DOI:
10.1039/D0RA10940A
(Paper)
RSC Adv., 2021,
11, 8444-8449
Light driven molecular lock comprises a Ru(bpy)2(hpip) complex and cucurbit[8]uril†
Received
30th December 2020
, Accepted 9th February 2021
First published on 23rd February 2021
Abstract
Here, complex 1 ([Ru(bpy)2(hpip)]2+–MV2+) and CB[8] can form a stable 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 inclusion complex in aqueous solution, resembling a U-shaped conformation. Upon light irradiation, two complex 1 were reversibly locked through the formation of a MV˙+ radical dimer that is stabilized in the cavity of CB[8] with Ru complexes as blockers, in which complex 1 was transformed from a U-shaped conformation to a interlocked complex. This study provided a feasible strategy for the fabrication of a photo-driven supramolecular machine resembling a “lock”.
:1 inclusion complex in aqueous solution, resembling a U-shaped conformation. Upon light irradiation, two complex 1 were reversibly locked through the formation of a MV˙+ radical dimer that is stabilized in the cavity of CB[8] with Ru complexes as blockers, in which complex 1 was transformed from a U-shaped conformation to a interlocked complex. This study provided a feasible strategy for the fabrication of a photo-driven supramolecular machine resembling a “lock”.
Introduction
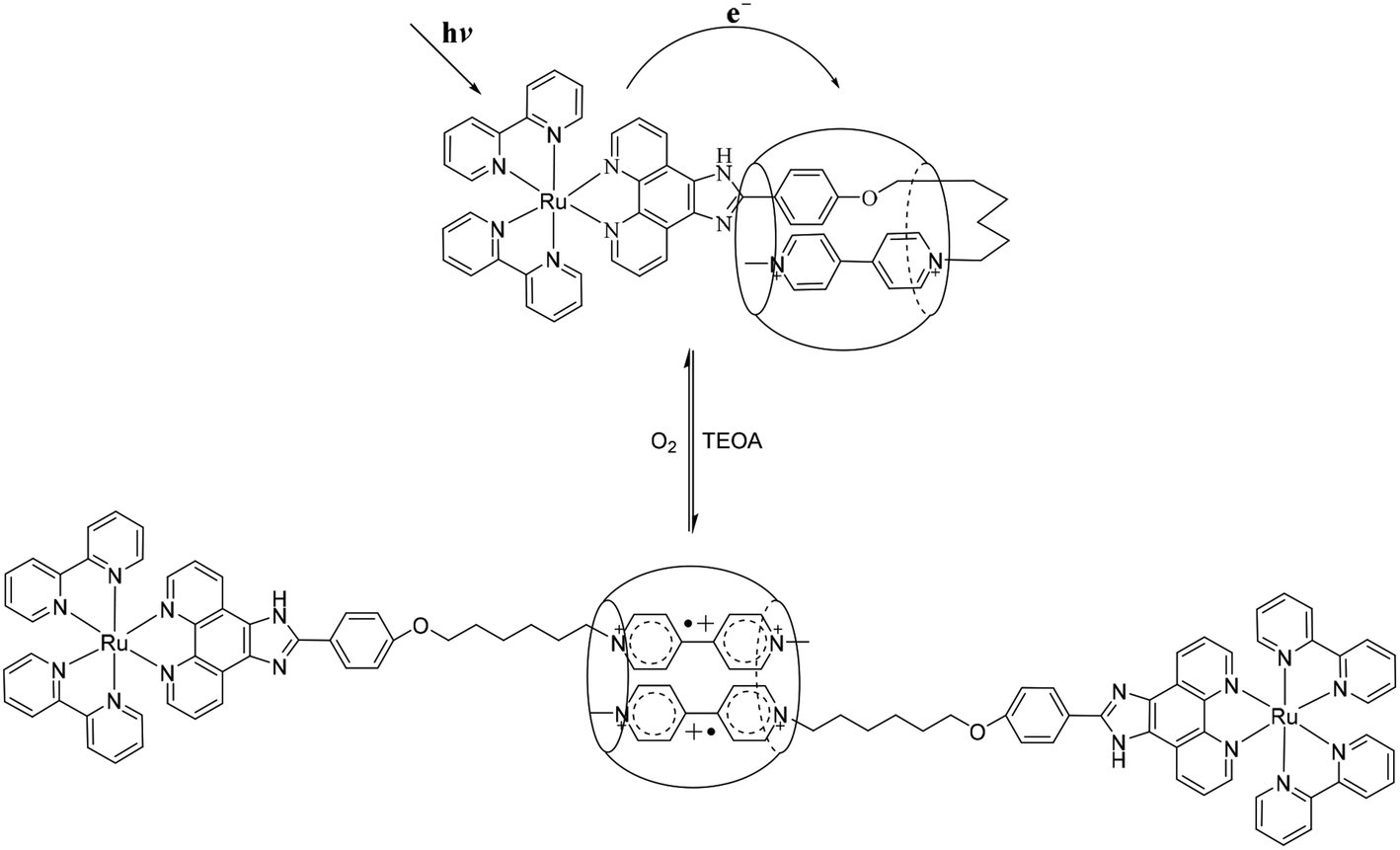
In recent years, great efforts have been made by chemists to construct and manipulate various interlocked supramolecular systems on the atomic level, such as molecular switches and molecular wires.1 As a relatively new class of host molecules, cucurbit[n]urils (CB[n], n = 5–8, 10), have attracted great attention due to their special structures and application in interactions with guest molecules.2 CB[8], one of the members of the cucurbituril host family, with 8 glycoluril motifs linked by methylene bridges forming two hydrophilic carbonylated portals and a hydrophobic cavity, has been shown to accommodate two identical3 or different4 aromatic guests in its cavity. For example, CB[8] can bind both methyl viologen (N,N-dimethyl-4,4-bipyridinium, MV2+) and methyl viologen radical (MV˙+) or radical dimer (MV˙+)2 inside its cavity.5 It is also noted that MV2+, a common electron acceptor, is often utilized together with electron donors such as ruthenium poly-bipyridyl complexes to construct donor–photosensitizer–acceptor (D–P–A) systems.6 And these host–guest systems have been used for the assembly of molecular loop-lock,7 molecular necklaces,8 and molecular machines. Kim and co-workers used [Ru(bpy)3]2+ as photosensitizer and MV2+ as electron acceptor to study light induced intermolecular electron-transfer (ET) and observed the formation of a MV˙+ radical dimer loop inside the CB[8] cavity.9 Sun and co-workers reported the formation of 1:1 inclusion between Ru(bpy)3–MV2+ and CB[8], and observed a long lived charge-separated state Ru(bpy)3–MV˙+–CB[8] upon light irradiation.10 They also reported that Ru(bpy)3–MV2+ and CB[8] could be used to create a light driven molecular “lock” through the formation of a stable dimer (MV˙+)2 inside one CB[8] cavity, and this “lock” can be “unlocked” by molecular oxygen.11 We have presented that Ru2+–MV2+-Np guest take a U-shaped conformation into the cavity of CB[8] and formed a stable 1:1 inclusion complex with CB[8]. Upon light irradiation, a MV˙+ radical cation stabilized in the cavity of CB[8] accompanied by the naphthalene residue has been observed.12 However, to the best of our knowledge, no work has been reported on the interaction between CB[8] and MV2+ together with an aromatic donor moiety that is coupled to a redox-active [Ru(bpy)2(hpip)]2+(hpip = 2-(4-hydroxyphenyl)-imidazo[4,5-f][1,10]phenanthroline, bpy = 2,2′-bipyridine) complex. Here, we report the formation of a 1:1 inclusion complex of CB[8] with complex 1, consisting of a [Ru(bpy)2(hpip)]2+ complex covalently linked to a MV2+ via a six carbon chain, in which the viologen residue is back-folded and inserted together with the phenol residue into the cavity of CB[8]. That is to say, complex 1 takes a U-shaped conformation inside the cavity of CB[8] with Ru complex as a blockers. Upon light irradiation, U-shaped conformation was broken, and a molecular “lock” was formed, in which a stable dimer of the (MV˙+)2 inside one CB[8] cavity.
Results and discussion
Interactions of complex 1 and CB[8]
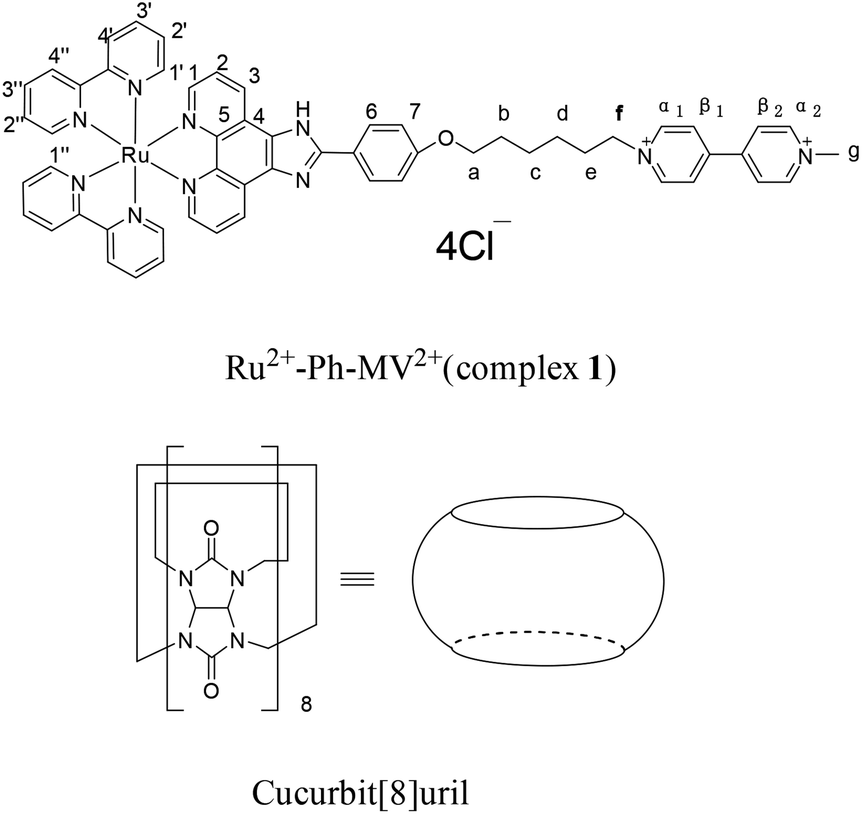
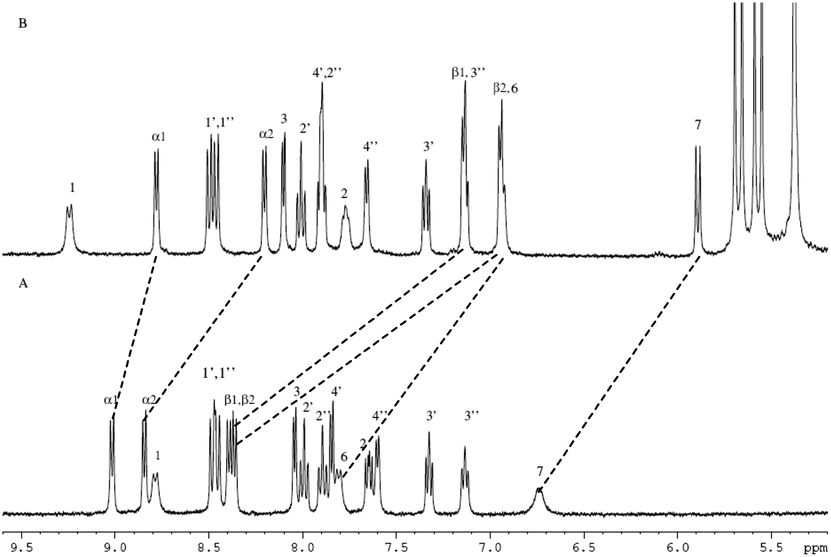
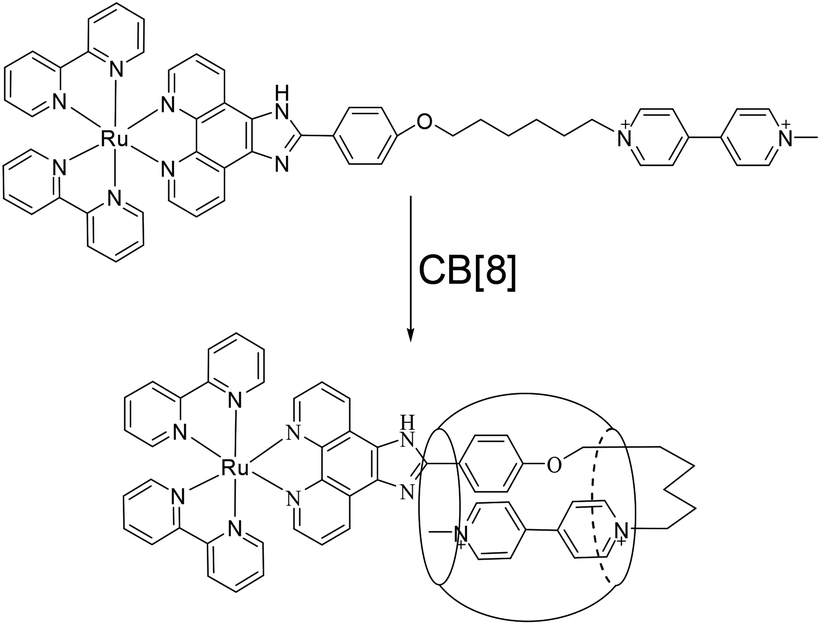
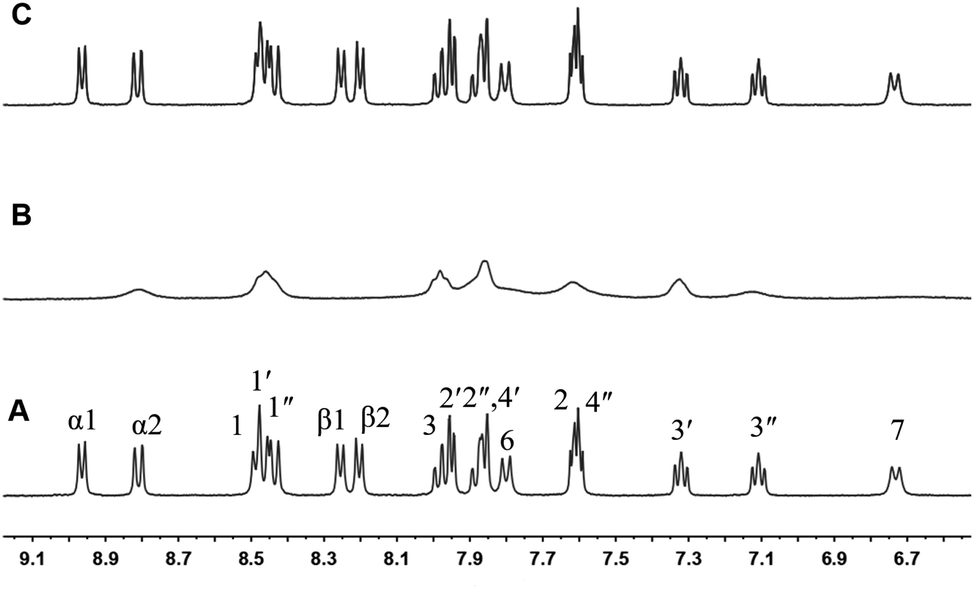
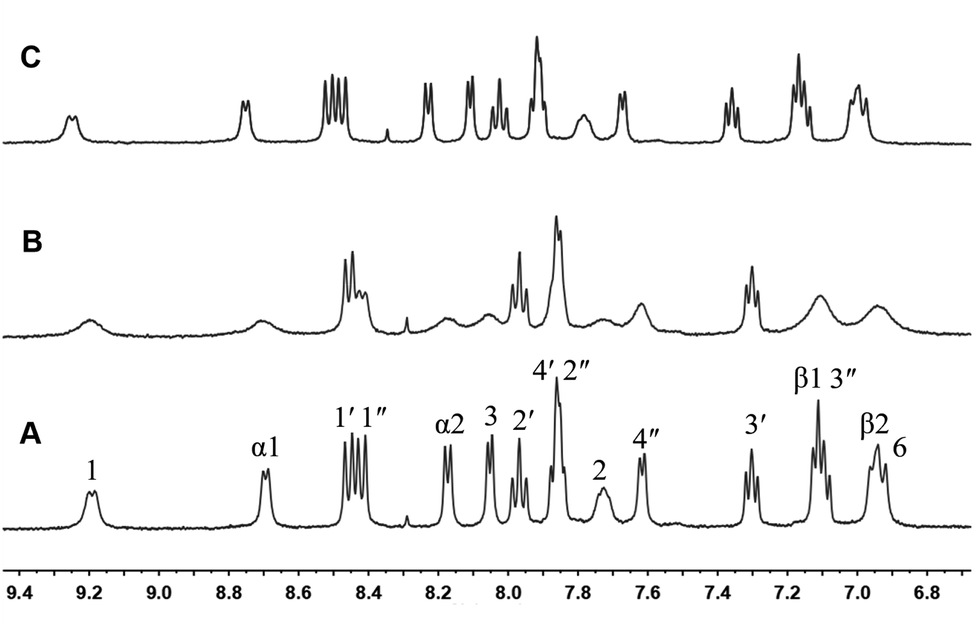
The structures of complex 1 used in this study are shown in Scheme 1. Complex 1 was synthesized and characterized by 1H NMR, 13C NMR, and mass spectra analysis (see ESI†). The binding interaction between complex 1 and CB[8] in the aqueous solution is determined by 1H NMR spectroscopy (see ESI†). In the presence of 0.5 equiv. of CB[8], the proton signals of both bound and free guests are clearly evidenced. Upon addtion of 1.0 equiv. of CB[8] to the solution of complex 1, the four α-protons of the viologen moiety in complex 1 shifted upfield from 9.02 to 8.78 (two protons) and 8.84 to 8.20 (two protons) (Fig. 1). The two β-protons shifted upfield from 8.40 to 7.14, and the other two β-protons shifted upfield from 8.36 to 6.95. In the same time, the four phenyl proton signals underwent upfield shifts. Whereas the protons of part of the six-carbon chain (b–f positions) underwent downfield shifts. This can be interpreted that the viologen moiety of complex 1 folds back to form an U-shaped conformation with phenyl moiety inside the hydrophobic cavity of CB[8] as shown in Scheme 2. This binding modes is similar with the system reported by Sun's group.12 1H NMR titration experiments supported the formation of a 1:1 host–guest complex 1 + CB[8]. It is also worth to mention that for the host, the peak splitting of the CB[8] protons into two sets indicates that the two portals of the CB[8] cavity are no longer in identical environment after including the complex 1.13
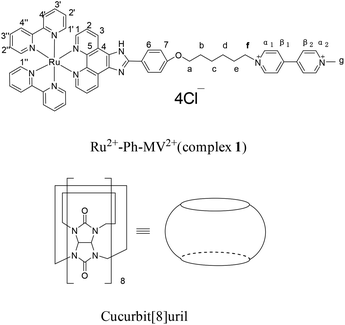 |
| | Scheme 1 Structures of Ru2+-Ph–MV2+ (complex 1) and CB[8]. | |
 |
| | Fig. 1 1H NMR spectra (400 MHz, D2O) of 1 in the absence (A) and in the presence (B) of 1 equiv. of CB[8]. | |
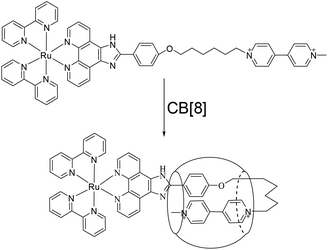 |
| | Scheme 2 Schematic illustration of the interaction of 1 with CB[8]. | |
Furthermore, we performed DOSY-NMR spectra of complex 1 and 1 + CB[8], showing that diffusion coefficient of 1 + CB[8] (1.55 × 10−10 m2 s−1) was slightly smaller than that of 1 (1.70 × 10−10 m2 s−1), as shown in Fig. S13 and S14 and Table S1.†
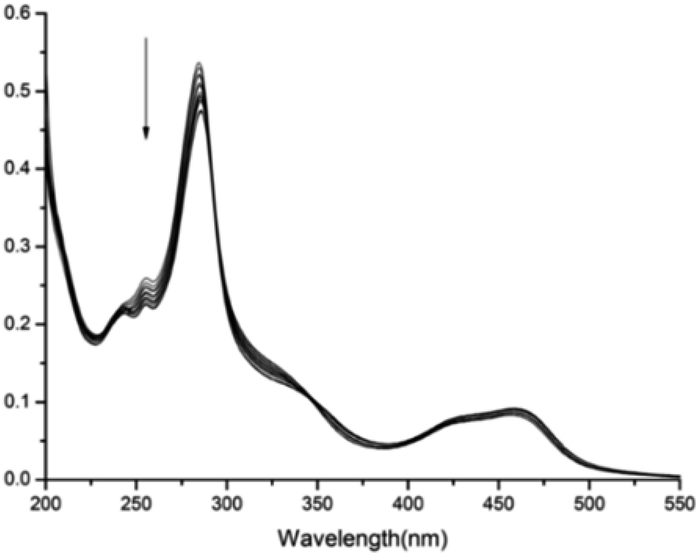
The formation of the stable 1:1 inclusion complex between complex 1 and CB[8] was further confirmed by ESI-MS. When equivalent amounts of complex 1 and CB[8] were dissolved in water, the ESI-MS spectrum in positive mode gave a quadruply charged peak at m/z 577.1781 (calculated for [1 + CB[8]–4Cl−]4+, 577.1779. see ESI†). No 2:1 or 2:2 inclusion complexes were observed. Both NMR and ESI-MS results provide strong evidence for the formation of a stable 1:1 host–guest inclusion complex between complex 1 and CB[8]. Fig. 2 displays the absorption spectra of complex 1 taken in the course of titration with CB[8]. On addition of CB[8] to the solution of complex 1, the intensity of the absorption maxima at 284 nm decreased, accompanied by a slightly red shift to 286 nm. The other strong absorption band around 460 nm can be attributed to the metal-to-ligand charge-transfer (MLCT) transition π* ← dπ(Ru) of the Ru(bpy)2(hpip) moiety.
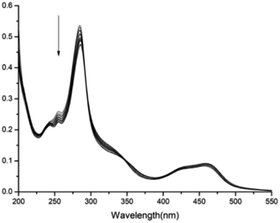 |
| | Fig. 2 Absorption spectra of complex 1 (6 μM) in aqueous solution in the presence of different amounts of CB[8] (0–1.6 equiv.). | |
The stoichiometry of the binding of complex 1 with CB[8] is further verified by titration curve. After addition of increasing equivalents of CB[8] into an aqueous solution of complex 1, the corresponding absorption spectra changes were recorded. Fig. S15† shows a plot of the absorbance as a function of total CB[8] concentration. The data can be easily fitted to a 1:1 binding model with a binding constant of 4.15 × 104 M−1.
Photochemistry
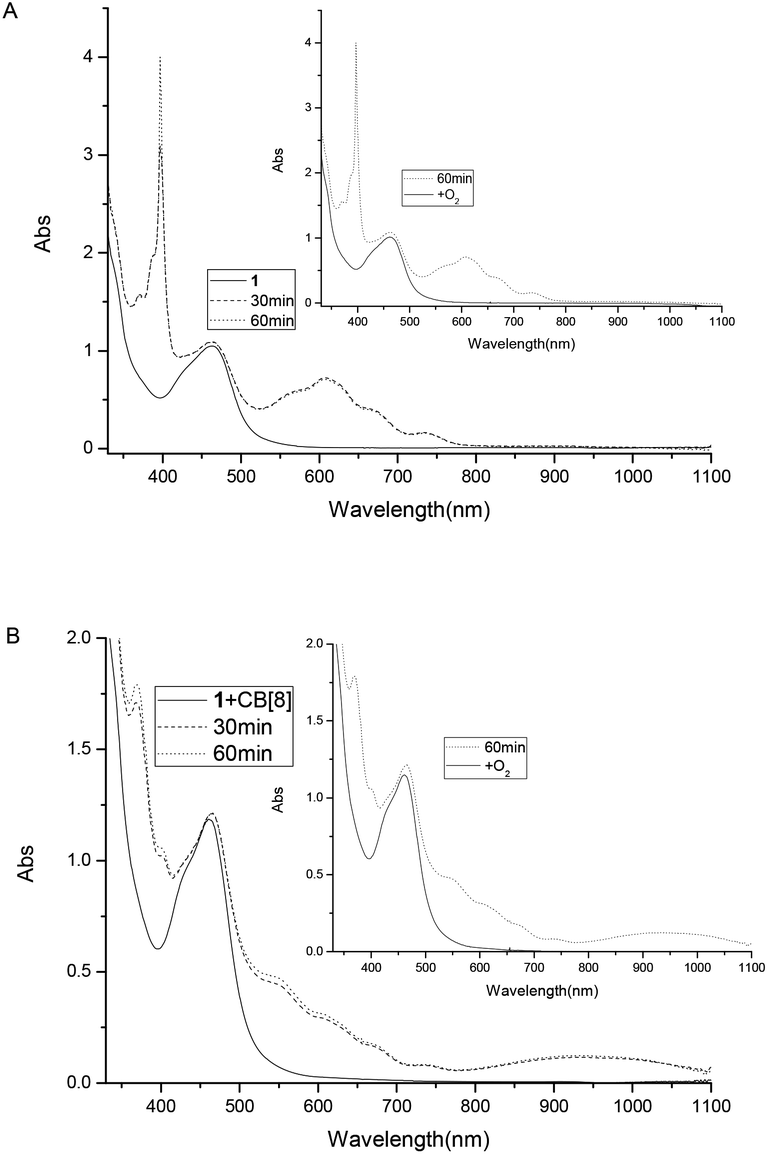
To study the possible photoinduced electron transfer processes in a system containing complex 1, excess sacrificial electron donor, triethanolamine (TEOA), was added to a cuvette containing complex 1, and UV-vis absorption was taken as shown in Fig. 3. For complex 1 alone, two absorption peaks around 397 and 607 nm can be easily monitored (Fig. 3A) after 30 min of irradiation, which was due to the formation of redox induced radical Ru2+-Ph–MV˙+. The color of the solution changed from yellow to dark green and the peak intensity increased as the irradiation time prolonged (Fig. 3A). With the addition of one equivalent of CB[8] to the solution of complex 1 and TEOA, no peaks for the free Ru2+-Ph–MV˙+ radical could be observed after irradiation (Fig. 3B). In this case, however, a very strong peak at 370 nm and a broad peak around 550 nm as well as a broad peak at 950 nm were observed and the color changed from yellow to brown. These three peaks are due to the formation of Ru2+-Ph–MV˙+ dimer. By comparison of the two systems, we found that two MV moieties are “locked” inside the cavity of one CB[8], forming Ru2+-Ph–(MV˙+)2(CB[8])–Ph-Ru2+ in the presence of CB[8] (Scheme 3). This photoinduced ET processes is in agreement with previously reported data.14
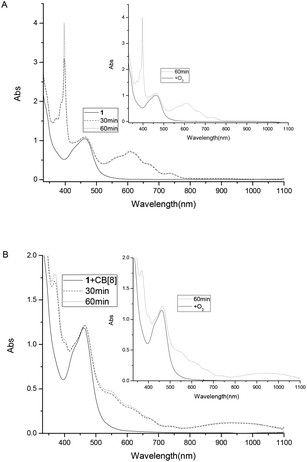 |
| | Fig. 3 Absorption spectra of complex 1 (8.0 × 10−5 M) and TEOA (0.1 M) in the absence of CB[8] (A) and in the presence (B) of 1 equiv. of CB[8]. | |
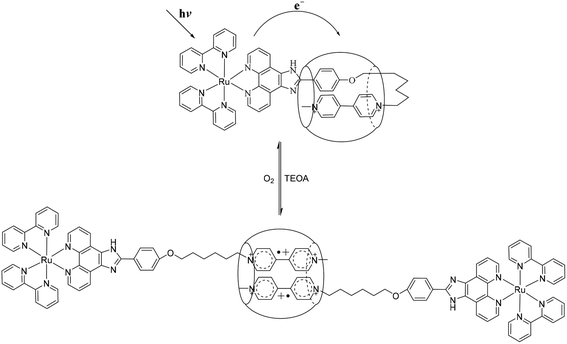 |
| | Scheme 3 Schematic representation of the light driven interaction between 1 and CB[8]. | |
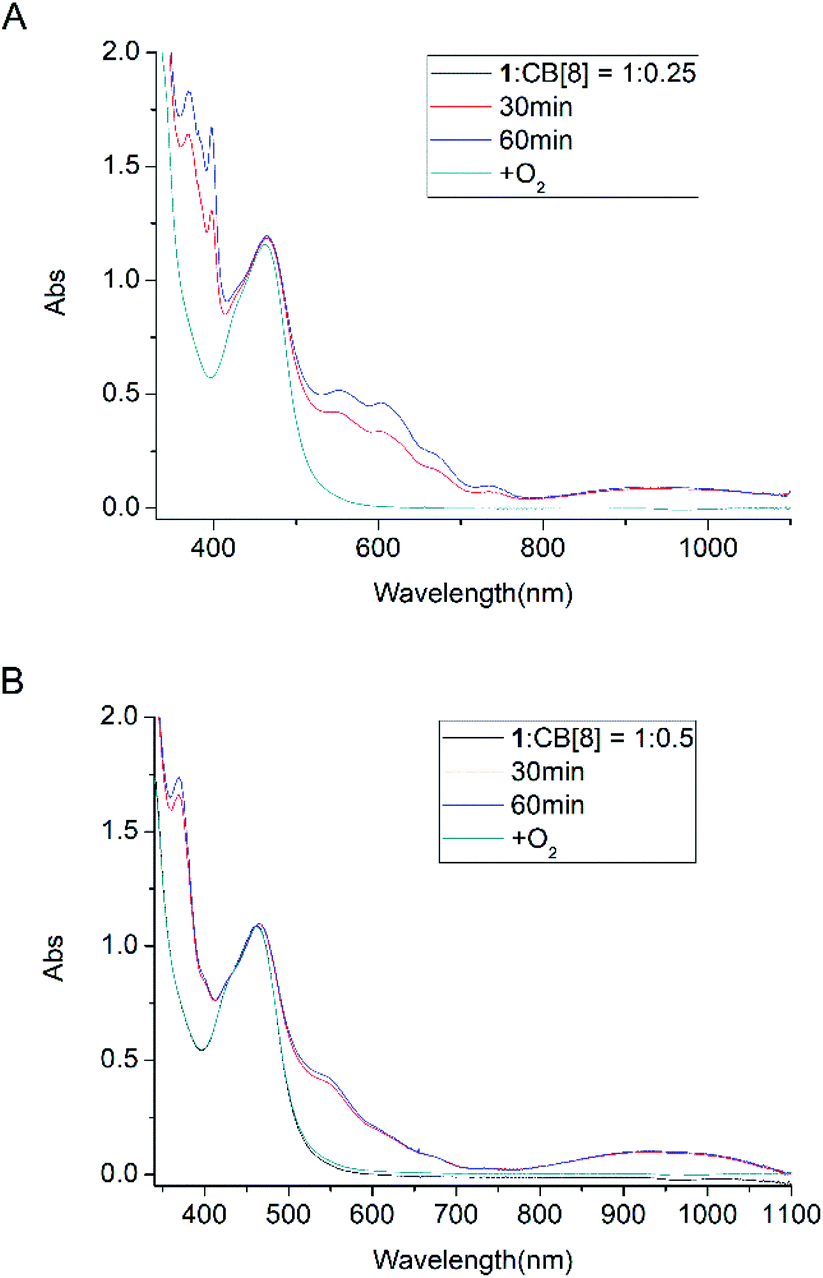
To establish that the [3]rotaxane was formed, UV-vis absorption of complex 1 with 0.25 or 0.5 equivalent of CB[8] were performed as taken in Fig. 4. In the presence of 0.25 equivalent of CB[8] in the complex 1, the characteristic absorption for both the viologen radical (two absorption peaks around 397 and 607 nm)and the radical dimer (three absorption peaks around 370, 550 and 950 nm) can be observed concomitantly after irradiation. However, in the case of 1:0.5 mixture of complex 1 and CB[8], only the characteristic absorption for the radical dimer was monitored after irradiation, no characteristic absorption for the viologen radical alone was observed. This indicates that all the viologen radical formed radical dimer inside the cavity of CB[8]. In other words, two MV moieties of complex 1 are “locked” inside the cavity of one CB[8], forming a [3]rotaxanes. This experiments supported the formation of Ru2+-Ph–(MV˙+)2(CB[8])-Ph–Ru2+ as shown in Scheme 3. When the light is turned off and oxygen goes into the cuvette, the initial absorption spectrum can be restored. The whole process can be repeated several times without significant photo degradation.
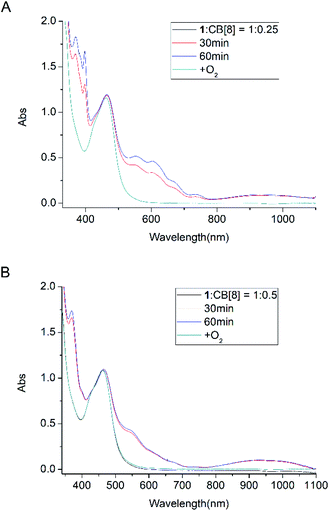 |
| | Fig. 4 Absorption spectra of complex 1 (8.0 × 10−5 M) and TEOA (0.1 M) in the presence of 0.25 equiv. (A) and 0.5 equiv. (B) of CB[8]: before and after light irradiation under Ar atmosphere. | |
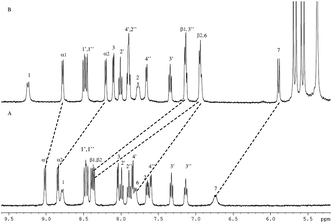
The light-induced formation of the stable Ru2+-Ph–MV+ dimer inside the cavity of one CB[8] can be further confirmed by NMR measurements. Upon 5 equivalents TEOA was added to a NMR tube containing complex 1, the reaction process was monitored by 1H NMR. When the NMR tube was degassed with argon and light-irradiated for 3 h, the color of the solution changed from orange to brown and the 1H NMR spectrum was taken again. As shown in Fig. 5, the peaks for the α1-and β-protons of the MV moiety shifted outside the normal NMR window due to its radical character. The peaks for the protons of the phenanthroline ligand, bipyridine ligand and phenol ring were also affected, becoming broad, which are due to the paramagnetic effect of the MV˙+ radical. When the light irradiation was stopped and the air (oxygen) was allowed go into the tube, the color of the solution changed back to orange and the 1H NMR spectrum was restored as shown in Fig. 5. This experiment strongly support that the Ru2+–MV˙+ radical was formed in complex 1.
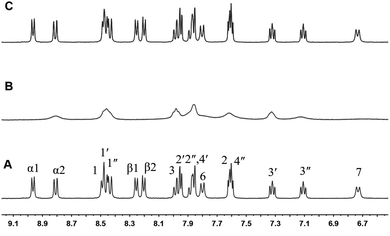 |
| | Fig. 5 1H NMR (400 MHz, D2O) of complex 1 with TEOA (1:5) (A), after 3 h of light irradiation (B), and after stopping light irradiation and exposing to air (O2) (C). | |
To investigate the formation of MV˙+ dimer in the presence of CB[8], TEOA was added to the 1:1 inclusion complex of 1 + CB[8], and the effect was followed by 1H NMR. On addition of 5 equiv. of TEOA to a D2O solution of complex 1 (1.0 equiv.) and CB[8] (1.0 equiv.), the NMR spectrum was taken again as shown in Fig. 6A. When the NMR tube was degassed with argon, and irradiated for 3 h by the light, the color of the solution changed to dark brown, and the 1H NMR was taken again as shown in Fig. 6B. The peaks corresponding to the α-, β-protons of the MV moiety become broad. This is typical behavior of the MV moiety after forming a stabilized radical dimer inside a CB[8] host. Moreover, the NMR peaks corresponding to the phenanthroline ligand, carbon linker and the N-methyl group also broadened while part of the peaks of the ruthenium moiety were clearly visible due to a paramagnetic effect. When the septum was removed from the NMR tube and the air (oxygen) went in, the color of the solution changed back from the dark brown to the original orange, and the 1H NMR spectrum was restored as shown in Fig. 6C. These results suggest the formation of Ru2+–MV+ dimer inside the cavity of one CB[8] as illustrated in Scheme 3 and supported by literature data on the behavior of MV2+ and related compounds.14
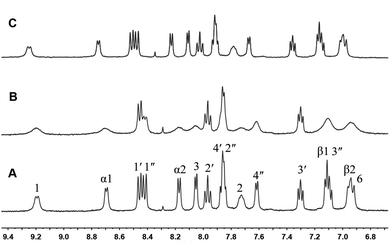 |
| | Fig. 6 1H NMR (400 MHz, D2O) of 1 + CB[8] with TEOA (1:5) (A), after 3 h of light irradiation (B), and after stopping light irradiation and exposing to air (O2) (C). | |
To investigate the formation of MV˙+ dimer in the presence of CB[8], TEOA was added to the 1:1 inclusion complex of 1 + CB[8], and the effect was followed by 1H NMR. On addition of 5 equiv. of TEOA to a D2O solution of complex 1 (1.0 equiv.) and CB[8] (1.0 equiv.), the NMR spectrum was taken again as shown in Fig. 6A. When the NMR tube was degassed with argon, and irradiated for 3 h by the light, the color of the solution changed to dark brown, and the 1H NMR was taken again as shown in Fig. 6B. The peaks corresponding to the α-, β-protons of the MV moiety become broad. This is typical behavior of the MV moiety after forming a stabilized radical dimer inside a CB[8] host. Moreover, the NMR peaks corresponding to the phenanthroline ligand, carbon linker and the N-methyl group also broadened while part of the peaks of the ruthenium moiety were clearly visible due to a paramagnetic effect. When the septum was removed from the NMR tube and the air (oxygen) went in, the color of the solution changed back from the dark brown to the original orange, and the 1H NMR spectrum was restored as shown in Fig. 6C. These results suggest the formation of Ru2+–MV+ dimer inside the cavity of one CB[8] as illustrated in Scheme 3 and supported by literature data on the behavior of MV2+ and related compounds.14
Electrochemistry
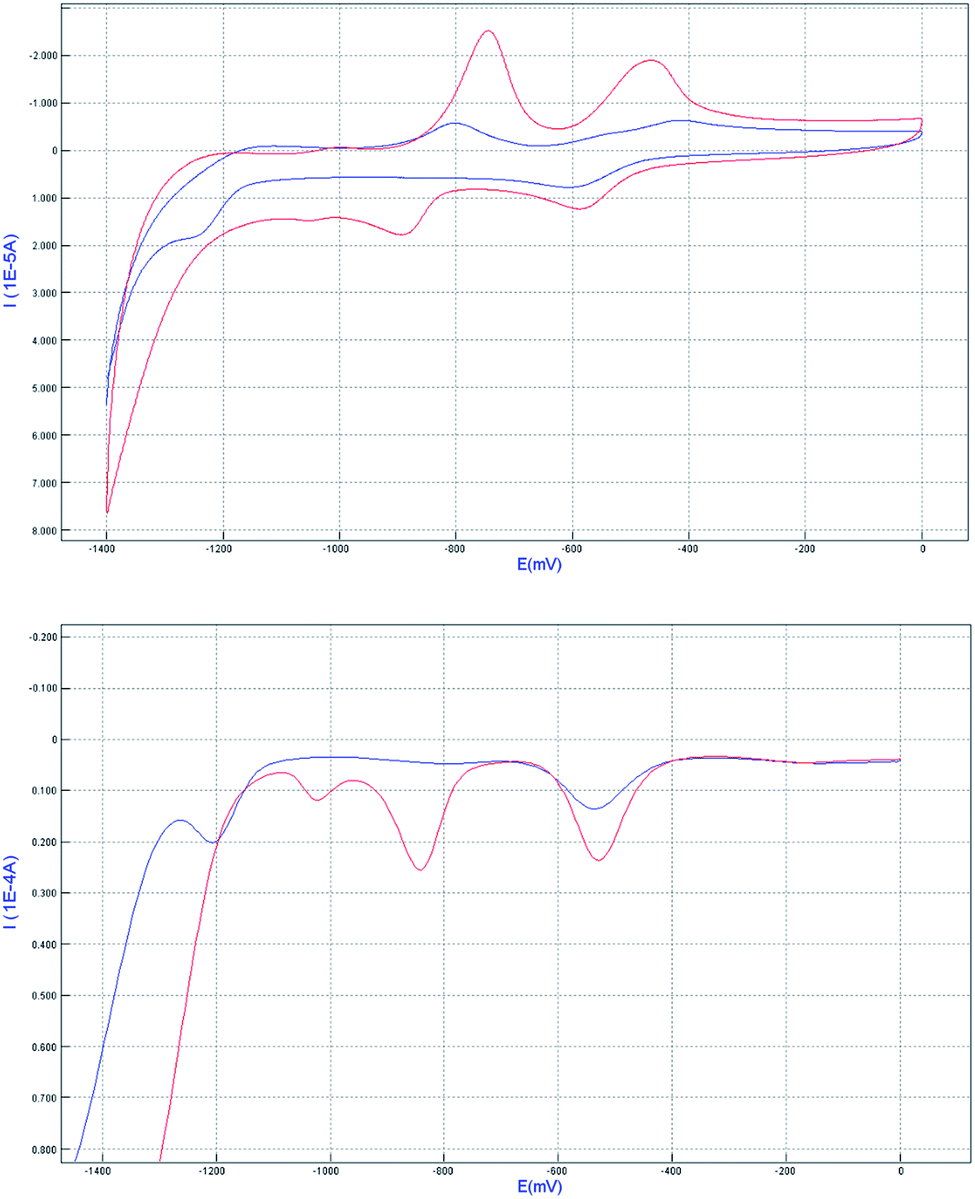
The redox properties of the complex 1 before and after the inclusion of CB[8] were studied by cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in 0.1 M phosphate buffer (pH 7.0) water solution (Fig. 7). According to the literature, for the complex 1 alone, the first wave at −0.53 V (for easy comparison, all the half-wave potentials in the context were determined from DPV peak potentials) corresponded to the reduction of MV2+ moiety to the cation-radical MV˙+, whereas the second wave at −0.84 V was attributed to the reduction of MV˙+ to MV0. By addition of 1 equiv. of CB[8] to the solution of complex 1, the peak potential related to the redox couple MV2+/MV˙+, MV˙+/MV0 were shifted to −0.54 and −1.20 V, respectively. The reason why MV2+ is harder to reduce is very likely that the formation of radical dimer within the cavity of CB[8]. This characteristic pattern for the formation of radical dimer is in good agreement with the literature precedent. In other words, the results further demonstrate the formation of Ru2+-Ph–(MV˙+)2(CB[8])–Ph-Ru2+ dimer in the system.
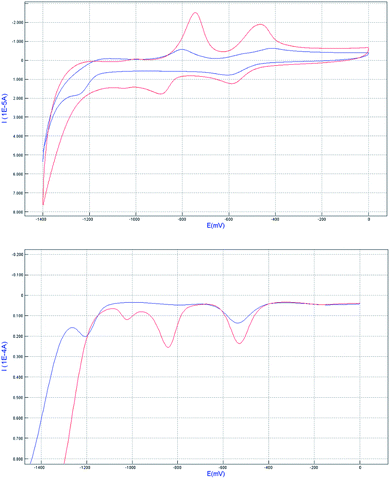 |
| | Fig. 7 Cyclic voltammetry (top) and differential pulse voltammetry (bottom) curves of related systems complex 1(red line) and 1 + CB[8] (blue line) in pH = 7 phosphate buffer solution. The concentration used is 1 mM for all the compounds, with glassy carbon disk as working electrode, and Ag/AgCl as reference electrode, the scan rate was 100 mV s−1. | |
Conclusions
In conclusion, complex 1 can form a stable 1:1 inclusion complex with CB[8] in aqueous solution through taking a U-shaped conformation, in which the viologen residue is back-folded and inserted together with the phenoxyl residue into the cavity of CB[8]. The formation of this stable 1:1 inclusion complex has been demonstrated by 1H NMR and ESI-MS. Upon light irradiation, two Ru(bpy)2(hpip) complexe were reversibly locked through the formation of a MV˙+ radical dimer stabilized in the cavity of CB[8] with Ru(bpy)2(hpip) as blockers, in which complex 1 from a U-shaped conformation transform to a interlocked complex. This process could be used to create a novel light driven molecular “lock”, which can be “unlock” by molecular oxygen. The light driven molecular “lock” and “unlock” processes can be repeated several times with good reversibility. This system may provide some new thinking for further design and synthesis of potential light-driven molecular devices.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (NSFC) (grant numbers 21702047 and 21702049), the Innovation Fund of Henan Agricultural University (grant number KJCX2017A20) for financial support.
Notes and references
-
(a) N. Yui and T. Ooya, Chem.–Eur. J., 2006, 12, 6730–6737 CrossRef CAS;
(b) B. L. Feringa, Acc. Chem. Res., 2001, 34, 504–513 CrossRef CAS;
(c) M. J. Frampton and H. L. Anderson, Angew. Chem., Int. Ed., 2007, 46, 1028–1064 CrossRef CAS.
-
(a) J. Kim, I. S. Jung, S. Y. Kim, E. Lee, J. K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef CAS;
(b) J. W. Lee, S. Samal, N. Selvapalam, H. J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS;
(c) J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS;
(d) G. Parvari, O. Reany and E. Keinan, Isr. J. Chem., 2011, 51, 646–663 CrossRef CAS;
(e) A. Ganguly, S. Ghosh and N. Guchhait, J. Phys. Chem. B, 2016, 120, 4421–4430 CrossRef CAS;
(f) M. Cao, F. Hu, X. Han, Y. Zhang, D. Wu, S. H. Liu and J. Yin, Chin. J. Chem., 2015, 33, 351–355 CrossRef CAS.
-
(a) K. Moon, J. Grindstaff, D. Sobransingh and A. E. Kaife, Angew. Chem., Int. Ed., 2004, 43, 5496–5499 CrossRef CAS;
(b) S. J. Barrow, S. Kasera, M. J. Rowland, J. Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS;
(c) R. Rabbani and E. Masson, Org. Lett., 2017, 19, 4303–4306 CrossRef CAS;
(d) J. Yin, Y. Hu, D. Wang, L. Yang, Z. Jin, Y. Zhang and G. Kuang, ACS Macro Lett., 2017, 6, 139–143 CrossRef CAS.
-
(a) U. Rauwald, J. Barrio, X. J. Loh and O. A. Scherman, Chem. Commun., 2011, 47, 6000–6002 RSC;
(b) J. Zhang, R. J. Coulston, S. T. Jones, J. Geng, O. A. Scherman and C. Abell, Science, 2012, 335, 690–694 CrossRef CAS;
(c) W. Jiang, Q. Wang, I. Linder, F. Klautzsch and C. A. Schalley, Chem.–Eur. J., 2011, 17, 2344–2348 CrossRef CAS;
(d) F. Tian, N. Cheng, N. Nouvel, J. Geng and O. A. Scherman, Langmuir, 2010, 26, 5323–5328 CrossRef CAS;
(e) S. Jiang, M. Liu, Y. Cui, D. Zou and Y. Wu, Eur. J. Org. Chem., 2013, 2591–2596 CrossRef CAS;
(f) S. Jiang, X. Yang, C. Yang, M. Tong, D. Zou and Y. Wu, Tetrahedron Lett., 2013, 54, 1638–1644 CrossRef;
(g) S. Li, X. Xu, Y. Zhou, Q. Zhao and Y. Liu, Org. Lett., 2017, 19, 6650–6653 CrossRef CAS.
-
(a) W. S. Jeon, H. J. Kim, C. Lee and K. Kim, Chem. Commun., 2002, 1828–1829 RSC;
(b) K. Kim, D. Kim, J. W. Lee, Y. H. Ko and K. Kim, Chem. Commun., 2004, 848–849 RSC;
(c) S. Andersson, D. Zou, R. Zhang, S. Sun, B. Åkermark and L. Sun, Eur. J. Org. Chem., 2009, 1163–1172 CrossRef CAS;
(d) T. Zhang, S. Sun, F. Liu, J. Fan, Y. Pang, L. Sun and X. Peng, Phys. Chem. Chem. Phys., 2009, 11, 11134–11139 RSC.
- S. Sun, Y. He, Z. Yang, Y. Pang, F. Liu, J. Fan, L. Sun and X. Peng, Dalton Trans., 2010, 39, 4411–4416 RSC.
- W. S. Jeon, E. Kim, Y. H. Ko, I. Hwang, J. W. Lee, S. Y. Kim, H. J. Kim and K. Kim, Angew. Chem., Int. Ed., 2005, 44, 87–91 CrossRef CAS.
- Y. H. Ko, K. Kim, J. K. Kang, H. Chun, J. W. Lee, S. Sakamoto, K. Yamaguchi, J. C. Fettinger and K. Kim, J. Am. Chem. Soc., 2004, 126, 1932–1933 CrossRef CAS.
- W. S. Jeon, A. Y. Ziganshina, J. W. Lee, Y. H. Ko, J. K. Kang, C. Lee and K. Kim, Angew. Chem., Int. Ed., 2003, 42, 4097–4100 CrossRef CAS.
- S. Sun, R. Zhang, S. Andersson, J. Pan, B. Åkermark and L. Sun, Chem. Commun., 2006, 4195–4197 RSC.
- S. Sun, R. Zhang, S. Andersson, J. Pan, D. Zou, B. Åkermark and L. Sun, J. Phys. Chem. B, 2007, 111, 13357–13363 CrossRef CAS.
- D. Zou, S. Andersson, R. Zhang, S. Sun, B. Åkermark and L. Sun, J. Org. Chem., 2008, 73, 3775–3783 CrossRef CAS.
- W. Ong, M. G. Kaifer and A. E. Kaifer, Org. Lett., 2002, 4, 1791–1794 CrossRef CAS.
-
(a) S. Andersson, D. Zou, R. Zhang, S. Sun and L. Sun, Org. Biomol. Chem., 2009, 7, 3605–3609 RSC;
(b) S. Sun, W. Gao, F. Liu, F. Li, J. Fan and X. Peng, Phys. Chem. Chem. Phys., 2011, 13, 570–575 RSC;
(c) S. Sun, S. Andersson, R. Zhang and L. Sun, Chem. Commun., 2010, 46, 463–465 RSC;
(d) T. K. Monhaphol, S. Andersson and L. Sun, Chem.–Eur. J., 2011, 17, 11604–11612 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra10940a |
| ‡ Equal contributors. |
|
| This journal is © The Royal Society of Chemistry 2021 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Puhui Xiea,
Guoxing Liu
*a,
Puhui Xiea,
Guoxing Liu