
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnzyme-functionalised, core/shell magnetic nanoparticles for selective pH-triggered sucrose capture†
Stephanie Fulaz‡
 ,
Carolina Scachetti and
Ljubica Tasic*
,
Carolina Scachetti and
Ljubica Tasic*
Laboratory of Chemical Biology, Institute of Chemistry, University of Campinas, Campinas 13083-970, Brazil. E-mail: ljubica@unicamp.br
First published on 25th January 2021
Abstract
Diabetes is a chronic metabolic disease which leads to high glucose levels in the blood, with severe consequences for human health. Due to the worldwide appeal for the reduction in calorie intake, this study presents the development of a nanomaterial able to capture sucrose selectively, thus providing a tool to remove naturally occurring sucrose from food, such as fruit juices, producing low-calorie juices for consumption. Magnetite nanoparticles (Fe3O4 NPs) coated with an inert material (SiO2) and functionalised with the enzyme invertase were designed to remove sucrose from solutions. Fe3O4 NPs were synthesised using the co-precipitation method, whereas the coating with a silica shell was done by the Stöber method. Its physicochemical characteristics were determined, with excellent stability over time. On the other hand, the invertase enzyme was extracted from dry Baker's yeast, purified and immobilised on the surface of the silica-coated Fe3O4 NPs. pH-triggered sucrose capture occurred at pH 3.0 once invertase with protonated catalytic residues was able just to bind with sucrose in a highly selective way. After a short, 1 min interaction, approximately 13.5 mmol L−1 of sucrose was captured per gram of nanomaterial and removed with the use of an external permanent magnet. The complex sucrose/nanomaterial was washed, and the released sucrose was put into buffered solution (pH = 4.8), where it underwent hydrolysis to yield inverted sugar. On the other side, sucrose-free nanomaterial was reused with no loss of enzymatic capability to capture sucrose at pH = 3.0 and maintained the invertase activity at pH 4.8 in ten consecutive rounds of re-use. As sucrose was recovered in the form of inverted sugar, not just low sugar beverage could be obtained, but also a high valued market product. Thus, the developed technology allows for the commercialisation of low-calorie food, offering healthier options to consumers and helping to fight diabetes and obesity.
1. Introduction
Diabetes is a chronic metabolic disease characterised by elevated levels of glucose in the blood, with severe consequences for human health.1 It is the fastest-growing disease in the world, and the most significant health challenge of this century,2 accounting as one of the leading causes of deaths.1 About 9% of the world's adult population has the disease, being 90% of these type 2 diabetes.1 In this case, the body develops insulin resistance or decreases its production. It is known that a poor diet and sedentary lifestyle are risk factors for type 2 diabetes. Recent studies show that the consumption of sweetened beverages is associated with an increased risk of contracting the disease.3 There is a global treaty to curb the growth of diabetes and obesity in the world by 2025.1Aiming to fight obesity and diabetes, there is a need to provide low-calorie food for the consumption of the population, amongst which we highlight low-calorie juices. Unsweetened fruit juices could be a healthier alternative to society if they had a reduced-calorie value. For instance, it is estimated that in a glass of orange juice (240 mL), there are naturally 25.8 g of sugar and 112 kcal.4 Thus, together with the worldwide appeal for reduced calorie intake, there is a need to reduce the caloric value of ready-to-drink beverages. To this end, it is proposed the development of a material tailored for the removal of naturally occurring sucrose. The developed technology is based on two main building blocks: insoluble and chemically inert support, based on a magnetite core capped by a silica shell, and a recognition part, composed of the enzyme invertase.
Separation techniques are prevalent in all kinds of industries, amongst them sedimentation, filtration, flocculation, crystallisation, centrifugation and chromatography are well established, and widespread.5,6 The use of magnetic separation started with the mining industry and have expanded to coal desulphurisation, steel production, biotechnology, wastewater treatment,7 bioremediation,8 pharmaceutical9 and biomedical applications.10–12 Magnetic separations are very promising for the food industry and downstream biotechnology processes, which can be explained by its main advantages: one-step capture and purification, due to its high affinity and selectivity; the possibility of high throughput and semi-continuous processes with low energy consumption. Therefore, magnetic separation can aid in increasing yields and productivity, while reducing costs of industrial processes.13,14
Magnetic separations are robust systems with a low running cost. They have been proposed for different sectors of the food industry, such as removal of yeast,15 haze, unwanted flavours and turbidity proteins during wine production,16 clarification of fruit juices17 and separation and purification of whey proteins in the dairy industry.18 The use of magnetic nanoparticles instead of microbeads is very promising, due to the nanoparticles higher surface area, higher surface area to volume ratio, easy separation and no mass transfer limitation for target molecule diffusion in the separation process.13 Considering the proposed application of the designed technology on the removal of naturally occurring sucrose from food, such as orange juices, the magnetic separation was the method of choice. The food industry often deals with complex mixtures from natural products, usually containing particulates of different sizes in suspension. The presence of particulates would preclude the use of membranes and columns as separation techniques. The use of non-magnetic nanoparticles would require the addition of an extra separation step, such as centrifugation. Thus, the magnetic separation process for the sucrose removal was chosen for this study.
Nanotechnology research has grown considerably in the last decades and has found countless applications ranging from material science,19–21 drug delivery,22,23 bioelectronic devices,24,25 biosensors,26–28 detectors29,30 to immunoassays.31,32 Amongst all nanoparticles, magnetic nanoparticles (MNPs) have been outstanding due to their different properties, such as magnetic behaviour, biocompatibility, and the option of surface modification.33,34 Magnetite was chosen as the basis for the development of this separation strategy, because apart from its magnetic response, iron oxide nanoparticles are generally considered as safe and are allowed as colour and food additives, and also have been approved for in vivo applications by the US Food and Drug Administration (FDA).35–37 Besides, iron oxide nanoparticles have low production costs, abundant precursors and high density.13 Elongated nanoparticles, such as nanorods, are very promising for different applications, due to their S-like hysteresis loop obtained when the magnetisation is applied parallel to the short-axis, and a square-like hysteresis loop when the magnetic field is applied along the long-axis.38 Nanorods are more commonly obtained through hydrothermal methods using shaping ligand39 or sacrificial template,40 solvothermal using template assisted41 or co-precipitation using a shaping ligand.42 It has also been reported that small polymers, small molecules containing halides and solvents can be used for coordination of certain crystal planes.38
The possibility of surface modification of magnetite nanoparticles includes enzyme immobilisation, which has been an active topic of research in enzyme technology and is crucial for enzyme application in industrial processes. Enzyme immobilisation onto supports, such as magnetic nanoparticles, offers a range of advantages, for example, the extension in enzyme stability and protection against degradation, leading to higher resistance to denaturants, temperature changes and organic solvents, it enables repeated use, it optimises and facilitates the separation step, allowing for a quicker recovery of the enzyme, and therefore, reducing operational costs.43–48
The enzyme invertase was used as the basis for the sucrose recognition part of the designed nanomaterial. Invertase (β-fructofuranosidase, EC 3.2.1.26) is one of the most studied enzymes and can be described as a highly efficient and specific enzyme for the hydrolysis of sucrose.49–51 Invertase is widely used in the chemical and food industry, mainly in the production of inverted sugar, a non-crystallisable syrup resulting from the mixture of fructose and glucose, characterised by the greater sweetness potential due to fructose.52 The hydrolysed sugar mixture obtained through the enzymatic process is a colourless product, in contrast with the coloured product resultant from the acid hydrolysis.53 Invertase's high industrial application has led to several immobilisation studies in different materials, such as magnetic polyvinyl alcohol microspheres,45 magnetic diatomaceous earth nanoparticles,44 bio-hybrid support comprising silica nanoparticles and Ocimum basilicum seed,54 polyamidoamine dendrimer-superparamagnetic nanoparticles,55 polyaniline-coated magnetic diatomite nanoparticles,56 silanised and polymer brush grafted magnetic nanoparticles57 and chitosan-coated γ-Fe2O3 magnetic nanoparticles.58
Invertase is a glycoprotein with maximum activity at pH 4.8 and 40 °C with KM of 5 mmol L−1.59 The mechanism of action of the invertase (Fig. 1) can be described as the oxygen protonation of the glycosidic bond by an acid–base reaction with the residue of glutamic acid. Then, negatively charged oxygen from the aspartate makes a nucleophilic attack on the fructose anomeric carbon-1, resulting in the formation of the enzyme–substrate complex. Glucose is released followed by hydrolysis of the bond between fructose carbon-1 and the aspartic acid residue, leading to the release of fructose and invertase reconstitution.60
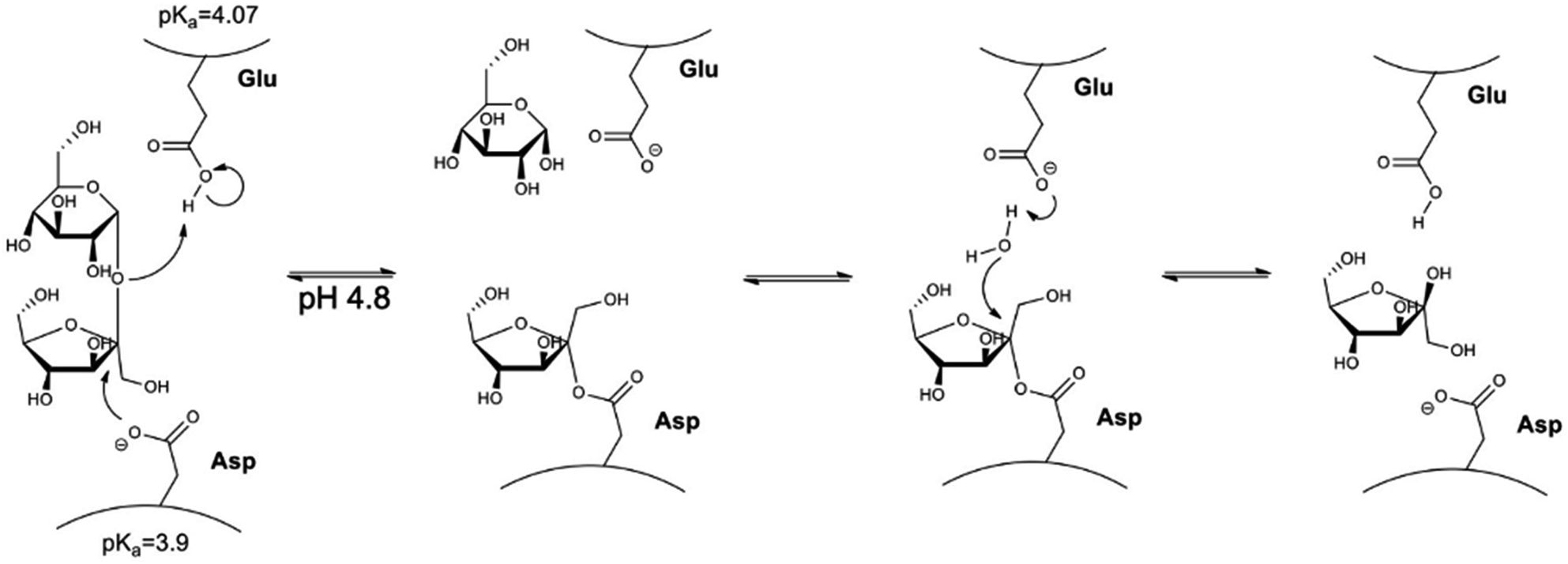 | ||
| Fig. 1 Representation of the sucrose hydrolysis reaction performed by the enzyme invertase. | ||
Herein, the proposed methodology is based on the importance of pH for invertase catalysed sucrose hydrolysis. At pH values lower than the pKa of invertase catalytic residues (glutamic acid and aspartate), invertase is inactive and can just interact with the sucrose that still is selectively recognised by the enzyme; however, the hydrolysis reaction cannot happen, once invertase catalytic residues are protonated. The precise pH control allowed for efficient sucrose capture from a solution, removal of the complex nanomaterial/sucrose without the occurrence of its hydrolysis. In another flask, sucrose was removed from the complex, the enzyme was recovered by providing a buffer solution at the enzyme's optimum pH (pH = 4.8), allowing for the production of inverted sugar as a valuable side product (Fig. 2).
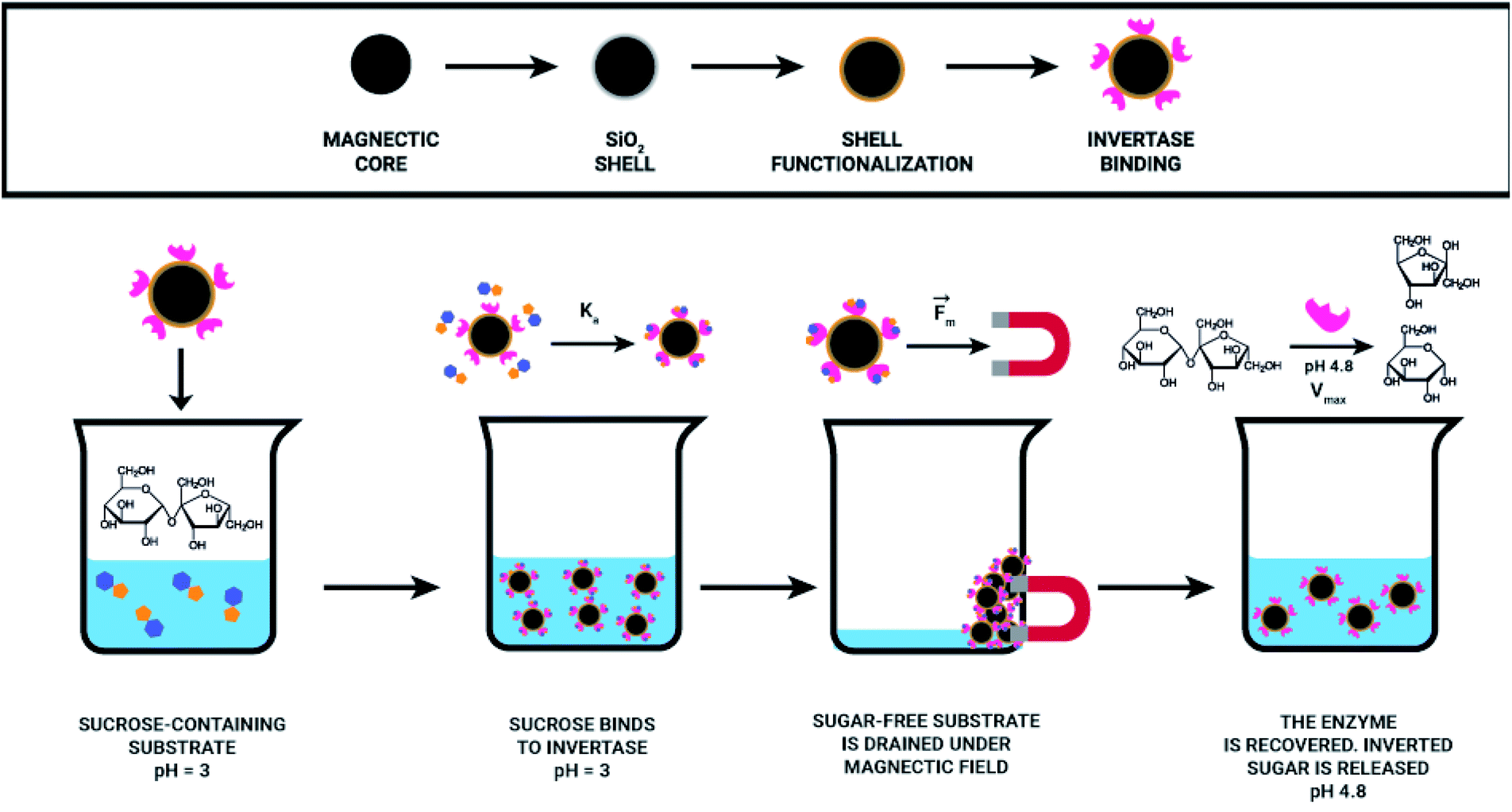 | ||
| Fig. 2 Representation of the magnetic separation process for sucrose capture. | ||
With the aim to design an effective magnetic separation process, it is crucial to think about the system and consider all relevant parameters. We decided to develop our technology envisaging its possible application for the removal of naturally occurring sucrose from orange juice characterised with low pH (3.0 or lower). By combining the invertase specificity against sucrose with the magnetic nanoparticles covered with an inert material (silica) and with invertase immobilised therein, a reusable, sucrose specific nanomaterial was developed, which can selectively remove sucrose from a solution.
2. Experimental
2.1 Materials
Iron(III) chloride hexahydrate (FeCl3·6H2O), ammonium iron(II) hexahydrate (Fe(NH4)2(SO4)2·6H2O), sodium hydroxide (NaOH), tetraethyl orthosilicate (TEOS), (3-aminopropyl)triethoxysilane (APTES), ammonia solution 25% (NH3), sodium bicarbonate (NaHCO3), disodium hydrogen phosphate (Na2HPO4), monosodium dihydrogen phosphate (NaH2PO4), 3,5-dinitro-2-hydroxybenzoic acid (DNS), sucrose and ethanol were obtained from Sigma Aldrich and used without further purification.2.2 Synthesis of Fe3O4 nanoparticles
The synthesis of Fe3O4 NPs was adapted from Mascolo et al.61 Briefly, 0.02 mol of FeCl3·6H2O (5.4 g) was added to 100 mL of water previously refluxed under argon. Subsequently, 0.01 mol of Fe(NH4)2(SO4)2·6H2O (3.92 g) was added, keeping the system under argon flow. 100 mL of 0.8 mol L−1 solution of NaOH was added dropwise, approximately 1 mL min−1, under vigorous stirring. Stirring was continued for 3 h at 25 °C. The nanoparticles were washed 3–4 times with water, dried under vacuum and kept in an evacuated desiccator until the next step.2.3 Surface modification with TEOS and APTES
Fe3O4 NP coating was adapted from Khatiria et al.62 Briefly, 1 g of Fe3O4 NPs was mixed with 20 mL of water and 80 mL of ethanol under argon. The suspension was dispersed in an ultrasonic bath for 30 min. A solution containing 2 g TEOS, 50 mL ethanol, and 6 mL ammonia (NH3, 25% v/v) was added. Vigorous stirring was maintained in a 25 °C water bath for 4 h. The Fe3O4@SiO2 were washed with water (3–4 times), twice with ethanol, and dried under vacuum at room temperature. For the amino functionalisation, 150 mg of Fe3O4@SiO2 were suspended in 30 mL of water, and 0.6 mL of APTES was added. The suspension was maintained at 70 °C for 16 h under vigorous stirring. The Fe3O4@SiO2–NH2 was washed with water (3–4 times), twice with ethanol, and dried under vacuum.2.4 Nanoparticles characterisation
Nanoparticles zeta potential and hydrodynamic diameter were measured using 0.01 mg mL−1 water suspensions in DST1070 folded cuvette in a Zetasizer Nano-ZS (Malvern). Size and shape were determined by Transmission Electron Microscopy in a Carl Zeiss CEM-902 with 80 keV using copper grids and parlodium film. The microscope was equipped with a Castaing-Henry-Ottensmeyer filter spectrometer used for the electron energy loss spectrometry (EELS). The crystal structure was determined by X-ray Diffraction (XRD) using a Shimadzu XRD7000 operating with Cu-Kα (λ = 0.154060 nm), in continuous mode with a 2° min−1 speed from 20° to 80°. Surface functionalisation was investigated by Infrared Spectroscopy equipped with Attenuated Total Reflectance (ATR-FTIR) using an Agilent Cary 630. X-Ray Photoelectron spectra were obtained using a SPECS system (SPECS GmbH) equipped with X-ray XR-50 with radiation Al Kα (hν = 1486.6 eV) and Phoibos 100 analyser with MCD-9 detector at the CCS Nano laboratory (Centro de Componentes Semicondutores, UNICAMP). Thermogravimetric Analysis (TGA) was performed using a STD q600 (TA Instruments), with a 10° min−1 speed in a nitrogen atmosphere.2.5 Invertase extraction
Dry Baker's yeast was used for invertase extraction in a procedure adapted from Timerman, 2012.59 Briefly, 125 g of dry yeast was suspended in 440 mL of sodium bicarbonate solution (NaHCO3, 0.1 mol L−1), divided into containers and kept at 35 °C for 16 h. The suspension was centrifuged at 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 30 min at 4 °C. The solid was discarded, and the supernatant followed for differential precipitation. Ethanol was added until 29% (v/v) concentration was reached. The suspension was incubated on ice for 15 min and centrifuged at 15000 rpm for 30 min at 4 °C. Pellet with contaminants was discarded, and more ethanol was added into the supernatant up to 40% (v/v). The suspension was incubated in the refrigerator until protein precipitation, centrifuged at 15000 rpm, 30 min, 4 °C discarding the supernatant. The invertase-containing solid was solubilised in sodium phosphate buffer A (5 mmol L−1 Na2HPO4 and 5 mmol L−1 NaH2PO4, pH 7.00).
000 rpm for 30 min at 4 °C. The solid was discarded, and the supernatant followed for differential precipitation. Ethanol was added until 29% (v/v) concentration was reached. The suspension was incubated on ice for 15 min and centrifuged at 15000 rpm for 30 min at 4 °C. Pellet with contaminants was discarded, and more ethanol was added into the supernatant up to 40% (v/v). The suspension was incubated in the refrigerator until protein precipitation, centrifuged at 15000 rpm, 30 min, 4 °C discarding the supernatant. The invertase-containing solid was solubilised in sodium phosphate buffer A (5 mmol L−1 Na2HPO4 and 5 mmol L−1 NaH2PO4, pH 7.00).
2.6 Invertase purification
Invertase purification was done in a sequence of steps based on the work of Timerman, and Rahman et al.59,63 First, the enzyme solution in buffer A was clarified by ultracentrifugation and then filtered using a 0.22 μm PVDF filter. This solution was purified by gel filtration chromatography using a manually packed (16 × 250 mm) Superdex 200 column and operated via AKTA Purifier 10 (GE) with Unicorn 5.2 program. The purification was performed using buffer A. The fractions that showed absorbance at 280 nm were evaluated for the presence of invertase based on the positive result for the activity test described in sequence (Section 2.7). Fractions containing invertase were pooled and stored in the refrigerator until the next purification step. The pre-purified invertase fractions were further purified by anion exchange chromatography using a DEAE (diethylaminoethyl) modified cellulose column packed manually (16 × 350 mm). Buffer A was used as the mobile phase and a linear gradient of NaCl promoted by buffer B (5 mmol L−1 Na2HPO4, 5 mmol L−1 of NaH2PO4, and 500 mmol L−1 NaCl, pH 7.00) which was progressively added (0–100% B) by the AKTA Purifier. Fractions were collected and checked regarding invertase presence (Section 2.7). All fractions containing invertase were dialysed against Buffer A using a Fisherbrand dialysis membrane with MWCO of 6000–8000 g mol−1. The last purification step was a cation exchange chromatography using an agarose column modified with carboxymethyl (CM) groups. Buffers A and B were used again as the mobile phases. The active fractions were dialysed as described above. After purification, the protein concentration was estimated by the Bradford method, using a calibration curve based on bovine serum albumin (BSA). The concentration can be calculated by the obtained equation, Cprotein = (Abs − (0.02 ± 0.01))/(0.027 ± 0.002); R2 = 0.994.2.7 Invertase activity test
The activity was monitored using the colorimetric test with 3,5-dinitro-2-hydroxybenzoic acid (DNS).64 25 μL of the solution containing the enzyme was incubated at 50 °C for 5 min in a dry bath. An aliquot of 200 μL of 10 mmol L−1 sucrose solution was added; the reaction was kept for 5 min at 50 °C. Then 300 μL of the DNS solution (0.2 mol L−1 NaOH, 23 mmol L−1 of DNS, and 0.53 mol L−1 tartrate of sodium and potassium) was added to complete the reaction. The mixture was boiled for 5 min, diluted with 2 mL of water, and the absorbance was measured at 540 nm. The activity was calculated using a calibration curve built with an equimolar mixture of glucose and fructose at the same test conditions. The concentration of glucose was estimated using the equation Cglucose = (Abs + (0.047 ± 0.03))/(0.00493 ± 0.00005); R2 = 0.9996.2.8 Invertase characterisation
All steps in invertase isolation were evaluated by SDS-PAGE 12% using MiniVE Vertical Electrophoresis System (GE Healthcare). Circular dichroism spectra were obtained for solutions with different enzyme concentrations; 0.15 mg mL−1 in buffer C (sodium acetate 40 mmol L−1, pH 4.8) and 0.03 mg mL−1 in sodium acetate buffer 8 mmol L−1, pH 4.8. Measurements were recorded in a Spectropolarimeter (Jasco, J720). Fluorescence spectra were recorded in a Spectrofluorometer (PerkinElmer, LS55) for invertase at 0.15 mg mL−1 in buffer C. For different pH values measurement, sodium-phosphate–citric acid 50 mmol L−1 buffers from pH 2.5 to pH 7.5 were used. For light rotation experiments, a 20 mg mL−1 sucrose solution in buffer C, was incubated with invertase at 0.15 mg mL−1. The observed light rotation was followed over time in a Polarimeter (PerkinElmer, 341). For the determination of kinetic parameters of the free enzyme and Fe3O4@SiO2–NH2-Inv, various concentration of sucrose (0–300 mmol L−1) in acetate buffer (sodium acetate 40 mmol L−1, pH 4.8) at 50 °C were used, and KM and VMAX values determined by Lineweaver–Burk plot.2.9 Immobilisation of invertase on nanoparticles surface
Invertase immobilisation on the surface of the Fe3O4@SiO2–NH2 was adapted from the procedure described by Abraham, et al.65 Briefly, a suspension of 5 mg mL−1 of Fe3O4@SiO2–NH2 in water was sonicated for 1 h. Then, a glutaraldehyde solution was added to the final concentration of 1 mol L−1 and stirred for 1 h. After this time, the NPs were washed with water thrice and once with buffer C. They were then suspended in a solution containing the invertase and kept under stirring for 2 h. Fe3O4@SiO2–NH2-Inv was washed with buffer C to remove the unbound enzyme and stored in the fridge.2.10 Sucrose capture and release test
100 μL of Fe3O4@SiO2–NH2-Inv was washed with buffer D (citrate–phosphate buffer 50 mmol L−1 pH 3.00) 3 times with the aid of a magnet and kept in a dry bath at 50 °C for 5 min. Then, 200 μL of a 5 mmol L−1 sucrose solution, also prepared in buffer D was added to Fe3O4@SiO2–NH2-Inv. After 1 min, with the aid of a magnet, the supernatant was removed and stored in another tube, and 200 μL of buffer C was added to Fe3O4@SiO2–NH2-Inv, which were maintained for 5 min at 50 °C. The supernatant was removed and added to 300 μL of DNS solution, which was then boiled, diluted with 2 mL of water, and had absorbance measured at 540 nm to quantify the sucrose removed. The Fe3O4@SiO2–NH2-Inv were washed three times with buffer D, and the cycle was repeated using the previously-stored sucrose solution.3. Results and discussion
3.1 Synthesis and characterisation of the nanomaterial
The synthesis of pristine Fe3O4 NPs was performed via a co-precipitation method and can be described by the chemical eqn (1) shown below.| Fe2+ + 2Fe3+ + 8OH− → Fe3O4 + 4H2O | (1) |
In this reaction, the complete precipitation of Fe3O4 is achieved at a pH between 8 and 14 in an oxygen-free environment. The use of an oxygen-free environment not only protects the oxidation of magnetite (Fe3O4) to maghemite (γFe2O3) but also leads to smaller particles if compared to methods in the presence of oxygen.66,67 The chemical method was preferred due to its low production cost, high yield and more straightforward scale-up. The as-synthesised Fe3O4 NPs are magnetics, as shown in Fig. 3.
 | ||
| Fig. 3 Representative image of the nanomaterial as-synthesised in the absence and the presence of a magnet. | ||
Bare iron oxide NPs tend to agglomerate, as seen by its hydrodynamic diameter (Table 1), due to the presence of strong magnetic attraction between the particles, high energy surface and attractive van der Waals forces.68–70 Furthermore, the high local concentration of Fe ions, released from Fe dissolution, can be toxic to organisms.71,72 These issues can be avoided by post-synthesis surface modification, such as NPs coating with a silica (SiO2) shell, which confers a hydrophilic surface, is chemically inert, biocompatible, easily functionalisable and improve NPs dispersion in water.73 The hydrodynamic sizes of the NPs were estimated by DLS, and, as expected, the diameter measurements indicated a significant overestimation of their size, when compared to the values obtained by other techniques, such as electron microscopy imaging. This overestimation can be explained by the influence of the surface coating and the hydration layer on the nanoparticles diffusion.74 As previously described, bare iron oxide NPs tend to agglomerate in solution, so silica capping helped to prevent this, thus, a smaller hydrodynamic diameter was measured. In sequence, it can be noted that additional functionalisation with amino groups, further decreased the hydrodynamic diameter. This can be explained by the reduction in hydrogen bonding between silanol groups on the NPs' surface.75 Therefore, the functionalisation steps helped to prevent the aggregation of the nanomaterial in water suspensions. The surface charge was also estimated through zeta potential measurements (Table 1), and the size and shape of the nanomaterial were evaluated by TEM, as shown in Fig. 4.
 | ||
| Fig. 4 Representative TEM images of nanomaterial samples: (a) Fe3O4, (b) Fe3O4@SiO2, (c) Fe3O4@SiO2–NH2, and (d) Fe3O4@SiO2–NH2-Inv. Bars represent 100 nm. | ||
Zeta potential measurements (Table 1) were performed to evidence different stages of the synthesis and surface grafting process. The alternating zeta potentials observed at each stage suggest that Fe3O4@SiO2–NH2-Inv has been successfully synthesised; besides, the high electrical potential confers elevated stability to the nanomaterial suspension. The as-synthesised Fe3O4 nanoparticles showed a positively-charged surface, differently to what is usually reported for magnetite, a negatively-charged surface formed by –OH groups. However, it is important to note the pH-dependent character of magnetite, an amphoteric solid, which has a positive charge in acidic pH and a negative charge in basic pH. Considering its point of zero charge previously reported at pH 7.9,76 and its isoelectric point reported at pH 7.3,77 the positive charge obtained by aqueous suspensions of the Fe3O4 nanoparticles can be understood. Upon surface capping with silica, the charge became negative, as expected, due to the presence of silanol groups on the nanoparticles' surfaces. Further, the amino-functionalisation changed the charge to positive values, owing to the protonation of the amine groups.
In the TEM images, it is clear that the nanomaterial obtained is composed of a mixed population of rods and spherical nanoparticles. The nanorods have 88 ± 22 nm in length and 14 ± 4 nm in diameter, with an aspect ratio of approximately 6. The spheres have a diameter of 27 ± 6 nm. Mixed morphology populations of iron oxides particles have been previously reported and will be further discussed.78,79 It is also important to highlight that the surface functionalisation did not significantly change the size of the nanomaterial, which show a capping layer of 6 ± 2 nm.
Shape anisotropy is an important characteristic of iron oxide particles. In elongated nanoparticles, magnetisation is reported to be easier along their long axis, rather than the short axis.38,80 The crystal shape can be controlled thermodynamically or kinetically. In a diffusion-limited regime, high concentration, the monomers are precipitated in the NPs' surface favouring monodisperse particles. On the other hand, in a reaction-limited regime, low concentration, the reaction on the surface limits the particle's growth and leads to different shapes.80 Despite being the most commonly used method, co-precipitation allows for limited control of particles morphology, size and composition, once the reaction is kinetically controlled.79,81 In the present work, the co-precipitation was performed with a Fe2+/Fe3+ ratio of 0.5, which has been previously reported to lead to the formation of a single oxide phase of either magnetite and maghemite. On the other hand, pH also plays a vital role in the formation of the particles, which a more acidic medium leads preferentially to the formation of goethite nanorods as reported by Blanco-Andujar et al.79 This initial goethite nanorods can act later as a seed for the formation of magnetite via a dissolution-precipitation mechanism, with the transformation from the oxyhydroxide phase to an oxide phase. In increasing pH values, due to the slow and continuous base addition, the higher concentration of hydroxyl groups favours the formation of iron oxides, with no remaining goethite after reaction complexion. Despite TEM images showing a mixed population, the nanomaterial obtained could be confirmed to be magnetite through XRD analysis (Fig. 5).
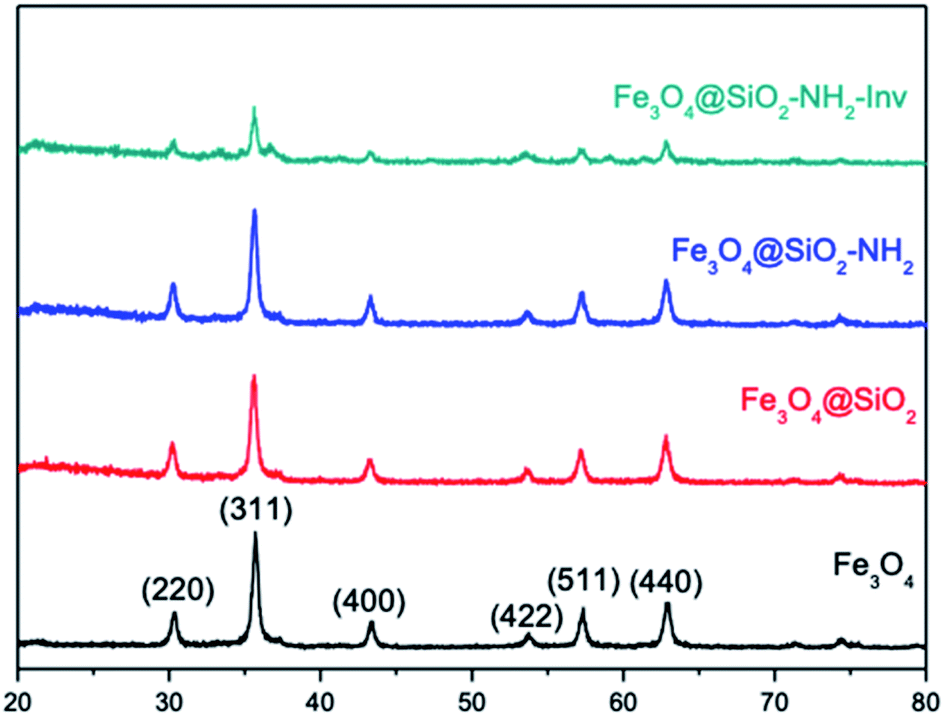 | ||
| Fig. 5 Characterisation of the nanomaterials using XRD. | ||
Magnetite (Fe2+Fe23+O4) is known by its crystallisation in the cubic structure of the inverse spinel, whose oxygen atoms form the face-centred cubic structure and the iron cations occupy the interstitial sites, where half of Fe3+ are in tetrahedral sites and the other half with Fe2+ occupying the octahedral sites. Fe3O4 NPs XRD showed the reflection planes (220), (311), (400), (422), (511) and (440)82 (Fig. 5). The results are consistent with those from the JCPDS card 19-0629. The absence of peaks at 21.22° and 33.15° is an indication goethite and hematite were not formed, besides the peak at 26.38° referent to Fe(OH)3, is also absent.83 The results suggest that magnetite was successfully synthesised. As can be noted in XRD spectra, the coating and functionalisation steps have not changed the crystallinity of Fe3O4. In addition, the characteristic diffraction peaks of Fe3O4 were attenuated in the pattern of Fe3O4@SiO2–NH2-Inv due to capping layers of silica and enzyme; however, the core structure was not altered in the process.
The coating and functionalisation were evaluated by ATR-FTIR (Fig. S1†). Stretching vibrations of Fe–O at 568 cm−1 (ν1) and 440 cm−1 (ν2) confirm the presence of Fe3O4.84 The bond Fe–O–Si is observed at 584 cm−1. The signals at 1058 and 960 cm−1 correspond to SiO–H and Si–O–Si, respectively, while the signals at 920 and 890 cm−1 correspond to Si–O–H signals. The N–H stretching and NH2 bending signals appeared at 1630 and 3407 cm−1. Silanol with hydrogen bonding also has a signal between 3200 and 3470 cm−1, the reason behind the enlargement of the signal.83,85,86 The composition and structure of the nanomaterial was also confirmed by electron spectroscopy images (ESI-TEM) combined with electron energy-loss spectroscopy (EELS) as shown in Fig. S2.† Homogeneous iron, oxygen and silicon atomic composition were observed for Fe3O4@SiO2 nanoparticles as expected. The core/shell structure is indicated by the presence of silicon atoms mostly in the shell region and the iron confined into the core region.
To further evaluate the functionalisation with the enzyme XPS spectra of the NPs (Fig. 6(a)) were recorded and showed the existence of the main elements' signals with peaks correspondent to Fe 2p (710 eV for Fe3+ and 724 eV for Fe2+), O 1s (529.7 eV) for Fe3O4 and N 1s (399 eV, amide) and C 1s (285 eV) for Fe3O4@SiO2–NH2-Inv. The Fe 2p peak was weaker in Fe3O4@SiO2–NH2-Inv than in Fe3O4, which can be explained by the shielding effect of the coating layer and enzyme-functionalisation.
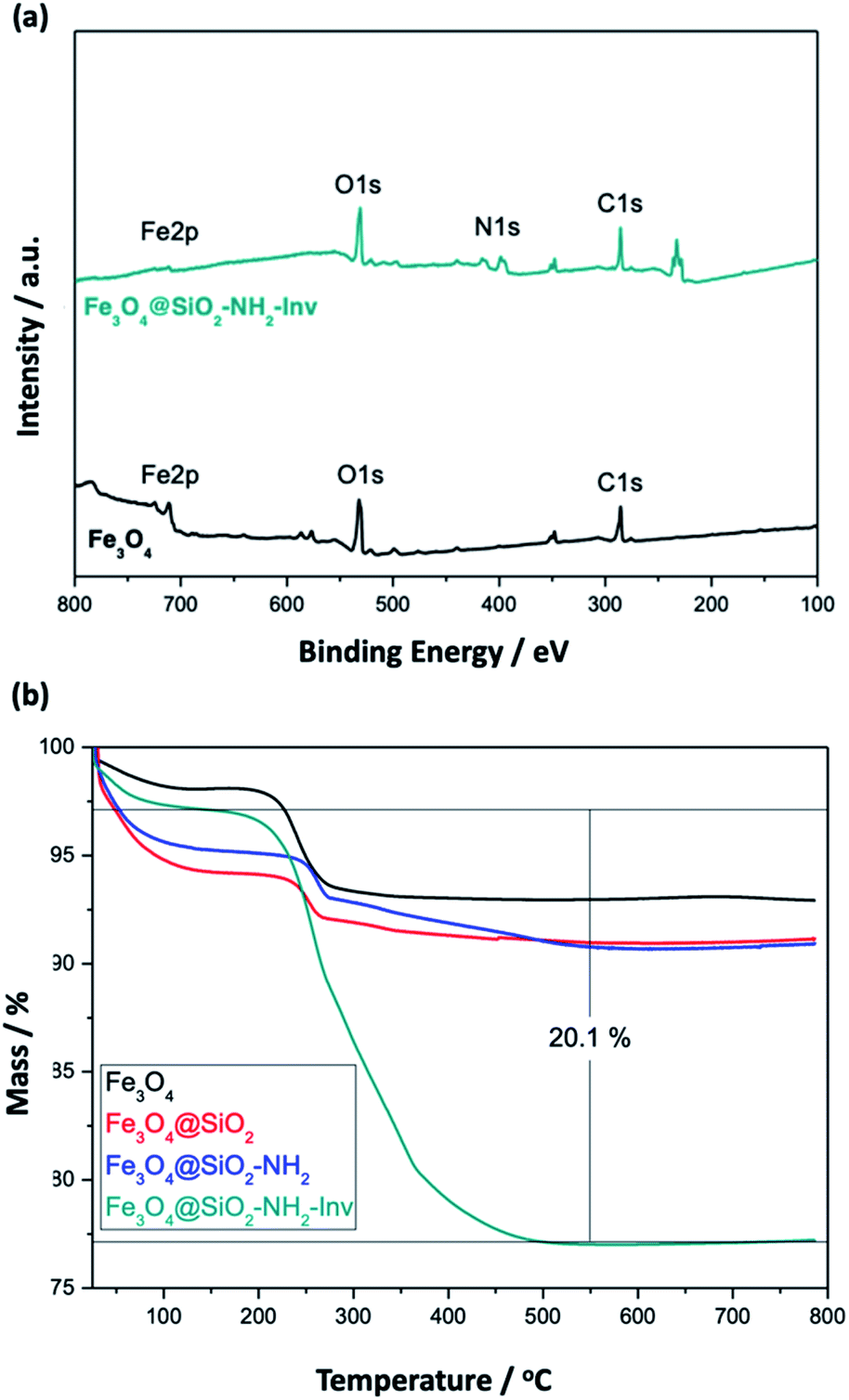 | ||
| Fig. 6 Characterisation of the nanomaterials using different techniques: (a) XPS and (b) TGA. | ||
TGA analysis (Fig. 6(b)) was used to confirm further and characterise the coating layer and for the quantitative analysis of enzyme content in the Fe3O4@SiO2–NH2-Inv. The correspondent weight loss and surface density can be observed in Table 2. Around 2.6% weight loss was observed at temperatures ranging from 50 to 100 °C, mainly due to the loss of physically adsorbed water on the products. The carbonisation of the organic matter led to a 4.7% weight loss for the nanomaterial before enzyme immobilisation. The decomposition of the enzyme coating layer led to a weight loss of 20.1%. Considering the surface density, approximately 0.04 (nanorods) and 0.05 (nanospheres) chains of invertase were immobilised per nm2, or around 155 invertase chains per nanorod (or 2.2 × 1018 chains per gram of nanomaterial) and 118 invertase chains per nanosphere (or 1.7 × 1018 chains per gram of nanomaterial).
| Nanoparticle | Surface molecule | Weight loss/% | Surface densitya/chain nm−2 | Surface densityb/chain nm−2 |
|---|---|---|---|---|
| a Calculated using the spheres surface area.b Calculated using the rods surface area. | ||||
| Fe3O4 | — | 2.66 | — | — |
| Fe3O4@SiO2 | Silica | 3.66 | 9.4 | 6.7 |
| Fe3O4@SiO2–NH2 | Amine-silane | 4.74 | 1.4 | 1.0 |
| Fe3O4@SiO2–NH2-Inv | Invertase | 20.11 | 0.05 | 0.04 |
3.2 Invertase extraction, purification and pH dependence
Invertase from Baker's yeast, Saccharomyces cerevisiae, is commonly found as a glycoprotein in the periplasmic space. Its primary function is to cleave the sucrose, forming monosaccharides that are then transported to the cytoplasm. The enzyme is composed of two glycosylated subunits of 135 kDa. Approximately 50% of the molecular mass is due to oligosaccharides.59,87,88 According to the structure deposited in the Protein Data Bank under PDB ID 4EQV,89 the biological unit consists of 8 protein chains, each having 512 amino acids, 544 Da, and a theoretical pI of 4.58, obtained by the ProtParam tool.90 Invertase extraction was performed using mild conditions that selectively ruptured the cell wall without breaking the plasma membrane, followed by differential precipitation and chromatography steps. The presence of the invertase in the collected fractions was evaluated by the activity test made spectroscopically using the DNS assay, as shown in Fig. S3.† Since sucrose is a non-reducing sugar, without the action of the invertase, i.e., negative reaction, there are no changes in colour. After the addition of the enzyme, the sucrose undergoes hydrolysis resulting in an equimolar mixture of glucose and fructose. Under experimental conditions, fructose rapidly undergoes isomerisation to glucose, and thus, all the glucose formed reduces the coloured reagent. Invertase concentration was estimated by the Bradford assay as 0.15 mg mL−1 while the sample was also evaluated by SDS-PAGE, Fig. S4.†Circular dichroism was used to estimate the relative amount of secondary structures in the purified invertase. The percentage of each secondary structure was estimated based on the primary sequence of the protein by APSSP2,91 and the deconvolution of the CD spectrum obtained by K2D3,92 and both results were compared to the Protein Data Bank (PDB) structure (PDB entry 4EQV)89 as can be seen in Table 3. The mathematical treatment indicates that the invertase obtained has a secondary structure very close to that expected by invertase's sequence and corresponds to that reported for its crystal structure (PDB entry 4EQV).89
| Secondary structure | Spectrum deconvolution K2D3/% | Primary structure prediction/% | PDB structure/% |
|---|---|---|---|
| α-Helix | 2.4 | 2.6 | 2 |
| β-Sheet | 35.3 | 38.8 | 47 |
| Random | 62.3 | 58.6 | — |
The intrinsic protein fluorescence, conferred by the three aromatic amino acid residues, tryptophan, tyrosine, and phenylalanine, taking into account that tryptophan is the dominant intrinsic fluorophore, offers the possibility to evaluate the nanoenvironment that involves these residues in the tertiary and quaternary structure of the protein.93 The fluorescence spectrum obtained is shown in Fig. S5(a).† Invertase from S. cerevisiae exhibits 16 Trp and 31 Tyr residues per chain, which account for the enzyme fluorescence emission spectrum.
In order to further confirm invertase activity, the inversion of the rotation of the polarised light was observed for a sucrose solution incubated with invertase, as shown in Fig. S5(b).† The inversion of the observed rotation from positive values (sucrose) to negative values (glucose and fructose) confirms the activity of the invertase extracted from Baker's yeast.
When we analyse the proposed mechanism of the enzymatic activity for invertase (Fig. 1), it is noted that the glutamic acid residue (Glu204) must be protonated, and the aspartate residue (Asp23) must be deprotonated in order for the catalysis to occur. Thus, invertase behaviour at different pH conditions was investigated using a sodium phosphate–citric acid buffer (50 mmol L−1). The fluorescence emission curves are shown in Fig. 7(a) and the enzymatic activity measured by the colourimetric test with DNS in Fig. 7(b).
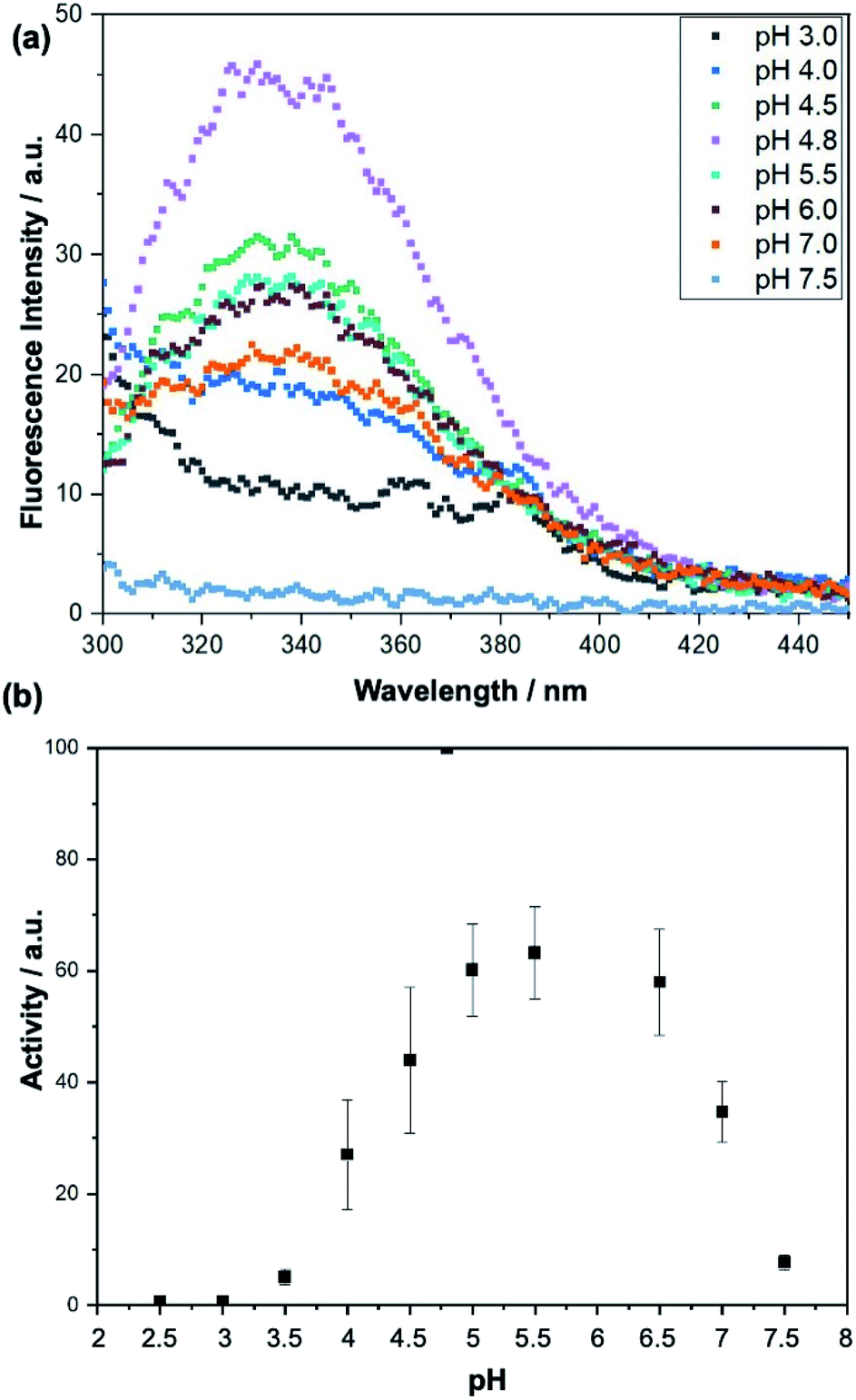 | ||
| Fig. 7 (a) Invertase emission fluorescence spectra excited at 280 nm at different pH conditions using a sodium phosphate–citric acid buffer (50 mmol L−1). (b) Curve showing activity of the invertase against a sucrose solution (10 mmol L−1) under different pH conditions. The activity was measured by the DNS test. | ||
As can be seen from the fluorescence spectra, the appropriate folding of the protein depends on the pH of the buffer in which it is stored. It is noted that at its optimum pH (pH = 4.80), the enzyme exhibits the highest fluorescence intensity at λmax = 338 nm. In contrast, at pH 7.50, it precipitated completely and showed no fluorescence when excited at 280 nm. Looking at the activity curve, we observed that at pH 4.80 invertase shows the highest activity. At pH values of 2.50 and 3.00, the enzyme showed no activity. At low pH values, where pH < pKa of the catalytic residues, both (Glu204 and Asp23) are protonated, and the reaction does not occur, while at higher pH values, pH > pKa of the two residues, both are deprotonated and once again the reaction is not observed. Therefore, the pH 3.00 of the solutions was chosen for the sucrose capture test.
3.3 Sucrose capture and release
Upon immobilization, enzyme conformation might be affected, what significantly impacts its affinity toward the substrate. To investigate how the immobilisation employed impacted invertase conformation, the kinetic parameters, KM and VMAX were determined for the free invertase and Fe3O4@SiO2–NH2-Inv, and compared to values reported in the literature, as shown in Table S1.† It can be noted a similar behaviour compared to invertase immobilised on other kinds of nanoparticles, where it is commonly observed an increase in KM values and a decrease in VMAX, which can be attributed to conformational changes of the enzyme after its immobilisation.In order to evaluate invertase enzyme efficiency before and after its functionalisation onto nanoparticles, an activity test was performed comparing free enzyme to Fe3O4@SiO2–NH2-Inv. The results can be seen in Fig. S5.† It can be observed the maintenance of 60% of the enzyme activity after its immobilisation onto nanoparticles' surface when used at first time, while an activity of approximately 40% is kept when the enzyme is continuously re-used, up to ten times. In previous reports on immobilised invertase, it was observed that significant invertase activity loss appeared after 10 cycles of its use,44 and an expressive reduction in immobilised invertase activity after 8 cycles.94 After 10 cycles of activity performed by the nanomaterial, the size and charge were measured again, and resulted in a reduction of the nanomaterial surface charge to +14 ± 3 mV and an increase in its size to 437 ± 45 nm. These changes in the nanomaterial structure could play a role in the slight decrease of its activity after continued use.
Using a suspension of 6 mg of Fe3O4@SiO2–NH2-Inv set at pH 3.00, sucrose was captured from a 5 mmol L−1 sucrose solution prepared in a sodium phosphate–citric acid buffer (50 mmol L−1 pH 3.00). Fe3O4@SiO2–NH2-Inv was first incubated in a sucrose solution at pH 3. In this condition occurred the formation of the nanomaterial–sucrose complex; however, the enzymatic reaction did not happen. The nanomaterial–sucrose complex was removed with the aid of a magnet, and then, placed into different flask, washed and incubated in a buffer solution. The pH-change (from pH 3.0 to sodium acetate 40 mmol L−1, pH 4.8), reactivated the immobilised invertase which catalysed sucrose hydrolysis. This way, the immobilised invertase was set free from sucrose and the nanomaterial-enzyme was recovered. Sucrose produced inverted sugar, which was used to quantify the concentration of sucrose removed, as explained in Fig. 2 and quantified in Fig. 8.
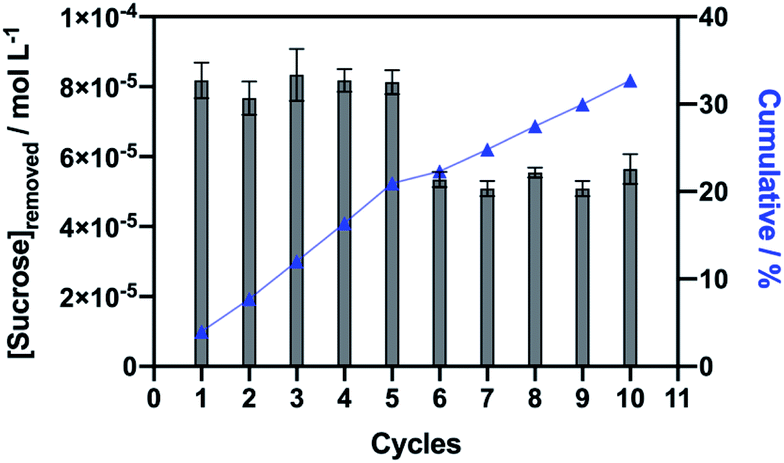 | ||
| Fig. 8 Illustration of removed sucrose in each of the ten cycles with re-used nanomaterial. | ||
The synthesised tailored nanomaterial, consisting of a magnetic core, an inert shell of silica and immobilised invertase, was stable throughout the capture of sucrose from a solution at pH 3.00 and it did not lose its binding ability and sucrose capture up to ten consecutive times of reuse and showed insignificant loss of the invertase activity. It is important to state that the further reuse of the nanomaterial led to a considerable decrease in the invertase activity but mostly because of the loss of magnetic nanoparticles during the washing process between the cycles, while enzyme specific activity was maintained.
The sucrose capture experiment was performed with a suspension of 6 mg of the nanomaterial. It is observed that each cycle resulted in the removal of approximately 81 μmol L−1 of sucrose from the solution (Fig. 8). The repetition of the sucrose capture cycles resulted in a cumulative effect of sucrose removal, with 40 mmol L−1 of sucrose removed at the end of 5 cycles, or 67 mmol L−1 of sucrose removed after 10 cycles. Furthermore, it can be estimated that 1 g of nanomaterial would be able to remove 13.5 mmol L−1 of sucrose from a solution per cycle of capture. Because invertase native structure assembles into an octamer, also described as a tetramer of dimers89 and comparing to our TGA data, we can estimate that each invertase assembly captures approximately five sucrose molecules per nanosphere or four sucrose molecules per nanorod.
4. Conclusions
Separation strategies rely on the target molecule, and the approach utilised should also consider the interaction between the target molecule and the magnetic material. Furthermore, the selection and optimisation of binding and elution steps are essential for the success of the whole process. The study reported herein was performed considering an application in the food industry, based on the sucrose removal from fruit juices. Thus, a nanomaterial with high specificity for sucrose, based on the use of the enzyme invertase, was designed. The high specificity is necessary considering the complex mixtures characteristic from natural products. Furthermore, the use of magnetic nanoparticles was particularly chosen, once natural products, such as fruit juices, have a myriad of particulates in suspension, which could preclude the use of columns and membranes.The development of the nanomaterial for sucrose capture involved two distinct steps: the production of the nanomaterial-based on magnetite NPs and the extraction and purification of the invertase from a natural source, Baker's yeast. The next step consisted of the immobilisation of the enzyme onto the surface of the nanomaterial resulting at the Fe3O4@SiO2–NH2-Inv production. The Fe3O4@SiO2–NH2-Inv nanomaterial allowed the capture of approximately 13.5 mmol L−1 of sucrose per gram of nanomaterial per cycle using a sodium phosphate–citric acid buffer (50 mmol L−1 pH 3.00). Fe3O4@SiO2–NH2-Inv could be used ten times without losing the ability to remove sucrose from the solution. The technology developed offers enormous scientific and economic potential. The use of nanotechnology to produce food with reduced caloric content could help curb the emergence of obesity and diabetes, offering healthier options. Using the example of the most consumed juice worldwide, orange juice, this nanomaterial is stable and optimised to capture naturally occurring sucrose at the natural juice pH (3.00), producing two distinct and with higher associated-value products; an orange juice with reduced sugar content, and also inverted sugar that can be used in the manufacture of other products in the food industry.
Envisioning the application of the described technology in the food industry, it is important to highlight, although iron oxides and silica nanoparticles are often considered as safe and have been previously approved for in vivo applications by the FDA, future studies also need to focus on the cytotoxicity of the material. With the increase of the number of proposed uses of nanotechnology is paramount to consider their impact on human health and the environment, and also focus on the development of new regulatory frameworks considering their release.
Novel separation processes are necessary to increase productivity and reduce time, energy and water consumptions in the industry. Magnetic separations are promising alternatives to conventional separation methods for food, pharma and biopharmaceutical industries. They can allow for cost reduction and increased productivity, owing mainly to the prospect of one-step capture and purification and the possibility for high throughput and semi-continuous processes. The plethora of functionalisation options of magnetic nanomaterials is a vital toolkit, which confers the opportunity to adapt the nanomaterial to the properties and characteristics of the target molecule and design appropriate conditions for the adsorption and desorption of any target molecule.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the support received from the Conselho Nacional de Desenvolvimento Científico e Tecnológico in the form of Stephanie Masters' scholarship (CNPq—Project No. 465389/201407) and CNPq and PIBIC for Carolina's undergraduate scholarship.References
- World Health Organization, Diabetes, https://www.who.int/health-topics/diabetes/#tab=tab_1, accessed, 17/06/2020, 2020 Search PubMed.
- L. M. Jaacks, K. R. Siegel, U. P. Gujral and K. M. V. Narayan, Best Pract. Res., Clin. Endocrinol. Metab., 2016, 30, 331–343 CrossRef.
- International Diabetes Federation, IDF Framework for Action on Sugar, https://www.idf.org/images/site1/content/Framework-for-Action-on-Sugar-010615.pdf Search PubMed.
- S. Lehman, Orange Juice Nutrition Facts, https://www.verywellfit.com/orange-juice-nutrition-facts-calories-and-health-benefits-4113143, accessed, 17/06/2020 Search PubMed.
- G. Carta and A. Jungbauer, in Protein Chromatography: Process Development and Scale-up, ed. G. Carta and A. Jungbauer, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 2010, pp. 1–55 Search PubMed.
- K. Bacon, A. Lavoie, B. M. Rao, M. Daniele and S. Menegatti, Acta Biomater., 2020, 112, 29–51 CrossRef CAS.
- D. G. Domingos, R. O. Henriques, J. A. Xavier, N. Libardi and R. H. R. da Costa, Water Sci. Technol., 2019, 79, 993–999 CrossRef CAS.
- R. Singh, M. Behera and S. Kumar, in Bioremediation of industrial waste for environmental safety, ed. R. Bharagava and G. Saxena, Springer, Singapore, 2019, pp. 165–182 Search PubMed.
- M. L. Bruschi and L. D. S. de Toledo, Magnetochemistry, 2019, 5, 50 CrossRef CAS.
- E. I. Anastasova, A. Y. Prilepskii, A. F. Fakhardo, A. S. Drozdov and V. V. Vinogradov, ACS Appl. Mater. Interfaces, 2018, 10, 30040–30044 CrossRef CAS.
- L. M. Zuo, Q. Qu, L. Li, X. Ran, J. W. Gui, Q. Wang, X. H. Cui and C. L. Jiang, Electroanalysis, 2018, 30, 910–920 CrossRef CAS.
- O. Hosu, M. Tertis and C. Cristea, Magnetochemistry, 2019, 5, 55 CrossRef CAS.
- S. P. Schwaminger, P. Fraga-Garca, M. Eigenfeld, T. M. Becker and S. Berensmeier, Front. Bioeng. Biotechnol., 2019, 7, 233 CrossRef.
- H. Fatima and K.-S. Kim, Korean J. Chem. Eng., 2017, 34, 589–599 CrossRef CAS.
- M. Berovic, M. Berlot, S. Kralj and D. Makovec, Biochem. Eng. J., 2014, 88, 77–84 CrossRef CAS.
- A. Mierczynska-Vasilev, P. Boyer, K. Vasilev and P. A. Smith, Food Chem., 2017, 232, 508–514 CrossRef CAS.
- L. Mosafa, M. Shahedi and M. Moghadam, J. Chin. Chem. Soc., 2014, 61, 329–336 CrossRef CAS.
- P. Nicolas, M. L. Ferreira and V. Lassalle, J. Food Eng., 2019, 246, 7–15 CrossRef CAS.
- C. M. Hussain and S. Palit, in Handbook of Ecomaterials, ed. L. Martínez, O. Kharissova and B. Kharisov, Springer, Cham, 2018, pp. 1–29 Search PubMed.
- C. S. Wood and M. M. Stevens, Nature, 2016, 539, 505–506 CrossRef CAS.
- D. Faivre and M. Bennet, Nature, 2016, 535, 235–236 CrossRef CAS.
- O. Esim, S. Kurbanoglu, A. Saves, S. A. Ozkan and Y. Ozkan, in New Developments in Nanosensors for Pharmaceutical Analysis, ed. S. A. Ozkan and A. Shah, Academic Press, 2019, pp. 273–301 Search PubMed.
- X. J. Lu, X. Y. Yang, Y. Meng and S. Z. Li, Chin. J. Polym. Sci., 2017, 35, 534–546 CrossRef CAS.
- Y. Lee, J. Kim, J. H. Koo, T. H. Kim and D. H. Kim, Korean J. Chem. Eng., 2018, 35, 1–11 CrossRef CAS.
- T. Zhang, J. L. Liu, C. Wang, X. Y. Leng, Y. Xiao and L. Fu, Biosens. Bioelectron., 2017, 89, 28–42 CrossRef CAS.
- K. Y. Goud, M. Satyanarayana, A. Hayat, K. V. Gobi and J. L. Marty, in Nanoparticles in Pharmacotherapy, ed. A. M. Grumezescu, William Andrew Publishing, 2019, pp. 195–216 Search PubMed.
- C. Z. Zhu, D. Du and Y. H. Lin, Biosens. Bioelectron., 2017, 89, 43–55 CrossRef CAS.
- L. Wang, Q. R. Xiong, F. Xiao and H. W. Duan, Biosens. Bioelectron., 2017, 89, 136–151 CrossRef CAS.
- Z. G. Liu, Q. Xue and Y. J. Guo, Biosens. Bioelectron., 2017, 89, 444–452 CrossRef CAS.
- L. Anfossi, F. Di Nardo, A. Russo, S. Cavalera, C. Giovannoli, G. Spano, S. Baumgartner, K. Lauter and C. Baggiani, Anal. Bioanal. Chem., 2019, 411, 1905–1913 CrossRef CAS.
- L. Liu, Y. Q. Hao, D. H. Deng and N. Xia, Nanomaterials, 2019, 9, 316 CrossRef CAS.
- W. Q. Lai, Q. H. Wei, M. D. Xu, J. Y. Zhuang and D. P. Tang, Biosens. Bioelectron., 2017, 89, 645–651 CrossRef CAS.
- X. Q. Li, H. Z. Xu, Z. S. Chen and G. F. Chen, J. Nanomater., 2011, 2011, 270974 Search PubMed.
- P. Singh, Y. J. Kim, D. B. Zhang and D. C. Yang, Trends Biotechnol., 2016, 34, 588–599 CrossRef CAS.
- N. T. K. Thanh, Magnetic Nanoparticles: From Fabrication to Clinical Applications, CRC Press, Boca Raton, Florida, 2012 Search PubMed.
- D. Bobo, K. J. Robinson, J. Islam, K. J. Thurecht and S. R. Corrie, Pharm. Res., 2016, 33, 2373–2387 CrossRef CAS.
- C. L. Ventola, Pharm. Ther., 2017, 42, 742–755 Search PubMed.
- R. G. D. Andrade, S. R. S. Veloso and E. M. S. Castanheira, Int. J. Mol. Sci., 2020, 21, 2455 CrossRef CAS.
- Y. L. Ding, F. T. Liu, Q. H. Jiang, B. Du and H. D. Sun, J. Inorg. Organomet. Polym. Mater., 2013, 23, 379–384 CrossRef CAS.
- C. L. Han, J. J. Ma, H. Z. Wu, Y. Wei and K. H. Hu, J. Chil. Chem. Soc., 2015, 60, 2799–2802 CrossRef CAS.
- G. C. Xi, C. Wang and X. Wang, Eur. J. Inorg. Chem., 2008, 2008, 425–431 CrossRef.
- F. Ling, J. Lei, Z. H. Mai and D. B. Zhu, J. Colloid Interface Sci., 2004, 278, 372–375 CrossRef.
- C. H. Liu, H. Y. Lai and W. C. Wu, Food Bioprod. Process., 2017, 102, 260–267 CrossRef CAS.
- M. P. Cabrera, C. R. D. Assis, D. F. M. Neri, C. F. Pereira, F. Soria and L. B. Carvalho Jr, Biotechnol. Rep., 2017, 14, 38–46 CrossRef.
- S. Akgol, Y. Kacar, A. Denizli and M. Y. Arica, Food Chem., 2001, 74, 281–288 CrossRef CAS.
- C. L. B. Reis, E. Y. A. de Sousa, J. D. Serpa, R. C. Oliveira and J. C. S. dos Santos, Quim. Nova, 2019, 42, 768–783 CAS.
- Y. K. Cen, Y. X. Liu, Y. P. Xue and Y. G. Zheng, Adv. Synth. Catal., 2019, 361, 5500–5515 CrossRef CAS.
- R. A. Wahab, N. Elias, F. Abdullah and S. K. Ghoshal, React. Funct. Polym., 2020, 152, 104613 CrossRef CAS.
- J. A. Barnett, Microbiology, 2003, 149, 557–567 CrossRef CAS.
- W. Ward, in Protein Purification, ed. R. Ahmad, InTech, Rijeka, 2012, pp. 1–28 Search PubMed.
- S. J. Romero-Gomez, C. Augur and G. Viniegra-Gonzalez, Biotechnol. Lett., 2000, 22, 1255–1258 CrossRef CAS.
- U. Andjelkovic, A. Milutinovic-Nikolic, N. Jovic-Jovicic, P. Bankovic, T. Bajt, Z. Mojovic, Z. Vujcic and D. Jovanovic, Food Chem., 2015, 168, 262–269 CrossRef CAS.
- L. M. O. Arruda and M. Vitolo, Appl. Biochem. Biotechnol., 1999, 81, 23–33 CrossRef CAS.
- A. Mishra, J. S. Melo, A. Agrawal, Y. Kashyap and D. Sen, Colloids Surf., B, 2020, 188, 110796 CrossRef CAS.
- K. Uzun, E. Cevik, M. Senel, H. Sozeri, A. Baykal, M. F. Abasiyanik and M. S. Toprak, J. Nanopart. Res., 2010, 12, 3057–3067 CrossRef CAS.
- M. P. Cabrera, T. F. da Fonseca, R. V. B. de Souza, C. R. D. de Assis, J. Q. Marcatoma, J. D. Maciel, D. F. M. Neri, F. Soria and L. B. de Carvalho, Appl. Surf. Sci., 2018, 457, 21–29 CrossRef CAS.
- G. Bayramoglu, T. Doz, V. C. Ozalp and M. Y. Arica, Food Chem., 2017, 221, 1442–1450 CrossRef CAS.
- P. P. Waifalkar, S. B. Parit, A. D. Chougale, S. C. Sahoo, P. S. Patil and P. B. Patil, J. Colloid Interface Sci., 2016, 482, 159–164 CrossRef CAS.
- A. Timerman, in Protein Purification, ed. R. Ahmad, InTech, Rijeka, 2012, pp. 29–52 Search PubMed.
- A. Reddy and F. Maley, J. Biol. Chem., 1996, 271, 13953–13958 CrossRef CAS.
- M. C. Mascolo, Y. B. Pei and T. A. Ring, Materials, 2013, 6, 5549–5567 CrossRef CAS.
- R. Khatiria, A. Reyhanic, S. Mortazavid and M. Hossainalipoura, presented in part at the 4th International Conference on Nanostructures, ICNS4, Sharif University of Technology, 2012 Search PubMed.
- S. Rahman, P. Sen, M. Hasan, M. Miah and M. Rahman, Pak. J. Biol. Sci., 2004, 7, 340–345 CrossRef.
- G. L. Miller, Anal. Chem., 1959, 31, 426–428 CrossRef CAS.
- R. E. Abraham, M. L. Verma, C. J. Barrow and M. Puri, Biotechnol. Biofuels, 2014, 7, 90 CrossRef.
- A. K. Gupta and S. Wells, IEEE Trans. Nanobiosci., 2004, 3, 66–73 CrossRef.
- D. K. Kim, Y. Zhang, W. Voit, K. V. Rao and M. Muhammed, J. Magn. Magn. Mater., 2001, 225, 30–36 CrossRef CAS.
- D. J. Dunlop and Ö. Özdemir, Rock Magnetism: Fundamentals and Frontiers, Cambridge University Press, Cambridge, 1997 Search PubMed.
- B. Vincent, J. Edwards, S. Emmett and A. Jones, Colloids Surf., 1986, 18, 261–281 CrossRef CAS.
- X. Wang, Q. Xiang, W. W. Cao, F. Jin, X. F. Peng, B. C. Hu and X. D. Xing, J. Biomater. Sci., Polym. Ed., 2016, 27, 1909–1925 CrossRef CAS.
- C. Hui, C. M. Shen, J. F. Tian, L. H. Bao, H. Ding, C. Li, Y. A. Tian, X. Z. Shi and H. J. Gao, Nanoscale, 2011, 3, 701–705 RSC.
- A. Ali, H. Zafar, M. Zia, I. U. Haq, A. R. Phull, J. S. Ali and A. Hussain, Nanotechnol., Sci. Appl., 2016, 9, 49–67 CrossRef CAS.
- S. N. Sun, C. Wei, Z. Z. Zhu, Y. L. Hou, S. S. Venkatraman and Z. C. Xu, Chin. Phys. B, 2014, 23, 037503 CrossRef.
- M. Kaasalainen, V. Aseyev, E. Haartman, D. Ş. Karaman, E. Mäkilä, H. Tenhu, J. Rosenholm and J. Salonen, Nanoscale Res. Lett., 2017, 12, 74 CrossRef.
- M. C. Ruiz-Canas, L. M. Corredor, H. I. Quintero, E. Manrique and A. R. R. Bohórquez, Molecules, 2020, 25, 2868 CrossRef CAS.
- E. Tombácz, A. Majzik, Z. S. Horvát and E. Illés, Rom. Rep. Phys., 2006, 58, 281–286 Search PubMed.
- R. A. Bini, R. F. C. Marques, F. J. Santos, J. A. Chaker and M. Jafelicci, J. Magn. Magn. Mater., 2012, 324, 534–539 CrossRef CAS.
- T. Ahn, J. H. Kim, H. M. Yang, J. W. Lee and J. D. Kim, J. Phys. Chem. C, 2012, 116, 6069–6076 CrossRef CAS.
- C. Blanco-Andujar, D. Ortega, Q. A. Pankhurst and N. T. K. Thanh, J. Mater. Chem., 2012, 22, 12498–12506 RSC.
- D. Lisjak and A. Mertelj, Prog. Mater. Sci., 2018, 95, 286–328 CrossRef CAS.
- W. S. Xie, Z. H. Guo, F. Gao, Q. Gao, D. Wang, B. S. Liaw, Q. Cai, X. D. Sun, X. M. Wang and L. Y. Zhao, Theranostics, 2018, 8, 3284–3307 CrossRef CAS.
- J. Wu, Y. J. Wang, W. Jiang, S. S. Xu and R. B. Tian, Appl. Surf. Sci., 2014, 321, 43–49 CrossRef CAS.
- M. Yamaura, R. L. Camilo, L. C. Sampaio, M. A. Macedo, M. Nakamura and H. E. Toma, J. Magn. Magn. Mater., 2004, 279, 210–217 CrossRef CAS.
- A. Dong, S. Lan, J. F. Huang, T. Wang, T. Y. Zhao, W. W. Wang, L. H. Xiao, X. Zheng, F. Q. Liu, G. Gao and Y. X. Chen, J. Colloid Interface Sci., 2011, 364, 333–340 CrossRef CAS.
- S. Zhang, Y. F. Zhou, W. Y. Nie and L. Y. Song, Cellulose, 2012, 19, 2081–2091 CrossRef CAS.
- E. Mazario, J. Sanchez-Marcos, N. Menendez, P. Herrasti, M. Garcia-Hernandez and A. Munoz-Bonilla, RSC Adv., 2014, 4, 48353–48361 RSC.
- N. P. Neumann and J. O. Lampen, Biochemistry, 1967, 6, 468–475 CrossRef CAS.
- J. O. Lampen, in The enzymes, ed. P. Boyer, Academic Press, 1971, vol. 5, pp. 291–305 Search PubMed.
- M. A. Sainz-Polo, M. Ramirez-Escudero, A. Lafraya, B. Gonzalez, J. Marin-Navarro, J. Polaina and J. Sanz-Aparicio, J. Biol. Chem., 2013, 288, 9755–9766 CrossRef CAS.
- E. Gasteiger, A. Gattiker, C. Hoogland, I. Ivanyi, R. D. Appel and A. Bairoch, Nucleic Acids Res., 2003, 31, 3784–3788 CrossRef CAS.
- G. Raghava, presented in part at the CASP5, 2002 Search PubMed.
- C. Louis-Jeune, M. A. Andrade-Navarro and C. Perez-Iratxeta, Proteins: Struct., Funct., Bioinf., 2012, 80, 2818 CrossRef CAS.
- J. R. Lakowicz, in Principles of fluorescence spectroscopy, ed. J. R. Lakowicz, Springer, New York, 2006, pp. 529–575 Search PubMed.
- L. Raj, G. S. Chauhan, W. Azmi, J. H. Ahn and J. Manuel, Bioresour. Technol., 2011, 102, 2177–2184 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: FT-IR, ESI-TEM and EEL spectra, representative image of the enzyme activity test, SDS-PAGE gel, fluorescence spectra, polarimetry, KM and VMAX values table and enzyme efficiency before and after immobilisation. See DOI: 10.1039/d0ra09259b |
| ‡ Present address: School of Bioprocess and Chemical Engineering, University College Dublin, Dublin D04 V1W8, Ireland. |
| This journal is © The Royal Society of Chemistry 2021 |