
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photooxidation of thiosaccharides mediated by sensitizers in aerobic and environmentally friendly conditions†
Miqueas G. Traverssiab,
Alicia B. Peñéñory ab,
Oscar Varelacd and
Juan P. Colomer*ab
ab,
Oscar Varelacd and
Juan P. Colomer*ab
aDepartamento de Química Orgánica, Universidad Nacional de Córdoba, Facultad Ciencias Químicas, Ciudad Universitaria, Edificio de Ciencias II, Córdoba, Argentina. E-mail: juanpablo@fcq.unc.edu.ar
bInstituto de Investigaciones en Fisico-Química de Córdoba (INFIQC), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), UNC, Argentina
cDepartamento de Química Orgánica, Universidad de Buenos Aires, Facultad Ciencias Exactas y Naturales, Ciudad Universitaria, Pab. 2, C1428EHA, Buenos Aires, Argentina
dCentro de Investigación en Hidratos de Carbono (CIHIDECAR), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET), UBA, Argentina
First published on 1st March 2021
Abstract
A series of β-D-glucopyranosyl derivates have been synthesized and evaluated in photooxidation reactions promoted by visible light and mediated by organic dyes under aerobic conditions. Among the different photocatalysts employed, tetra-O-acetyl riboflavin afforded chemoselectively the respective sulfoxides, without over-oxidation to sulfones, in good to excellent yields and short reaction times. This new methodology for the preparation of synthetically useful glycosyl sulfoxides constitutes a catalytic, efficient, economical, and environmentally friendly oxidation process not reported so far for carbohydrates.
Introduction
Sulfoxides play important roles as organic synthetic intermediates1 and biologically active compounds employed in the pharmaceutical industry.2 Therefore, the development of new chemoselective oxidation methodologies of sulfides to sulfoxides, avoiding the formation of sulfones, has been increased.3–5One of the most powerful glycosylation methods discovered by Kahne and coworkers employs anomeric glycosyl sulfoxides as glycoside donors.6 Since then, several reports describing the study and synthesis of a wide range of glycosides7–12 and oligosaccharides13,14 using this methodology have been reported. Moreover, glycosyl sulfoxides have demonstrated several biological applications such as the proliferation inhibition of selected tumor cell lines,15 oral antithrombotic activity,16 and the capability of binding to proteins.17 Additionally, in previous works, our research group has synthesized sulfoxide derivates of thiodisaccharides and established the configuration at the sulfur stereocenter by a procedure developed by us employing high resolution 1D and 2D NMR techniques.18,19 Furthermore, some of these diastereomeric thiodisaccharides S-oxides (with different configurations at the sulfur stereocenter) have demonstrated to inhibit the activity of specific glycosidases, showing different reactivity towards enzymatic hydrolysis according to the S-configuration.18,20
An important disadvantage of the methodologies described for the oxidation of thiomonosaccharides employing conventional oxidation agents, is the extremely low reaction temperatures required to obtain the respective sulfoxides, without over-oxidation to sulfone.6,21–23 Also, it is important to highlight that these conditions are difficult to achieve and control.
Several reagents employed in the sulfide oxidations are toxic, generate by-products difficult to separate from the desired product, and/or contain heavy metals that produce hazardous goods. Furthermore, some of them are very efficient but also very expensive and must be employed stoichiometrically.24 In regard to some peroxy acids, certain limitations arise due to the instability of the pure compounds. This is the case of m-chloroperbenzoic acid, which is rarely available with a purity higher than 77%.25 Furthermore, the use of these compounds should be avoided in production processes since they offer a low atom economy. For all the reasons described above, the development of an energy-saving, catalytic, atom-economical, environmentally friendly, and highly selective oxidation process from thioethers to sulfoxides is required.
Among the different oxidants, molecular oxygen is a flawless reagent since it is “practically unlimited” and “free”, as is light. Photosensitized sulfides oxidation occur according to two main mechanisms (type I and type II).26,27 In type I mechanism, an electron transfer between the sulfide and the excited sensitizer takes place, giving rise to sulfide radical cation (RSR′˙+) that could follow different pathways:28 One of them affords the respective sulfoxide and another one, is the fragmentation to yield a thiyl radical ˙SR′ and alkyl cation R+ which evolves to different products. However, the C–S bond cleavage is less common for alkyl sulfides than for aromatic sulfides, and the nature of cleavage products depends on the stabilization of the cation intermediate.29,30
On the other hand, in a type II mechanism, an energy transfer process occurs to generate singlet oxygen (1O2) that is the responsible for the oxidation of the sulfide into a peroxysulfoxide intermediate. This intermediate reacts with another molecule of sulfide to afford the respective sulfoxide.31
Several photooxidation methodologies of thioethers to sulfoxides are described in the bibliography.26,32–37 Despite the important role of glycosyl sulfoxides in glycosylation reactions and the potential biological applications mentioned above, we were not able to find any study involving the photochemical oxidation of glycosyl sulfides. Therefore, as continuation of our research on the oxidation of thioglycosides, we report here the photosensitized oxidation of thiosaccharides under aerobic conditions. As catalytic amounts of organic dyes, oxygen, and light are employed, instead of the stoichiometric amounts of usually toxic oxidants, generally used for the oxidation of thiosaccharides, this is considered to be a catalytic, efficient, economical, and environmentally friendly method no reported so far for carbohydrates.
Results and discussion
To study the photosensitized oxidation methodology, the thiosaccharides 3a–e were synthesized and used as substrates. Thus, penta-O-acetyl-D-glucopyranose 1 was stereoselectively transformed into the β-thioaldose 2.38 Subsequently, 2 was treated with dimethyl sulfate as methylating agent to afford the thioglucoside 3a in 96% isolated yield after simple extractions from the reaction mixture. Other thiomonosaccharides were also synthesized performing the reaction between 2 as glycosyl acceptor and several alkyl/allyl bromides in acetonitrile and in the presence of triethylamine. As shown in Scheme 1, all the obtained products 3a–d maintain the β configuration of the glycosyl acceptor precursor and the isolated yields range from good to excellent (72–99%). The β configuration at the anomeric centers of the alkyl/allyl thioglucose derivates was established according to the large coupling constant value (J1,2 ≈ 9–10 Hz), determined from the 1H NMR spectra.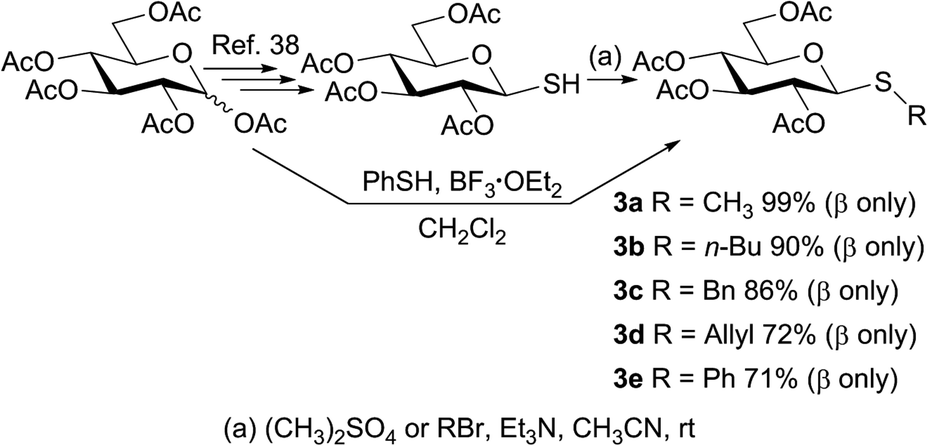 | ||
| Scheme 1 Synthesis of thiomonosaccharides 3a–e. | ||
With the objective of determining the scope of the reaction studied, an aryl thioglycoside was also synthesized employing the procedure described in bibliography.39 The phenyl thiosaccharide 3e, having the β-configuration at the anomeric center, was obtained after column chromatography purification in 71% isolated yield (Scheme 1).
With the thiomonosaccharides in hand, we proceeded to perform the photochemical oxidation studies employing diverse photocatalysts under aerobic conditions. To optimize the reaction settings, different variables as photocatalysts, solvents, atmospheres, and time were evaluated using the thiomonosaccharide 3a as a model substrate.
Several organic dyes are remarkably effective photosensitizers under visible light, since they possess triplet states of proper energies for sensitization of oxygen.40,41 This photosensitization is capable to generate singlet oxygen which is one of the species capable of oxidizing sulfides to sulfoxides. The structures of some of these organic dyes are shown in Chart 1. After each reaction, conversion was determined by 1H NMR using the integrals of the 5-H signal of the thiosaccharides (sulfide and sulfoxide). The ratio of the diastereomeric sulfoxides obtained was calculated from specific signals of the respective products.
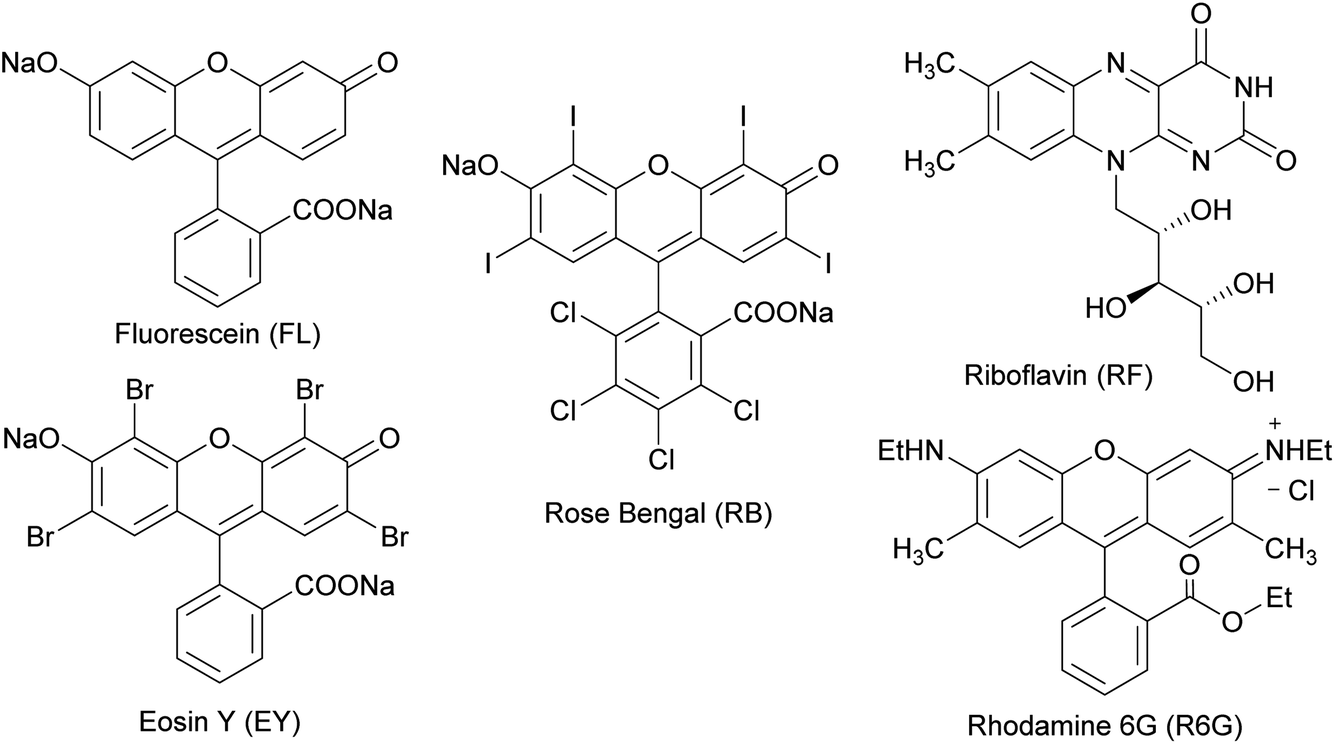 | ||
| Chart 1 Structures of some organic dyes that are effective photosensitizers capable to generate singlet oxygen under visible light. | ||
As starting conditions, the organic dyes were used as photocatalyst (1 mol%) in an oxygen atmosphere, in aprotic or protic polar solvents (acetonitrile and isopropanol, respectively), previously saturated with O2. The LED selection to irradiate the organic dyes was performed considering the maximum absorption wavelengths of each sensitizer. When fluorescein was employed, the reaction mixtures were irradiated under blue LED light (467 nm) for 48 h, but the substrate 3a remains unalterable (Table 1, entries 1 and 2).
|
||||
|---|---|---|---|---|
| Entrya | Dye (mol%) | hν (nm) | Solvent | Yield 4ab (%) |
| a Reaction conditions: 3a (0.05 M), solvent (2 mL), 45 °C, irradiated with blue LED (467 nm) or green LED (522 nm), oxygen atmosphere, 48 h.b Determined by 1H NMR. N.R. = no reaction. | ||||
| 1 | FL (1) | 467 | MeCN | N.R. |
| 2 | FL (1) | 467 | i-PrOH | N.R. |
| 3 | FL−2 (1) | 467 | MeCN | N.R. |
| 4 | FL−2 (1) | 467 | i-PrOH | N.R. |
| 5 | EY (1) | 522 | MeCN | 16 |
| 6 | EY (1) | 522 | i-PrOH | 15 |
| 7 | RB (1) | 522 | MeCN | 28 |
| 8 | RB (1) | 522 | i-PrOH | 37 |
| 9 | R6G (1) | 522 | MeCN | 84 |
| 10 | R6G (1) | 522 | i-PrOH | 25 |
Subsequently, the reactions were repeated in the presence of NaHCO3 to generate the basic form of the photocatalyst, since the production of singlet oxygen by some dye photosensitization was found to be very sensitive to pH changes.42 Nevertheless, the thiosaccharide 3a remained also unmodified (Table 1, entries 3 and 4). We have selected this base as is nontoxic and does not affect the substrate (otherwise O-deacetylation may take place).
Afterward, the experiments carried out with Eosin Y (EY), Rose Bengal (RB) or rhodamine 6G (R6G) gave the desired diastereomeric sulfoxides 4a in varied yields (Table 1, entries 5–10). The best result was obtained employing rhodamine 6G as photocatalyst, which led to the diastereomeric sulfoxides 4a in 84% yield.
Once selected the best photocatalyst, a solvent screening was evaluated to determine the more efficient medium to perform the photooxidation reaction, as depicted in Table 2.
| Entrya | Dye (mol%) | Solvent | Yield 4ab (%) |
|---|---|---|---|
| a Reaction conditions: 3a (0.05 M), solvent (2 mL), 45 °C, irradiated with green LED (522 nm), oxygen atmosphere, 48 h.b Determined by 1H NMR. N.R. = no reaction. | |||
| 1 | R6G (1) | MeCN | 84 |
| 2 | R6G (1) | i-PrOH | 25 |
| 3 | R6G (1) | MeOH | 11 |
| 4 | R6G (1) | Me2CO | 30 |
| 5 | R6G (1) | MeCN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (9:1) :MeOH (9:1) |
81 |
| 6 | R6G (1) | CH2Cl2 | 25 |
| 7 | R6G (1) | PhMe | N.R. |
| 8 | R6G (1) | PEG | N.R. |
The photooxidation reaction works better in polar solvents as MeCN and in a mixture MeCN:MeOH (9:1) (entries 1 and 5, Table 2). Despite the particularly good conversions and high chemoselectivities obtained towards the sulfoxides, we were unsatisfied with the long reaction times required (48 h), without complete consumption of the substrate, probably due to the photocatalyst bleaching under such long periods of irradiation.
As efficient photooxidations of sulfides to sulfoxides, under visible light,26,34,35 mediated by riboflavin derivates are reported,37,43 we decided to evaluate the photooxidation reactions employing tetra-O-acetyl riboflavin (RFTA) as photocatalyst. First, riboflavin (RF) was peracetylated employing a protocol described in the bibliography.44 Then, the photooxidation reactions were performed under various conditions, as shown in Table 3. When MeCN, MeOH, EtOH or H2O were employed as solvents, the degree of conversion was low (entries 1–4). Surprisingly, in MeCN:H2O (85:15) an excellent yield (99%) and chemoselectivity were achieved in considerably lower reaction times (entry 5). This result was in agreement with the obtained by Neveselý et al. for oxidation reactions performed in such solvent mixture to avoid catalyst aggregation.43 Replacement of MeCN:H2O (85:15) with EtOH:H2O (95:5) as solvent mixture led to a lower isolated yield (57%) for the same reaction time (entry 6). Since no bleaching of the RFTA was observed, the reaction was repeated extending the irradiation time to 6 h, leading to complete conversion with excellent chemoselectivity (entry 7). Subsequently, a few control experiments were carried out at longer reaction times. In the absence of an oxygen atmosphere, photocatalyst or an irradiation source, the sulfoxide 4a was not produced and the substrate 3a remains unchanged (entries 8–10). In contrast, when the photooxidation reaction was carried out in MeCN:H2O (85:15) and air atmosphere excellent conversion and selectivity was also obtained in 6 h (entry 11). For comparison, the air atmosphere was also tested in EtOH:H2O (95:5) leading to incomplete conversion even after 24 h of irradiation (64%, entry 12).
| Entrya | Dye (mol%) | Solvent | Additive | hν (nm) | Atm | Time (h) | Yield 4ab (%) |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: 3a (0.05 M), solvent (2 mL), 45 °C.b Determined by 1H NMR. N.R. = no reaction. | |||||||
| 1 | RFTA (2) | MeCN | — | 467 | O2 | 24 | 24 |
| 2 | RFTA (2) | MeOH | — | 467 | O2 | 24 | 14 |
| 3 | RFTA (2) | EtOH | — | 467 | O2 | 24 | 25 |
| 4 | RFTA (2) | H2O | — | 467 | O2 | 24 | 13 |
| 5 | RFTA (2) | MeCN:H2O (85:15) |
— | 467 | O2 | 2 | 99 |
| 6 | RFTA (2) | EtOH:H2O (95:5) |
— | 467 | O2 | 2 | 57 |
| 7 | RFTA (2) | EtOH:H2O (95:5) |
— | 467 | O2 | 6 | 99 |
| 8 | RFTA (2) | MeCN:H2O (85:15) |
— | 467 | N2 | 24 | N.R. |
| 9 | RFTA (2) | MeCN:H2O (85:15) |
— | Dark | O2 | 24 | N.R. |
| 10 | — | MeCN:H2O (85:15) |
— | 467 | O2 | 24 | N.R. |
| 11 | RFTA (2) | MeCN:H2O (85:15) |
— | 467 | Air | 6 | >99 |
| 12 | RFTA (2) | EtOH:H2O (95:5) |
— | 467 | Air | 24 | 64 |
| 13 | RFTA (2) | MeCN:H2O (85:15) |
NaN3 | 467 | O2 | 24 | N.R. |
| 14 | RFTA (2) | MeCN:H2O (85:15) |
DABCO | 467 | O2 | 24 | N.R. |
To evidence the formation of singlet oxygen in the media some additional control reactions were performed. As sodium azide and 1,4-diazabicyclo[2.2.2]octane (DABCO) are specific and efficient quenchers of singlet oxygen,45–48 the reactions were conducted in the presence of these additives. As expected, formation of the respective sulfoxides 4a was completely inhibited (entries 13 and 14).
Finally, to determine the scope of the photooxidation reaction, different thiosaccharides 3a–f were oxidized under the optimized experimental conditions (Table 4).
|
|||||
|---|---|---|---|---|---|
| Entrya | Sulfide | Time (h) | Conversion (%) | Isolated yield 4b (%) | Diastereomeric ratioc SS/SR |
| a Reaction conditions: 3 (0.05 M), solvent (2 mL), 45 °C, oxygen atmosphere (balloon).b isolated yield.c determined by 1H NMR. N.R. = no reaction. | |||||
| 1 | 3a | 2 | 100 | 99 | 1.6/1.0 |
| 2 | 3b | 2 | 100 | 93 | 2.0/1.0 |
| 3 | 3c | 6 | 94 | 57 | 1.5/1.0 |
| 4 | 3d | 6 | 84 | 60 | 1.5/1.0 |
| 5 | 3e | 6 | 0 | 0 | N.R. |
| 6 | 3f | 6 | 0 | 0 | N.R. |
| 7 | 3f | 24 | 0 | 0 | N.R. |
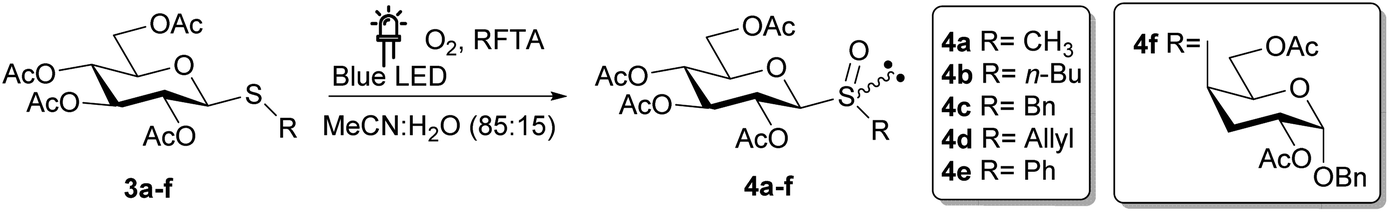
As summarized in Table 4, an excellent total isolated yield (99%) was obtained for the oxidation of 3a to sulfoxide 4a (entry 1). This was in fact a diastereomeric mixture, which could be chromatographically separated affording the SS and SR sulfoxides in 61% and 37% isolated yields, respectively. The photooxidation reaction of 3b was also highly efficient, affording the diastereomeric mixture of sulfoxides 4b after 2 h (isolated yield 93%). On the other hand, the conversion of substrates 3c and 3d was incomplete after 6 h of irradiation, and the isolated yields fell to 57% and 60% respectively (entries 3 and 4).
These results were not surprising since it was described that C–S bond cleavage can occur when benzyl and allyl sulfides undergo photooxidation under singlet oxygen conditions, leading to lower sulfoxide yields.49–51 However, it is important to highlight that the photooxidation reaction of the allyl sulfide 3d was completely chemoselective, and the sulfoxide 4d was obtained without oxidation of the vinyl group.
Unfortunately, no evidence of an oxidation reaction was obtained for the sulfide 3e, as this substrate was recovered unchanged after 6 h under irradiation (entry 5). Probably the excited state of the photocatalyst is quenched prior to the formation of singlet oxygen by some interaction with 3e.
To evaluate if the studied photochemical reaction could be applied to oxidize thiodisaccharides, the compound 3f was synthesized using a protocol described by our research group.52 The fact that no chemical changes of the thiodisaccharide were observed during the photooxidation, even after 24 h of irradiation, indicated that no reaction took place (Table 4, entries 6 and 7). This result may be explained considering the mechanism proposed in the bibliography.26,27 The peroxysulfoxide intermediate generated from the thiodisaccharide 3f needs to react with another molecule of this compound to afford the desired sulfoxide. The large steric hindrance produced by the sugar groups could prevent the peroxysulfoxide intermediate to evolve to the sulfoxide and consequently returns to the substrate. Displeased with these results, the removal of the acetyl protecting group of 3a, 3b, 3c, 3e, and 3f was performed (Scheme 2), since the oxidation potential of some carbohydrates can be modified by changing the protecting groups attached to these molecules.53 The de-O-acetylation reactions were carried out under mild conditions employing a mixture of MeOH/Et3N/H2O (4:1:5)54–56 to afford the free thiosaccharides 3g–k in very good to excellent isolated yields (90–97%).
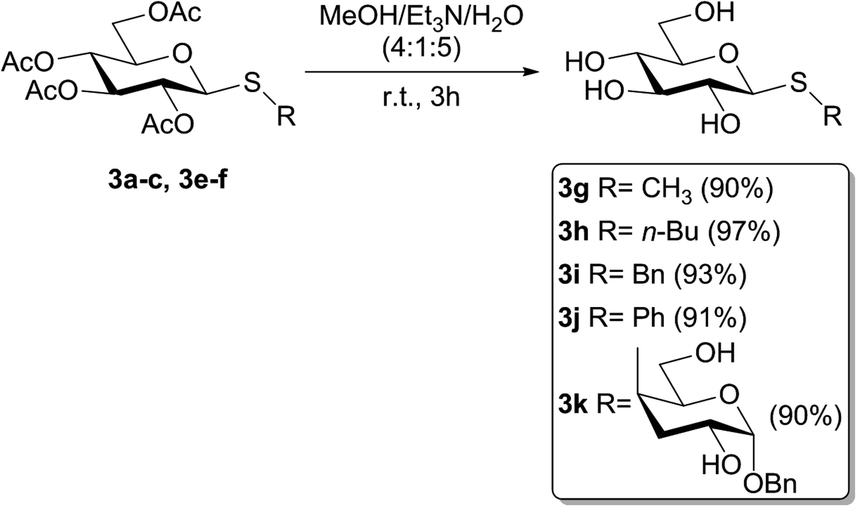 | ||
| Scheme 2 De-O-acetylation of thiosaccharides 3a–c, 3e–f. | ||
With the free thioglycosides in hand, the photooxidation reactions were conducted under the optimized conditions. Similar to the photooxidation of its analogue 3a, the sulfide 3g gave excellent results, as the diastereomeric mixture of sulfoxides 4g was obtained in 88% isolated yield (ratio SS/SR = 1.5/1) under irradiation for only 30 min. A mixture of α and β anomers of D-glucopyranose was also isolated as a minor product (5%). The diastereomeric mixture of free sulfoxides 4h and 4i were also obtained in lower reaction times than their peracetylated analogues, although in lower yields (66 and 53% yield, respectively). In addition, a major amount of D-glucopyranose was also obtained (Scheme 3).
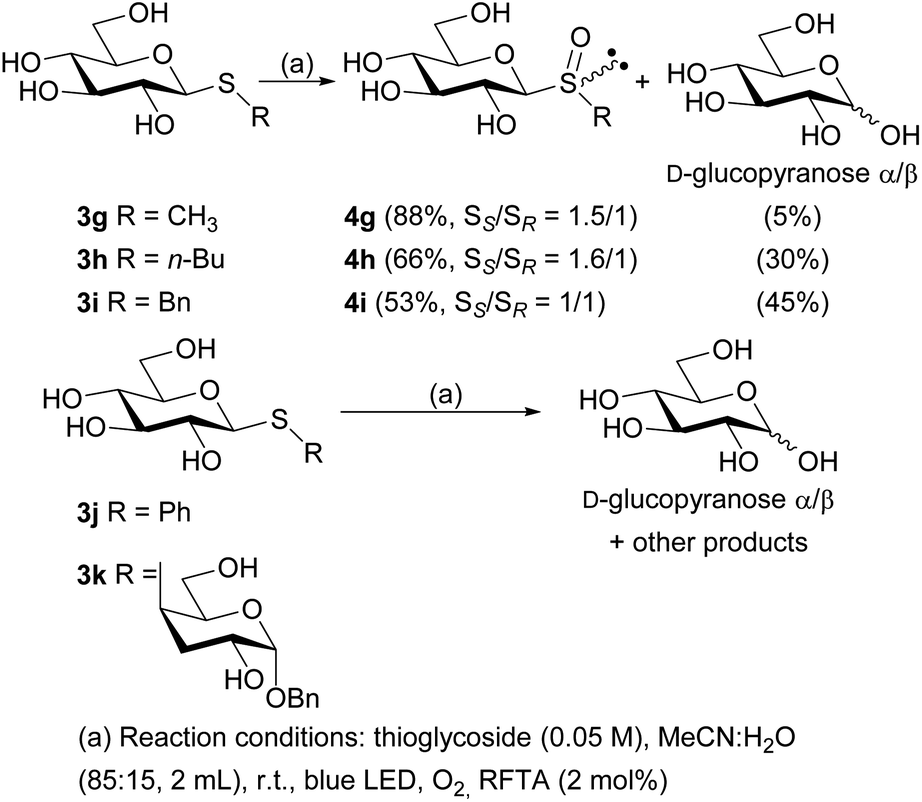 | ||
| Scheme 3 Photooxidation reactions of free thiosaccharides 3g–k. | ||
Unfortunately, the photooxidation reaction of sulfide 3j showed incomplete consumption of the starting material after 8 h and the corresponding diastereomeric sulfoxides were not obtained. Instead, a complex mixture was formed, probably as result of radical pathways in the oxidative medium. Most of these products remained unidentified, although from the reaction crude, a mixture of both anomers of D-glucopyranose was isolated as main product (yield 35%, ratio α/β = 1:1.6) (Scheme 3). Similarly, when the free thiodisaccharide 3k was subjected to the photooxidation a complex mixture was obtained. With the purpose of determining the structure of such products, the reaction crude was peracetylated employing pyridine and acetic anhydride (1:1), and subsequently subjected to column chromatography purification. The 1H NMR spectrum of one of the fractions displayed characteristic signals evidencing the presence of peracetylated glucose (ratio α/β = 1:1.2). These facts demonstrate that a competitive fragmentation reaction took place in these cases probably via a radical cation intermediate generated by the oxidation of the thiosaccharides 3g–k through an electron transfer reaction (type I mechanism), which is the main reaction for 3j and 3k.
The stereocontrol in the oxidation reactions was provided by the asymmetric induction of the glucosyl residue, which favors the formation of the sulfoxides with SS configuration in almost all cases.57,58 These results agree with previous reports describing that thioglycosides with α-configuration lead predominantly to sulfoxides with the SR absolute configuration at the sulfur atom, while their β-anomers lead to diastereomeric mixtures of SS (major) and SR sulfoxides.21,22,59
The configuration at the sulfur stereocenter of each sulfoxide obtained was established employing the methodology developed by our research group.18,19 In this protocol, the anisotropic effect of the S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group on the chemical shift of specific protons in the 1H NMR spectra must be considered. To perform this type of analysis, it is necessary to determine the rotamers, formed by rotation of the thioglycosidic linkage, present in the conformational equilibrium. As β-glucopyranosides populate almost exclusively the 4C1 conformation, each rotamer is characterized by specific NOE interactions observed in the corresponding 2D-NOESY spectra.
O group on the chemical shift of specific protons in the 1H NMR spectra must be considered. To perform this type of analysis, it is necessary to determine the rotamers, formed by rotation of the thioglycosidic linkage, present in the conformational equilibrium. As β-glucopyranosides populate almost exclusively the 4C1 conformation, each rotamer is characterized by specific NOE interactions observed in the corresponding 2D-NOESY spectra.
As depicted in Fig. 1, in the sulfoxides with the SR configuration, the rotamer gauche g− is favored because disposes the residue CH2R in a position with less steric hindrance and it is stabilized by the exo-anomeric effect.
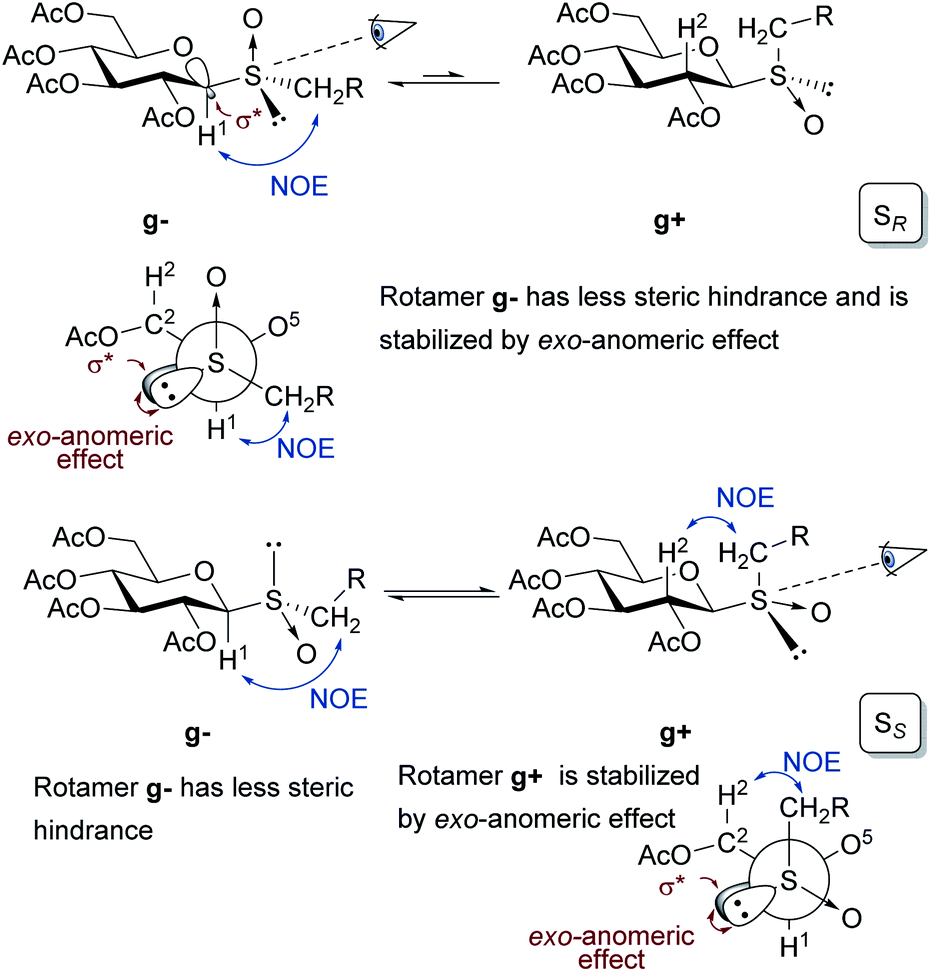 | ||
| Fig. 1 Conformations displayed by rotation of the anomeric linkage of β-glucopyranosyl sulfoxides with SR or SS configuration depicted for the 4C1 chair and in Newman projections. | ||
This was justified by interresidue NOE contacts between the sugar-H1 and the methylene protons of the CH2R observed in the 2D-NOESY spectra. The lack of the NOE contact between sugar-H2–CH2R suggested the almost exclusive presence of the g− conformer. On the other hand, in the sulfoxides with the SS configuration, both rotamers g− and g+ are stabilized by different factors. The first one (g−), arranges the residue CH2R in a position with less steric hindrance, while the second one (g+) presents stabilization by the exo-anomeric effect. This fact explains the coexistence of these two rotamers in equilibrium and was demonstrated by observing the NOE contacts between sugar-H1–CH2R protons for the g− rotamer, and the spatial interaction between sugar-H2–CH2R for the g+ rotamer. These results are in agreement with those reported by Sanhueza et al. in their study on the stereochemical properties of structurally related glucosyl sulfoxides.60
Once determined the preferential rotamers, the differences in the chemical shift for signals of specific protons in the 1H NMR spectra were analyzed for each sulfoxide. The shielding and deshielding effects were explained considering the position of the SO group and the sulfur lone pair relative to the protons H1 and H2 of the thiomonosaccharides 4a–d, 4g–i. The sulfoxides with the SR configuration in the most populated rotamer g− arranges the lone pair of electrons of the sulfur in the direction of the H1, generating a shielding effect, while H2 should be deshielded as result of the 1,3-diaxial interaction between the C2–H2 bond and the SO group. To perform this kind of analysis in the sulfoxides with the SS configuration both rotamers g− and g+ must be considered. In the rotamer g− the H1 is located in the SO anisotropic deshielding region, although this effect could be partially countered by the protection effect generated by the proximity to the sulfur lone pair in the g+ rotamer. The shielding effect observed for the H2 protons could be explained by the 1,3-diaxial interaction with the sulfur lone pair in the g− rotamer. Furthermore, an additional shielding contribution should be generated by the proximity of the sulfur lone pair to H2 in the g+ rotamer, as evidenced in the Newman projection. As consequence of all these effects, the signal of proton H1 in SR sulfoxides are expected to be shifted upfield compared to that of H1 in SS sulfoxides, while the protons H2 are deshielded (higher δ value) in the sulfoxides SR sulfoxides compared to SS sulfoxides (Table 5).
| Sulfoxide | δH1 (ppm) | δH2 (ppm) |
|---|---|---|
| 4a(SR) | 4.15 | 5.44 |
| 4a(SS) | 4.38 | 5.06 |
| 4b(SR) | 4.16 | 5.44 |
| 4b(SS) | 4.34 | 5.21 |
| 4c(SR) | 3.83 | 5.44 |
| 4c(SS) | 4.05 | 5.24 |
| 4d(SR) | 4.16 | 5.43 |
| 4d(SS) | 4.33 | 5.34 |
| 4g(SR) | 4.25 | 3.74 |
| 4g(SS) | 4.63 | 3.66 |
| 4h(SR) | 4.28 | 3.74 |
| 4h(SS) | 4.62 | 3.71 |
| 4i(SR) | 4.07 | 3.76 |
| 4i(SS) | 4.58 | 3.64 |
Some glycosyl sulfoxides, such as 4aS and 4aR, have been previously synthesized.57 The fact that their physical and spectral data agree with those of the same products obtained in this work, and which configurations at the sulfur stereocenter has been assigned using the procedure described above, serve as validation of this methodology developed by us.
Only the peracetylated glucosyl sulfoxides containing R = CH3, could be separated by column chromatography, and all the other sulfoxide derivates were obtained as their diastereomeric mixtures. Nevertheless, applications of glycosyl sulfoxides as glycosyl donors allows the use of the diastereomeric mixture, since the stereochemical outcome in glycosylation reactions is independent of the sulfoxide stereochemistry (SR or SS).61
Experimental
Materials and methods
All photocatalysts were acquired commercially, except for tetra-O-acetyl riboflavin which was synthesized by a known procedure44 from commercially available riboflavin. Column chromatography was carried out with silica gel 60 (230–400 mesh). Analytical thin-layer chromatography (TLC) was carried out on silica gel 60 F254 aluminium-backed plates (layer thickness 0.2 mm). The spots were visualized by charring with a solution of (NH4)6Mo7O24·4H2O 25 g L−1, (NH4)4Ce(SO4)4·2H2O 10 g L−1 and 10% H2SO4 in H2O. Nuclear magnetic resonance (NMR) spectra were recorded at 400 MHz (1H) or 100 MHz (13C). Chemical shifts were calibrated to tetramethylsilane or to a residual solvent peak (CHCl3: 1H: δ = 7.26 ppm, 13C: δ = 77.2 ppm or H2O: δ = 4.79 ppm, respectively). Assignments of 1H and 13C NMR spectra were assisted by 2D 1H-COSY and 2D 1H–13C HSQC and HMBC experiments. For the assignment of the NMR signals, the H and C atoms of the aglycone residue have been labeled as depicted for each individual compound in the ESI.†The coupling constants values are reported in Hz and resonance multiplicities abbreviated as follows: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, sx = sextet, m = multiplet, br = broad. High-resolution mass spectra (HRMS) were obtained using the electrospray ionization (ESI) technique and Q-TOF detection.
Synthetic procedures
Methyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (3a). Following the general procedure, the synthesis of 3a was performed adding dimethyl sulfate (52 μL, 0.55 mmol) as the electrophile to the basic solution of 2. Upon reaction completion, the reaction mixture was diluted with acetonitrile, extracted with hexane (20 mL × 3) to remove excess of dimethyl sulfate, and then treated as was mentioned above. The thiosaccharide 3a62–64 (205.6 mg, 99%) was obtained as a white solid, m.p. at 86.8 °C (dec.). Rf = 0.60, hexane/EtOAc (1
:1). [α]24D = −11.9 (c = 0.8, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.20 (t, J2,3 = J3,4 = 9.4 Hz, 1H, 3-H), 5.05 (t, J3,4 = J4,5 = 9.7 Hz, 1H, 4-H), 5.04 (t, J1,2 = J2,3 = 9.6 Hz, 1H, 2-H), 4.37 (d, J1,2 = 10.0 Hz, 1H, 1-H), 4.22 (dd, J6a,6b = 12.4, J5-6a = 4.8 Hz, 1H, 6a-H), 4.12 (dd, J6a,6b = 12.4, J5-6b = 1.9 Hz, 1H, 6b-H), 3.71 (ddd, J4,5 = 9.9, J5,6a = 4.5, J5,6b = 2.1 Hz, 1H, 5-H), 2.14 (s, 3H, CH3S), 2.05, 2.04, 2.00, 1.98 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.8, 170.3, 169.6 (×2) (COCH3), 83.0 (C-1), 76.1 (C-5), 74.0 (C-3), 69.2 (C-2), 68.4 (C-4), 62.2 (C-6), 20.9, 20.8, 20.7 (×2) (COCH3), 11.4 (CH3S) ppm. HRMS (ESI): calcd for C15H22NaO9S 401.0877 [M + Na]+; found 401.0862.
Butyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (3b). Following the general procedure, the synthesis of 3b63,65,66 was performed adding n-butyl bromide (59 μL, 0.55 mmol) to the basic solution of 2. The thiosaccharide 3b (224 mg, 97%) was obtained as a white solid, m.p. 69.1–70.0 °C. Rf = 0.75, hexane/EtOAc (1
:1). [α]24D = −26.9 (c = 0.9, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.21 (t, J2,3 = J3,4 = 9.4 Hz, 1H, 3-H), 5.07 (t, J3,4 = J4,5 = 9.9 Hz, 1H, 4-H), 5.02 (dd, J1,2 = 10.1, J2,3 = 9.4 Hz, 1H, 2-H), 4.47 (d, J1,2 = 10.0 Hz, 1H, 1-H), 4.23 (dd, J6a,6b = 12.4, J5,6a = 5.0 Hz, 1H, 6a-H), 4.13 (dd, J6a,6b = 12.3, J5,6b = 2.4 Hz, 1H, 6b-H), 3.70 (ddd, J4,5 = 10.0, J5,6a = 4.9, J5,6b = 2.4 Hz, 1H, 5-H), 2.73–2.60 (m, 2H, a-H), 2.07, 2.05, 2.02, 2.00 (4s, 12H, COCH3), 1.60–1.53 (m, 2H, b-H), 1.39 (sx, Jb,c = Jc,d = 7.4 Hz, 2H, c-H), 0.90 (t, Jc,d = 7.3 Hz, 3H, d-H) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.8, 170.3, 169.6, 169.5 (COCH3), 83.8 (C-1), 76.0 (C-5), 74.1 (C-3), 70.0 (C-2), 68.5 (C-4), 62.3 (C-6), 31.8 (C-b), 29.8 (C-a), 22.0 (C-c), 20.9 (×2), 20.7 (×2) (COCH3), 13.7 (C-d) ppm. HRMS (ESI): calcd for C18H28NaO9S 443.1346 [M + Na]+; found 443.1357.
Benzyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (3c). Following the general procedure, the synthesis of 3c was performed adding benzyl bromide (65 μL, 0.55 mmol) to the basic solution of 2. The thiosaccharide 3c63,66,67 (215 mg, 86%) was obtained as a white solid, m.p. at 93.8 °C (dec.). Rf = 0.65, hexane/EtOAc (1
:1). [α]24D = −86.5 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 7.34–7.29 (m, 5H, PhCH2O), 5.16–5.04 (m, 3H, 3-H, 2-H, 4-H), 4.29 (d, J1,2 = 9.7 Hz, 1H, 1-H), 4.23 (dd, J6a,6b = 12.4, J5,6a = 5.1 Hz, 1H, 6a-H), 4.13 (dd, J6a,6b = 12.3, J5,6b = 2.2 Hz, 1H, 6b-H), 3.94 (d, Jgem = 12.9 Hz, 1H, a-H), 3.83 (d, Jgem = 12.9 Hz, 1H, a-H), 3.59 (ddd, J4,5 = 9.6, J5,6a = 5.0, J5,6b = 2.3 Hz, 1H, 5-H), 2.11, 2.01 (×2), 1.99 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.7, 170.3, 169.5 (×2) (COCH3), 137.0, 129.2, 128.8, 127.6 (C-aromatics), 82.2 (C-1), 76.0 (C-5), 74.0 (C-3), 70.0 (C-2), 68.6 (C-4), 62.4 (C-6), 34.0 (C-a), 20.9, 20.8, 20.7 (×2) (COCH3) ppm. HRMS (ESI): calcd for C21H26NaO9S 477.1190 [M + Na]+; found 477.1222.
Allyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (3d). Following the general procedure, the synthesis of 3d was carried out adding allyl bromide (47 μL, 0.55 mmol) to the basic solution of 2. The thiosaccharide 3d68 (160 mg, 72%) was obtained as a colorless syrup. Rf = 0.67, hexane/EtOAc (1
:1). [α]25D = −15.9 (c = 0.9, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.81–5.71 (m, 1H, b-H), 5.18 (t, J2,3 = J3,4 = 9.3 Hz, 1H, 3-H), 5.13–5.08 (m, 2H, c-H, c′-H), 5.02 (t, J3,4 = J4,5 = 9.7 Hz, 1H, 4-H), 5.01 (dd, J1,2 = 9.9, J2,3 = 9.4 Hz, 1H, 2-H), 4.45 (d, J1,2 = 10.1 Hz, 1H, 1-H), 4.19 (dd, J6a,6b = 12.3, J5,6a = 5.2 Hz, 1H, 6a-H), 4.09 (dd, J6a,6b = 12.3, J5,6b = 2.3 Hz, 1H, 6b-H), 3.62 (ddd, J4,5 = 10.0, J5,6a = 5.1, J5,6b = 2.3 Hz, 1H, 5-H), 3.35 (dd, Jgem = 13.5, Ja,b = 8.4 Hz, 1H, a-H), 3.19 (dd, Jgem = 13.5, Ja,b = 6.1 Hz, 1H, a-H), 2.04, 2.00, 1.98, 1.96 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.6, 170.2, 169.4 (×2) (COCH3), 133.5 (C-b), 118.0 (C-c), 82.0 (C-1), 75.8 (C-5), 74.0 (C-3), 70.0 (C-2), 68.6 (C-4), 62.3 (C-6), 32.9 (C-a), 20.7 (×2), 20.6 (×2) (COCH3) ppm. HRMS (ESI): calcd for C17H24NaO9S 427.1033 [M + Na]+; found 427.1014.
Phenyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (3e). This compound was synthesized employing a procedure previously described.39 To a solution of penta-O-acetyl glucopyranose 1 (975 mg, 2.5 mmol) in anhydrous CH2Cl2 (5.0 mL) was added thiophenol (0.3 mL, 3 mmol), followed by the dropwise addition of BF3·OEt2 (1.6 mL, 12.5 mmol) under argon atmosphere. The reaction mixture was stirred 4 h until complete consumption of the starting materials. The mixture was diluted to 50 mL in CH2Cl2 and it was extracted with water (2 × 50 mL), sodium bicarbonate (1 × 50 mL), and brine (1 × 50 mL). The organic phase was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. Column chromatography of the residue using pentane/EtOAc (4
:1 → 1:1) afforded 3e63,67,69,70 (782 mg, 71%) as a white solid, m.p. 116.3–117.9 °C. Rf = 0.47, pentane/EtOAc (2:1). 1H NMR (400 MHz, CDCl3): δ = 7.51–7.48 (m, 2H, aromatic), 7.32–7.30 (m, 3H, aromatic), 5.22 (t, J2,3 = J3,4 = 9.3 Hz, 1H, 3-H), 5.04 (t, J3,4 = J4,5 = 9.8 Hz, 1H, 4-H), 4.97 (dd, J1,2 = 10.0, J2,3 = 9.3 Hz, 1H, 2-H), 4.71 (d, J1,2 = 10.1 Hz, 1H, 1-H), 4.22 (dd, J6a,6b = 12.3, J5-6a = 5.0 Hz, 1H, 6a-H), 4.18 (dd, J6a,6b = 12.3, J5,6b = 2.6 Hz, 1H, 6b-H), 3.72 (ddd, J4,5 = 10.0, J5,6a = 5.0, J5,6b = 2.7 Hz, 1H, 5-H), 2.08 (×2), 2.01, 1.99 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.7, 170.3, 169.5, 169.4 (COCH3), 133.3, 131.8, 129.1, 128.6 (aromatic), 85.9 (C-1), 76.0 (C-5), 74.1 (C-3), 70.1 (C-2), 68.4 (C-4), 62.3 (C-6), 20.9, 20.8, 20.7 (×2) (COCH3) ppm. HRMS (ESI): calcd for C20H24NaO9S 463.1033 [M + Na]+; found 463.0989.
Benzyl 3-deoxy-2,6-di-O-acetyl-4-S-(2,3,4,6-tetra-O-acetyl-β-D-glucopyranosyl)-4-thio-α-D-xylo-hexopyranoside (3f). Thiodisaccharide 3f was obtained following a protocol described by our research group52 as a white solid, m.p. at 110 °C (dec.). Rf = 0.53 (pentane/EtOAc, 1
:1). [α]25D = +28.5 (c = 1.2, CHCl3) 1H NMR (400 MHz, CDCl3): δ = 7.36–7.29 (m, 5H, aromatic), 5.21 (m, 2H, 2-H, 3′-H), 5.08 (t, J3′,4′ = J4′,5′ = 9.7 Hz, 1H, 4′-H), 5.01 (t, J1′,2′ = J2′,3′ = 9.6 Hz, 1H, 2′-H), 5.01 (d, J1,2 = 3.2 Hz, 1H, 1-H), 4.74 (d, Jgem = 12.0 Hz, 1H, PhCH2O), 4.61 (d, J1′,2′ = 10.0 Hz, 1H, 1′-H), 4.52 (d, Jgem = 12.0 Hz, 1H, PhCH2O), 4.36 (m, 1H, 5-H), 4.21 (dd, J6′a,6′b = 12.5, J5′,6′a = 4.6 Hz, 1H, 6′a-H), 4.16 (m, 2H, 6a-H, 6b-H), 4.13 (dd, J6′a,6′b = 12.5, J5′,6′b = 2.3 Hz, 1H, 6′b-H), 3.67 (ddd, J4′,5′ = 9.9, J5′,6′a = 4.4, J5′,6′b = 2.4 Hz, 1H, 5′-H), 3.39 (br d, J = 2.1 Hz, 1H, 4-H), 2.34 (td, J2,3a = J3a,3b = 12.6, J3a,4 = 3.6 Hz, 1H, 3a-H), 2.14 (m, 1H, 3b-H), 2.07, 2.06 (×2), 2.03, 2.01, 1.99 (COCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.7, 170.6, 170.2 (×2), 169.7, 169.4 (COCH3), 137.3, 128.6, 128.1, 128.0 (C-Ph), 94.9 (C-1), 82.8 (C-1′), 76.1 (C-5′), 73.9 (C-3′), 70.0 (C-2′), 69.2 (PhCH2O), 68.3 (×2, C-4′, C-5), 66.9 (C-2), 65.4 (C-6), 62.0 (C-6′), 42.9 (C-4), 30.9 (C-3), 21.1, 20.9, 20.8, 20.7 (×3) (COCH3). HRMS (ESI): calcd for C31H40NaO15S 707.1980 [M + Na]+; found 707.1959.
Methyl 1-thio-β-D-glucopyranoside (3g). The thiosaccharide 3a (37.8 mg, 0.1 mmol) was dissolved in MeOH/Et3N/H2O (4
:1:5, 0.55 mL) and the reaction mixture was stirred at 30 °C for 2 h. When TLC showed complete consumption of the starting material, the mixture was concentrated under reduced pressure. Column chromatography using CH2Cl2/MeOH (4:1 → 2:1) afforded 3g71–73 (18.9 mg, 90%) as a colorless syrup. Rf = 0.40, CH2Cl2/MeOH (4:1). [α]23D = −17.2 (c = 1.1, H2O) 1H NMR (400 MHz, D2O): δ = 4.49 (d, J1,2 = 9.8 Hz, 1H, 1-H), 3.96 (dd, J6a,6b = 12.4, J5,6a = 2.1 Hz, 1H, 6a-H), 3.77 (dd, J6a,6b = 12.5, J5,6b = 5.6 Hz, 1H, 6b-H), 3.55 (t, J2,3 = J3,4 = 8.8 Hz, 1H, 3-H), 3.54–3.50 (m, 1H, 5-H), 3.46 (dd, J3,4 = 8.9, J4,5 = 9.6 Hz, 1H, 4-H), 3.41 (dd, J1,2 = 9.7, J2,3 = 8.8 Hz, 1H, 2-H), 2.27 (s, 3H, CH3S) ppm. 13C NMR (100 MHz, D2O): δ = 85.6 (C-1), 79.9 (C-5), 77.2 (C-3), 71.7 (C-2), 69.6 (C-4), 60.9 (C-6), 11.4 (CH3S) ppm. HRMS (ESI): calcd for C7H14NaO5S 233.0454 [M + Na]+; found 233.0447.
Butyl 1-thio-β-D-glucopyranoside (3h). The thiosaccharide 3b (538.0 mg, 1.28 mmol) was dissolved in MeOH/Et3N/H2O (4
:1:5, 7.2 mL) and the reaction mixture was stirred at 30 °C for 3 h. When TLC showed complete consumption of the starting material, the mixture was concentrated under reduced pressure. Column chromatography using CH2Cl2/MeOH (8:1) afforded 3h (312 mg, 97%) as a colorless syrup. Rf = 0.78, CH2Cl2/MeOH (4:1). [α]23D = −40.1 (c = 1.2, MeOH) 1H NMR (400 MHz, D2O): δ = 4.55 (d, J1,2 = 9.9 Hz, 1H, 1-H), 3.93 (dd, J6a,6b = 12.4, J5,6a = 1.6 Hz, 1H, 6a-H), 3.73 (dd, J6a,6b = 12.4, J5,6b = 5.5 Hz, 1H, 6b-H), 3.51 (t, J2,3 = J3,4 = 8.6 Hz, 1H, 3-H), 3.48 (ddd, J4,5 = 9.4, J5,6b = 5.4, J5,6a = 1.6 Hz, 1H, 5-H), 3.43 (t, J3,4 = J4,5 = 9.3 Hz, 1H, 4-H), 3.34 (t, J1,2 = J2,3 = 9.3 Hz, 1H, 2-H), 2.85–2.72 (m, 2H, a-H), 1.65 (p, Ja,b = Jb,c = 7.4 Hz, 2H, b-H), 1.43 (sx, Jb,c = Jc,d = 7.4 Hz, 2H, c-H), 0.92 (t, Jc,d = 7.4 Hz, 3H, d-H) ppm. 13C NMR (100 MHz, D2O): δ = 85.4 (C-1), 79.9 (C-5), 77.3 (C-3), 72.4 (C-2), 69.6 (C-4), 61.0 (C-6), 31.5 (C-b), 29.7 (C-a), 21.3 (C-c), 12.9 (C-d) ppm. HRMS (ESI): calcd for C10H20NaO5S 275.0924 [M + Na]+; found 275.0929.
Benzyl 1-thio-β-D-glucopyranoside (3i). The thiosaccharide 3c (260.0 mg, 0.57 mmol) was dissolved in MeOH/Et3N/H2O (4
:1:5, 3.2 mL) and the reaction mixture was stirred at 30 °C for 3 h. When TLC showed complete consumption of the starting material, the mixture was concentrated under reduced pressure. Column chromatography using CH2Cl2/MeOH (8:1) afforded 3i (152.3 mg, 93%) as a colorless syrup. Rf = 0.82, CH2Cl2/MeOH (4:1). [α]23D = −129.4 (c = 1.0, MeOH) 1H NMR (400 MHz, D2O): δ = 7.45–7.35 (m, 5H, aromatic), 4.34 (d, J1,2 = 9.4 Hz, 1H, 1-H), 4.05 (d, Jgem = 13.2 Hz, 1H, a-H), 3.96 (d, Jgem = 13.3 Hz, 1H, a-H), 3.88 (dd, J6a,6b = 12.5, J5,6a = 2.1 Hz, 1H, 6a-H), 3.71 (dd, J6a,6b = 12.5, J5,6b = 5.6 Hz, 1H, 6b-H), 3.44–3.34 (m, 4H, 4-H, 3-H, 2-H, 5-H) ppm. 13C NMR (100 MHz, D2O): δ = 138.0, 129.1, 128.8, 127.4 (aromatic), 84.0 (C-1), 79.8(C-5), 77.3(C-3), 72.2 (C-2), 69.5(C-4), 60.9 (C-6), 33.6 (C-a) ppm. HRMS (ESI): calcd for C13H18NaO5S 309.0767 [M + Na]+; found 309.0770.
Phenyl 1-thio-β-D-glucopyranoside (3j). The thiosaccharide 3e (44.1 mg, 0.1 mmol) was dissolved in MeOH/Et3N/H2O (4
:1:5, 0.55 mL) and the reaction mixture was stirred at 30 °C for 3 h. After TLC showed complete consumption of the starting material, the mixture was concentrated under reduced pressure and the residue was purified by column chromatography using CH2Cl2/MeOH (4:1 → 2:1), affording 3j74,75 (24.5 mg, 91%) as a white solid, m.p. at 128.5 °C (dec.). Rf = 0.66, CH2Cl2/MeOH (4:1). [α]24D = +49.2 (c = 1.0, EtOH) 1H NMR (400 MHz, D2O): δ = 7.65–7.63 (m, 3H, aromatic), 7.50–7.44 (m, 3H, aromatic), 4.85 (d, J1,2 = 9.9 Hz, 1H, 1-H), 3.95 (dd, J6a,6b = 12.5, J5-6a = 2.2 Hz, 1H, 6a-H), 3.77 (dd, J6a,6b = 12.5, J5-6b = 5.6 Hz, 1H, 6b-H), 3.58 (t, J2,3 = J3,4 = 8.9 Hz, 1H, 3-H), 3.56–3.51 (m, 1H, 5-H), 3.47 (dd, J4,5 = 9.7, J3,4 = 9.0 Hz, 1H, 4-H), 3.41 (dd, J1,2 = 9.8, J2,3 = 9.0 Hz, 1H, 2-H) ppm. 13C NMR (100 MHz, D2O): δ = 132.0, 131.7, 129.4, 128.1 (aromatic), 87.3 (C-1), 79.9 (C-5), 77.3 (C-3), 71.8 (C-2), 69.4 (C-4), 60.9 (C-6) ppm. HRMS (ESI): calcd for C12H16NaO5S 295.0611 [M + Na]+; found 295.0619.
Benzyl 3-deoxy-4-S-(β-D-glucopyranosyl)-4-thio-α-D-xylo-hexopyranoside (3k). The thiodisaccharide 3f (68.4 mg, 0.1 mmol) was dissolved in MeOH/Et3N/H2O (4
:1:5, 0.84 mL) and the reaction mixture was stirred at 30 °C for 3 h. When TLC showed complete consumption of the starting material into a more polar product (Rf = 0.74, BuOH/EtOH/H2O (10:4:4)), the mixture was concentrated under reduced pressure. Subsequently, purification of the residue by column chromatography using CH2Cl2/MeOH (4:1 → 2:1) afforded 3k (40.2 mg, 93%) as a colorless syrup. 1H NMR (400 MHz, D2O): δ = 7.55–7.44 (m, 5H, aromatic), 5.02 (d, J1,2 = 3.9 Hz, 1H, 1-H), 4.84 (d, Jgem = 11.7 Hz, 1H, PhCH2O), 4.71 (d, Jgem = 11.8 Hz, 1H, PhCH2O), 4.64 (d, J1′,2′ = 9.8 Hz, 1H, 1′-H), 4.26–4.16 (m, 2H, 5-H, 2-H), 3.95 (dd, J6′a,6′b = 12.3 Hz, J5′,6′a = 2.0 Hz, 1H, 6′a-H), 3.77 (dd, J6a,6b = 11.8, J5,6a = 5.0 Hz, 1H, 6a-H), 3.75 (dd, J6′a,6′b = 12.4, J5′,6′b = 5.4 Hz, 1H, 6′b-H), 3.63 (dd, J6a,6b = 11.8, J5,6b = 7.4 Hz, 1H, 6b-H), 3.58–3.43 (m, 4H, 4-H, 3′-H, 4′-H, 5′-H), 3.36 (dd, J1′,2′ = 9.7, J2′,3′ = 8.9 Hz, 1H, 2′-H), 2.29–2.16 (m, 2H, 3a-H, 3b-H) ppm. 13C NMR (100 MHz, D2O): δ = 137.3, 128.8, 128.7, 128.4 (aromatic), 97.5 (C-1), 84.9 (C-1′), 80.0 (C-4′), 77.3 (C-3′), 72.6 (C-2′), 70.6 (C-5), 69.8 (PhCH2O), 69.6 (C-5′), 64.2 (C-2), 62.5 (C-6), 61.0 (C-6′), 42.9 (C-4), 32.7 (C-3) ppm. HRMS (ESI): calcd for C19H28NaO9S 455.1346 [M + Na]+; found 455.1369.
:1). For sulfoxides 4a–d, the reaction mixtures were purified by column chromatography with hexane/EtOAc (8:1 → 1:1), while sulfoxides 4g–i were purified using MeCN/H2O (9:1 → 4:1) or CH2Cl2/MeOH (8:1 → 4:1).
Methyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (S)-S-oxide (4aS). The major product of the oxidation of thiosaccharide 3a was sulfoxide 4aS57 (24 mg, 61%) obtained as a colorless syrup. Rf = 0.25, EtOAc. [α]23D = −12.2 (c = 0.9, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.31 (t, J2,3 = J3,4 = 9.3 Hz, 1H, 3-H), 5.11 (dd, J4,5 = 9.9, J3,4 = 9.6 Hz, 1H, 4-H), 5.07 (dd, J1,2 = 10.0, J2,3 = 9.4 Hz, 1H, 2-H), 4.38 (d, J1,2 = 10.2 Hz, 1H, 1-H), 4.32 (dd, J6a,6b = 12.6, J5,6a = 4.5 Hz, 1H, 6a-H), 4.21 (dd, J6a,6b = 12.6, J5,6b = 2.2 Hz, 1H, 6b-H), 3.84 (ddd, J4,5 = 10.1, J5,6a = 4.5, J5,6b = 2.2 Hz, 1H, 5-H), 2.68 (s, 3H, CH3SO), 2.09, 2.07, 2.04, 2.02 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.6, 170.1, 169.9, 169.5 (COCH3), 90.9 (C-1), 77.0 (C-5), 73.3 (C-3), 68.4 (C-2), 67.8 (C-4), 61.5 (C-6), 33.0 (CH3SO), 20.8, 20.7 (×3) (COCH3) ppm. HRMS (ESI): calcd for C15H22NaO10S 417.0826 [M + Na]+; found 417.0756.
Methyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (R)-S-oxide (4aR). The minor product of the oxidation of thiosaccharide 3a was sulfoxide 4aR57 (14.6 mg, 36%) obtained as a colorless syrup. Rf = 0.20, EtOAc. [α]23D = −64.7 (c = 0.8, CHCl3). 1H NMR (400 MHz, CDCl3): δ = 5.44 (t, J1,2 = J2,3 = 9.6 Hz, 1H, 2-H), 5.36 (t, J2,3 = J3,4 = 9.3 Hz, 1H, 3-H), 5.15 (dd, J4,5 = 9.9, J3,4 = 9.4 Hz, 1H, 4-H), 4.27 (dd, J6a,6b = 12.7, J5,6a = 5.1 Hz, 1H, 6a-H), 4.23 (dd, J6a,6b = 12.7, J5,6b = 3.0 Hz, 1H, 6b-H), 4.15 (d, J1,2 = 9.8 Hz, 1H, 1-H), 3.82 (ddd, J4,5 = 10.1, J5,6a = 4.9, J5,6b = 2.9 Hz, 1H, 5-H), 2.69 (s, 3H, CH3SO), 2.08, 2.06, 2.05, 2.03 (4s, 12H, COCH3) ppm. 13C NMR (100 MHz, CDCl3): δ = 170.7, 170.6, 169.3, 169.1 (COCH3), 87.6 (C-1), 77.0 (C-5), 73.8 (C-3), 67.9 (C-4), 67.0 (C-2), 62.0 (C-6), 33.2 (CH3SO), 20.8 (×2), 20.7 (×2) (COCH3) ppm. HRMS (ESI): calcd for C15H22NaO10S 417.0826 [M + Na]+; found 417.0798.
Butyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (R,S)-S-oxide (4bR,S). The oxidation reaction of thiosaccharide 3b gave the diastereomeric mixture of sulfoxides 4bR,S that could not be separated by column chromatography. The mixture (40.5 mg, 93%, ratio S/R, 2
:1) was obtained as a white solid and showed a single spot by TLC analysis. Rf = 0.17, hexane/EtOAc (1:1). 1H NMR (400 MHz, CDCl3) data for S isomer: δ = 5.29 (t, J2,3 = J3,4 = 9.2 Hz, 1H, 3-HS), 5.21 (t, J1,2 = J2,3 = 9.6 Hz, 1H, 2-HS), 5.09 (dd, J4,5 = 9.7, J3,4 = 9.4 Hz, 1H, 4-HS overlapping with 4-HR of 4bR), 4.34 (d, J = 9.9 Hz, 1H, 1-HS), 4.27 (dd, J6a,6b = 12.6, J5,6a = 4.6 Hz, 1H, 6a-HS), 4.17 (dd, J6a,6b = 12.6, J5,6b = 2.3 Hz, 1H, 6b-HS), 3.82–3.78 (m, 1H, 5-HS,overlapping with 5-HR of 4bR), 2.96–2.89 (m, 1H, a-HS), 2.83–2.76 (m, 1H, a-HS), 2.07–2.01 (4s, 12H, COCH3 overlapping with COCH3 of 4bR), 1.79–1.73 (m, 2H, b-HS overlapping with b-HR of 4bR), 1.57–1.43 (m, 2H, c-HS overlapping with c-HR of 4bR), 0.96 (t, Jc,d = 7.3 Hz, 3H, d-HS overlapping with d-HR of 4bR) ppm. 13C NMR (100 MHz, CDCl3) data for S isomer: δ = 170.6–168.9 (COCH3 overlapping with COCH3 of 4bR), 90.4 (C-1S), 77.0 (C-5S), 73.3 (C-3S), 68.5 (C-2S), 67.8 (C-4S), 61.6 (C-6S), 47.2 (C-aS), 24.2 (C-bS), 22.1 (C-cS), 20.8–20.7 (COCH3 overlapping with COCH3 of 4bR), 13.8 (C-dS overlapping with C-dR of 4bR) ppm. 1H NMR (400 MHz, CDCl3) data for R isomer: δ = 5.44 (dd, J1,2 = 9.8, J2,3 = 9.4 Hz, 1H, 2-HR), 5.35 (t, J2,3 = J3,4 = 9.3 Hz, 1H, 3-HR), 5.12 (dd, J4,5 = 10.0, J3,4 = 9.5 Hz, 1H, 4-HR overlapping with 4-HS of 4bS), 4.27–4.21 (m, 2H, 6a-HR, 6b-HR), 4.16 (d, J1,2 = 9.9 Hz, 1H, 1-HR), 3.82–3.78 (m, 1H, 5-HR,overlapping with 5-HS of 4bS), 3.16–3.09 (m, 1H, a-HR), 2.74–2.67 (m, 1H, a-HR), 2.07–2.01 (4s, 12H, COCH3 overlapping with COCH3 of 4bS), 1.79–1.73 (m, 2H, b-HR overlapping with b-HS of 4bS), 1.57–1.43 (m, 2H, c-HR overlapping with c-HS of 4bS), 0.96 (t, Jc,d = 7.3 Hz, 3H, d-HR overlapping with d-HS of 4bS) ppm. 13C NMR (100 MHz, CDCl3) data for R isomer: δ = 170.6–168.9 (COCH3 overlapping with COCH3 of 4bS), 87.0 (C-1R), 77.0 (C-5R), 73.9 (C-3R), 68.0 (C-4R), 67.0 (C-2R), 62.1 (C-6R), 47.1 (C-aR), 24.9 (C-bR), 22.3 (C-cR), 20.8–20.7 (COCH3 overlapping with COCH3 of 4bS), 13.8 (C-dR overlapping with C-dS of 4bS) ppm. HRMS (ESI): calcd for C18H28NaO10S 459.1295 [M + Na]+; found 459.1271.
Benzyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (R,S)-S-oxide (4cR,S). The oxidation reaction of thiosaccharide 3c gave the diastereomeric mixture of sulfoxides 4cR,S76 that could not be separated by column chromatography. The mixture (26.8 mg, 57%, ratio S/R, 1.5
:1) was obtained as a white solid and showed a single spot in TLC analysis. Rf = 0.12, hexane/EtOAc (1:1). 1H NMR (400 MHz, CDCl3) data for S isomer: δ = 7.41–7.32 (m, 5H, aromaticS overlapping with aromaticR of 4cR), 5.28 (t, J1,2 = J2,3 = 9.3 Hz, 1H, 2-HS), 5.24 (t, J2,3 = J3,4 = 9.1 Hz, 1H, 3-HS), 5.14–5.07 (m, 1H, 4-HS overlapping with 4-HR of 4cR), 4.33–4.26 (m, 1H, 6a-HS overlapping with 6b-HR, a-HR), 4.24 (dd, J6a,6b = 12.6, J5,6b = 2.4 Hz, 1H, 6b-HS), 4.16 (d, Jgem = 13.0 Hz, 1H, a-HS), 4.16 (d, J1,2 = 9.8 Hz, 1H, 1-HS), 4.07 (d, Jgem = 13.0 Hz, 1H, a-HS), 3.77 (ddd, J4,5 = 9.9, J5,6a = 4.6, J5,6b = 2.3 Hz, 1H, 5-HS), 2.16–2.00 (4s, 12H, COCH3 overlapping with COCH3 of 4cR) ppm. 13C NMR (100 MHz, CDCl3) data for S isomer: δ = 170.6–168.7 (COCH3 overlapping with COCH3 of 4cR), 130.7–128.7 (aromaticS overlapping with aromaticR of 4cR), 88.9 (C-1S), 76.9 (C-5S), 73.2 (C-3S), 68.6 (C-2S), 67.9 (C-4S), 61.7 (C-6S), 53.8 (C-aS), 20.9–20.6 (COCH3 overlapping with COCH3 of 4cR) ppm. 1H NMR (400 MHz, CDCl3) data for R isomer: δ = 7.41–7.31 (m, 5H, aromaticR overlapping with aromaticS of 4cS), 5.44 (dd, J1,2 = 10.1, J2,3 = 9.3 Hz, 1H, 2-HR), 5.22 (t, J2,3 = J3,4 = 9.3 Hz, 1H, 3-HR), 5.14–5.07 (m, 1H, 4-HR overlapping with 4-HS of 4cS), 4.41 (d, Jgem = 12.3 Hz, 1H, a-HR), 4.35 (dd, J6a,6b = 12.5, J5,6a = 2.5 Hz, 1H, 6a-HR), 4.33–4.26 (m, 2H, 6b-HR, a-HR overlapping with 6a-HS of 4cS), 3.83 (d, J1,2 = 10.2 Hz, 1H, 1-HR), 3.73 (ddd, J4,5 = 10.0, J5,6a = 6.1, J5,6b = 2.3 Hz, 1H, 5-HR), 2.16–2.00 (4s, 12H, COCH3 overlapping with COCH3 of 4cS) ppm. 13C NMR (100 MHz, CDCl3) data for R isomer: δ = 170.6–168.7 (COCH3 overlapping with COCH3 of 4cS), 130.7–128.7 (aromaticR overlapping with aromaticS of 4cS), 84.6 (C-1R), 77.1 (C-5R), 73.9 (C-3R), 68.1 (C-4R), 66.5 (C-2R), 62.6 (C-6R), 53.5 (C-aR), 20.9–20.6 (COCH3 overlapping with COCH3 of 4cS) ppm. HRMS (ESI): calcd for C21H26NaO10S 493.1139 [M + Na]+; found 493.1115.
Allyl 2,3,4,6-tetra-O-acetyl-1-thio-β-D-glucopyranoside (R,S)-S-oxide (4dR,S). The oxidation reaction of thiosaccharide 3d gave the diastereomeric mixture of sulfoxides 4dR,S that could not be separated by column chromatography. The mixture (25.3 mg, 60%, ratio S/R, 1.5
:1) was obtained as a white solid and showed a single spot by TLC. Rf = 0.10, hexane/EtOAc (1:1). 1H NMR (400 MHz, CDCl3) data for S isomer: δ = 6.00–5.89 (m, 1H, b-HS), 5.51–5.41 (m, 2H, c-HS, c′-HS overlapping with c-HR, c′-HR, 2-HR of 4dR), 5.34 (t, J1,2 = J2,3 = 9.2 Hz, 1H, 2-HS), 5.29 (t, J2,3 = J3,4 = 9.1 Hz, 1H, 3-HS), 5.08 (t, J3,4 = J4,5 = 9.2 Hz, 1H, 4-HS), 4.33 (d, J1,2 = 9.4 Hz, 1H, 1-HS), 4.28–4.22 (m, 1H, 6a-HS overlapping with 6a-HR, 6b-HR, 1-HR of 4dR), 4.19 (dd, J6a,6b = 12.6, J5,6b = 2.1 Hz, 1H, 6b-HS), 3.80–3.76 (m, 1H, 5-HS overlapping with 5-HR of 4dR), 3.66 (dd, Jgem = 13.3, Ja,b = 7.0 Hz, 1H, a-HS), 3.57 (dd, Jgem = 13.1, Ja,b = 8.0 Hz, 1H, a-HS), 2.09–2.02 (4s, 12H, COCH3 overlapping with COCH3 of 4dR) ppm. 13C NMR (100 MHz, CDCl3) data for S isomer: δ = 170.5–168.8 (COCH3 overlapping with COCH3 of 4dR), 125.5 (C-bS), 124.3 (C-cS), 89.0 (C-1S), 76.9 (C-5S), 73.3 (C-3S), 68.6 (C-2S), 67.8 (C-4S), 61.7 (C-6S), 52.0 (C-aS), 20.8–20.6 (COCH3 overlapping with COCH3 of 4dR) ppm. 1H NMR (400 MHz, CDCl3) data for R isomer: δ = 5.84–5.73 (m, 1H, b-HR), 5.51–5.41 (m, 3H, c-HR, c′-HR, 2-HR overlapping with c-HS, c′-HS of 4dS), 5.35 (t, J2,3 = J3,4 = 9.3 Hz, 1H, 3-HR), 5.11 (t, J3,4 = J4,5 = 9.7 Hz, 1H, 4-HR), 4.28–4.22 (m, 3H, 6a-HR, 6b-HR, 1-HR overlapping with 6a-HS of 4dS), 3.83 (dd, Jgem = 12.7, Ja,b = 9.0 Hz, 1H, a-HR), 3.80–3.76 (m, 1H, 5-HR overlapping with 5-HS of 4dS), 3.75 (dd, Jgem = 12.6, Ja,b = 7.0 Hz, 1H, a-HR), 2.09–2.02 (4s, 12H, COCH3 overlapping with COCH3 of 4dS) ppm. 13C NMR (100 MHz, CDCl3) data for R isomer: δ = 170.5–168.8 (COCH3 overlapping with COCH3 of 4dS), 125.6 (C-bR), 124.3 (C-cR), 85.5 (C-1R), 77.0 (C-5R), 73.3 (C-3R), 68.1 (C-4R), 66.7 (C-2R), 62.3 (C-6R), 51.8 (C-aR), 20.8–20.6 (COCH3 overlapping with COCH3 of 4dS) ppm. HRMS (ESI): calcd for C17H24NaO10S 443.0982 [M + Na]+; found 443.0980.
Methyl 1-thio-β-D-glucopyranoside (R,S)–S-oxide (4gR,S). The oxidation reaction of the free thiosaccharide 3g gave the diastereomeric mixture of sulfoxides 4gR,S that could not be separated by column chromatography. The mixture (19.9 mg, 88%, ratio S/R, 1.5
:1) was obtained as a white solid and showed a single spot by TLC analysis. Rf = 0.38, MeCN/H2O (4:1). 1H NMR (400 MHz, D2O) data for S isomer: δ = 4.63 (d, J1,2 = 9.7 Hz, 1H, 1-HS), 4.00 (dd, J6a,6b = 12.6, J5,6a = 2.1 Hz, 1H, 6a-HS), 3.83 (dd, J6a,6b = 12.6, J5,6b = 5.9 Hz, 1H, 6b-HS), 3.71–3.61 (m, 3H, 3-HS, 2-HS, 5-HS overlapping with 3-HR, 5-HR of 4gR), 3.50 (t, J3,4 = J4,5 = 9.3 Hz, 1H, 4-HS), 2.85 (s, 3H, CH3SOS) ppm. 13C NMR (100 MHz, D2O) data for S isomer: δ = 91.0 (C-1S), 80.8 (C-5S), 77.2 (C-3S), 69.4 (C-2S), 69.0 (C-4S), 60.9 (C-6S), 30.7 (CH3SOS) ppm. 1H NMR (400 MHz, D2O) data for R isomer: δ = 4.25 (d, J1,2 = 9.6 Hz, 1H, 1-HR), 4.03 (dd, J6a,6b = 12.7, J5,6a = 2.2 Hz, 1H, 6a-HR), 3.88 (dd, J6a,6b = 12.6, J5,6b = 4.9 Hz, 1H, 6b-HR), 3.74 (dd, J1,2 = 9.5, J2,3 = 9.2 Hz, 1H, 2-HR), 3.71–3.61 (m, 2H, 3-HR, 5-HR overlapping with 3-HS, 2-HS, 5-HS of 4gS), 3.55 (t, J3,4 = J4,5 = 9.2 Hz, 1H, 4-HR), 2.86 (s, 3H, CH3SOR) ppm. 13C NMR (100 MHz, D2O) data for R isomer: δ = 89.3 (C-1R), 80.3 (C-5R), 77.0 (C-3R), 68.9 (C-4R), 68.0 (C-2R), 60.6 (C-6R), 31.7 (CH3SOR) ppm. HRMS (ESI): calcd for C7H14NaO6S 249.0403 [M + Na]+; found 249.0395.
Butyl 1-thio-β-D-glucopyranoside (R,S)-S-oxide (4hR,S). The oxidation reaction of the free thiosaccharide 3h afforded after purification by column chromatography the diastereomeric mixture of sulfoxides 4hR,S. The mixture (19.1 mg, 66%, ratio S/R, 1.6
:1) was obtained as a colorless syrup and showed a single spot by TLC analysis. Rf = 0.58, CH2Cl2/MeOH (4:1). 1H NMR (400 MHz, D2O) data for S isomer: δ = 4.62 (d, J1,2 = 9.7 Hz, 1H, 1-HS), 3.96 (dd, J6a,6b = 12.6, J5,6a = 2.1 Hz, 1H, 6a-HS), 3.79 (dd, J6a,6b = 12.8, J5,6b = 6.1 Hz, 1H, 6b-HS), 3.71 (t, J1,2 = J2,3 = 9.3 Hz, 1H, 2-HS), 3.64 (t, J2,3 = J3,4 = 9.0 Hz, 1H, 3-HS), 3.59 (ddd, J4,5 = 9.7, J5,6b = 5.9, J5,6a = 2.1 Hz, 1H, 5-Hs), 3.46 (br t, J3,4 = J4,5 = 9.4 Hz, 1H, 3-Hs), 3.30–3.23 (m, 1H, a-Hs overlapping with a-HR of 4hR), 3.02–2.93 (m, 1H, a-Hs overlapping with a-HR of 4hR), 1.85–1.66 (m, 2H, b-Hs overlapping with b-HR of 4hR) 1.60–1.45 (m, 2H, c-Hs overlapping with c-HR of 4hR), 0.97(t, Jc,d = 7.3 Hz, 3H, d-Hs) ppm. 13C NMR (100 MHz, D2O) data for S isomer: δ = 90.8 (C-1S), 80.8 (C-5S), 77.2 (C-3S), 69.1 (C-2S), 68.9 (C-4S), 60.8 (C-6S), 44.8 (C-aS), 24.1 (C-bs overlapping with C-bR), 21.2 (C-cS), 12.9 (C-ds) ppm. 1H NMR (400 MHz, D2O) data for R isomer: δ = 4.28 (d, J1,2 = 9.8 Hz, 1H, 1-HR), 3.97 (dd, J6a,6b = 12.7, J5,6a = 2.2 Hz, 1H, 6a-HR), 3.83 (dd, J6a,6b = 13.0, J5,6b = 5.1 Hz, 1H, 6b-HR), 3.74 (t, J1,2 = J2,3 = 9.7 Hz, 1H, 2-HR), 3.66 (t, J2,3 = J3,4 = 8.7 Hz, 1H, 3-HR), 3.64–3.59 (m, 1H, 5-HR), 3.52 (br t, J3,4 = J4,5 = 9.4 Hz, 1H, 3-HR), 3.30–3.23 (m, 1H, a-HR overlapping with a-HS of 4hS), 3.02–2.93 (m, 1H, a-HR overlapping with a-HS of 4hS), 1.85–1.66 (m, 2H, b-HR overlapping with b-HS of 4hS) 1.60–1.45 (m, 2H, c-HR overlapping with c-HS of 4hS), 0.97(t, Jc,d = 7.4 Hz, 3H, d-HR) ppm. 13C NMR (100 MHz, D2O) data for R isomer: δ = 88.2 (C-1R), 80.3 (C-5R), 77.0 (C-3R), 68.7 (C-4R), 67.8 (C-2R), 60.5 (C-6R), 45.7 (C-aR), 24.1 (C-bR overlapping with C-bS), 21.3 (C-cR), 12.9 (C-dR) ppm HRMS (ESI): calcd for C10H20NaO6S 291.0873 [M + Na]+; found 291.0879.
Benzyl 1-thio-β-D-glucopyranoside (R,S)-S-oxide (4iR,S). The oxidation reaction of the free thiosaccharide 3i afforded the diastereomeric mixture of sulfoxides 4iR,S after purification by column chromatography. The mixture (18.4 mg, 53%, ratio S/R, 1
:1) was obtained as a white solid and showed a single spot by TLC analysis. Rf = 0.56, CH2Cl2/MeOH (4:1). 1H NMR (400 MHz, D2O) data for S isomer: δ = 7.53–7.46 (m, 5H, aromaticS overlapping with aromaticR of 4iR), 4.58 (d, J1,2 = 9.7 Hz, 1H, 1-HS), 4.48 (d, Jgem = 13.2 Hz, 1H, a-HS), 4.41 (d, Jgem = 13.1 Hz, 1H, a-HS), 4.00 (dd, J6a,6b = 12.6, J5,6a = 2.0 Hz, 1H, 6a-HS), 3.84 (dd, J6a,6b = 12.6, J5,6b = 5.8 Hz, 1H, 6b-HS), 3.66–3.45 (m, 4H, 2-HS, 3-HS, 5-HS, 4-HS overlapping with 3-HR, 5-HR, 4-HR of 4iR) ppm. 13C NMR (100 MHz, D2O) data for S isomer: δ = 130.6–128.9 (aromaticS overlapping with aromaticR of 4iR), 90.8 (C-1S), 80.9 (C-5S), 77.2 (C-3S), 69.4 (C-2S), 68.9 (C-4S), 60.8 (C-6S), 51.7 (C-aS) ppm. 1H NMR (400 MHz, D2O) data for R isomer: δ = 7.53–7.46 (m, 5H, aromaticR overlapping with aromatics of 4iS), 4.53 (d, Jgem = 12.6 Hz, 1H, a-HR), 4.42 (d, Jgem = 12.5 Hz, 1H, a-HR), 4.07 (d, J1,2 = 10.0 Hz, 1H, 1-HR), 4.07 (dd, J6a,6b = 12.6, J5,6a = 2.0 Hz, 1H, 6a-HR), 3.90 (dd, J6a,6b = 12.7, J5,6b = 4.8 Hz, 1H, 6b-HR), 3.76 (dd, J1,2 = 9.9, J2,3 = 8.7 Hz, 1H, 2-HR), 3.66–3.45 (m, 3H, 3-HR, 5-HR, 4-HR overlapping with 2-HS, 3-HS, 5-HS, 4-HS of 4iS) ppm. 13C NMR (100 MHz, D2O) data for R isomer: δ = 130.6–128.9 (aromaticR overlapping with aromaticS of 4iS), 86.8 (C-1R), 80.4 (C-5R), 76.9 (C-3R), 68.7 (C-4R), 67.6 (C-2R), 60.6 (C-6R), 51.6 (C-aR) ppm. HRMS (ESI): calcd for C13H18NaO6S 325.0716 [M + Na]+; found 325.0720.
Conclusions
Photooxidation reactions of thiomonosaccharides under aerobic conditions were performed employing different organic dyes as sensitizers. Among the photocatalysts evaluated tetra-O-acetyl riboflavin provided good to excellent yields for the oxidation of alkyl, benzyl, and vinyl thioglycosides in considerably short reaction times. In addition, outstanding chemoselectivity towards the glycosyl sulfoxides without over-oxidation to sulfone was achieved. Furthermore, high selectivity was observed for the photooxidation of alkyl, allyl and benzyl thioglycosides as, under the same controlled conditions, phenyl thioglycosides are not oxidized. In the photooxidation reactions the formation of the sulfoxides with the SS configuration was favored in almost all cases, due to the stereoselectivity generated by the asymmetric induction of the glucosyl residue.The absolute configuration at the sulfur stereocenter of each sulfoxide was determined considering the shielding/deshielding effects generated by the anisotropy of the SO bond in the chemical shift on the chemical shift of the 1H NMR signals of specific protons, by the anisotropy of the SO bond. These effects were analyzed for the preferential rotational conformers (g+ and/or g−) for each diastereoisomer, which were confirmed by the presence of characteristic NOE contacts in the NOESY spectra. On the basis of all these data the SS or SR configurations were assigned.
This photosensitized oxidation reaction, employing visible light under aerobic conditions, constitutes a remarkably simple, efficient, and economical methodology to obtain glycosyl sulfoxides. In addition, it is an environmentally friendly process since high atom economy is achieved. The desired sulfoxides were isolated after a rather simple purification process due to the use of catalytic amounts of organic dyes together with the high chemoselectivity observed.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are indebted to the Secretaría de Ciencia y Tecnología (SECyT), Universidad Nacional de Córdoba (UNC), Universidad de Buenos Aires (UBA), Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET) and Fondo para la investigación Científica y Tecnológica Argentina (FONCyT) for financial support. M. G. T. gratefully acknowledges the receipt of a fellowship from CONICET. J. P. C., A. B. P. and O. V. are research members from CONICET. The authors want to thank Dr Guillermo A. Oliveira Udry for his collaboration in the bibliographic search of previously reported compounds.References
- M. Carmen Carreño, G. Hernández-Torres, M. Ribagorda and A. Urbano, Chem. Commun., 2009, 6129–6144 RSC.
- J. Legros, J. R. Dehli and C. Bolm, Adv. Synth. Catal., 2005, 347, 19–31 CrossRef CAS.
- S. Rayati, F. Nejabat and S. Zakavi, Inorg. Chem. Commun., 2014, 40, 82–86 CrossRef CAS.
- B. Karimi, M. Ghoreishi-Nezhad and J. H. Clark, Org. Lett., 2005, 7, 625–628 CrossRef CAS.
- V. G. Shukla, P. D. Salgaonkar and K. G. Akamanchi, J. Org. Chem., 2003, 68, 5422–5425 CrossRef CAS.
- D. Kahne, S. Walker, Y. Cheng and D. Van Engen, J. Am. Chem. Soc., 1989, 111, 6881–6882 CrossRef CAS.
- J. Zeng, Y. Liu, W. Chen, X. Zhao, L. Meng and Q. Wan, Top. Curr. Chem., 2018, 376, 27 CrossRef.
- M. A. Fascione, R. Brabham and W. B. Turnbull, Chem.–Eur. J., 2016, 22, 3916–3928 CrossRef CAS.
- M. C. Aversa, A. Barattucci and P. Bonaccorsi, Tetrahedron, 2008, 64, 7659–7683 CrossRef CAS.
- D. Crich and L. B. L. Lim, in Organic Reactions, 2004, pp. 115–251 Search PubMed.
- C. S. Callam, R. R. Gadikota, D. M. Krein and T. L. Lowary, J. Am. Chem. Soc., 2003, 125, 13112–13119 CrossRef CAS.
- D. Crich and S. Sun, J. Am. Chem. Soc., 1997, 119, 11217–11223 CrossRef CAS.
- P. J. Garegg, in Adv Carbohydr. Chem. Biochem., ed. D. Horton, Academic Press, 1997, vol. 52, pp. 179–205 Search PubMed.
- L. Yan, C. M. Taylor, R. Goodnow and D. Kahne, J. Am. Chem. Soc., 1994, 116, 6953–6954 CrossRef CAS.
- Z. J. Witczak, P. Kaplon and P. Markus Dey, Carbohydr. Res., 2003, 338, 11–18 CrossRef CAS.
- É. Bozó, Á. Demeter, A. Rill and J. Kuszmann, Tetrahedron: Asymmetry, 2002, 12, 3423–3433 CrossRef.
- I. Cumpstey, C. Ramstadius, T. Akhtar, I. J. Goldstein and H. C. Winter, Eur. J. Org. Chem., 2010, 1951–1970 CrossRef CAS.
- J. P. Colomer, M. Á. Canales Mayordomo, B. Fernández de Toro, J. Jiménez-Barbero and O. Varela, Eur. J. Org. Chem., 2015, 1448–1455 CrossRef CAS.
- J. P. Colomer, V. E. Manzano and O. Varela, Eur. J. Org. Chem., 2013, 7343–7353 CrossRef CAS.
- J. P. Colomer, B. Fernández de Toro, F. J. Cañada, F. Corzana, J. Jiménez-Barbero, A. Canales and O. Varela, Eur. J. Org. Chem., 2016, 5117–5122 CrossRef CAS.
- N. Khiar, Tetrahedron Lett., 2000, 41, 9059–9063 CrossRef CAS.
- N. Khiar, I. Alonso, N. Rodriguez, A. Fernandez-Mayoralas, J. Jimenez-Barbero, O. Nieto, F. Cano, C. Foces-Foces and M. Martin-Lomas, Tetrahedron Lett., 1997, 38, 8267–8270 CrossRef CAS.
- R. Kakarla, R. G. Dulina, N. T. Hatzenbuhler, Y. W. Hui and M. J. Sofia, J. Org. Chem., 1996, 61, 8347–8349 CrossRef CAS.
- E. G. Mata, Phosphorus, Sulfur Silicon Relat. Elem., 1996, 117, 231–286 CrossRef CAS.
- Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/aldrich/273031, 2020.
- C. Ye, Y. Zhang, A. Ding, Y. Hu and H. Guo, Sci. Rep., 2018, 8, 2205 CrossRef.
- V. Latour, T. Pigot, M. Simon, H. Cardy and S. Lacombe, Photochem. Photobiol. Sci., 2005, 4, 221–229 CrossRef CAS.
- R. Glass, in Topics in Current Chemistry, Springer-Verlag, Berlin, Heidelberg, 1999, pp. 2–87 Search PubMed.
- W. Adam, J. E. Argüello and A. B. Peñéñory, J. Org. Chem., 1998, 63, 3905–3910 CrossRef CAS.
- N. Somasundaram and C. Srinivasan, J. Photochem. Photobiol., A, 1998, 115, 169–173 CrossRef CAS.
- S. M. Bonesi, I. Manet, M. Freccero, M. Fagnoni and A. Albini, Chem.–Eur. J., 2006, 12, 4844–4857 CrossRef CAS.
- H. Guo, H. Xia, X. Ma, K. Chen, C. Dang, J. Zhao and B. Dick, ACS Omega, 2020, 5, 10586–10595 CrossRef CAS.
- D.-Y. Zheng, E.-X. Chen, C.-R. Ye and X.-C. Huang, J. Mater. Chem. A, 2019, 7, 22084–22091 RSC.
- L.-P. Li and B.-H. Ye, Inorg. Chem., 2019, 58, 7775–7784 CrossRef.
- X. Liang, Z. Guo, H. Wei, X. Liu, H. Lv and H. Xing, Chem. Commun., 2018, 54, 13002–13005 RSC.
- X. Gu, X. Li, Y. Chai, Q. Yang, P. Li and Y. Yao, Green Chem., 2013, 15, 357–361 RSC.
- J. Dad'ová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620–623 CrossRef.
- N. Floyd, B. Vijayakrishnan, J. R. Koeppe and B. G. Davis, Angew. Chem., Int. Ed., 2009, 48, 7798–7802 CrossRef CAS.
- A. R. Paramewar, A. Imamura and A. V. Demchenko, in Carbohydrate Chemistry Proven Synthetic Methods, ed. P. Kovac, CRC Press, Boca Raton, 2012, ch. 20, vol. 1, pp. 1–12 Search PubMed.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- R. Bonneau, R. Pottier, O. Bagno and J. Joussot-Dubien, Photochem. Photobiol., 1975, 21, 159–163 CrossRef CAS.
- T. Neveselý, E. Svobodová, J. Chudoba, M. Sikorski and R. Cibulka, Adv. Synth. Catal., 2016, 358, 1654–1663 CrossRef.
- R. A. Larson, P. L. Stackhouse and T. O. Crowley, Environ. Sci. Technol., 1992, 26, 1792–1798 CrossRef CAS.
- A. Casado-Sánchez, R. Gómez-Ballesteros, F. Tato, F. J. Soriano, G. Pascual-Coca, S. Cabrera and J. Alemán, Chem. Commun., 2016, 52, 9137–9140 RSC.
- K. Fatima, N. Masood and S. Luqman, Biomed. Res. Ther., 2016, 3, 514–527 CrossRef.
- M. Bancirova, Luminescence, 2011, 26, 685–688 CrossRef CAS.
- C. Ouannes and T. Wilson, J. Am. Chem. Soc., 1968, 90, 6527–6528 CrossRef CAS.
- S. M. Bonesi, R. Torriani, M. Mella and A. Albini, Eur. J. Org. Chem., 1999, 1723–1728 CrossRef CAS.
- W. Ando, Sulfur Rep., 1981, 1, 147–207 CrossRef CAS.
- E. J. Corey and C. Ouannés, Tetrahedron Lett., 1976, 17, 4263–4266 CrossRef.
- J. P. Colomer, A. B. Penenory and O. Varela, RSC Adv., 2017, 7, 44410–44420 RSC.
- G. Balavoine, A. Gref, J.-C. Fischer and A. Lubineau, Tetrahedron Lett., 1990, 31, 5761–5764 CrossRef CAS.
- V. E. Manzano, M. L. Uhrig and O. Varela, J. Org. Chem., 2008, 73, 7224–7235 CrossRef CAS.
- Z. J. Witczak, D. Lorchak and N. Nguyen, Carbohydr. Res., 2007, 342, 1929–1933 CrossRef CAS.
- R. M. Srivastava, F. J. S. Oliveira, L. P. da Silva, J. R. de Freitas Filho, S. P. Oliveira and V. L. M. Lima, Carbohydr. Res., 2001, 332, 335–340 CrossRef CAS.
- C. A. Sanhueza, A. C. Arias, R. L. Dorta and J. T. Vázquez, Tetrahedron: Asymmetry, 2010, 21, 1830–1832 CrossRef CAS.
- D. Crich, J. Mataka, L. N. Zakharov, A. L. Rheingold and D. J. Wink, J. Am. Chem. Soc., 2002, 124, 6028–6036 CrossRef CAS.
- N. Khiar, I. Fernández, C. S. Araújo, J.-A. Rodríguez, B. Suárez and E. Álvarez, J. Org. Chem., 2003, 68, 1433–1442 CrossRef CAS.
- C. A. Sanhueza, R. L. Dorta and J. T. Vázquez, J. Org. Chem., 2011, 76, 7769–7780 CrossRef CAS.
- J. F. Moya-López, E. Elhalem, R. Recio, E. Álvarez, I. Fernández and N. Khiar, Org. Biomol. Chem., 2015, 13, 1904–1914 RSC.
- M. Sakata, M. Haga and S. Tejima, Carbohydr. Res., 1970, 13, 379–390 CrossRef CAS.
- Z. Pakulski, D. Pierożyński and A. Zamojski, Tetrahedron, 1994, 50, 2975–2992 CrossRef CAS.
- A. Tatami, Y.-S. Hon, I. Matsuo, M. Takatani, H. Koshino and Y. Ito, Biochem. Biophys. Res. Commun., 2007, 364, 332–337 CrossRef CAS.
- W. Pilgrim and P. V. Murphy, J. Org. Chem., 2010, 75, 6747–6755 CrossRef CAS.
- M. Černý and J. Pacák, Collect. Czech. Chem. Commun., 1959, 24, 2566–2570 CrossRef.
- S.-S. Weng, Y.-D. Lin and C.-T. Chen, Org. Lett., 2006, 8, 5633–5636 CrossRef CAS.
- D. Crich, V. Krishnamurthy, F. Brebion, M. Karatholuvhu, V. Subramanian and T. K. Hutton, J. Am. Chem. Soc., 2007, 129, 10282–10294 CrossRef CAS.
- J. Bogusiak and W. Szeja, Pol. J. Chem., 1985, 59, 293–298 CAS.
- M. S. Cheng, Q. L. Wang, Q. Tian, H. Y. Song, Y. X. Liu, Q. Li, X. Xu, H. D. Miao, X. S. Yao and Z. Yang, J. Org. Chem., 2003, 68, 3658–3662 CrossRef CAS.
- W. Schneider, J. Sepp and O. Stiehler, Ber. Dtsch. Chem. Ges., 1918, 51, 220–234 CrossRef CAS.
- P. V. Murphy, C. McDonnell, L. Hämig, D. E. Paterson and R. J. K. Taylor, Tetrahedron: Asymmetry, 2003, 14, 79–85 CrossRef CAS.
- L. N. Lameijer, J. le Roy, S. van der Vorm and S. Bonnet, J. Org. Chem., 2018, 83, 12985–12997 CrossRef CAS.
- Z. Marton, V. Tran, C. Tellier, M. Dion, J. Drone and C. Rabiller, Carbohydr. Res., 2008, 343, 2939–2946 CrossRef CAS.
- R. W. Gantt, P. Peltier-Pain, W. J. Cournoyer and J. S. Thorson, Nat. Chem. Biol., 2011, 7, 685–691 CrossRef CAS.
- A. K. Singh, V. Tiwari, K. B. Mishra, S. Gupta and J. Kandasamy, Beilstein J. Org. Chem., 2017, 13, 1139–1144 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ra09534f |
| This journal is © The Royal Society of Chemistry 2021 |