cn; 2180702049@cnu.edu.cn
bDepartment Beijing Synchrotron Radiation Facility, Institute of High Energy Physics, Chinese Academy of Sciences, Beijing 100049, P. R. China
First published on 24th February 2021
Abstract
Herein, we report a novel method to synthesize Fe/N/C composites from a carbon-supported iron (II) coordination complex of 2,3-dicyanotetraazabenzotriphenylene (2,3-DCTBT) ligands towards oxygen reduction reaction (ORR) in alkaline media. We investigated the influence of different temperatures during the thermal carbonization process on the performance of the catalyst, and Fe/N/C-900 stood out among all other samples because of the existence of Fe–Nx active sites. The as-obtained Fe/N/C-900 exhibited excellent electrochemical performance (Eonset is 0.98 V vs. RHE in 0.1 M KOH) for the oxygen reduction reaction (ORR) compared with the commercial 20% Pt/C. Moreover, this catalyst possessed good stability and high tolerance to methanol, and followed the four-electron mechanism (n = 3.97). Remarkably, the use of 2,3-DCTBT for the first time as a precursor has opened new avenues to prepare electrocatalysts for ORR.
Introduction
Benefits such as high energy density, environmentally friendly byproducts and high efficiency are the reasons why fuel cells are the most promising energy production devices.1,2 However, large-scale commercial production of fuel cells has been hampered because of the oxygen reduction reaction (ORR) with sluggish kinetics.3,4 Although platinum (Pt)-based catalysts are the most utilized materials for ORR,5 there is still a bottleneck as Pt-based catalysts have relatively high cost for applications.6 Hence, the development of new materials is necessary for fuel cells.Many efforts are devoted to earth-abundant-metal (for example, iron and cobalt)-based electrocatalysts,7 especially electrocatalysts with transition metal–Nx (M–Nx) active sites.8–10 The formation of stable M–Nx metal centers before pyrolysis helps to reduce the loss of active sites during pyrolysis. Although a number of studies have focused on metal porphyrins11,12 and phthalocyanines,13,14 which have been proven to have good catalytic activity towards ORR, there is still considerable interest in other complex tetraaza macrocyclic compounds. For example, Zhang, Z. et al. demonstrated that nitrogen-containing ligands with bisnitrogen-containing groups on various benzene structures could effectively work like crabs to clamp Fe ions via coordination to avoid Fe aggregations. The derived two-dimensional Fe/N codoped catalyst had good catalytic activity (a half-wave potential of 0.82 V and a kinetics current density of 5.41 mA cm−2).15 Liu, R. et al. designed novel triangular trinuclear metal–N4 complexes, which possess large planar conjugation and an unprecedented high density of active sites that prompted the O–O cleavage. Liu, J. et al. loaded 1,10-phenanthroline-cobalt(II) metal-complex onto reduced graphene oxide (rGO) surfaces by π–π interaction to fabricate a new class of catalyst.16 These catalysts obtained from the above macrocyclic precursors showed better methanol resistance and stability, nevertheless, their electrocatalytic activity still needs to be improved.
2,3-Dicyanotetraazatriphenylene (2,3-DCTBT) is a nitrogen-containing organic compound with a conjugated structure. This electron-deficient structure, due to four electronegative nitrogen atoms, has a range of electron acceptor characteristics. The nitrogen atom on the pyrazine ring in the molecule can coordinate with the metal and increase the electron density on the central metal, which could facilitate the adsorption and cleavage of the O–O bond.17 In general, these electrocatalysts suffer from low activity because of bad conductivity. As a result, carbon support materials such as carbon black,18,19 carbon nanotubes (CNTs)20 and graphene21,22 are often synthesized with transition metal catalysts that have the capacity to promote charge transport during electrocatalysis. To obtain catalysts with better electrocatalytic performance for ORR, pyrolysing precursor compounds turns into a strategy. In addition, numerous reports have shown that the formation of active sites relates to the pyrolysing temperature.23,24
In this work, we synthesized Fe/N/C composites by hydrothermal treatment to obtain a carbon-supporting iron(II) coordination complex of 2,3-dicyanotetraazabenzotriphenylene (2,3-DCTBT) ligands. To study the influence of the pyrolysis temperature on the oxygen reduction performance of the catalyst, Fe/N/C series was prepared at different pyrolysis temperatures. The resulting Fe/N/C samples were characterized by transmission electron microscopy (TEM), high resolution transmission electron microscopy (HRTEM), X-ray diffraction (XRD), Raman spectroscopy and X-ray photoelectron spectroscopy (XPS). The electrochemical performance of the catalyst in alkaline media was investigated using electrochemical techniques such as cyclic voltammetry (CV), linear sweep voltammetry (LSV), and chronoamperometry (CA). Especially, X-ray absorption near-edge structure (XANES) and X-ray absorption fine structure (EXAFS) techniques were used to further prove the coordination structure and active sites of the catalysts for ORR.
Experimental
Chemicals
XC-72 (Vulcan), FeSO4·7H2O (Shanghai Sinopharm Chemical Reagent Co Ltd), N,N-dimethylformamide (Beijing Chemical Works), Nafion (5%) (Sigma) and absolute ethyl alcohol (Beijing Chemical Works) were purchased. All the chemicals were of AR grade and used as received without further purification. Commercial 20 wt% Pt/C electrocatalyst was supplied by Sigma-Aldrich.Synthesis of Fe–N–C catalyst
2,3-DCTBT was prepared by a typical synthesis process according to a published work.40 Subsequently, 0.3320 g (0.001 mol) 2,3-DCTBT was dispersed in 10 mL N,N-dimethylformamide (DMF) under the protection of N2. 0.2780 g FeSO4·7H2O was dissolved in 5 mL ultra-pure water and was added to the above DMF solution. The mixture was heated and refluxed for 3 h and then 0.1 g carbon black (XC-72) was added under continuous stirring for 2 h. Next, the obtained powder was placed in a tubular furnace and pyrolyzed at 900 °C for 2 h with a heating rate of 3 °C min−1 in an Ar atmosphere. After cooling to room temperature, the product was soaked in 50 mL 3 mol L−1 sulfuric acid to remove intermediates and impurities, including the Fe species. The precipitate was dried overnight under vacuum at 80 °C and the obtained sample was recorded as Fe/N/C-900. Further, following the above synthesis method, products were also prepared at different pyrolysis temperatures (800 °C, 1000 °C), and the obtained samples were recorded as Fe/N/C-800 and Fe/N/C-1000, respectively.Material characterization
The morphology of the sample was studied using transmission electron microscopy (TEM, Hitachi, Japan) and high-resolution transmission electron microscopy (HR-TEM, FEI Tecnai F20, USA). The TEM sample was dispersed in ethanol and then added dropwise onto carbon-supported films. X-ray diffraction (XRD) patterns were obtained on a D8 Advance/Bruker diffractometer (Germany) employing Cu Kα radiation source (λ = 0.1541 nm). The specific surface area was evaluated using the surface area and pore size analyzer (NOVA 1000e); X-ray photoelectron spectroscopy (XPS) (ESCALAB 250, UK) was performed using a monochromatic Al Kα radiation as the excitation source. X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) constituting XAFS spectroscopy studies were performed at the Beijing Synchrotron Radiation Facility. The Fe K-edge absorption spectra were collected in the fluorescence mode using a double-crystal Si (111) monochromator.Electrochemical measurements
All electrochemical analyses were performed using a three-electrode system controlled by an SP-200 electrochemistry station (Bio-logic SA, France). The reference electrode was a saturated calomel electrode (SCE) and the counter electrode was a platinum sheet electrode. The working electrode was a rotating disk electrode (RDE) (Pine Research Instrument, USA). The disk electrode was a glassy carbon disk with a diameter of 5.0 mm and the area of the disk electrode was 0.196 cm−2.The working electrode was prepared following a drop-casting method. Typically, the catalyst ink was prepared as follows: 3 mg electrocatalyst and 10 μL of 5 wt% Nafion were added to 490 μL anhydrous ethanol. The ink was kept under ultrasonic irradiation for at least 2 h. Then, 10 μL of the catalyst ink was dropped onto the surface of the RDE and dried at room temperature. The reference electrocatalyst of Pt/C for ORR was prepared using the same method with uniform areal mass loading. The measurement was carried out in 0.1 M KOH, which was saturated with O2/N2. All the samples were activated with a scan window from −0.2 to 0.8 V vs. SCE and a scan rate of 200 mV s−1 before electrochemical measurements. The ORR reactivity was evaluated using cyclic voltammetry (CV) from −0.20 to 0.8 V vs. SCE at a scan rate of 20.0 mV s−1 and linear sweep voltammetry (LSV) was recorded at 400, 625, 900, 1225 and 1600 rpm at a potential scan rate of 5 mV s−1.
Durability testing was conducted using the chronoamperometric technique in O2-saturated 0.1 M KOH electrolyte with a rotation rate of 1600 rpm. Potential versus SCE was converted to the RHE scale according to E (vs. RHE) = E (vs. SCE) + 0.0591 pH + 0.244. Electrochemical impedance spectroscopy (EIS) measurements were conducted from 1 Hz to 100 MHz, and the magnitude of the modulation signal was 5 mV amplitude. The transfer electron number (n) of the ORR was calculated using the Koutecky–Levich equation as given below:
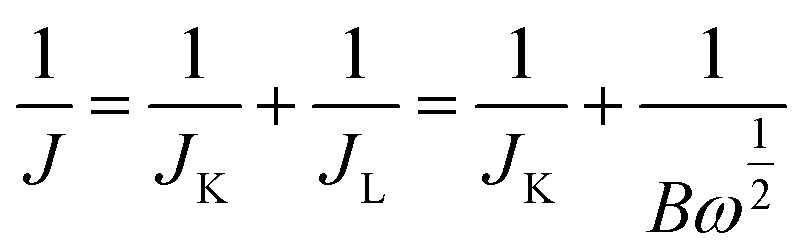 | (1) |
| B = 0.62nFC0(D0)2/3ν−1/6 | (2) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1), C0 is the concentration of oxygen (1.2 × 10−3 mol cm−3), D0 indicates the diffusion coefficient of oxygen (1.9 × 10−5 cm2 s−1), ν is the kinematic viscosity of the electrolyte (ν = 1.07 × 10−2 cm2 s−1). The tolerance to methanol of the as-made Fe–N/C catalyst and Pt/C was estimated by using amperometric current–time chronoamperometric response measurement. Firstly, the i–t chronoamperometric curve was obtained in O2-saturated 0.1 M KOH solution for 600 s at 1600 rpm, followed by the sequential addition of 3 M methanol. Then, the i–t chronoamperometric curve was obtained for another 600 s.
485 C mol−1), C0 is the concentration of oxygen (1.2 × 10−3 mol cm−3), D0 indicates the diffusion coefficient of oxygen (1.9 × 10−5 cm2 s−1), ν is the kinematic viscosity of the electrolyte (ν = 1.07 × 10−2 cm2 s−1). The tolerance to methanol of the as-made Fe–N/C catalyst and Pt/C was estimated by using amperometric current–time chronoamperometric response measurement. Firstly, the i–t chronoamperometric curve was obtained in O2-saturated 0.1 M KOH solution for 600 s at 1600 rpm, followed by the sequential addition of 3 M methanol. Then, the i–t chronoamperometric curve was obtained for another 600 s.
Results and discussion
As shown in Scheme 1, 2,3-DCTBT has four negatively charged nitrogen atoms, which cause this electron-deficient structure to have a range of electron acceptor characteristics. The IR spectrum of 2,3-DCTBT is shown in Fig. S2,† which is consistent with previous reports. The nitrogen atom of pyrazine can coordinate with the Fe after reflux and increase the electron density of the central metal,25 thereby forming a precursor with a clear Fe–N coordination, stacking on the XC-72 surface by π–π conjugation. During the carbonization under different temperature pyrolysis, Fe/N/C electrocatalysts were obtained with various catalytic active species (Fe–Nx, oxidized-N, pyrrole-N, pyridinic-N, graphite-N). Finally, acid leaching removes unstable materials from the material and increases the exposure of active sites.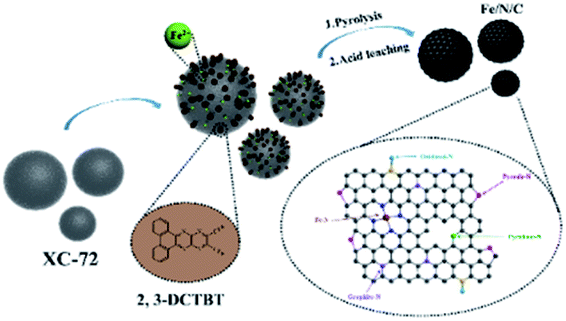 | ||
| Scheme 1 Schematic illustration of the synthetic process of the Fe/N/C electrocatalyst. | ||
The morphologies of the as-synthesized Fe/N/C catalysts were characterized by TEM and SEM (Fig. S3–S5†). Fig. 1a–c depict the TEM images of Fe/N/C-800, Fe/N/C-900, and Fe/N/C-1000 samples, respectively. The Fe/N/C-800 sample reveals an irregular spherical architecture, which confirms that XC-72 has a 2D-layered structure. The Fe/N/C-900 and Fe/N/C-1000 samples exhibit similar morphologies to that of the Fe/N/C-800 sample. An evident crystalline lattice can be observed between the carbon layers from the high-resolution TEM (HRTEM) images in Fig. 1d. The inset of Fig. 1d further displays that the crystalline lattice has a distance of 0.21 nm,26,27 which is slightly larger than the lattice spacing (0.203 nm) of the Fe due to N doping into the lattice gap of Fe during pyrolysis. Some studies reported that trace amounts of iron could promote the improvement of ORR performance. Fe/N/C-900 shows irregular morphology with layered distribution, as shown from the SEM image in Fig. S6a.† The corresponding elemental mapping of C (Fig. S6c†), N (Fig. S6d†), and Fe (Fig. S6e†) confirmed the effective doping of Fe into the materials.
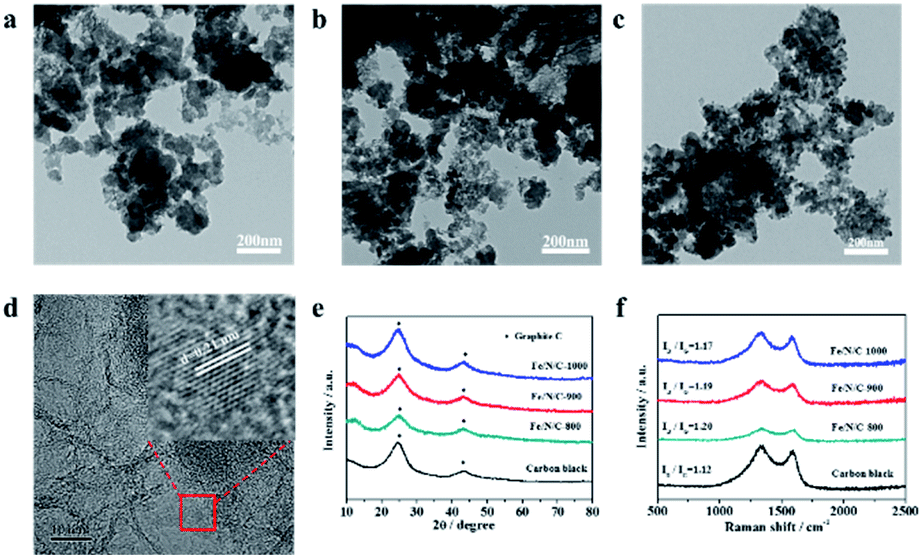 | ||
| Fig. 1 TEM images of (a) Fe/N/C-800, (b) Fe/N/C-900 and (c) Fe/N/C-1000, the HRTEM image of (d) Fe/N/C-800 (inset: the high-magnification image). (e) XRD patterns of Fe/N/C-800, Fe/N/C-900, Fe/N/C-1000 and carbon black. (f) Raman spectra of Fe/N/C-800, Fe/N/C-900, Fe/N/C-1000 and carbon black. | ||
The crystallinity of the as-prepared Fe/N/C series was investigated by XRD. Fig. 1e shows the XRD spectra of Fe/N/C catalysts heated at different annealing temperatures. Regardless of the annealing temperature, the XRD patterns of all samples exhibit two characteristic peaks at 2θ values of 24.6° and 43.1°, which may be assigned to the (002) and (101) planes of graphitic carbon. It is known that the sharpness of the diffraction peak can reflect the degree of graphitization of the catalysts. It is clearly seen that these two peaks become narrower and stronger with increasing temperature, indicating that more ordered graphitic carbon was formed at higher annealing temperature.28,29 Raman spectroscopy was performed to investigate the degree of carbon graphitization and defect of sp2 carbon, which have a great influence on the performance of the catalyst. The D (≈1356 cm−1) and G (≈1580 cm−1) bands are ascribed to the disordered carbon (sp3) and ordered graphitic carbon (sp2), respectively. The Raman spectrum of Fe/N/C is shown in Fig. 1f; the value of ID/IG can be used to analyze the structure intensity of graphene. No other peak was found except two distinct characteristic peaks at 1340 cm−1 and 1590 cm−1. The intensity ratios ID/IG are 1.20, 1.19 and 1.17 for Fe/N/C-800, Fe/N/C-900, and Fe/N/C-1000, respectively. Compared to carbon black (ID/IG = 1.12), the values for the Fe/N/C samples have increased significantly, indicating that defects have occurred after pyrolysis at high temperatures. This may be due to other atoms being doped into the carbon skeleton. The value of ID/IG for the Fe/N/C series decreases as the temperature increases, indicating that the higher the temperature, the higher is the degree of graphitization of the catalyst.30 As a result, the content of carbon in the catalyst increases as the temperature increases, which is in agreement with the XPS analysis mentioned.
X-ray photoelectron spectroscopy (XPS) was used to further characterize the chemical state of nitrogen in the framework. Obviously, Fig. S7† indicates the presence of Fe, N, C, and O, which proves that Fe and N are doped into the catalyst successfully. The chemical state and the content of nitrogen in N-doped catalysts are considered as part of the active sites, which is closely related to the electrochemical catalysis performance. The N 1s spectra of the Fe/N/C catalysts are depicted in Fig. 2a–c, and the results are listed in Table 1. As the pyrolysis temperature was increased, the total nitrogen content decreased. The N 1s spectra of Fe/N/C catalysts can be fitted into five sub-peaks, including pyridinic nitrogen at 398.4 ± 0.1 eV, Fe–N at 399.9 ± 0.1 eV, pyrrolic nitrogen at 400.7 ± 0.1 eV, graphitic nitrogen at 401.4 ± 0.1 eV, and oxidized-N at 402.6 ± 0.1 eV. The presence of Fe–Nx bond results from Fe ions coordinated to N.31
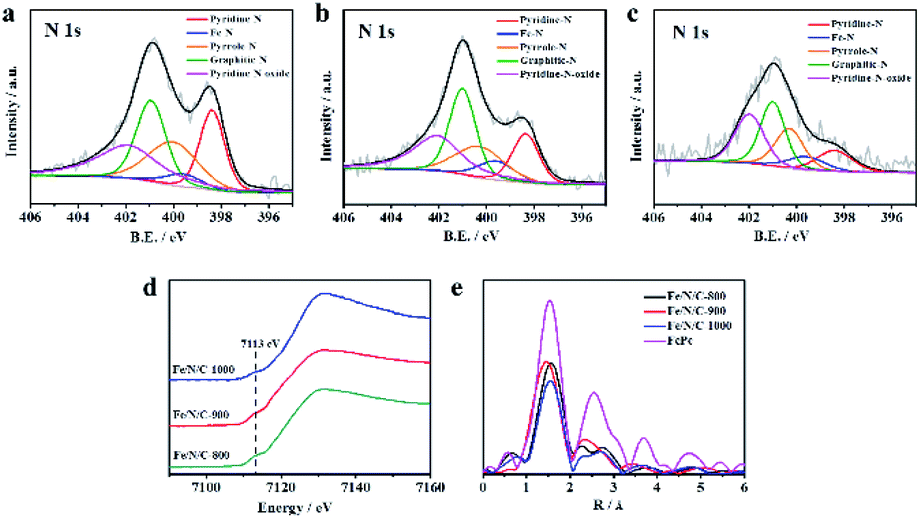 | ||
| Fig. 2 (a) High-resolution XPS N 1s spectra of (b) Fe/N/C-800, (c) Fe/N/C-900 and (d) Fe/N/C-1000. (d) Fe K-edge XANES spectra of Fe/N/C-800, Fe/N/C-900 and Fe/N/C-1000 and (e) Fe K-edge Fourier transform EXAFS spectra of Fe/N/C-800, Fe/N/C-900 and Fe/N/C-1000. | ||
| Sample | Total N (at%) | Pyridinic-N (at%) | Pyrrolic-N (at%) | Graphitic-N (at%) | Pyrrolic-N-oxidized (at%) | Fe–N (at%) |
|---|---|---|---|---|---|---|
| Fe/N/C-800 | 3.12 (100%) | 0.64 (20.56%) | 0.70 (22.47%) | 0.78 (24.93%) | 0.85 (27.15%) | 0.15 (4.89%) |
| Fe/N/C-900 | 2.08 (100%) | 0.34 (16.53%) | 0.40 (19.17%) | 0.59 (28.49%) | 0.56 (26.94%) | 0.18 (8.87%) |
| Fe/N/C-1000 | 0.82 (100%) | 0.10 (12.78%) | 0.19 (23.37%) | 0.23 (28.29%) | 0.22 (26.74%) | 0.07 (8.82%) |
Interestingly, the content of Fe–N in Fe/N/C-900 is higher than that in other catalysts. Therefore, we speculate that the N-coordinated Fe sites may be active sites in electrocatalysis towards ORR. Additionally, the Fe/N/C-900 catalyst has good electrocatalytic performance that is also dependent on the relatively high content of pyridinic nitrogen, pyrrolic nitrogen and graphitic nitrogen, which can accelerate the electron transfer from the carbon electronic bands to the antibonding orbitals of oxygen.32–34 Due to the electron donor properties, some pyridinic-N species provide the position of coordination with Fe atoms for the formation of Fe–N. Therefore, changes in pyridinic-N and Fe–N contents in pyrolysis samples at different temperatures are related to each other. The change in pyrrolic-N and graphitic-N contents is mainly affected by temperature. The high-resolution Fe 2p spectra and C 1s spectra were fitted for Fe/N/C-800, Fe/N/C-900 and Fe/N/C-1000 and are shown in Fig. S8.† Next, the relationship between the coordination structure of Fe in the catalyst obtained at different temperatures and the oxygen reduction catalytic performance of the material was observed from XANES and EXAFS. As shown in Fig. 2d, a weak pre-edge peak at about 7113 eV appeared, which was ascribed to the 1s → 4pz transition with a simultaneous ligand-to-metal charge transfer, indicating that Fe/N/C-900 samples contained N-coordinated Fe as the axial coordination structure. The result is consistent with the high-resolution N 1s peak in XPS, demonstrating the presence of Fe–N bonds. In Fig. 2e, the first strongest peak in the spectrum represents the distance between the coordinating atom and the central atom of the first shell. All the catalysts have a radius of the first coordination layer that is very close to that of iron phthalocyanine, which further proves the presence of N-coordinated Fe. Compared with the peak values of Fe/N/C-1000 (1.53 Å) and Fe/N/C-800 (1.57 Å), the peak value of Fe/N/C-900 (1.45 Å) is the smallest, indicating the shortest coordination distance between Fe–N, which in turn means the highest electron transfer rate in the ORR process; this may be one of the reasons why Fe/N/C-900 has the best electrocatalytic performance.35–37
The nitrogen adsorption–desorption analysis results of nanomaterials in Fig. 3 show that Fe/N/C-800, Fe/N/C-900 and Fe/N/C-1000 exhibit a hysteresis loop of type IV isotherm, indicating the existence of a mesoporous structure. Fe/N/C-900 shows the highest surface area and pore volume (Table S1, ESI†). The size distribution of Barrett–Joyner–Halenda (BJH) pores is concentrated in the range of 2–6 nm, which is attributed to the formed mesopores. The porous structure and high surface area contribute to the electron transfer and diffusion, and maintain oxygen on the cathode surface. The content of substances with catalytic activity for ORR is also a decisive factor that cannot be ignored.
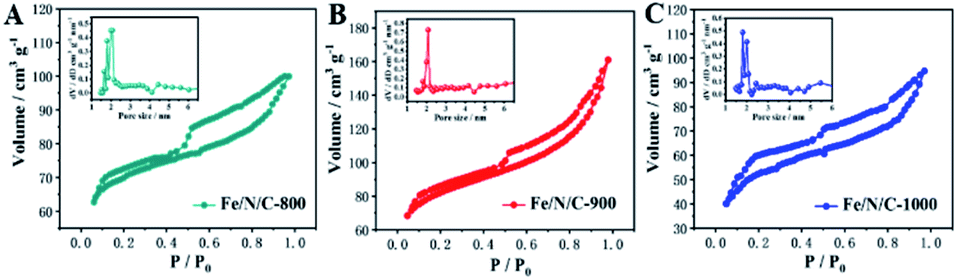 | ||
| Fig. 3 N2 adsorption–desorption isotherms for (A) Fe/N/C-800, (B) Fe/N/C-900 and (C) Fe/N/C-1000. | ||
Fig. 4a shows CV scans of Fe/N/C-800, Fe/N/C-900 and Fe/N/C-1000 in N2- and O2-saturated solutions. All the electrocatalytic data display an irreversible ORR peak when they are in O2-saturated solutions, which prove that Fe/N/C samples have a catalytic effect on the reduction of O2 in alkaline media. Interestingly, a sharp cyclic voltammetric peak of Fe/N/C-900 was obtained at 0.81 V vs. RHE, which is more positive than that of Fe/N/C-800 (0.79 V vs. RHE) and Fe/N/C-1000 (0.80 V vs. RHE). Meanwhile, Fe/N/C-900 also has the largest current density of 2.83 mA cm−2. The LSV at 1600 rpm of the Fe/N/C series and 20% commercial Pt/C for comparison were tested under the same conditions and are shown in Fig. 4b. With a comparably higher limiting current density of 5.36 mA cm−2, Fe/N/C-900 also showed a significantly more positive onset potential (Eonset) and half-wave potential (E1/2) than other Fe/N/C electrocatalysts. The Eonset of Fe/N/C-900 is 0.98 V vs. RHE, which is close to the Eonset of the 20% commercial Pt/C catalyst we tested (1.02 V vs. RHE). These results are superior to most of the relevant catalysts, as shown in Table S2.† As shown in Fig. 4c, the limiting current density rises with increasing rotating rate as a result of accelerated mass transport and the shortened diffusion distance. The corresponding Koutecky–Levich (K–L) plots represent a linear tendency for J−1 and ω1/2 (inset of Fig. 3C).38,39 The number of transferred electrons evaluated by the K–L equation is 3.97, indicating a four-electron pathway. The comparison of the Eonset and E1/2 among all samples and 20% commercial Pt/C is also discussed in Fig. 4d. These results confirm that the ORR performance of Fe/N/C-900 stands out from other prepared electrocatalysts and display favorable behavior even compared to the 20% commercial Pt/C.
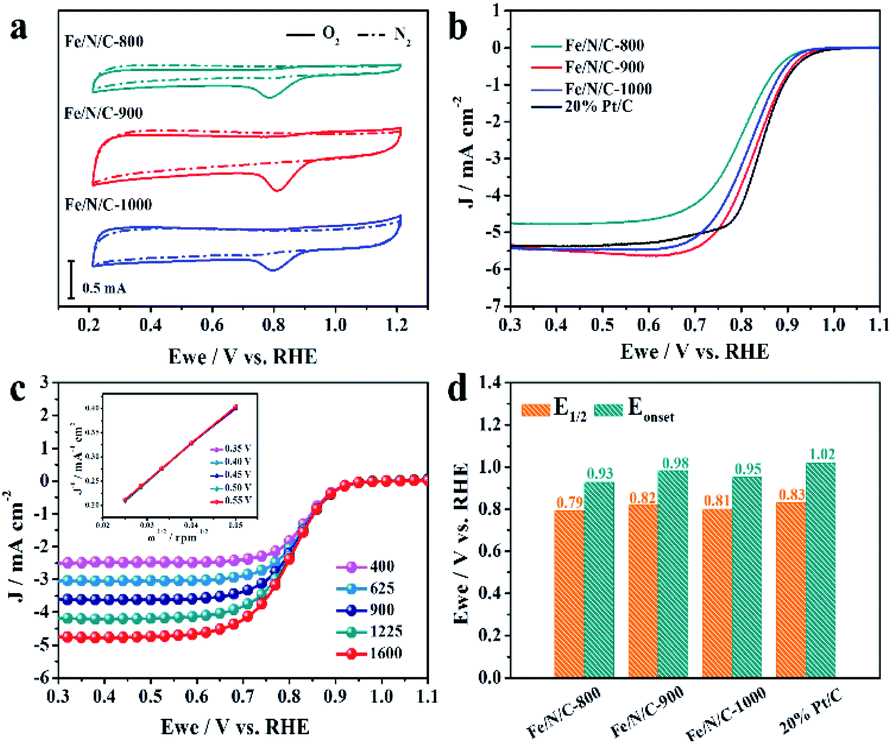 | ||
| Fig. 4 (a) The cyclic voltammetry curves in 0.1 M KOH N2-saturated (dotted lines) and O2-saturated solutions (solid lines) of Fe/N/C-800, Fe/N/C-900 and Fe/N/C-1000. (b) The LSV curves of Fe/N/C-900 and Pt/C in O2 saturated 0.1 M KOH solution. Rotation speed: 1600 rpm. (c) The LSV curves of Fe/N/C-900 in O2 saturated 0.1 M KOH solution and the Koutecky–Levich plots for oxygen reduction on Fe/N/C-900 in O2 saturated 0.1 M KOH solution. Chronoamperometric response for the ORR at Fe/N/C-900 and 20% Pt/C electrodes; (d) the comparison of the Eonset and E1/2 among all samples and the 20% commercial Pt/C. | ||
Tafel plots in Fig. 5a manifest that the ORR kinetics is significantly improved as the pyrolysis temperature increases; Fe/N/C-900 electrocatalyst (97 mV dec−1) over Fe/N/C-1000 (117 mV dec−1) and Fe/N/C-800 (146 mV dec−1). Nevertheless, Fe/N/C-900 affords a small increase in the Tafel slope of 97 mV dec−1 compared with 88 mV dec−1 of Pt/C. Comprehensively, the ORR performance of Fe/N/C-900 is acceptable considering the obvious improvement in comparison with others. We use poisoning experiments to further clarify the effective active sites of the Fe/N/C-900 catalyst. The electrochemical signal was recollected under the same conditions, except for the introduction of 5 mM KSCN. After the introduction of SCN−, the half-wave potential shifted to the negative direction and the limit diffusion current of the catalyst decreased as shown in Fig. 5b. This proved that the electrocatalytic performance of the Fe/N/C-900 obviously weakens towards ORR, which is mainly because effective Fe–N active sites were blocked. Finally, we carried out chronoamperometric (i–t) measurements to evaluate the ORR stability and resistance to methanol, which are of particular importance for ORR applications in fuel cells. The ORR performance of the Fe/N/C-900 catalyst and the commercial 20% Pt/C catalyst were tested in O2-saturated 0.1 M KOH solution at a rotation rate of 1600 rpm. As shown in Fig. 5c, the Fe/N/C-900 catalyst showed high current retention (87.3%) after 20000 s while the ORR current on Pt/C decreased significantly under the same conditions. The Fe/N/C-900 catalyst exhibits better long-term stability for ORR than the commercial 20% Pt/C. After injection of methanol into the electrolyte at 400 s, the relative current of the Fe/N/C-900 catalyst retained 91.2% after 1000 s, as shown in Fig. 5d. The relative current of commercial 20% Pt/C drops obviously. As shown in Fig. 5e, Fe/N/C-900 shows a minor deterioration with only 13 mV negative shift of E1/2 after 10000 s continuous potential cycles, which is related to the fact that the atomically dispersed Fe atoms are firmly anchored to the highly stable multilayer carbon. During the ORR, the Nyquist diagram was obtained at 0.82 V relative to RHE (Fig. 5f). The ORR process on the catalyst is controlled by the charge transfer process. Therefore, compared with Fe/N/C-800 and Fe/N/C-1000, Fe/N/C-900 with smaller charge transfer resistance has better catalytic activity in the catalytic process. Considering these results, the Fe/N/C-900 catalyst is promising for the replacement of commercial Pt/C for ORR.
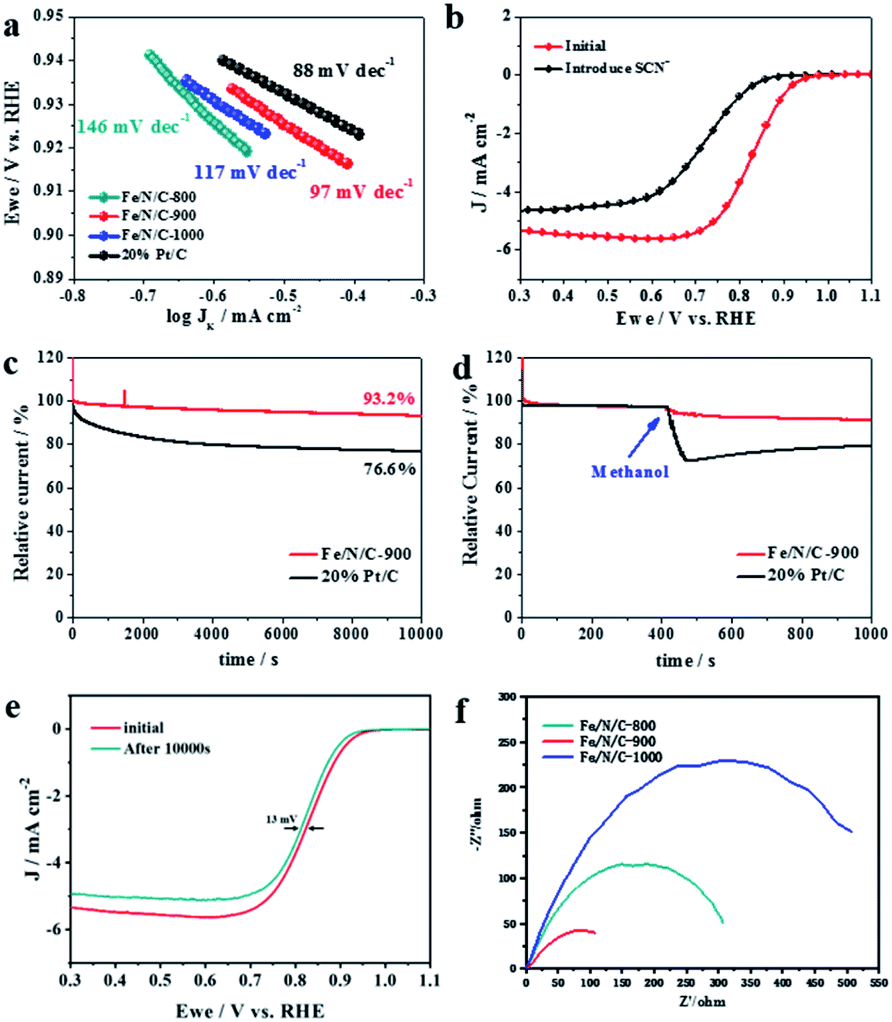 | ||
| Fig. 5 (a) The Tafel plots of Fe/N/C-800, Fe/N/C-900 and Fe/N/C-1000 electrocatalysts. (b) LSV of Fe/N/C in O2-saturated 0.1 M KOH at a rotating speed of 1600 rpm with and without KSCN; (c) durability evaluation of Fe/N/C-900 and 20% Pt/C electrodes for 10000 s at a rotation rate of 1600 rpm, in O2-saturated 0.1 M KOH; (d) methanol tolerance test; (e) ORR polarization curves of Fe/N/C-900 before and after durability evaluation; (f) Nyquist plots of Fe/N/C-800, Fe/N/C-900, and Fe/N/C-1000. | ||
Conclusions
We report a new method using 2,3-DCTBT with four negatively charged nitrogen atoms as a precursor to synthesize an ORR catalyst Fe/N/C-900 for the first time. The as-obtained Fe/N/C-900 (Eonset is 0.98 V vs. RHE in 0.1 M KOH) exhibits excellent electrochemical catalytic performance for the ORR compared to the commercial 20% Pt/C catalyst (Eonset = 1.02 V and E1/2 = 0.83 V vs. RHE). Moreover, the catalyst possesses good stability and high tolerance to methanol, and follows a four-electron mechanism (n = 3.97). The main reason for this excellent performance may be due to the coordination of 2,3-DCTBT with metal Fe forming active site Fe–Nx, which is still retained even after pyrolysis as proved by EXAFS and XPS studies. Finally, from the results of poisoning experiments, it can be seen that Fe–Nx played a key role during catalysis. 2,3-DCTBT as a precursor provides a unique perspective for the design of Fe/N/C catalysts towards oxygen reduction reaction.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Beijing Municipality (no. 2192010 and 2182012), the National Natural Science Foundation (no. 11179033), and Capacity Building for Sci-Tec Innovation-Fundamental Scientific Research Funds (025185305000/210).References
- V. R. Stamenkovic, D. Strmcnik, P. P. Lopes and N. M. Markovic, Nat. Mater., 2016, 16, 57–69 CrossRef.
- D. Gielen, F. Boshell and D. Saygin, Nat. Mater., 2016, 15, 117–120 CrossRef CAS.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS.
- J. Liang, R. F. Zhou, X. M. Chen, Y. H. Tang and S. Z. Qiao, Adv. Mater., 2014, 26, 6074–6079 CrossRef CAS.
- M. Liu, Z. Zhao, X. Duan and Y. Huang, Adv. Mater., 2019, 31, e1802234 CrossRef.
- F. Twisk, J. Rehabil. Res. Dev., 2013, 50, vii–viii Search PubMed.
- S. Reis-Dennis, Monash Bioeth. Rev., 2020, 38, 83–86 CrossRef.
- L. Wu, X. Cao, W. Hu, Y. Ji, Z.-Z. Zhu and X.-F. Li, ACS Appl. Energy Mater., 2019, 2, 6634–6641 CrossRef CAS.
- J. Liu, H. Zhang, M. Qiu, Z. Peng, M. K. H. Leung, W.-F. Lin and J. Xuan, J. Mater. Chem. A, 2020, 8, 2222–2245 RSC.
- Y. Yu, J. Ma, C. Chen, Y. Fu, Y. Wang, K. Li, Y. Liao, L. Zheng and X. Zuo, ChemCatChem, 2019, 11, 1722–1731 CrossRef CAS.
- W. Liu, K. Wang, C. Wang, W. Liu, H. Pan, Y. Xiang, D. Qi and J. Jiang, J. Mater. Chem. A, 2018, 6, 22851–22857 RSC.
- R. Sassi, R. R. Bond, A. Cairns, D. D. Finlay, D. Guldenring, G. Libretti, L. Isola, M. Vaglio, R. Poeta, M. Campana, C. Cuccia and F. Badilini, J. Electrocardiol., 2017, 50, 776–780 CrossRef.
- J.-D. Yi, R. Xu, G.-L. Chai, T. Zhang, K. Zang, B. Nan, H. Lin, Y.-L. Liang, J. Lv, J. Luo, R. Si, Y.-B. Huang and R. Cao, J. Mater. Chem. A, 2019, 7, 1252–1259 RSC.
- X. Guo, C. Qian, R. Shi, W. Zhang, F. Xu, S. Qian, J. Zhang, H. Yang, A. Yuan and T. Fan, Small, 2019, 15, e1804855 CrossRef.
- Z. Zhang, S. Yang, M. Dou, J. Ji and F. Wang, Catal. Sci. Technol., 2017, 7, 1529–1536 RSC.
- C. Ren, H. Li, R. Li, S. Xu, D. Wei, W. Kang, L. Wang, L. Jia, B. Yang and J. Liu, RSC Adv., 2016, 6, 33302–33307 RSC.
- Y. Fu, D. Xu, Y. Wang, X. Li, Z. Chen, K. Li, Z. Li, L. Zheng and X. Zuo, ACS Sustainable Chem. Eng., 2020, 8, 8338–8347 CrossRef CAS.
- Y. Peng, Z. Li, D. Xia, L. Zheng, K. Liao, K. Li and X. Zuo, J. Power Sources, 2015, 291, 20–28 CrossRef CAS.
- A. Ly, T. Asset and P. Atanassov, J. Power Sources, 2020, 478, 228516–228524 CrossRef CAS.
- S. Zeng, F. Lyu, L. Sun, Y. Zhan, F.-X. Ma, J. Lu and Y. Y. Li, Chem. Mater., 2019, 31, 1646–1654 CrossRef CAS.
- W. Li, C. Min, F. Tan, Z. Li, B. Zhang, R. Si, M. Xu, W. Liu, L. Zhou, Q. Wei, Y. Zhang and X. Yang, ACS Nano, 2019, 13, 3177–3187 CrossRef CAS.
- B. C. Lund, T. E. Abrams and A. A. Gravely, J. Rehabil. Res. Dev., 2011, 48, vii–ix Search PubMed.
- S. Yuan, M. Weng, D. Liu, X. He, L. L. Cui and T. Asefa, ACS Sustainable Chem. Eng., 2019, 7, 18912–18925 CrossRef CAS.
- M. Mao, J. Deng, T. Yan, J. Shen, J. Zhang, L. Shi and D. Zhang, ACS Sustainable Chem. Eng., 2019, 7, 19268–19276 CrossRef CAS.
- R. Venegas, F. J. Recio, J. Riquelme, K. Neira, J. F. Marco, I. Ponce, J. H. Zagal and F. Tasca, J. Mater. Chem. A, 2017, 5, 12054–12059 RSC.
- S. Carozzi, M. G. Nasini, C. Schelotto, P. M. Caviglia, S. Barocci, A. Cantaluppi and M. Salit, Adv. Peritoneal Dial., 1990, 6, 110–113 CAS.
- Y. Qiao, P. Yuan, Y. Hu, J. Zhang, S. Mu, J. Zhou, H. Li, H. Xia, J. He and Q. Xu, Adv. Mater., 2018, 30, 1804504–1804512 CrossRef.
- D. Xia, X. Yang, L. Xie, Y. Wei, W. Jiang, M. Dou, X. Li, J. Li, L. Gan and F. Kang, Adv. Funct. Mater., 2019, 29, 1906174–1906183 CrossRef CAS.
- F. Razmjooei, J.-H. Yu, H.-Y. Lee, B.-J. Lee, K. P. Singh, T.-H. Kang, H.-J. Kim and J.-S. Yu, ACS Appl. Energy Mater., 2020, 3, 11164–11176 CrossRef CAS.
- J. Ma, D. Xiao, C. L. Chen, Q. Luo, Y. Yu, J. Zhou, C. Guo, K. Li, J. Ma, L. Zheng and X. Zuo, J. Power Sources, 2018, 378, 491–498 CrossRef CAS.
- Y. Qian, P. Du, P. Wu, C. Cai and D. F. Gervasio, J. Phys. Chem. C, 2016, 120, 9884–9896 CrossRef CAS.
- X. Fu, Y. Liu, X. Cao, J. Jin, Q. Liu and J. Zhang, Appl. Catal., B, 2013, 130–131, 143–151 CrossRef CAS.
- D. Zhou, L. Yang, L. Yu, J. Kong, X. Yao, W. Liu, Z. Xu and X. Lu, Nanoscale, 2015, 7, 1501–1509 RSC.
- D. Liu, J. C. Li, Q. Shi, S. Feng, Z. Lyu, S. Ding, L. Hao, Q. Zhang, C. Wang, M. Xu, T. Li, E. Sarnello, D. Du and Y. Lin, ACS Appl. Mater. Interfaces, 2019, 11, 39820–39826 CrossRef CAS.
- C. Yang, S. Tao, N. Huang, X. Zhang, J. Duan, R. Makiura and S. Maenosono, ACS Appl. Nano Mater., 2020, 3, 5481–5488 CrossRef CAS.
- Y. Peng, L. Cui, S. Yang, J. Fu, L. Zheng, Y. Liao, K. Li, X. Zuo and D. Xia, Electrochim. Acta, 2015, 154, 102–109 CrossRef CAS.
- X. Ao, W. Zhang, Z. Li, J. G. Li, L. Soule, X. Huang, W. H. Chiang, H. M. Chen, C. Wang, M. Liu and X. C. Zeng, ACS Nano, 2019, 13, 11853–11862 CrossRef CAS.
- W. Liu, Y. Hou, H. Pan, W. Liu, D. Qi, K. Wang, J. Jiang and X. Yao, J. Mater. Chem. A, 2018, 6, 8349–8357 RSC.
- A. Mulyadi, Z. Zhang, M. Dutzer, W. Liu and Y. Deng, Nano Energy, 2017, 32, 336–346 CrossRef CAS.
- G. J. Richards, J. P. Hill, K. Okamoto, A. Shundo, M. Akada, M. R. Elsegood, T. Mori and K. Ariga, Langmuir, 2009, 25, 8408–8413 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09730f |
| This journal is © The Royal Society of Chemistry 2021 |